
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEnantioselective aromatic Claisen rearrangement of allyl 2-naphthyl ethers catalyzed by π–Cu(II) complexes†
Lu
Yao
a,
Kazuki
Takeda
a,
Kaori
Ando
 *b and
Kazuaki
Ishihara
*a
*b and
Kazuaki
Ishihara
*a
aGraduate School of Engineering, Nagoya University, B2-3(611) Furo-cho, Chikusa, Nagoya 464-8603, Japan. E-mail: ishihara@cc.nagoya-u.ac.jp
bDepartment of Chemistry and Biomolecular Science, Faculty of Engineering, Gifu University, Gifu 501-1193, Japan. E-mail: ando@gifu-u.ac.jp
First published on 20th January 2023
Abstract
The first catalytic enantioselective aromatic Claisen rearrangement of allyl 2-naphthyl ethers using 5–10 mol% of π–copper(II) complexes is reported. A Cu(OTf)2 complex with an L-α-homoalanine amide ligand gave (S)-products in up to 92% ee. Conversely, a Cu(OSO2C4F9)2 complex with an L-tert-leucine amide ligand gave (R)-products in up to 76% ee. Density-functional-theory (DFT) calculations suggest that these Claisen rearrangements proceed stepwise via tight-ion-pair intermediates, and that (S)- and (R)-products are enantioselectively obtained via the staggered transition states for the cleavage of the C–O bond, which is the rate-determining step.
Introduction
We have previously developed a chiral π–Cu(II) complex formed from Cu(OTf)2 and chiral ligand 1a that catalyzes the enantioselective [1,3] O-to-C rearrangement of various methyl 2-(cinnamyloxy)-1-naphthoates (2) and methyl 3-(cinnamyloxy)-4-substituted-2-naphthoates (5) (Scheme 1a).1,2 Optically active dearomatized products 3 and 6 can be produced in high enantioselectivity (Scheme 1a), which is induced by the π–Cu(II) interaction in the active intermediate 4. Independently, alternative methods have been developed by You et al.3a,b as well as Zeng and Zhong et al.3c based on the catalytic enantioselective allylic dearomatization of 1,3-dialkyl-2-naphthols (7) to give optically active dearomatized products (8) which are structurally similar to 6 (Scheme 1b).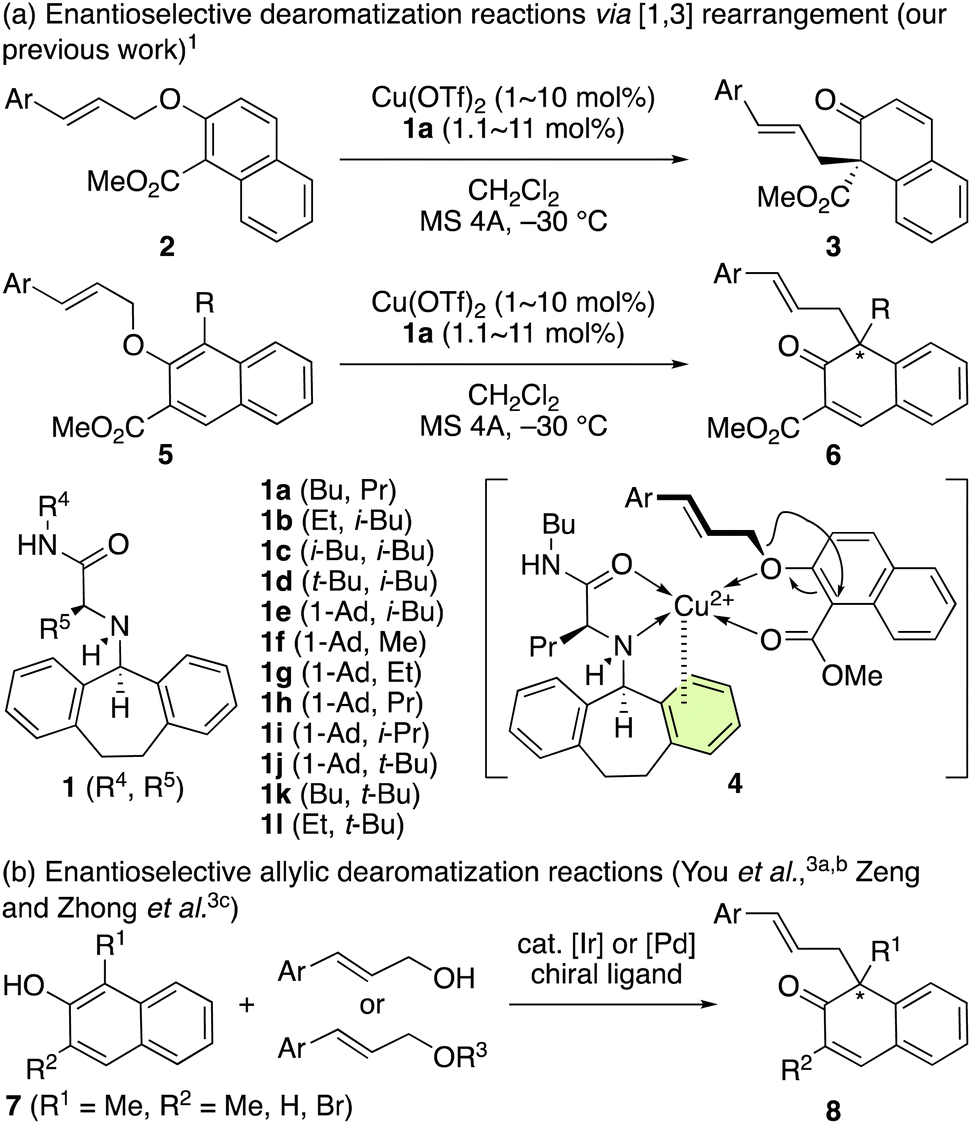 | ||
| Scheme 1 (a) Enantioselective dearomatization of 2-naphthol derivatives via [1,3] rearrangement, and (b) dehydrative coupling. | ||
