
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCascade autohydrolysis of Alzheimer's Aβ peptides†
Martin
Wolfram
 *a,
Manish K.
Tiwari‡
a,
Tue
Hassenkam
b,
Ming
Li
c,
Morten J.
Bjerrum
a and
Morten
Meldal
*a
*a,
Manish K.
Tiwari‡
a,
Tue
Hassenkam
b,
Ming
Li
c,
Morten J.
Bjerrum
a and
Morten
Meldal
*a
aDepartment of Chemistry, University of Copenhagen, Universitetsparken 5, 2100 Copenhagen, Denmark. E-mail: martin.wolfram@nano.ku.dk; meldal@chem.ku.dk; Tel: +45 27202355 Tel: +45 21308299
bGlobe Institute, Section for Geobiology, Copenhagen University, Øster Voldgade 5-7, 1350 Copenhagen K, Denmark
cTechnical University of Denmark, The Danish Hydrocarbon Research and Technology Centre, Elektrovej, 2800 Kongens Lyngby, Denmark
First published on 12th April 2023
Abstract
Protein/peptide self-assembly into amyloid structures associates with major neurodegenerative disorders such as Alzheimer's disease (AD). Soluble assemblies (oligomers) of the Aβ peptide and their aggregates are perceived as neurotoxic species in AD. While screening for synthetic cleavage agents that could break down such aberrant assemblies through hydrolysis, we observed that the assemblies of Aβ oligopeptides, containing the nucleation sequence Aβ14–24 (H14QKLVFFAEDV24), could act as cleavage agents by themselves. Autohydrolysis showed a common fragment fingerprint among various mutated Aβ14–24 oligopeptides, Aβ12–25-Gly and Aβ1–28, and full-length Aβ1–40/42, under physiologically relevant conditions. Primary endoproteolytic autocleavage at the Gln15–Lys16, Lys16–Leu17 and Phe19–Phe20 positions was followed by subsequent exopeptidase self-processing of the fragments. Control experiments with homologous D-amino acid enantiomers Aβ12–25-Gly and Aβ16–25-Gly showed the same autocleavage pattern under similar reaction conditions. The autohydrolytic cascade reaction (ACR) was resilient to a broad range of conditions (20–37 °C, 10–150 μM peptide concentration at pH 7.0–7.8). Evidently, assemblies of the primary autocleavage fragments acted as structural/compositional templates (autocatalysts) for self-propagating autohydrolytic processing at the Aβ16–21 nucleation site, showing the potential for cross-catalytic seeding of the ACR in larger Aβ isoforms (Aβ1–28 and Aβ1–40/42). This result may shed new light on Aβ behaviour in solution and might be useful in the development of intervention strategies to decompose or inhibit neurotoxic Aβ assemblies in AD.
Introduction
Proteolytic enzymes have evolved to cleave small peptides and proteins with high precision and efficacy.1 The complex interplay of multiple proteolytic enzymes is vital to the regulation of physiological function by controlling protein maturation, folding, assembly and degradation. During aging the proteostatic network can become defective and collapse.2 Proteostatic collapse is characterized by dysregulation of e. g. intrinsically disordered proteins (IDPs). Such dysregulation may lead to protein misfolding, polymorphous aggregation and self-propagating amyloid formation.3–6 Accumulating evidence suggests that the soluble oligomeric assemblies of the Aβ peptide are the neurotoxic species in AD.7–13 In solution, Aβ forms dynamic oligomers which in turn self-assemble into globular oligomers (spherical aggregates) and extended β-sheet structures in distinct folding pathways.10,14–18 Unsuccessful attempts of clinical intervention effecting Aβ clearance involved the design of antibodies that should promote Aβ uptake into cells with subsequent digestion through proteolytic enzymes.19 As an alternative, metal-based cleavage agents with the ability for specific molecular recognition and hydrolytic activity towards dysfunctional proteins have been proposed as potential drug candidates for disease intervention.20–22 Cobalt(III) complexes of macrocyclic polyamines, in particular, have been found to mediate direct specific cleavage of Aβ assemblies and thereby disrupt their aggregates.23,24 Initially, palladium peptide catalysts were investigated for the cleavage of Aβ14–24(HQKLVFFAEDV) peptides (ESI Fig. S2C†). During this process, we observed hydrolysis even in the absence of a palladium catalyst and decided to investigate this peculiar metal-independent behaviour of Aβ peptides in more detail.While autoproteolytic processing by proteins is considered an evolutionarily conserved mechanism in protein regulation and activation,25–29 autoprocessing in the self-assembly of small peptides is a rare phenomenon, not well characterized. One example is the spontaneous hydrolysis of the vasoactive intestinal peptide (VIP).30 The VIP is a neuropeptide,31 presenting substantial neuroprotective activities against Aβ25–35 neurotoxicity in relation to AD.32 Intriguingly, recent reports showed that Aβ oligomers possess intrinsic autoproteolytic properties with autohydrolysis occurring within hours to days.33,34 In contrast, non-catalyzed peptide bond hydrolysis in neutral water at 25 °C shows half-times of 300–500 years,35 with the exception of specific acceleration with e. g. F–G and D–G containing sequences.36 In their recent report, Mondal and Mandal37 presented a construct with the Aβ nucleation site LVFFA Aβ17–21 fused to a peptide mimicking serine protease triad that induced hydrolysis of Aβ1–40 along its nucleation site. Furthermore, the peptide YKGSGFRMI can promote hydrolysis in Aβ self-assemblies.38 However, the mechanism of autocatalytic Aβ self-degradation and thereby, the critical role of fragments containing the Aβ nucleation site for the mechanism have not been examined, yet probably relevant to Alzheimer's disease.
Here we show evidence for autohydrolysis occurring intrinsically with a characteristic fragment fingerprint to a variety of Aβ peptides, under physiologically relevant conditions and at room temperature (20 °C). Given the complex dynamic behaviour of amyloidogenic peptides in solution, in a manner that reflects their aggregation potential, we decided to follow an exploratory approach for finding the conditions that drive Aβ14–24 congener autohydrolysis. From a large number of independent experiments, we identified general sequence selective patterns in Aβ autohydrolysis. We also observed recurring aggregation pathways that lead to these self-assemblies, active in the autohydrolytic cascade reaction (ACR). LC-MS was used to investigate the key fragmentation steps and kinetic properties of the ACR. Atomic force microscopy (AFM), fluorescence techniques and circular dichroism (CD) spectroscopy were applied to study the structural polymorphism and conformational changes that can precede or occur with autohydrolytic reaction mixtures of Aβ14–24 containing peptides and Aβ1–40/42. Our findings establish the autohydrolytic behavior of Aβ14/16–24/25 congeners and their potential for cross-seeding (of self-templating) cascade hydrolysis in larger Aβ isoforms. In various aggregation states with diverse secondary structure, Aβ congeners can become susceptible to cascade hydrolysis by changes in the environment and through cross-seeding catalysis of autohydrolytic active fragment compositions.
Results
Peptide substrates and initial catalysis screenings
Quenched fluorescence resonance energy transfer (FRET) substrates containing 3-nitrotyrosine (Y(NO2) or Y*) and 2-aminobenzoic amide (Abz) have previously been used to monitor the progress of enzymatic peptide hydrolysis by the increase of fluorescence intensity (IF) with progressing peptide cleavage.39 During initial studies of combinatorial catalyst libraries for putative Pd-assisted hydrolysis, the Aβ14–24 congener Abz-Pra-[HQKLVFFAEDV]-Y(NO2)G (1a) was synthesized by Fmoc-based solid-phase peptide synthesis (SPPS) and characterized by LC-MS (Fig. S1†). As is well documented for Aβ congeners aggregation states depend strongly on the mode of preparation of stock solutions.16,40 This became obvious during incubation of solutions of the FRET substrate 1a at 37 °C for up to 180 h. The observed dynamic changes in IF were complex and varied with pH, ionic strength, and temperature. Surprisingly, some samples with increased IF did not show the expected substrate cleavage, rather it reflected changes in aggregation states. Upon ten-fold dilution in 70% acetonitrile/water, the substrate eventually hydrolysed while the undiluted samples remained intact (Fig. S2A†). The hydrolysis experiments with 1a were repeated several times under slightly varied conditions with the same result of delay and specificity of cleavage also upon dilution in water in the absence of a palladium catalyst (Fig. S2C†). These results indicated that we were observing an autohydrolytic cascade reaction (ACR), inherent to short Alzheimer's Aβ-derived peptides (Fig. 1). To investigate the formation of the proteolytically active peptide self-assemblies, we, in addition to 1a, synthesized amyloidogenic peptide 2 (Bz-Pra-[HQKLVFFAEDV]-YG), the natural sequence homologue Aβ12–25–Gly-OH (3), D-amino acid analogue 4 and acquired Aβ1–28 (5) as well as Aβ1–40/42 (6 & 7) (Table 1). We also synthesized 1b, 1c and 1d as permutations of 1a as well as selected their autocatalytic fragments for ACR experiments (Fig. S1 and S3A†). | ||
| Fig. 1 Synthesized Aβ peptide sequences and their autohydrolytic self-fragmentation pattern. Color coded amino acid sequence of Aβ1–28 is presented for the extended Aβ14–24 nucleation site (blue, basic (+); orange, H-bonding and polar; green, hydrophobic; red, acidic (−)). Blue arrows indicate primary scissile bonds. Horizontal arrows indicate cleavage fragments (f0–f6) and f0*–f4*. The f7*/f7-scissile bond represents an autohydrolysis pathway distinct from f0-relayed-processing. | ||
| Peptide | Aβ sequence | f0 fragmentb | f0 sequence | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| a Abbreviations: Abz, 2-aminobenzoyl; Pra, propargylglycine (for peptide immobilization); Y*, Tyr(3-NO2); Bz, benzoyl. b Primary autocleavage fragment of the Aβ–ACR. c Aβ peptides with additional C-terminal nitro-tyrosine and glycine. d Derivatives to test the influence of termini and lysine: 1a = C-terminal acid; 1b = 1a (Lys16(Tfa)), 1c = 1a (C-terminal propylamide) and 1d = 1a (C-terminal 2-ethanol amide). e D-Amino acid peptides. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 1a–d , | Abz-Pra-[Aβ14–24]-Y*G | [Aβ16–24]-Y*G | KLVFFAEDVY*G | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 2 | Bz-Pra-[Aβ14–24]-YG | [Aβ16–24]-YG | KLVFFAEDVYG | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 3 | [Aβ12–25]-G | [Aβ16–25]-G | KLVFFAEDVGG | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 4 | [Aβ12–25]-G | [Aβ16–25]-G | KLVFFAEDVGG | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 5 | Aβ1–28 | Aβ16–28 | KLVFFAEDVGSNK | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 6 | Aβ1–40 | Aβ16–40 | KLVFFAEDVGSNK- | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| GAIIGLMVGGVV | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 7 | Aβ1–42 | Aβ16–42 | KLVFFAEDVGSNK- | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| GAIIGLMVGGVVIA | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Aβ14–24 congeners are susceptible to intrinsic autohydrolysis
To ensure that hydrolysis was neither due to proteases or microbial contamination, experiments were conducted in sterilized Milli-Q water in the presence of sodium azide and a protease inhibitor cocktail. We observed autohydrolytic cascade of 1a–3 with a common fragment fingerprint (Fig. 1, 2B and S3–S6†), which was reproducible under a variety of experimental conditions (Tables 2, S1 and S2†). This result indicated that the unnatural modifications in peptide 1a (Abz/Y*) and 2 (Bz/Y) were not pivotal for the autocatalysis. The initial step of the ACR in Aβ14–24 congeners 1–3 was the endoproteolytic cleavage between Gln15 and Lys16 as well as at the natural α-secretase cleavage site Lys16–Leu17 (Fig. 1, 2A and C). However, the specificity seemed to be quite promiscuous and initial cleavage was followed by several steps of aminopeptidase-like relay processing of C-terminal fragments f0 and f1 (Aβ16/17–24 congeners in Fig. 1) along the hydrophobic core-sequence Aβ17–21 toward fragment f4 (Aβ20–24 congener) and a final processing towards fragment f6 for peptides 1a–1d (Fig. 2B and S6A–C†).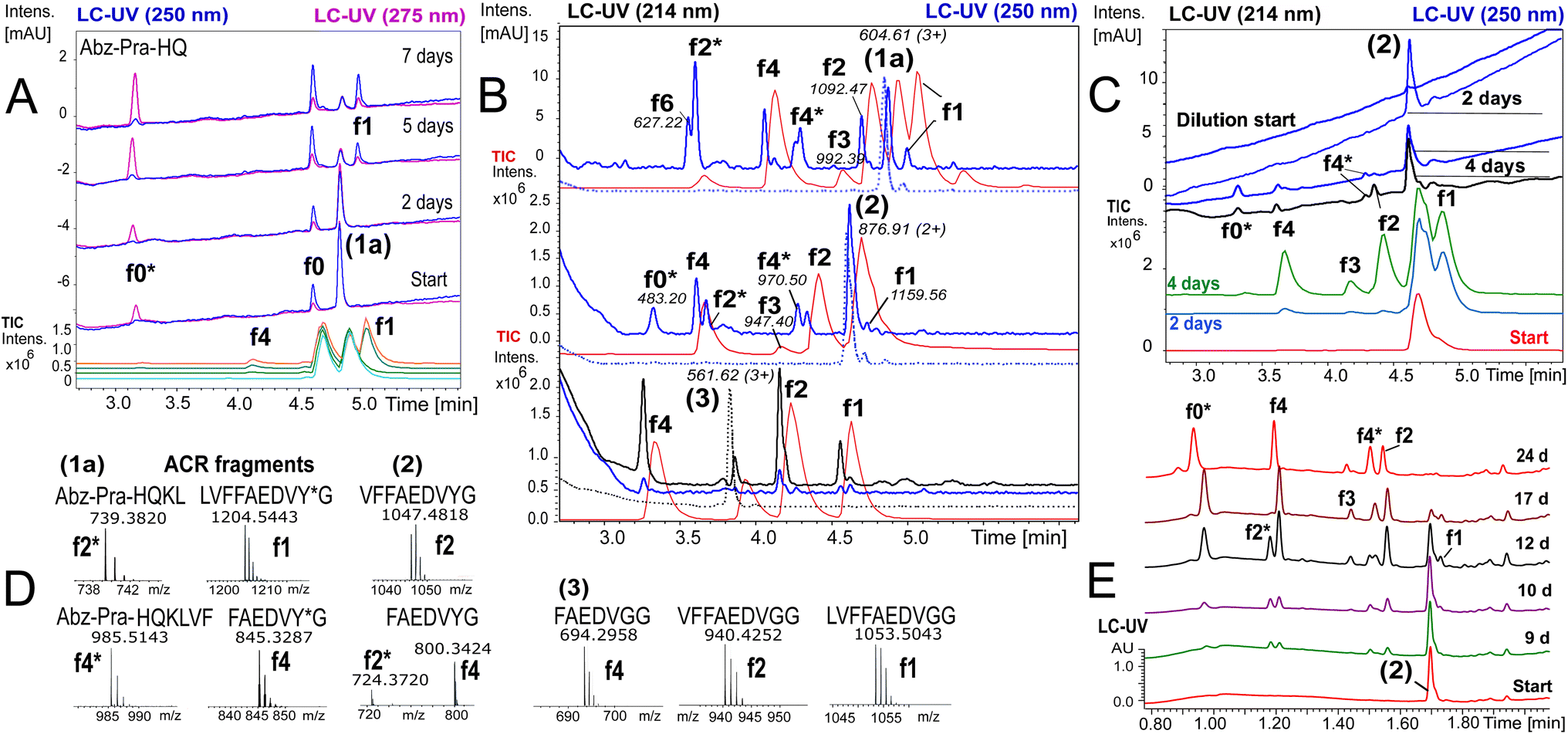 | ||
Fig. 2 Autoprocessing of truncated Aβ-peptides. LC-MS chromatograms presenting autohydrolysis of Aβ14–24 congeners 1a, 2 and 3 with the assignment of cleavage fragments. (A) Hydrolysis kinetics for 1a upon dilution of reaction stock (Table 2, entry 1 and Fig. S7A†) over 7 d at 37 °C: cyan, green and orange (MS-TIC); blue (UV-250 nm); magenta (UV-275 nm). Repeat experiments are presented in Fig. S7B and G.† (B) LC-MS monitoring of the ACR upon 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4.5 dilution of reaction stock solutions (dotted traces, conditions Table 2, entry 2) of 1a, 2 and 3, respectively, after 6 d at 20 °C. Blue traces (UV-250 nm) and red traces (MS-TIC) represent fragmentation (as annotated) after 9 d. Repeat samples are presented in Fig. S9A, B and E.† (C) Hydrolysis upon 1:4.5 dilution of aged aggregated samples (Table 2, entry 3 and Table S1,† stock entry XIX) at 20 °C indicated that disaggregation correlates with the onset of the ACR: green, red and light blue traces (MS-TIC), black (UV-214 nm) and blue (UV-250 nm) traces are stacked according to the time of acquisition. (D) Identification of ACR cleavage fragments by MS with annotation of the fragment sequences. (E) Independent 24 d kinetics of the autohydrolysis of peptide 2 (Table 2, entry 4). The kinetic analysis is presented in Fig. S8†(see also repeat reactions in Fig. S7C and E†). :4.5 dilution of reaction stock solutions (dotted traces, conditions Table 2, entry 2) of 1a, 2 and 3, respectively, after 6 d at 20 °C. Blue traces (UV-250 nm) and red traces (MS-TIC) represent fragmentation (as annotated) after 9 d. Repeat samples are presented in Fig. S9A, B and E.† (C) Hydrolysis upon 1:4.5 dilution of aged aggregated samples (Table 2, entry 3 and Table S1,† stock entry XIX) at 20 °C indicated that disaggregation correlates with the onset of the ACR: green, red and light blue traces (MS-TIC), black (UV-214 nm) and blue (UV-250 nm) traces are stacked according to the time of acquisition. (D) Identification of ACR cleavage fragments by MS with annotation of the fragment sequences. (E) Independent 24 d kinetics of the autohydrolysis of peptide 2 (Table 2, entry 4). The kinetic analysis is presented in Fig. S8†(see also repeat reactions in Fig. S7C and E†). | ||
| Entry | Peptide | [A]/μM | [B]/mM | pH | % DMSO | t (37)/d | t (20)/d | 37 °C | 20 °C | t lag/h | t 1/2/h | Figure ref | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
a [A] or [A]/[f0]: concentrations of the parent peptide (and f0-seed) in reaction stock solutions; [B]: buffer concentration (PB); t(37): primary incubation time at 37 °C; t(20): secondary incubation time at 20 °C.
b Extent of hydrolysis: (+++) >95%, (++) 40–95%, (+) 20–40%, and (−) 0%. na not applicable. tlag: latency for detected onset of the ACR. The half-life t1/2 is the average value estimated from several experiments (Table S2).
c Conditions: ACR progress upon 1:4.5 downstream dilution and continuous incubation at 37 °C.
d Conditions: spontaneous ACR of 1a–3 at 20 °C upon 1:4.5 dilution of preincubated reaction stock solutions with water.
e f0(1a) cross-seeding ACR in Aβ1–28 after 4 days at 20 °C.