Although significant progress has been made in this area,4 there is still room for the development of other asymmetric catalytic rearrangements. Here we report an enantioselective Claisen rearrangement of methyl 3-(cinnamyloxy)-2-naphthoates (9; R3 = Me) catalyzed by a π–Cu(II) complex to give optically active aromatized products (10) (Scheme 2). To the best of our knowledge, this is the first example of a catalytic enantioselective Claisen rearrangement5,6 of allyl naphthyl ethers and it should be noted here that even examples of non-enantioselective Claisen rearrangements of allyl naphthyl ethers remain scarce to date.7
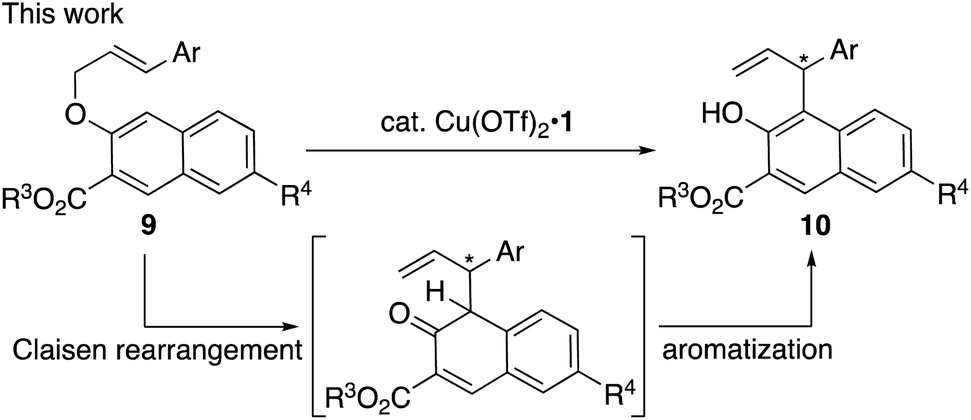 | ||
| Scheme 2 Catalytic enantioselective aromatic Claisen rearrangement of methyl 3-(cinnamyloxy)-2-naphthoates (9, R3 = Me). | ||
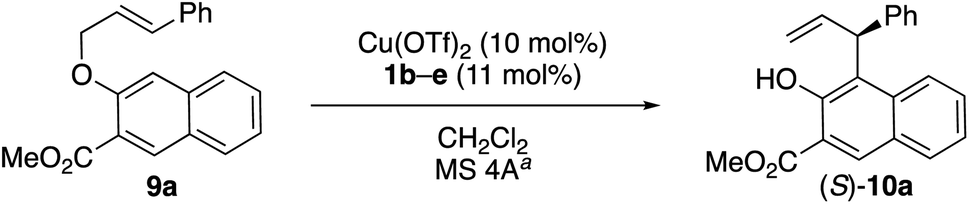
Based on our previous report on [1,3] rearrangements,1 we began by examining the N-5-dibenzosuberyl-L-leucine-derived amides 1b–e as chiral ligands in combination with Cu(OTf)2 for the enantioselective Claisen rearrangement of 9a in dichloromethane (Table 1). The enantioselectivity was increased from 29% ee to 60% ee by using derivatives of 1 with more sterically bulky R4 groups (entries 1–4) at lower temperature (entries 5–7). Interestingly, the enantioselectivity was further increased to 70% ee by using chloroform as the solvent instead of dichloromethane (entry 5 vs. entry 6).
|
|
||||
|---|---|---|---|---|
| Entry | Ligand 1 (R4, R5) | Temp. [°C], time [h] | Product 10a | |
| Conv. [%] | Eeb [%] | |||
| a MS 4A means the zeolite A type, known as LTA (Linde Type A), with 4 Å pore diameter. b The absolute configuration was determined in analogy to that of 10f (Fig. 1). c rt = room temperature. d 1-Ad = 1-adamantyl. e CHCl3 stabilized with 0.3–1.0% ethanol was used instead of CH2Cl2. | ||||
| 1 | 1b (Et, i-Bu) | rt,c 0.42 | >99 | 29 |
| 2 | 1c (i-Bu, i-Bu) | rt,c 0.67 | >99 | 30 |
| 3 | 1d (t-Bu, i-Bu) | rt,c 0.75 | >99 | 42 |
| 4 | 1e (1-Ad, i-Bu)d | rt,c 1 | >99 | 45 |
| 5 | 1e (1-Ad, i-Bu)d | −20, 5 | >99 | 60 |
| 6e | 1e (1-Ad, i-Bu)d | −20, 24 | >99 | 70 |
| 7 | 1e (1-Ad, i-Bu)d | −60, 48 | 95 | 66 |
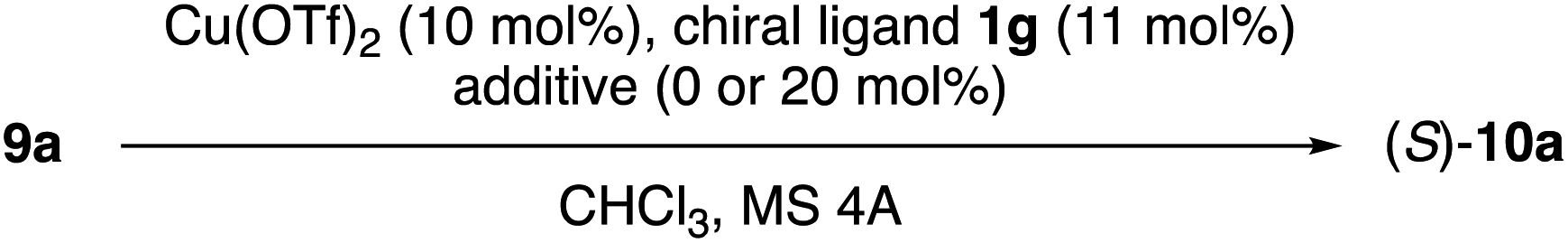
Next, N-5-dibenzosuberyl-L-amino acid-derived N-(1-adamantyl)amides 1e–j were examined as chiral ligands in combination with Cu(OTf)2 for the enantioselective Claisen rearrangement of 9a in chloroform at −20 °C (Table 2). With respect to the enantioselectivity, common primary alkyl groups such as Et and Pr were found to be more suitable R5 groups than Me, i-Bu and i-Pr (entries 1–5). Interestingly, the absolute stereochemistry of 9a was reversed for R5 = t-Bu (entry 6).
|
|
||||
|---|---|---|---|---|
| Entry | Ligand 1 (R4, R5) | Temp. [°C], time [h] | Product 10a | |
| Conv. [%] | Eeb [%] | |||
| a CHCl3 stabilized with 0.3–1.0% ethanol was used as the solvent. MS 4A means the zeolite A type, known as LTA (Linde Type A), with 4 Å pore diameter. b The absolute configuration was determined in analogy to that of 10f (Fig. 1). c rt = room temperature. | ||||
| 1 | 1f (1-Ad, Me) | −20, 24 | >99 | 59 (S) |
| 2 | 1g (1-Ad, Et) | −20, 24 | >99 | 73 (S) |
| 3 | 1h (1-Ad, Pr) | −20, 24 | >99 | 73 (S) |
| 4 | 1e (1-Ad, i-Bu) | −20, 24 | >99 | 70 (S) |
| 5 | 1i (1-Ad, i-Pr) | −20, 24 | >99 | 64 (S) |
| 6 | 1j (1-Ad, t-Bu) | rt,c 1 | >99 | 28 (R) |

In Tables 1 and 2, the experiments in chloroform were carried out using chloroform stabilized with 0.3–1.0% ethanol. To investigate the effect of ethanol on the Claisen rearrangement of 9a, chloroform stabilized with 0.015% 2-methyl-2-butene was used as a solvent. As shown in Table 3, the enantioselectivity decreased from 72% ee (entry 2) to 60% ee (entry 1) in the absence of ethanol, albeit that the reactivity increased. The addition of 20 mol% of i-PrOH and t-BuOH was also effective for increasing the enantioselectivity without any significant suppression of the reactivity (entries 3 and 5). When Cu(OSO2C4F9)2 was used instead of Cu(OTf)2, the enantioselectivity did not increase (entry 3 vs. entry 4). Thus, the desired product (10a) was obtained in 96% conversion with 81% ee under the optimal conditions (entry 6).
|
|
||||
|---|---|---|---|---|
| Entry | Additive [mol%] | Temp. [°C], time [h] | Product 10a | |
| Conv. [%] | Eeb [%] | |||
| a Unless otherwise noted, CHCl3 stabilized with 0.015% 2-methyl-2-butene was used as the solvent. MS 4A means the zeolite A type, known as LTA (Linde Type A), with 4 Å pore diameter. b The absolute configuration was determined in analogy to that of 10f (Fig. 1). c CHCl3 stabilized with 0.3–1.0% ethanol was used as the solvent. d Cu(OSO2C4F9)2 was used instead of Cu(OTf)2. | ||||
| 1 | — | −10, 1 | >99 | 60 |
| 2c | — | −10, 24 | >99 | 72 |
| 3 | i-PrOH (20) | −10, 1.5 | >99 | 73 |
| 4d | i-PrOH (20) | −10, 24 | >99 | 70 |
| 5 | t-BuOH (20) | −10, 1 | >99 | 73 |
| 6 | i-PrOH (20) | −35, 24 | 96 | 81 |
| 7 | i-PrOH (20) | −40, 24 | 62 | 82 |
The substrate scope of the enantioselective aromatic Claisen rearrangement of 9a–m catalyzed by Cu(OTf)2·1g in chloroform is shown in Table 4. Methyl ester 9a was obtained in a higher enantioselectivity than ethyl ester 9b (entry 1 vs. entry 2). Higher enantioselectivity was observed in the rearrangement of those substrates bearing electron-withdrawing groups (9c–i) (entries 3–10). In contrast, a substrate bearing an electron-donating group (9j) was more reactive but furnished 10j in a lower ee (entry 11). 3-(3-Thienyl)allyl naphthyl ether 9k could also be used as a substrate (entry 12). Interestingly, the 7-bromo group of 9l decreased both the reactivity and enantioselectivity (entry 1 vs. entry 13), whereas, the 7-methoxy group of 9m increased both the reactivity and enantioselectivity (entry 8 vs. entry 14).
|
|
||||
|---|---|---|---|---|
| Entry | Substrate 9 (Ar, R3, R4) | Temp. [°C], time [h] | Product 10 | |
| Yieldb [%] | Eec [%] | |||
| a Unless otherwise noted, CHCl3 stabilized with 0.015% 2-methyl-2-butene was used as the solvent. MS 4A means the zeolite A type, known as LTA (Linde Type A), with 4 Å pore diameter. b Isolated yield. c The absolute configuration was determined in analogy to that of 10f (Fig. 1). d Cu(OTf)2 (20 mol%) and 1g (22 mol%) were used without i-PrOH. | ||||
| 1 | 9a (Ph, Me, H) | −35, 24 | 92 | 81 (S) |
| 2 | 9b (Ph, Et, H) | −35, 48 | 80 | 77 (S) |
| 3 | 9c (2-ClC6H4, Me, H) | −20, 48 | 80 | 82 (R) |
| 4 | 9d (3-ClC6H4, Me, H) | −20, 48 | 76 | 84 (S) |
| 5 | 9e (3,5-(CF3)2C6H3, Me, H) | −10, 48 | 77 | 89 (S) |
| 6d | 9e (3,5-(CF3)2C6H3, Me, H) | −20, 48 | 95 | 92 (S) |
| 7 | 9f (3,5-Br2C6H3, Me, H) | −20, 48 | 44 | 84 (S) |
| 8 | 9g (4-ClC6H4, Me, H) | −20, 24 | 95 | 81 (S) |
| 9 | 9h (4-BrC6H4, Me, H) | −20, 24 | 85 | 82 (S) |
| 10 | 9i (4-CF3C6H4, Me, H) | −20, 48 | 67 | 87 (S) |
| 11 | 9j (4-MeC6H4, Me, H) | −20, 24 | 90 | 65 (S) |
| 12 | 9k (3-thienyl, Me, H) | −20, 24 | 80 | 73 (R) |
| 13 | 9l (Ph, Me, Br) | −20, 24 | 70 | 77 (S) |
| 14 | 9m (4-ClC6H4, Me, OMe) | −20, 12 | 95 | 85 (S) |
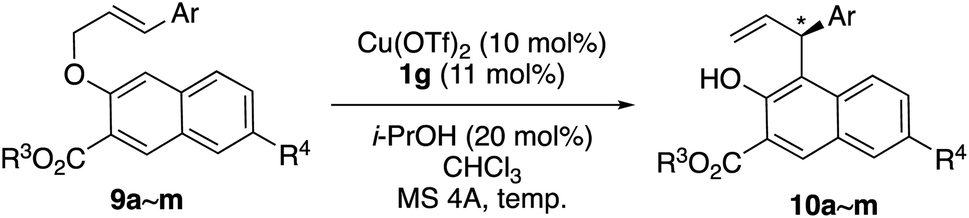
Using single-crystal X-ray diffraction analysis, the absolute configuration of 10f (entry 7, Table 4) was determined to be (S) (Fig. 1). Thus, the absolute configuration of the other products 10 obtained in Tables 1–4 was determined in analogy to that of 10f.