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 1 | 1a | 165 | 10 | 7.6 | 3.2 | 14 | 0 | ++c | na | 60 | 66 | Fig. 2A | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 2d | 1a, 2, 3 | 100–117 | 10 | 7.6 | 5 | 28 | 9 | — | ++ | 160 | 36 –54 | Fig. 2B | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 3d | 2 | 55 | 10 | 7.6 | 0 | 7 | 16 | — | + | 48 | 60 | Fig. 2C | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 4 | 2 | 100 | 5 | 7.6 | 5 | 24 | 0 | +++ | na | 192 | 60 | Fig. 2E | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 5 | f0(1a) | 50 | 0.9 | 7.6 | 0 | 1 | 20 | — | +++ | 216 | 76 | Fig. 3A | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 6e | Aβ1–28/f0(1a) | 30/8 | 10 | 7.6 | 0 | 0 | 18 | na | +++ | 72 | 60 | Fig. 3B | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 7d | 3 | 60 | 10 | 7.6 | 0 | 7 | 16 | — | ++ | 92 | 36 | Fig. 4A | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 8d | 2 | 120 | 5 | 7.6 | 3.7 | 7 | 7 | + | ++ | 140 | na | Fig. 4C | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 9 | f0(1a) | 110 | 10 | 7.4 | 0 | 14 | 0 | ++ | na | 140 | na | Fig. 4H | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 10 | 1a/f0 | 45/12 | 10 | 7.4 | 0 | 16 | 10 | + | ++ | 200 | na | Fig. 4I | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 11 | Aβ1–40/f0(1a) | 30/5 | 10 | 7.33 | 0 | 12 | 12 | +++ | +++ | 120 | na | Fig. 5A | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
The N-terminal fragments (R1: peptide) R1-HQKL (f2*) and R1-HQKLVF (f4*) were reproducibly detected both in ACR assays with peptides 1a and 2 (Fig. 2B–E and S7C†) as well as with Aβ1–28 (Fig. S16†) and Aβ1–40. This result suggests a second pathway for primary endoproteolytic cleavage which generates the corresponding C-terminal fragments [Aβ18–24]-R2 (f2) and fragment [Aβ20–24]-R2 (f4) independently from the f1 → f4 aminopeptidase-like processing route (Fig. 1). The increase of f4*/f4 cleavage was accompanied by progressive disappearance of the f2* fragment and increase of the f0* fragment (Fig. 2E). This result suggests an autocatalytic carboxypeptidase-like reaction pathway of f4* → f0* (R1-HQKLVF → R1-HQ) processing. In addition, the N-terminal f0*-fragment (R1-HQ) is prone to subsequent carboxypeptidase-like processing toward R1 (fragments f8* and f9* in Fig. 1, for LC-MS analysis see ESI Fig. S3D, E, S16, S17B and F†). The identity of cleavage fragments was confirmed by their high-resolution mass spectra and MS/MS sequencing (Fig. 2D and S4†). Derived from hydrolysis samples of peptides 1a and 2, MS/MS sequencing of the primary autocleavage fragment f0 (m/z = 666.8262+ and m/z = 644.3182+, respectively) confirmed the identity of the respective C-terminal fragment sequence [Aβ16–24]Y*G (Fig. S7D and F†). This result is consistent with the primary scissile bond Gln15–Lys16 observed in autoproteolysis of Aβ1–42 oligomers, as previously reported by Rudinskiy et al.34
Influence of environmental parameters on Aβ-ACR
Autohydrolysis samples showed a lag-phase, followed by a period with rapidly increased hydrolytic activity (Fig. 2A–C and E). However, the times of the onset of catalysis and rate of autohydrolysis were condition-dependent (Table 2) and particularly influenced by the stock recipe and co-solvent content (Tables S1 and S2†). At 37 °C, the onset for the ACR occurred with tlag = 180–240 h and an approximate t1/2 (f1 → f4/f6) of 48–72 h (Fig. 2E, S7C and E†). Surprisingly, dilution and subsequent incubation at 20–22 °C increased rates to a t1/2 of 20–48 h for the conversion of the starting material (Table S2 and Fig. S9†). Samples of 1a (100 μM) and 2 (158 μM) containing 1.7–3.2 vol% DMSO (stock conditions in Table S1,† entry III) showed 15–40% selective accumulation of f0-fragment KLVFFAEDVY*G at 37 °C over 7 d (Fig. S7A†), which only continued towards autohydrolysis fragments f1/f4 upon sample dilution in Milli-Q water (Fig. 2A). Stock solutions of 1a, 2 and 3 in 0.2% 0.1 M NaOH/DMSO, diluted (1:20) in phosphate buffer (PB) to 5% final DMSO, did not provide onset of the ACR after 4–5 weeks of incubation at 37 °C. However, all samples of 1a, 2 and 3 being subsequently further diluted 1:4.5 in Milli-Q water showed a spontaneous ACR at 20 °C with a common fragment fingerprint after 6–9 days (Fig. 2B and S9A–D†). Similar samples prepared after 2 days from the same stock to 5% DMSO also hydrolysed at 37 °C with a lag-phase of 8 d (Table 2, entry 4 and Fig. 2E) indicating changes in this stock. A spontaneous ACR also occurred upon longer periods of incubation in undiluted samples at 20 °C (Fig. S6A†). Peptide 2 disaggregated upon 1:4.5 downstream dilution as indicated by increased intensity in the LC-UV-trace and presented f1 fragment release and onset of the ACR within 4 days at 20 °C (Fig. 2C). This was observed under a variety of conditions with all Aβ14–24 congeners, 1a–3, showing similar lag-times of 48–60 h and conversion to f4 with a t1/2 of 20–48 h (Fig. 4A and S9A–E†). At 37 °C, the aminopeptidase- and carboxypeptidase-like activity following initial endoproteolytic cleavages has a t1/2 of ∼48 h producing mainly f4 and f0* as end products (Fig. 2E and S8†).
Aβ16–24 congeners possess intrinsic autohydrolytic properties
To further elucidate the nature of the ACR, we investigated the autohydrolytic properties of the synthesized primary autocleavage fragment KLVFFAEDVY*G (f0) of peptide 1a (Table 1). In a series of experiments, we reproducibly observed f0 → f4 → f6 autoprocessing (Fig. 3A), under a variety of conditions at 20 °C as well as 37 °C (Fig. S10 and S25B†). A half time t1/2 of 70–76 h was observed for the cleavage steps in the autoprocessing at 20 °C (Fig. 3C). Furthermore, processing to f1 showed a different kinetic behaviour than that to f4. Once the autohydrolytic complex of f0(1a) formed a catalytic seed (tlag = 7–11 d), f0 converted initially to f1, followed by relay-processing to f4/f6 (Fig. 3C), suggesting that truncated fragments contribute significantly to the autocatalysis. Irrespective of changes in the conditions (concentration, pH, and T), repeat experiments showed similar half-times t1/2 of 48–84 h for f0 → f1 primary cleavage (Fig. S11†) and further autoprocessing with a t1/2 (f4) of 30–72 h after an initial tlag of 100–200 h (Fig. S10†). To clarify on the intrinsic nature of autohydrolysis in Aβ16–24 congeners, we performed ACR screening assays using the D-amino acid analog 4 of L-peptide 3 ([Aβ12–25]–Gly, vhhqklvffaedvgg) and the respective f0 fragment [Aβ16–25]–Gly (Fig. S12–S14†). Working without a co-solvent afforded autohydrolysis samples of f0 with 10–60% conversion toward the f2 fragment after 9–15 d at 37 °C (Fig. S13†). The stability and conditional fluctuation in aggregation states influenced the rate of hydrolysis of 4 even in the presence of traces of f0(4) resulting in rather slow hydrolysis that eventually provided the f4 fragment (Fig. S14D†) over a prolonged period. After 22 weeks, the f0 fragment showed complete conversion towards f1, f3 and f4 fragments as well as the truncated C-terminal Aβ19–25 fragment (Fig. S14A and E†).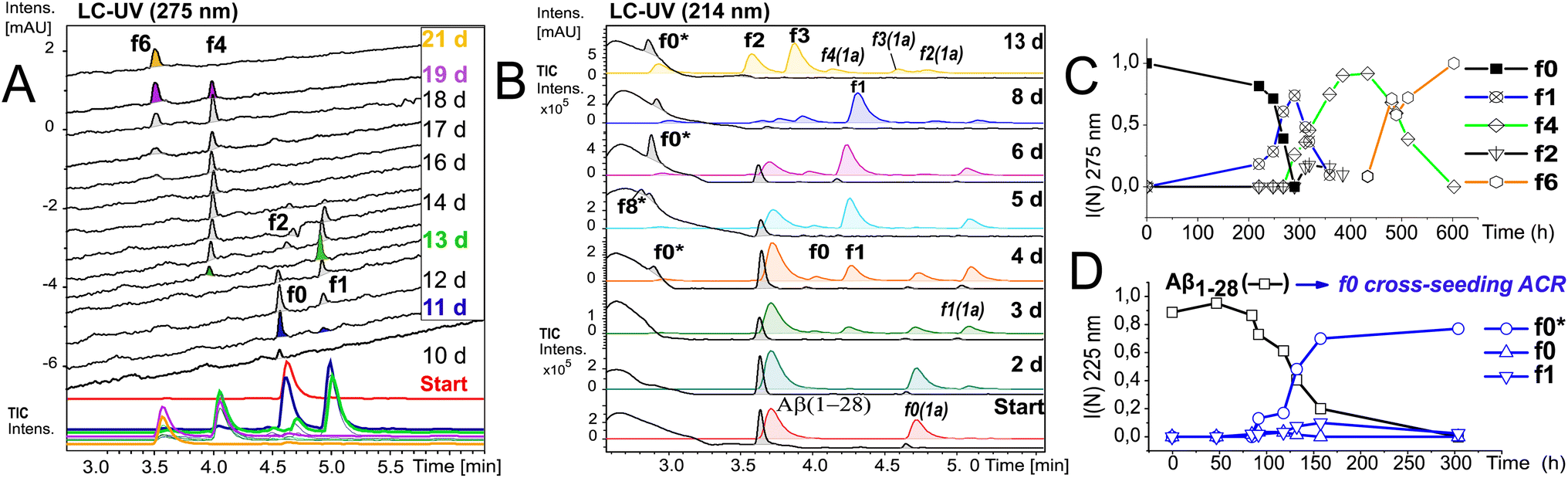 | ||
| Fig. 3 Kinetics of f0-ACR and of cross-seeding with f0(1a) to Aβ1–28. (A) Autohydrolysis of f0(1a) (Table 2, entry 5 and Table S1,† stock entry XII) was monitored by LC-MS after 9 d at 20 °C followed by 1 d at 37 °C: black traces (UV-275 nm) and colored traces (MS-TIC). (B) Cross-catalysis with autohydrolytic assemblies of f0(1a) (Fig. S10B†) that unlock quantitative self-processing in Aβ1–28 at 20 °C (conditions Table 2, entry 6): black traces (UV-214 nm) and colored traces (MS-TIC). Repeat experiments are presented in Fig. S15A, D and E.† (C) Kinetic plot (UV-275 nm) of integral intensities for data in (A), and (D) kinetic plot (UV-214 nm) of integral intensities for data in (B) showing both the lag time and rate of Aβ1–28 hydrolysis by cross seeding. | ||
Cross-catalytic ACR seeding in Aβ1–28
To discern the cross-catalytic ability of the f0 fragment, we investigated seeding the ACR in Aβ1–28 with [Aβ16–24]–Y*G (f0(1a)). We screened several stock solutions of f0(1a) (100–300 μM and pH 7.6–8.2) for subsequent ACR activation (Table S1,† stock entries VIII & XII) upon dilution (pH 7.4–7.8). When f0(1a) was pre-activated separately in that way (Fig. 3A and S10†), cross-seeding into samples of Aβ1–28 induced the onset of autohydrolysis within 3–5 d at 20 °C (Fig. 3B and S15A, D and E(iii)†). The observed half-life (t1/2 ∼ 76 h) for template-directed f0*/f0 autocleavage of Aβ1–28 (Fig. 3D) was similar to that of several independent repeat samples only containing f0(1a) (Fig. S10, S11B and C†). Repeat experiments for f0-cross-seeding catalysis showed enhanced reaction rates (t1/2 < 24 h) after preincubation at 37 °C and afforded further Aβ1–28 autoprocessing toward fragments f9*, f2 and f3 at 20 °C (Fig. S15A–C and S16†). Negative controls with no activated f0-cross-seeding remained intact (as shown in LC-MS (i) and (iv) in Fig. S15D†). Independent of catalyst addition, Aβ1–28 showed intrinsic autohydrolysis fragments Aβ1–24/25 as well as Aβ1–26 and Aβ18/20–26 next to Aβ1–16 (f1*) or Aβ1–15/17 (f0*/f2*) (see ESI Fig. S15E, S17B, C and F†). Both experiments confirmed f0*/f0 (Gln15–Lys16), f1*/f1, f2*/f2 and f4*/f4 (Phe19–Phe20) as intrinsic scissile bonds in assembly-driven autohydrolysis of Aβ1–28 (Fig. 1 and S16†), which is in agreement with observations by Rudinskiy et al. for autoproteolysis of Aβ1–42.34Aggregation and structural behaviour of Aβ14–24 peptides in relation to their autohydrolysis
The fluorogenic properties of 1a allowed for qualitative monitoring of structural changes in the aggregation process that could associate with the onset and progression of the ACR (Fig. S18A and E). To elucidate further on the possible relation between β-sheet self-assembly and the occurrence of autohydrolysis fragments, we screened 1a, 2 and 3 under different stock solvent and buffer conditions via Thioflavin T (ThT) aggregation assay.41 To confirm largely monomeric (unstructured) starting conditions for a set of repeat reactions, CD (Fig. S18C and 25D†) and dynamic light scattering (DLS) studies were performed on diluted stock aliquots (Fig. S19†).40 Fibril formation was most prominent with 3 (Fig. S20A and D†), but also occurred in 2 and 1a (Fig. S18B, S21A and S22A†), which is in agreement with previous reports on the aggregation behaviour of similar Aβ peptides.42,43 Fibrillation samples of 3 remained stable at 37 °C but showed f0-autocleavage after prolonged incubation at 20 °C (Fig. S20B†). Furthermore, several independent fibrillation assays provided ThT-positive samples that did show accelerated downstream autohydrolysis at 20 °C upon dilution (Fig. S18D, S20E and F†).During screenings for autohydrolysis, we observed a pattern of aggregation in diluted LC-MS samples at 20 °C, with the disappearance of the LC-UV trace (Fig. 4A, day 2). In the subsequent LC-MS analysis, a pronounced LC-UV signal reappeared indicating the disaggregation, fragment release and ACR of f1 to f4 within 2–4 days. This was consistent with spontaneous higher-order amyloid aggregation upon dilution and subsequent reorganization into more soluble amyloid self-assemblies may accompany the ACR in Aβ14–24 congeners (Fig. S9D and F†). We applied AFM to study the possible morphologies of insoluble aggregates that could relate to the loss of the UV-absorbance signal in the LC-MS chromatogram (Fig. S23†). Incubation samples of 1a showed different sized lumps, granules and biomolecular condensate or coacervate structures upon the disappearance of the UV-signal (Fig. S23A–C and S24†). Upon dilution, wet samples of peptide 2 (Fig. 4B and C) showed spherical-shaped oligomers (0.8 ± 0.4 nm average height, Fig. 4D) and molten granules (1.06 ± 0.36 nm average height, Fig. 4F) at different stages of autohydrolysis by AFM. The abundance and size distribution of these aggregates changed over time and presented bundled fibrils next to larger spherulites (2.4 ± 0.7 average height) in the least reactive sample (Fig. 4B and E), while at higher dilution (Fig. 4C) AFM of the particles was consistent with that of flattened spherical structures (1.65 ± 0.75 nm average height and 7–18 nm wide). This result indicated dynamic changes of globular aggregates and spontaneous fibril formation can coexist in autohydrolytic amyloid assembly. Furthermore, the identified f3* fragment Bz-Pra-HQKLV (m/z = 412.23(2+)) confirmed carboxypeptidase-like activity in the ACR from f4* to f0* (Fig. 4C).