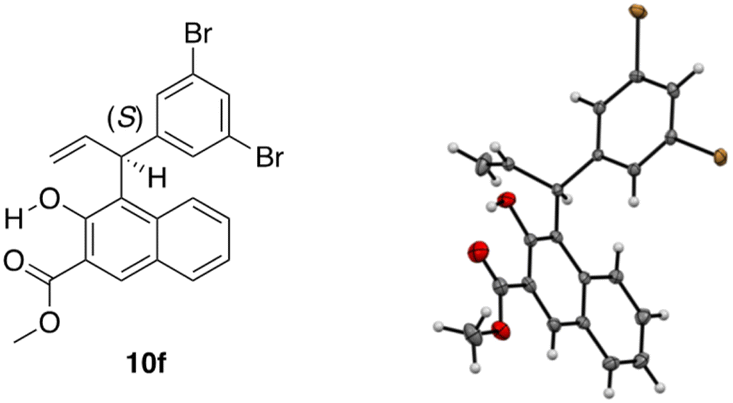 | ||
| Fig. 1 X-ray structure of product 10f. | ||
This catalytic Claisen rearrangement is scalable. On the gram scale, the rearrangement of 9a was complete within 7 h in the presence of 5 mol% of Cu(OTf)2·1g at −20 °C to give (S)-10a in 95% isolated yield with 77% ee (eqn (1)).
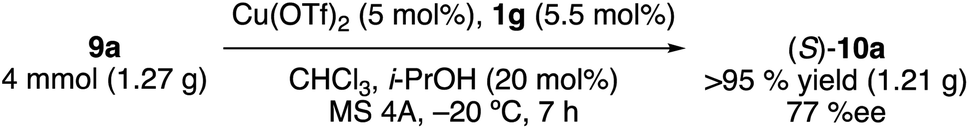 | (1) |
Next, we focused on the inverse asymmetric induction observed in entry 6 of Table 2. To improve the enantioselectivity, the R4 group of ligand 1 was optimized to improve the enantioselectivity (Table 5). Primary alkyl groups such as Et and Bu were found to be more suitable than bulkier groups such as Ad (entries 1–3). In terms of the enantioselectivity, chloroform was better than dichloromethane (entry 2 vs. entry 4). The addition of i-PrOH decreased the catalytic activity and did not influence the enantioselectivity (entry 5 vs. entry 6). An enantioselectivity of 67% ee was observed when using Cu(OTf)2·1k in chloroform at −30 °C (entry 6). The enantioselectivity increased to 71% ee when Cu(OSO2C4F9)2 was used instead of Cu(OTf)2 (entry 7).
|
|
||||
|---|---|---|---|---|
| Entry | Ligand 1 (R4) | Solvent, temp. [°C], time [h] | Product 10a | |
| Conv. [%] | Eeb [%] | |||
| a MS 4A means the zeolite A type, known as LTA (Linde Type A), with 4 Å pore diameter. b The absolute configuration was determined in analogy to that of 10f (Fig. 1). c 1-Ad = 1-adamantyl. d rt = room temperature. e CHCl3 stabilized with 0.015% 2-methyl-2-butene was used. f 20 mol% of i-PrOH was added. g Cu(OSO2C4F9)2 was used instead of Cu(OTf)2. | ||||
| 1 | 1j (1-Ad)c | CH2Cl2, rt,d 1 | >99 | 28 |
| 2 | 1k (Bu) | CH2Cl2, rt,d 0.5 | >99 | 45 |
| 3 | 1l (Et) | CH2Cl2, rt,d 0.5 | >99 | 43 |
| 4e | 1k (Bu) | CHCl3, −10, 6 | >99 | 61 |
| 5e | 1k (Bu) | CHCl3, −30, 48 | 97 | 68 |
| 6e,f | 1k (Bu) | CHCl3, −30, 48 | 33 | 68 |
| 7e,g | 1k (Bu) | CHCl3, −30, 48 | 95 | 71 |

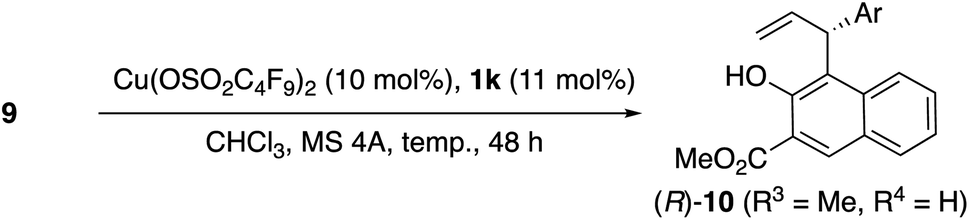
As shown in Table 6, substrates 9g, 9j, and 9a, were also transformed to 10 with moderate enantioselectivity in the presence of Cu(OSO2C4F9)2·1k.
|
|
||||
|---|---|---|---|---|
| Entry | Substrate 9 (Ar, R3, R4) | Temp. [°C] | Product 10 | |
| Yieldb [%] | Eec [%] | |||
| a CHCl3 stabilized with 0.015% 2-methyl-2-butene was used. MS 4A means the zeolite A type, known as LTA (Linde Type A), with 4 Å pore diameter. b Isolated yield. c The absolute configuration was determined by analogy with that of 10f (Fig. 1). | ||||
| 1 | 9a (Ph, Me, H) | −30 | 95 | 71 (R) |
| 2 | 9g (4-Cl-C6H4, Me, H) | −20 | 64 | 76 (R) |
| 3 | 9j (4-MeC6H4, Me, H) | −30 | 88 | 72 (R) |
Finally, we turned our attention to the mechanism of the reaction. For that purpose, a crossover experiment using a mixture of substrates 9g and 9b in the presence of 10 mol% of Cu(OTf)2·1g in chloroform was conducted (Scheme 3). The intramolecular rearrangements of 9g and 9b proceeded smoothly and no crossover products were obtained. Therefore, it was ascertained that this reaction proceeds via a concerted or tight-ion-pair pathway.
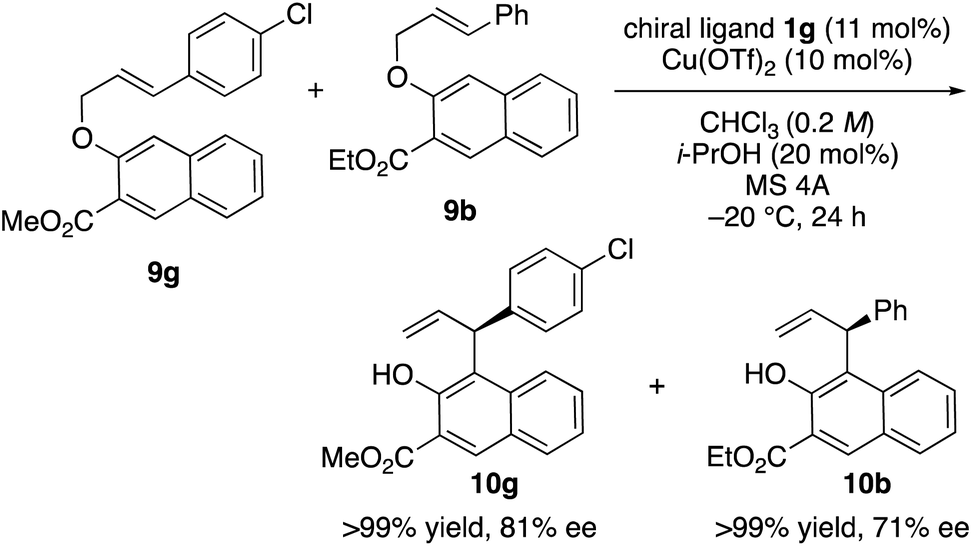 | ||
| Scheme 3 Crossover experiment. | ||
To understand the origin of the enantioselectivity of this reaction, we performed density-functional-theory (DFT) calculations at the B3LYP/6-31G(d) and LANL2DZ for Cu(II) level (Gaussian 16 (ref. 8)). At first, we studied the complexation of substrate 9a with catalyst Cu(OTf)2·1. There are two possible geometrical chelate structures for the complex between 9a and Cu(OTf)2·1. In the case of Cu(OTf)2·1g, one triflate anion is released from Cu(II) by chelation of 9a (Fig. 2). The trans structure of 9a·Cu(OTf)+·1g, wherein the ether oxygen is located trans to the nitrogen of 1g, is 9.3 kcal mol−1 more stable than the cis structure, which is unstable due to steric hindrance between the axial hydrogen of N-5-dibenzosuberyl of 1g and one of the OCH2 hydrogens of 9a.9 Here, the H–H distance (2.28 Å) is shorter than the sum of their van der Waals radii (2.40 Å).
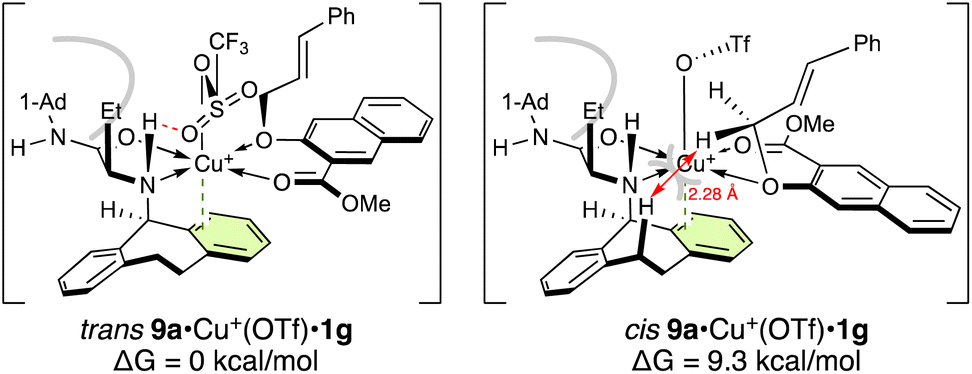 | ||
| Fig. 2 Two geometrical structures of 9a·Cu(OTf)+·1g. For computational details, see the ESI.† | ||
Next, we studied the complexation of 9a with Cu(OTf)2·1k (Scheme 4). Interestingly, cis9a·Cu+(OTf)·1k is predominantly formed via monocoordinated complex 11a. On the other hand, trans9a·Cu+(OTf)·1k is not obtained from 11a due to the steric demand of the t-butyl group. When the ether oxygen of 11a is moved closer to Cu(II), one of the triflates is easily eliminated to give 11b. Subsequently, the ether oxygen of 11b approaches Cu(II) and the triflate is repelled to the apical position to form cis9a·Cu+(OTf)·1k (for details, see the ESI†).