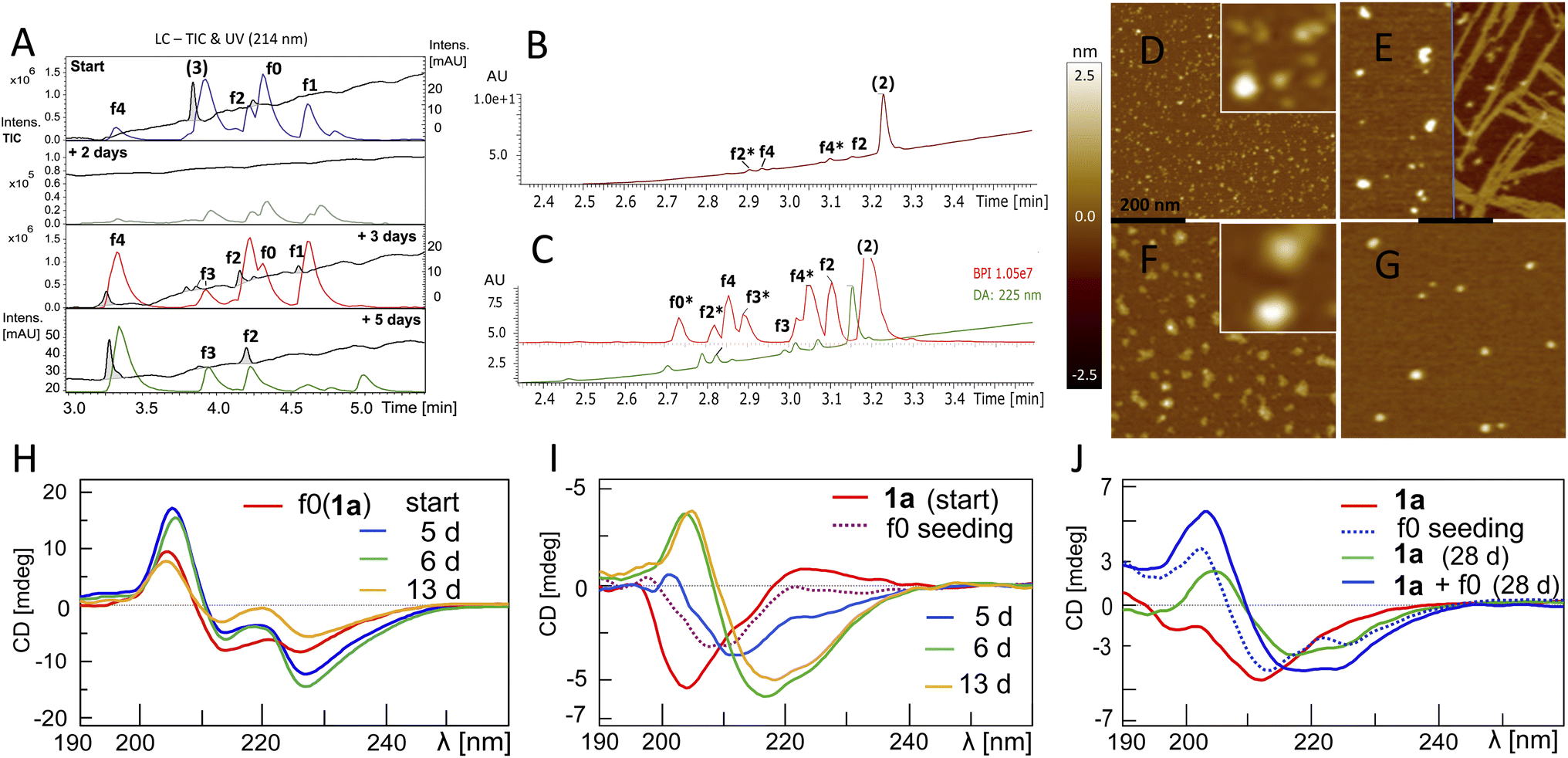 | ||
| Fig. 4 Aggregation behaviour and structural properties of autohydrolytic Aβ14–24 peptides. (A) Stacked LC-MS (colored traces (MS-TIC), black traces (UV-214 nm)) of peptide 3 (stock entry XIX, Table S1†) upon 1:4.5 dilution after 23 d (see entry 7, Table 2 and Fig. S20A and F†). (B) LC-MS spectra of peptide 2 (for conditions see entry 8, Table 2 and S1,† stock entry V) were recorded 7 days after 1:2 dilution at 20 °C subsequent to 7 days incubation at 37 °C. (C) LC-MS of a duplicate sample at 1:4 dilution. (D) AFM image (black bar represents 200 nm) of a hydrolysis sample (B). The white square inset shows 5-fold magnification. (E) AFM image after 4 additional days shows enlarged aggregates and fibrils (black bar, 200 nm). (F) AFM image (black bar, 200 nm) of the hydrolysis sample (C) shows depletion of aggregates by hydrolysis of the sample in (G) after 4 additional days (black bar, 200 nm). (H) CD spectra of f0(1a) (Table 2, entry 9) upon 13 d at 37 °C (ACR onset on day 5, see Fig. S10D–F† and for repeat experiments see Fig. S25A and B†). (I) Transformation of the 1a random coil structure (Table 2, entry 10) to β-sheets over 13 d upon seeding with f0(1a) (ACR observed after ∼10 d at 37 °C, for LC-MS see Fig. S25H†). (J) CD of 1a (50 μM, 7 d at 37 °C) before (red) and after (blue dotted) seeding with f0(1a) (20 μM). CD of 1a (green) and 1a + f0(1a) (blue) show β-sheets after 28 d at 37 °C (for cross-seeding repeat experiments see Fig. S25D and G†). All samples showed the same fragment fingerprint with 30–60% turn-over (Fig. S25C†). | ||
A variety of repeat experiments with Pd and Cu as trace metals showed no direct correlation with the onset of the ACR (e.g. Fig. S17A–D, S20A, B, S22F and S25C†). Furthermore, Inductively Coupled Plasma-Mass Spectrometry (ICP-MS) analysis of selected buffers and of autohydrolytically active samples indicated only low concentrations of metal ions (Cr, Ni, Cu, Zn, and Pd). With added EDTA (50–220 μM), we observed f0-primary autocleavage (Fig. S20B†) and successive cascade hydrolysis over a prolonged period of time (Fig. S22D†) as well as upon f0(1a) cross-seeding to Aβ1–28 (Fig. S26E and F†). These results suggest that metallated species do not play a major role in the ACR as outlined in the ESI.† However, the metal binding chelate ligand EDTA showed an inhibitory effect on the ACR at 10–200 μM (Fig. S9E†). Occasionally, we observed moderate autocleavage between Glu23 and Val24 (fragment R1–HQKLVFFAED (f7*), Fig. 1 and S2B†), correlating with β-sheet formation, particularly in the presence of high concentrations of Cu(II) and EDTA, at pH 7.8–8.0 of peptide 1a, 1d and 2 (Fig. S21 and S22A–C†), which confirms previous reports by others observing Aβ1–23 from autohydrolysis of Aβ1–40 and Aβ1–42 in the early aggregation phase.33,34
Conformational transitions accompanying autohydrolytically active amyloid assemblies of Aβ14–24 congener 1a
The autohydrolysis presented here raised the question whether the progression of the ACR coincides with conformational changes? Conformational transitions were investigated for 1a during autohydrolysis at 37 °C, both with and without seeding with f0(1a). 1a showed enhanced solubility and increased conformational dynamics as compared to the native analog [Aβ12–25]–G (3). CD of f0(1a) showed enrichment of β-sheet conformation in aqueous buffer at pH 7.1–7.8 (Fig. 4H). Despite the caveat that the unnatural side-chains may influence the CD simulations, secondary structure analysis was performed with the methods Dichroweb CDSSTR and Bestsel,44–46 suggesting extended structures, dominated by β-sheets and turns with a transient appearance of α-helical structures during the f0 → f1 → f2 processing. Seeding with β-sheet-rich f0(1a) on freshly prepared or preincubated solution of disordered peptide 1a induced a slow conformational shift toward β-sheet dominated assemblies (Fig. 4I and J), which was accompanied by subsequent progression of autohydrolysis over the course of 2–4 weeks (Fig. S25C, G and H†). This result indicated the potential of f0(1a) for structural influence on 1a by cross-seeding. However, due to the favorable properties of 1a, the ACR was also observed within 2–3 weeks starting from disordered 1a at 37 °C as well as 20 °C (Fig. S6A and S25D–F†). The actual ACR of 1a seemed to show no main conformational assembly preference for catalysis by 1a and f0(1a), and catalysis rather depends on minor populations of structured active clusters not detectable by CD. Thus, a structurally distinct population of peptides is not a prerequisite for the ACR. The selectivity of the fragmentation reaction suggests that well-structured peptide assemblies are key to the autohydrolytic process, showing the ability to self-replicate from the precursor peptides.Cascade autohydrolysis of Aβ1–40/42
To confirm that self-templating autohydrolysis at the nucleation site is a general property of Aβ peptides, we performed cross-seeding experiments with activated assemblies of the f0(1a) fragment on Aβ1–40 (20–30 μM). Several repeat experiments showed autohydrolysis with the typical ACR fragment fingerprint (f9*/f0*/f2*/f4*) and Aβ16–40 (f0) → Aβ22–40 (f6) upon seeding (Fig. 5A and S28–S34†). In addition, we observed a variety of terminally truncated isoforms of these fragments e.g. Aβ12–28, Aβ20–27, Aβ20–34 and its terminally truncated congeners (Fig. S30†). However, Aβ1–40 also showed the ACR with the common fragment fingerprint under various conditions without active fragment cross-seeding (Fig. S27, S28A and S29†). Nonetheless, the substoichiometric addition of f0/f1/f4(1a) (15–30 mol%) to aggregation samples of Aβ1–40 did neither inhibit nor decrease its fibrillation in the ThT assay at 37 °C (Fig. S31A and B†). However, fragment-seeded ThT-fibrillation samples showed a notable number of autocleavage fragments f0*/f2* already after 3 days of incubation (with periodic agitation), while ThT-containing controls without seeding remained intact (Fig. S31C–E†). This result confirmed that the ACR and fibril formation can coexist as confirmed by AFM imaging (Fig. S28B, C and S31F†). The hydrolysis of ACR-seeded fibrillation samples proceeded at 20 °C over the course of 6 days showing fragments f0/f1/f2/f3/f4 but notably Aβ20–27 among other truncated Aβ fragment isoforms (Fig. S28 and S31C–E†). | ||
| Fig. 5 Cross-seeding autohydrolysis in Aβ1–40 and characterisation of aggregation states. (A) Stacked LC-MS (colored traces (MS-TIC), black traces (UV-214–320 nm)) of Aβ1–40 (30 μM) replicates PB (10 mM) with 0.02 w% NaN3 and protease inhibitor (*) at pH 7.33 after 12 d at 25 °C (i) & (iii) and 12 d at 37 °C (ii) & (iv). Samples (iii) and (iv) received additional f0(1a) cross-seeding (Table S2,† stock entry XII and Fig. S26B(ii)†) upon initial sample preparation. f0(1a)-seeded samples (ii) and (iii) showed full degradation of Aβ1–40 with the common ACR fragment fingerprint (f0*/f2*/f4* and f2/f4) (Fig. S32†) including a variety of truncated isoforms such as Aβ12–18 (m/z = 859.457(1+), Aβ20–33 (m/z = 689.264(2+)), Aβ20–34 (m/z = 745.899(2+)) and Aβ22–34 (m/z = 636.845(2+)) (Fig. S30 and S33†). High-resolution mass analysis with a list of the main fragments identified in these mixtures are presented in Fig. S29, 30 and S34.† (B) AFM images after drying of the wet mica surface upon exposure to the sample (iv) (black bar, 200 nm). Right panel shows a zoomed-in image of ring-shaped oligomers. (C) AFM images after drying of the mica exposed to sample (ii) (black bar, 200 nm). Right panel shows zoomed-in images of ring-shaped oligomers next to fibrils. | ||
In parallel, we used AFM imaging to investigate the aggregate morphologies of autohydrolytic active samples incubated without ThT and no periodic agitation. Compared to ThT-fibrillation samples, we observed a lower abundance of fibrils in these samples but also a variety of spherical particles as well as ring-segmented oligomers and annular protofibrils with a discrete size distribution (20–80 nm in diameter) in both non-hydrolytic and ACR-active samples (Fig. 5B and S27D†). This result is consistent with previous reports on the appearance ring-shaped oligomers and annular aggregates in Aβ1–40 self-assembly.47–49
Preincubation at 25 °C (5–9 d) followed by incubation at 37 °C repeatedly provided autohydrolytic active samples of Aβ1–40 that showed ring-shaped structures, particles and amorphous aggregates besides fibrils (Fig. S27D and S28†). This result suggests that the initial conditions favoring oligomer formation at 25 °C over fibril nucleation/elongation (37 °C)16,48,50 do increase the probability for inducing the ACR in Aβ1–40 (Fig. S27A†). Assays with Aβ1–42 were more challenging due to its increased aggregation propensity in phosphate buffer. However, preincubation at 25 °C followed by f0-cross-seeding and incubation at 37 °C provided notable autocleavage after 14 d at 37 °C with increased autohydrolytic activity as compared to non-seeded replicates (Fig. S35D and S37†). Upon dilution Aβ1–42 fibrils with added ThT and f0(1a)-seed solution showed gradual f9*/f0*/f2* autocleavage after 3–4 weeks of incubation at 25 °C (Fig. S35A–C and S36†). These results confirmed that cross-seeding catalysis can unlock autohydrolysis in complex aggregation mixtures of full-length Aβ1–42. As the peptide solubility is critical for the formation of ACR active oligomers/aggregates, the extent of fibrillation is in balance with the ACR and may account for the differences observed between the Aβ1–42, Aβ1–40 and Aβ1–28 autohydrolysis.