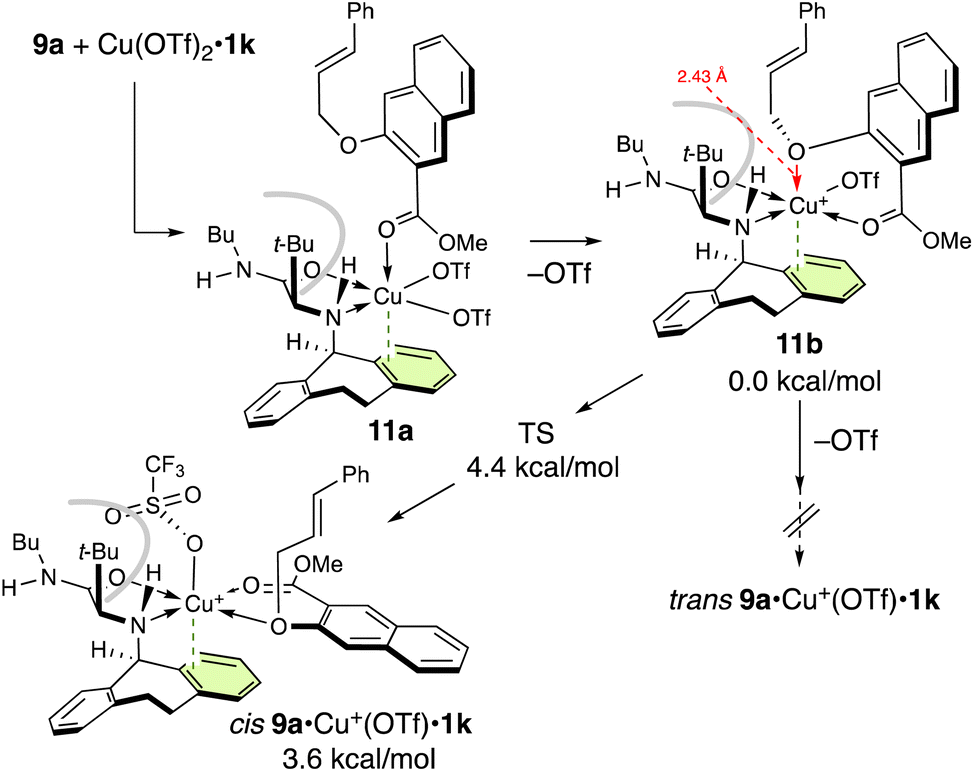 | ||
| Scheme 4 Potential energy profile at 25 °C and the complexation of 9a with Cu(OTf)2·1k.a aFor computational details, see the ESI.† | ||
Interestingly, the Claisen rearrangement of 9a proceeds stepwise via a tight-ion-pair intermediate. Both potential energy profiles that lead to enantiomeric products (S)-10a and (R)-10a are shown in Schemes 5 and 6. Substrate 9a predominantly chelates to Cu(OTf)2·1g in a trans manner between the nitrogen atom of 1g and the ether oxygen atom of 9a due to steric and electronic effects (for details, see the ESI†). One triflate anion is released from Cu(II) due to the π–Cu(II) interaction to generate [9a·Cu+(OTf)·1g][−OTf]. Another triflate group occupies the apical position of the octahedral Cu(II) complex, avoiding the bulky N-1-adamantyl group of 1g. Both the transition structures A1 and B1 for the cleavage of the C–O bond are stabilized by hydrogen bonding between the allylic protons and the triflate oxygens. Staggered transition state (TS) A1 is more stable than eclipsed TS B1 due to torsional effect (mainly electronic repulsion). Thus, (S)-10a is obtained as the major enantiomer via TS A1. Although the energy values of TSs A2 and B2 for the formation of the C–C bond are slightly higher than those of A1 and B1, the lack of crossover products suggests that cleavage of the C–O bond is the rate-limiting step. This energy difference between TSs A1 and B1 is 0.97 kcal mol−1 at 25 °C and 1.17 kcal mol−1 at −35 °C, corresponding to 84% ee at −35 °C. This energy difference is in good agreement with the experimental results (entry 1 in Table 4).
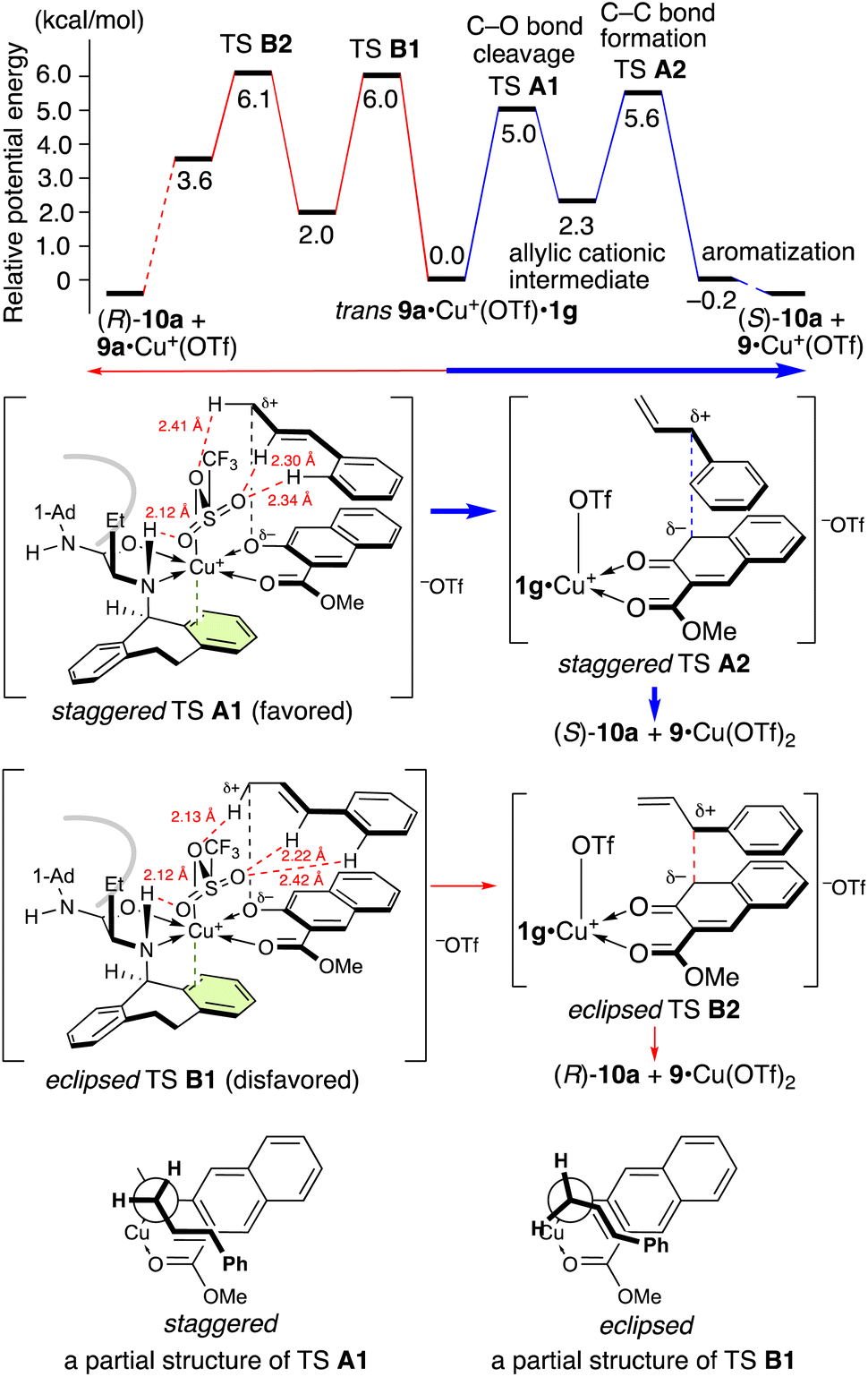 | ||
| Scheme 5 Potential energy profile at 25 °C and transition states for the aromatic Claisen rearrangement of 9a catalyzed by Cu(OTf)2·1g.a aFor computational details, see the ESI.† | ||
 | ||
| Scheme 6 Potential energy profile at 25 °C and the transition states for the aromatic Claisen rearrangement of 9a catalyzed by Cu(OTf)2·1k.a aFor computational details, see the ESI.† | ||
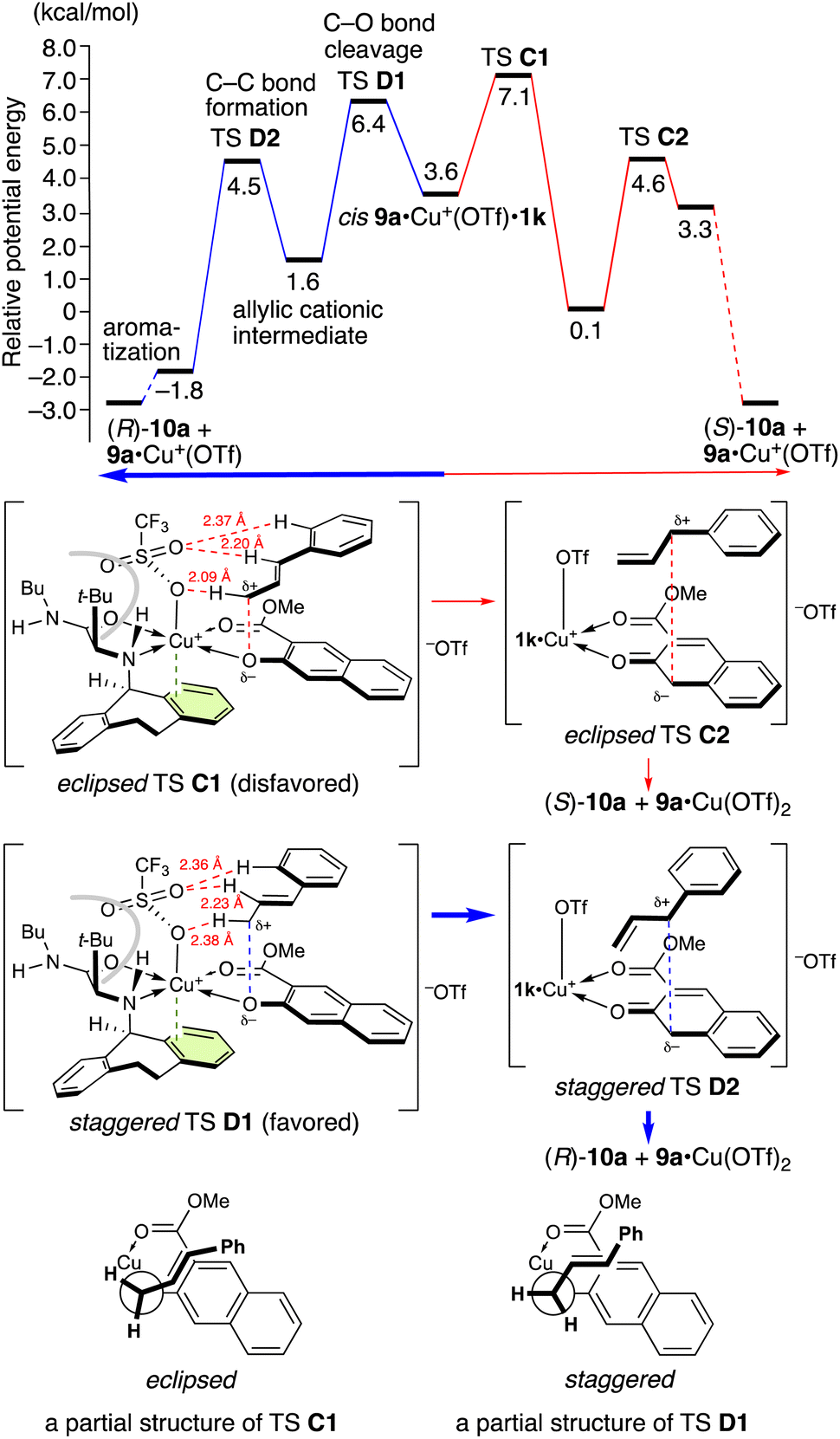
In contrast, when 1k is used, cis9a·Cu+(OTf)·1k is preferred (Scheme 4). The apical triflate group is positioned to avoid steric hindrance of the t-butyl group and cannot be stabilized by a hydrogen bonding with the amino protons (TS C1 and TS D1 in Scheme 6). The staggered TS D1 is favored compared to the eclipsed TS C1 due to the torsional effect. Thus, (R)-10a is obtained as a major enantiomer via TS D1. The energy difference between TSs D1 and C1 is 0.72 kcal mol−1 at 25 °C and 0.80 kcal mol−1 at −30 °C, corresponding to 68% ee at −30 °C. This energy difference is in good agreement with the experimental results (entry 5 in Table 5).