Discussion
We demonstrated that assemblies of truncated Aβ peptides, containing the extended nucleation sequence Aβ14–24 (HQKLVFFAEDV) possess intrinsic and specific autohydrolytic properties. The autocleavage fragment fingerprint (Fig. 1) recurred under a broad spectrum of conditions (Table S2†). The results show that primary C-terminal cleavage fragments f0/f1 as well as f4, derived through endoproteolytic activity in Aβ14–24 congeners, are in turn autohydrolytically active components within an autocatalytic reaction network that extends to aminopeptidase-like processing along the Aβ nucleation site KLVFFA. The N-terminal counterpart shows respective enzyme-like carboxypeptidase and/or endoproteolytic processing in the cleavage sequence f4* → f2*(f1*) → f0* → (f0*)–HQ. The assemblies of the isolated primary autocleavage fragment Aβ16–24 congener (f0) can transmit the ACR towards intact assemblies of parent peptides and unlock cascade hydrolysis in Aβ1–28 (Fig. 3B) as well as in Aβ1–40 (Fig. 5A and S28F†). This result indicates the self-templating nature of the ACR and its cross-catalytic properties. Hence, the self-organization of autohydrolytic amyloids shows nonlinear kinetic behaviour and mutually cooperative effects in the autocatalytic reaction network.51–57 The autohydrolysis observed upon dilution of stable peptide aggregates (Fig. 2B, C and 4A–C) involves dynamic assembly remodeling probably caused by monomer release, cleavage and re-incorporation forming increasingly active assemblies within the collaborative molecular network. Less stable aggregates leading to increased rates are formed at lower temperature (cold denaturation).58,59Autohydrolysis emerged among various types of β-sheet rich assemblies (Fig. 4H–J), which illustrates that there are a variety of conformational states within the larger peptide clusters that participate in cascade hydrolysis. We suspect a possible role in this complex process of autohydrolysis of molten globules (dynamic peptide cluster containing a pool of defined secondary structures) formed transiently during the aggregation process – as widely observed among amyloids, including Aβ16–22 peptides.60–62 Structured supramolecular Aβ complexes probably act as enzyme-like autocatalytic seeds being capable to self-replicate (Fig. 6). The propensity of self-recognition in Aβ–peptides, which is the root cause of fibril formation, may also facilitate the formation of molten globules with specific cleavage activity toward assemblies or free-floating monomers and maybe even unrelated proteins. From this perspective, the formation of amphiphilic fragments f0–f4 with autocatalytic properties could be considered as transmitting compositional and even structural information (Fig. 6).63
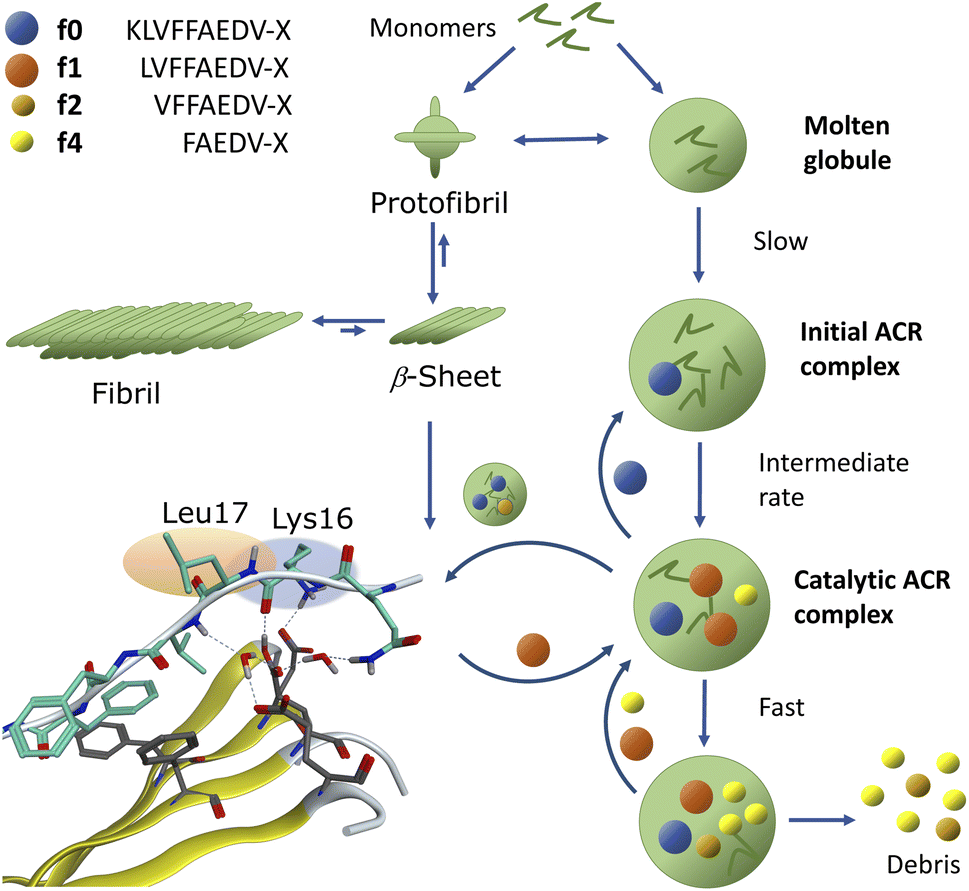 | ||
| Fig. 6 Suggested pathways via globular oligomers and protofibrils for the autocatalytic growth mechanism of the ACR. Aβ self-assembly and progression into cascade hydrolysis is indicated. The figure shows that parent peptide monomers can assemble into either fibrils or catalytically active globular oligomers/aggregates (all displayed in green) providing fragments: f0, f1, f2 and f4 (with f2 and f3 (not shown) formed from f1). The results indicate a slightly more complex scenario where two primary peptide-dependent cleavage sites yielding either f0*/f0 or f2*/f2 combined with f4*/f4. This is followed by aminopeptidase-like processing of f0, f2 and f4 fragments and carboxypeptidase-like processing of f0*, f2* and f4* fragments to end with a mixture of f0* and f4 fragments. All fragment species seem to feed into a grid of an autocatalytic reaction network including early fibrils. As an illustration of a reaction prone β-sheet formation prior to catalysis, four in-register parallel β-strands of the Aβ14–24 segment in water were modelled (MOE®) and interacted with the scissile bond of a fifth antiparallel strand within a water droplet. | ||
The assembly-driven autoproteolysis of disease related amyloids has been previously reported for α-synuclein (Parkinson's disease)64 and tau-protein (AD)65 as well as Aβ.33,34 We were able to characterise the complete patterns of autohydrolytic pathways of Aβ self-processing which lead to successive cleavage of both hydrophobic core regions Aβ17–21 and Aβ30–40/42 (Fig. 5A and S29–S36†). Our results demonstrate that catalytic activity self-propagates through incorporation of autocleavage fragments into larger oligomer complexes of the parent peptide (Fig. 6). This is consistent with observations by Rudinskiy et al.34 Increasing evidence shows that amyloid fibrils act as a reservoir of toxic oligomers and increasingly over time in aging, senile plaques.66 The pathways towards annular oligomers and Aβ fibrils may be evenly important in AD etiology.48,67 Cascade autohydrolysis may be governed by collective adaptation between pathways and could influence the formation and propagation of neurotoxic oligomers. The protease-like properties of assemblies of Aβ peptides described here could be an underappreciated feature of the neurotoxicity observed around Aβ-plaques.34,66,68–70 It remains yet to be defined what governs the soluble state of Aβ1–42 oligomers and if their fragment compositions could promote their self-replication and in particular if they impair other neuronal proteins. Aβ17–42 (f1) and its homologous f0/f2 isoforms are the major constituents in cerebellar pre-amyloid in Down syndrome71 and could exert neurotoxic behaviour in AD etiology through neuronal membrane disruption.72 Unlocking of templated ACR-processing may potentially open a way to abolish these toxic Aβ fragment assemblies. Furthermore, certain Aβ fragments could also play a role in Aβ regulation, because the observed intrinsic scissile bonds of the ACR are also specific to digestion by Aβ-degrading enzymes (e.g. neprilysin) in the brain.73
Conclusions and future outlook