Conclusions
In summary, we have accomplished the first catalytic enantioselective aromatic Claisen rearrangement of allyl 2-naphthyl ethers 9 by using a chiral π–Cu(II) catalyst. The Cu(OTf)2 complex with L-α-homoalanine amide ligand 1g gave (S)-10 products in up to 92% ee. Conversely, the Cu(OSO2C4F9)2 complex with L-tert-leucine amide ligand 1k gave (R)-10 products in up to 71% ee. DFT calculations suggest that the preference of these catalysts for (S)- and (R)-products might be ascribed to a preference of the reactions to proceed via a staggered rather than an eclipsed transition state.Data availability
All experimental procedures, spectral data and computational calculations are available in the ESI.†Author contributions
K. I. conceived and directed the project. L. Y. and K. T. carried out the experiments and collected data. K. A. performed and analyzed the DFT calculations. K. I. wrote the manuscript with contributions from all authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was financially supported by Academic Research & Industry-Academia-Government Collaboration, THERS.Notes and references
- L. Yao and K. Ishihara, Chem. Sci., 2019, 10, 2259–2263 RSC.
- (a) K. Ishihara and M. Fushimi, Org. Lett., 2006, 8, 1921–1924 CrossRef CAS PubMed; (b) K. Ishihara, M. Fushimi and M. Akakura, Acc. Chem. Res., 2007, 40, 1049–1055 CrossRef CAS PubMed; (c) K. Ishihara and M. Fushimi, J. Am. Chem. Soc., 2008, 130, 7532–7533 CrossRef CAS PubMed; (d) A. Sakakura, M. Hori, M. Fushimi and K. Ishihara, J. Am. Chem. Soc., 2010, 132, 15550–15552 CrossRef CAS PubMed; (e) A. Sakakura and K. Ishihara, Chem. Soc. Rev., 2011, 40, 163–172 RSC; (f) M. Hori, A. Sakakura and K. Ishihara, J. Am. Chem. Soc., 2014, 136, 13198–13201 CrossRef CAS PubMed; (g) K. Ishihara, K. Nishimura and K. Yamakawa, Angew. Chem., Int. Ed., 2020, 59, 17641–17647 CrossRef CAS PubMed; (h) K. Nishimura, Y. Wang, Y. Ogura, J. Kumagai and K. Ishihara, ACS Catal., 2022, 12, 1012–1017 CrossRef CAS; (i) K. Nishimura and K. Ishihara, Synlett, 2022, 33, 585–588 CrossRef CAS; (j) K. Nishimura, Y. Ogura, K. Takeda, W. Guo and K. Ishihara, Org. Lett., 2022, 24, 7685–7689 CrossRef CAS PubMed.
- (a) C.-X. Zhuo and S.-L. You, Angew. Chem., Int. Ed., 2013, 52, 10056–10059 CrossRef CAS PubMed; (b) H.-F. Tu, C. Zheng, R.-Q. Xu, X.-J. Liu and S.-L. You, Angew. Chem., Int. Ed., 2017, 56, 3237–3241 CrossRef CAS PubMed; (c) D. Shen, Q. Chen, P. Yan, X. Zeng and G. Zhong, Angew. Chem., Int. Ed., 2017, 56, 3242–3246 CrossRef CAS PubMed.
- (a) L. Wang, P. Zhou, Q. Lin, S. Dong, X. Liu and X. Feng, Chem. Sci., 2020, 11, 10101–10106 RSC; (b) H. Zheng, Y. Wang, C. Xu, X. Xu, L. Lin, X. Liu and X. Feng, Nat. Commun., 2018, 9, 1968 CrossRef PubMed.
- Taguchi et al. have reported an enantioselective aromatic Claisen rearrangement using 150 mol% of chiral Lewis acid; for details see: H. Ito, A. Sato and T. Taguchi, Tetrahedron Lett., 1997, 38, 4815–4818 CrossRef CAS.
- For the examples of asymmetric Claisen rearrangements of propargyl ethers, see: (a) Y. Liu, H. Hu, H. Zheng, Y. Xia, X. Liu, L. Lin and X. Feng, Angew. Chem., Int. Ed., 2014, 53, 11579–11582 CrossRef CAS PubMed; (b) L. Wang, Y. Zhou, Z. Su, F. Zhang, W. Cao, X. Liu and X. Feng, Angew. Chem., Int. Ed., 2022, 61, e202211785 CAS.
- T. Ollevier and T. M. Mwene-Mbeja, Tetrahedron Lett., 2006, 47, 4051–4055 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega,G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision C.01, Gaussian, Inc., Wallingford CT, 2019 Search PubMed.
- The energy difference of 9.3 kcal mol−1 is very large. To clarify this result, we performed optimization and energy calculations of complexes trans-9a·Cu+(OTf)·1′ and cis-9a·Cu+(OTf)·1′ using sterically less hindered N-methylglycine N-methylamide (1′) instead of 1g. trans-9a·Cu+(OTf)·1′, wherein the ether oxygen located trans to the amino nitrogen, is 5.5 kcal mol−1 more stable than cis-9a·Cu+(OTf)·1′. Given that no steric hindrance is observed in cis-9a·Cu+(OTf)·1′, the instability of cis-9a·Cu+(OTf)·1′ was attributed to the electronic effect of nitrogen.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 20190517. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2sc06771d |
| This journal is © The Royal Society of Chemistry 2023 |