Assembly-driven cascade autohydrolysis is an inherent property of Aβ peptides and their truncated isoforms carrying the nucleation sequence Aβ14–24 (HQKLVFFAEDV). The general fragmentation pattern extends to Aβ1–40 with congruent self-processing in both hydrophobic regions Aβ17–21 and Aβ30–40. The onset of autohydrolytic self-processing can be induced through environmental changes in concentration, pH and/or temperature and can even be seeded from active assemblies. An important conclusion is that there always exists a balance between the extend of precipitation and the formation of proteolytic Aβ. This could explain the release and the slow processing observed in concentrated samples and in plaques in vivo. The present work proposes a mechanistic model for self-propagating autohydrolysis in Aβ aggregates. Assemblies of Aβ autocleavage fragments may function as neurotoxins through their proteolytic properties or membrane-disrupting pore formation. Their environment controlled self-degradation could also present an opportunity to intercept Aβ neurotoxicity and plaque formation. Future characterization of transient, highly dynamic aggregation states is a complex affair and will require powerful analytical tools to identify key species that facilitate cascade hydrolysis. Exhaustive mutation studies on various Aβ congeners may elucidate critical residues in autohydrolytic active-site formation. In perspective, autohydrolytic peptide self-assembly could furthermore provide new insight into the emerging field of systems chemistry.51,57,60,74–80Data availability
All additional data are in the ESI† associated with this publication.Author contributions
M. Wolfram and M. Meldal conceived the project, M. Wolfram and M. Meldal synthesized all compounds, T. Hassenkam performed the AFM studies, M. Wolfram performed hydrolysis studies and CD, DLS and ThT assays, M. Wolfram and M. Meldal performed LC-MS analysis, M. Wolfram and M. Li performed MS–MS analysis, all co-authors contributed to the collective data analysis and conclusions on results, M. Wolfram, M. K. Tiwari and M. Meldal drafted the manuscript and all co-authors contributed to reviewing and editing the manuscript into its final format.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful to Lars Øgendal for DLS assistance, to David Teze, Sophia Häfner, Lars Hemmingsen and Sanne Schoffelen for discussions, to Theis-Brock Nannenstad for MS assistance, to Peter W. Thulstrup for access to a CD instrument and to Afra Fekri, Adrian Schiefler and Christian Koefod for LC-MS support. The Lundbeck Foundation (R95-2012-9008 to MW and R231-2016-3276 to MKT), DFF (DFF-4005-00082 to MJB and MKT), VILLUM FONDEN (00028295 to MW) and UCPH (CECB – Lighthouse grant) are gratefully acknowledged for their financial support.Notes and references
- H. Neurath, Science, 1984, 224, 350–357 CrossRef CAS PubMed.
- W. E. Balch, R. I. Morimoto, A. Dillin and J. W. Kelly, Science, 2008, 319, 916–919 CrossRef CAS PubMed.
- J.-P. Colletier, A. Laganowsky, M. Landau, M. Zhao, A. B. Soriaga, L. Goldschmidt, D. Flot, D. Cascio, M. R. Sawaya and D. Eisenberg, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 16938–16943 CrossRef CAS PubMed.
- M. Jucker and L. C. Walker, Nature, 2013, 501, 45–51 CrossRef CAS PubMed.
- T. P. J. Knowles, M. Vendruscolo and C. M. Dobson, Nat. Rev. Mol. Cell Biol., 2014, 15, 384–396 CrossRef CAS PubMed.
- R. Riek and D. S. Eisenberg, Nature, 2016, 539, 227 CrossRef PubMed.
- M. P. Lambert, A. K. Barlow, B. A. Chromy, C. Edwards, R. Freed, M. Liosatos, T. E. Morgan, I. Rozovsky, B. Trommer, K. L. Viola, P. Wals, C. Zhang, C. E. Finch, G. A. Krafft and W. L. Klein, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 6448–6453 CrossRef CAS PubMed.
- D. M. Hartley, D. M. Walsh, C. P. Ye, T. Diehl, S. Vasquez, P. M. Vassilev, D. B. Teplow and D. J. Selkoe, J. Neurosci., 1999, 19, 8876–8884 CrossRef CAS PubMed.
- M. Bucciantini, E. Giannoni, F. Chiti, F. Baroni, L. Formigli, J. Zurdo, N. Taddei, G. Ramponi, C. M. Dobson and M. Stefani, Nature, 2002, 416, 507–511 CrossRef CAS PubMed.
- R. Kayed, E. Head, J. L. Thompson, T. M. McIntire, S. C. Milton, C. W. Cotman and C. G. Glabe, Science, 2003, 300, 486–489 CrossRef CAS PubMed.
- C. Haass and D. J. Selkoe, Nat. Rev. Mol. Cell Biol., 2007, 8, 101–112 CrossRef CAS PubMed.
- E. Y. Hayden and D. B. Teplow, Alzheimer's Res. Ther., 2013, 5, 60 CrossRef PubMed.
- E. N. Cline, M. A. Bicca, K. L. Viola and W. L. Klein, J. Alzheimer's Dis., 2018, 64, S567–s610 CAS.
- M. Hoshi, M. Sato, S. Matsumoto, A. Noguchi, K. Yasutake, N. Yoshida and K. Sato, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 6370–6375 CrossRef CAS PubMed.
- S. Barghorn, V. Nimmrich, A. Striebinger, C. Krantz, P. Keller, B. Janson, M. Bahr, M. Schmidt, R. S. Bitner, J. Harlan, E. Barlow, U. Ebert and H. Hillen, J. Neurochem., 2005, 95, 834–847 CrossRef CAS PubMed.
- M. Necula, R. Kayed, S. Milton and C. G. Glabe, J. Biol. Chem., 2007, 282, 10311–10324 CrossRef CAS PubMed.
- S. Matsumura, K. Shinoda, M. Yamada, S. Yokojima, M. Inoue, T. Ohnishi, T. Shimada, K. Kikuchi, D. Masui, S. Hashimoto, M. Sato, A. Ito, M. Akioka, S. Takagi, Y. Nakamura, K. Nemoto, Y. Hasegawa, H. Takamoto, H. Inoue, S. Nakamura, Y.-i. Nabeshima, D. B. Teplow, M. Kinjo and M. Hoshi, J. Biol. Chem., 2011, 286, 11555–11562 CrossRef CAS PubMed.
- F. Hasecke, T. Miti, C. Perez, J. Barton, D. Schölzel, L. Gremer, C. S. R. Grüning, G. Matthews, G. Meisl, T. P. J. Knowles, D. Willbold, P. Neudecker, H. Heise, G. Ullah, W. Hoyer and M. Muschol, Chem. Sci., 2018, 9, 5937–5948 RSC.
- C. H. van Dyck, Biol. Psychiatry, 2018, 83, 311–319 CrossRef CAS PubMed.
- N. M. Milović, J. D. Badjić and N. M. Kostić, J. Am. Chem. Soc., 2004, 126, 696–697 CrossRef PubMed.
- J. Suh and W. S. Chei, Curr. Opin. Chem. Biol., 2008, 12, 207–213 CrossRef CAS PubMed.
- A. M. Embaby, L. P. W. M. Lelieveldt, F. Diness and M. Meldal, Chem. - Eur. J., 2018, 24, 17424–17428 CrossRef CAS PubMed.
- J. Suh, S. Yoo, M. Kim, K. Jeong, J. Ahn, M.-s. Kim, P. Chae, T. Lee, J. Lee, J. Lee, Y. Jang and E. Ko, Angew. Chem., Int. Ed., 2007, 46, 7064–7067 CrossRef CAS PubMed.
- J. S. Derrick, J. Lee, S. J. C. Lee, Y. Kim, E. Nam, H. Tak, J. Kang, M. Lee, S. H. Kim, K. Park, J. Cho and M. H. Lim, J. Am. Chem. Soc., 2017, 139, 2234–2244 CrossRef CAS PubMed.
- J. A. Brannigan, G. Dodson, H. J. Duggleby, P. C. E. Moody, J. L. Smith, D. R. Tomchick and A. G. Murzin, Nature, 1995, 378, 416–419 CrossRef CAS PubMed.
- P. Chen and M. Hochstrasser, Cell, 1996, 86, 961–972 CrossRef CAS PubMed.
- M. S. Wolfe, W. Xia, B. L. Ostaszewski, T. S. Diehl, W. T. Kimberly and D. J. Selkoe, Nature, 1999, 398, 513–517 CrossRef CAS PubMed.
- J. S. Rosenblum and G. Blobel, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 11370–11375 CrossRef CAS PubMed.
- Y. Liao, J. Pei, H. Cheng and N. V. Grishin, J. Mol. Biol., 2014, 426, 3935–3945 CrossRef CAS PubMed.
- R. Mody, A. Tramontano and S. Paul, Int. J. Pept. Protein Res., 1994, 44, 441–447 CrossRef CAS PubMed.
- S. I. Said and V. Mutt, Science, 1970, 169, 1217–1218 CrossRef CAS PubMed.
- I. Gozes, A. Bardea, A. Reshef, R. Zamostiano, S. Zhukovsky, S. Rubinraut, M. Fridkin and D. E. Brenneman, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 427–432 CrossRef CAS PubMed.
- W. Hosia, W. J. Griffiths and J. Johansson, J. Mass Spectrom., 2005, 40, 142–145 CrossRef CAS PubMed.
- N. Rudinskiy, C. Fuerer, D. Demurtas, S. Zamorano, C. De Piano, A. G. Herrmann, T. L. Spires-Jones, P. Oeckl, M. Otto, M. P. Frosch, M. Moniatte, B. T. Hyman and A. W. Schmid, Alzheimer's Dementia, 2016, 12, 996–1013 CrossRef PubMed.
- A. Radzicka and R. Wolfenden, J. Am. Chem. Soc., 1996, 118, 6105–6109 CrossRef CAS.
- D. Kahne and W. C. Still, J. Am. Chem. Soc., 1988, 110, 7529–7534 CrossRef CAS.
- T. Mondal and B. Mandal, Chem. Commun., 2020, 56, 2348–2351 RSC.
- Y. Hatakawa, R. Nakamura, M. Konishi, T. Sakane, M. Saito and T. Akizawa, Heliyon, 2019, 5, e02454 CrossRef CAS PubMed.
- M. Meldal and K. Breddam, Anal. Biochem., 1991, 195, 141–147 CrossRef CAS PubMed.
- D. B. Teplow, Preparation of Amyloid β-Protein for Structural and Functional Studies, in Methods in Enzymology, Academic Press, 2006, vol. 413, pp. 20–33 Search PubMed.
- H. Naiki, K. Higuchi, M. Hosokawa and T. Takeda, Anal. Biochem., 1989, 177, 244–249 CrossRef CAS PubMed.
- P. E. Fraser, J. T. Nguyen, W. K. Surewicz and D. A. Kirschner, Biophys. J., 1991, 60, 1190–1201 CrossRef CAS PubMed.
- L. O. Tjernberg, D. J. E. Callaway, A. Tjernberg, S. Hahne, C. Lilliehöök, L. Terenius, J. Thyberg and C. Nordstedt, J. Biol. Chem., 1999, 274, 12619–12625 CrossRef CAS PubMed.
- N. Sreerama and R. W. Woody, Anal. Biochem., 2000, 287, 252–260 CrossRef CAS PubMed.
- A. Micsonai, F. Wien, L. Kernya, Y.-H. Lee, Y. Goto, M. Réfrégiers and J. Kardos, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, E3095–E3103 CrossRef CAS PubMed.
- A. Micsonai, F. Wien, v. Bulyáki, J. Kun, v. Moussong, Y.-H. Lee, Y. Goto, M. Réfrégiers and J. Kardos, Nucleic Acids Res., 2018, 46, W315–W322 CrossRef CAS PubMed.
- M. Zhu, S. Han, F. Zhou, S. A. Carter and A. L. Fink, J. Biol. Chem., 2004, 279, 24452–24459 CrossRef CAS PubMed.
- R. Kayed, A. Pensalfini, L. Margol, Y. Sokolov, F. Sarsoza, E. Head, J. Hall and C. Glabe, J. Biol. Chem., 2009, 284, 4230–4237 CrossRef CAS PubMed.
- H. Choi, W. Lee, G. Lee, D. S. Yoon and S. Na, ACS Chem. Neurosci., 2019, 10, 3830–3838 CrossRef CAS PubMed.
- Y. Kusumoto, A. Lomakin, D. B. Teplow and G. B. Benedek, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 12277–12282 CrossRef CAS PubMed.
- S. A. Kauffman, The Origins of Order: Self Organization and Selection in Evolution, Oxford University Press, 1993 Search PubMed.
- D. H. Lee, K. Severin, Y. Yokobayashi and M. R. Ghadiri, Nature, 1997, 390, 591–594 CrossRef CAS PubMed.
- G. Ashkenasy, R. Jagasia, M. Yadav and M. R. Ghadiri, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 10872–10877 CrossRef CAS PubMed.
- R. Plasson, A. Brandenburg, L. Jullien and H. Bersini, Artif. Life, 2011, 17, 219–236 CrossRef PubMed.
- W. Hordijk and M. Steel, Biosystems, 2017, 152, 1–10 CrossRef CAS PubMed.
- J. R. Nitschke, Nature, 2009, 462, 736–738 CrossRef CAS PubMed.
- A. I. Hanopolskyi, V. A. Smaliak, A. I. Novichkov and S. N. Semenov, ChemSystemsChem, 2021, 3, e2000026 CrossRef CAS.
- M. Jaremko, u. Jaremko, H.-Y. Kim, M.-K. Cho, C. D. Schwieters, K. Giller, S. Becker and M. Zweckstetter, Nat. Chem. Biol., 2013, 9, 264–270 CrossRef CAS PubMed.
- T. Ikenoue, Y.-H. Lee, J. Kardos, M. Saiki, H. Yagi, Y. Kawata and Y. Goto, Angew. Chem., Int. Ed., 2014, 53, 7799–7804 CrossRef CAS PubMed.
- J. Greenwald and R. Riek, J. Mol. Biol., 2012, 421, 417–426 CrossRef CAS PubMed.
- Y. Liang, D. G. Lynn and K. M. Berland, J. Am. Chem. Soc., 2010, 132, 6306–6308 CrossRef CAS PubMed.
- R. F. Rengifo, A. Sementilli, Y. Kim, C. Liang, N. X. Li, A. K. Mehta and D. G. Lynn, ChemSystemsChem, 2020, 2, e2000007 CAS.
- D. Segré, D. Ben-Eli and D. Lancet, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 4112–4117 CrossRef PubMed.
- C. Vlad, K. Lindner, C. Karreman, S. Schildknecht, M. Leist, N. Tomczyk, J. Rontree, J. Langridge, K. Danzer, T. Ciossek, A. Petre, M. L. Gross, B. Hengerer and M. Przybylski, Chembiochem, 2011, 12, 2740–2744 CrossRef CAS PubMed.
- J. G. Moe, E. J. Davidowitz and P. Lopez, Tau protease composition and methods of use, US Pat.US20140286954, May 18, 2012 Search PubMed.
- A. Bigi, R. Cascella, F. Chiti and C. Cecchi, BioEssays, 2022, 44, 2200086 CrossRef CAS PubMed.
- S. A. Kotler, P. Walsh, J. R. Brender and A. Ramamoorthy, Chem. Soc. Rev., 2014, 43, 6692–6700 RSC.
- B. Lyons, M. Friedrich, M. Raftery and R. Truscott, Anal. Chem., 2016, 88, 2675–2684 CrossRef CAS PubMed.
- J. Dunys, A. Valverde and F. Checler, J. Biol. Chem., 2018, 293, 15419–15428 CrossRef CAS PubMed.
- N. C. Wildburger, T. J. Esparza, R. D. LeDuc, R. T. Fellers, P. M. Thomas, N. J. Cairns, N. L. Kelleher, R. J. Bateman and D. L. Brody, Sci. Rep., 2017, 7, 9520 CrossRef PubMed.
- M. Lalowski, A. Golabek, C. A. Lemere, D. J. Selkoe, H. M. Wisniewski, R. C. Beavis, B. Frangione and T. Wisniewski, J. Biol. Chem., 1996, 271, 33623–33631 CrossRef CAS PubMed.
- H. Jang, F. T. Arce, S. Ramachandran, R. Capone, R. Azimova, B. L. Kagan, R. Nussinov and R. Lal, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 6538–6543 CrossRef CAS PubMed.
- N. N. Nalivaeva, L. R. Fisk, N. D. Belyaev and A. J. Turner, Curr. Alzheimer Res., 2008, 5, 212–224 CrossRef CAS PubMed.
- O. Taran, C. Chen, T. O. Omosun, M.-C. Hsieh, A. Rha, J. T. Goodwin, A. K. Mehta, M. A. Grover and D. G. Lynn, Philos. Trans. R. Soc. London, Ser. A, 2017, 375, 20160356 CrossRef PubMed.
- C. P. J. Maury, Cell. Mol. Life Sci., 2018, 75, 1499–1507 CrossRef CAS PubMed.
- B. J. Cafferty, A. S. Y. Wong, S. N. Semenov, L. Belding, S. Gmür, W. T. S. Huck and G. M. Whitesides, J. Am. Chem. Soc., 2019, 141, 8289–8295 CrossRef CAS PubMed.
- L. R. Marshall, M. Jayachandran, Z. Lengyel-Zhand, C. M. Rufo, A. Kriews, M.-C. Kim and I. V. Korendovych, ChemBioChem, 2020, 21, 2611–2614 CrossRef CAS PubMed.
- M. Frenkel-Pinter, M. Samanta, G. Ashkenasy and L. J. Leman, Chem. Rev., 2020, 120, 4707–4765 CrossRef CAS PubMed.
- F. Sheehan, D. Sementa, A. Jain, M. Kumar, M. Tayarani-Najjaran, D. Kroiss and R. V. Ulijn, Chem. Rev., 2021, 121, 13869–13914 CrossRef CAS PubMed.
- D. H. Lee, K. Severin, Y. Yokobayashi and M. R. Ghadiri, Nature, 1997, 390, 591–594 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: The experimental procedures, solid-phase peptide synthesis and analytical methods. Mass spectra, chromatographic data, MS/MS fragment sequencing, results from Aβ-peptide aggregation assays, CD time laps analysis, AFM images, analysis of hydrolysis screenings and tables describing conditions for repeat experiments. See DOI: https://doi.org/10.1039/d2sc06668h |
| ‡ Present address: Novozymes Innovation Campus, Novozymes A/S, Biologiens Vej 2, 2800 Kongens Lyngby. |
| This journal is © The Royal Society of Chemistry 2023 |