
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceChemical signal regulated injectable coacervate hydrogels†
Bohang
Wu
ab,
Reece W.
Lewis
 b,
Guotai
Li
b,
Yifan
Gao
a,
Bowen
Fan
b,
Benjamin
Klemm
b,
Jianan
Huang
a,
Junyou
Wang
a,
Martien A.
Cohen Stuart
a and
Rienk
Eelkema
*b
b,
Guotai
Li
b,
Yifan
Gao
a,
Bowen
Fan
b,
Benjamin
Klemm
b,
Jianan
Huang
a,
Junyou
Wang
a,
Martien A.
Cohen Stuart
a and
Rienk
Eelkema
*b
aEast China University of Science and Technology, Department of Chemical Engineering, Meilong Road 130, 200237 Shanghai, China
bDelft University of Technology, Department of Chemical Engineering, Van der Maasweg 9, 2629 HZ Delft, The Netherlands. E-mail: R.Eelkema@tudelft.nl
First published on 19th January 2023
Abstract
In the quest for stimuli-responsive materials with specific, controllable functions, coacervate hydrogels have become a promising candidate, featuring sensitive responsiveness to environmental signals enabling control over sol–gel transitions. However, conventional coacervation-based materials are regulated by relatively non-specific signals, such as temperature, pH or salt concentration, which limits their possible applications. In this work, we constructed a coacervate hydrogel with a Michael addition-based chemical reaction network (CRN) as a platform, where the state of coacervate materials can be easily tuned by specific chemical signals. We designed a pyridine-based ABA triblock copolymer, whose quaternization can be regulated by an allyl acetate electrophile and an amine nucleophile, leading to gel construction and collapse in the presence of polyanions. Our coacervate gels showed not only highly tunable stiffness and gelation times, but excellent self-healing ability and injectability with different sized needles, and accelerated degradation resulting from chemical signal-induced coacervation disruption. This work is expected to be a first step in the realization of a new class of signal-responsive injectable materials.
Introduction
Soft materials that can respond to signals from the environment, display promising applications in therapeutic delivery.1–3 Programmable sol–gel transitions are required for many therapeutic applications, e.g., where materials could be injected into the body as a gel, but then degrade in response to external triggers.4,5 Among various materials which are broadly applied, hydrogels formed by strong electrostatic interactions (coacervation assembly), have been of great interest due to their inherent responsiveness to external signals.6–8 These signal-sensitive (pH, temperature or salt) coacervate materials do, however, show limited responsiveness under physiological conditions.9–12 To overcome the restrictions above, it is of great significance to develop materials which are responsive to specific signals, enabling precise regulation of material properties.Inspired by biological systems, where biochemical signals play an important role to control the growth and behavior of supramolecular structures, chemical reaction networks (CRNs) have been developed as a useful tool to control the properties of synthetic supramolecular materials.13–17 Typically, non-interacting building blocks are activated into an assembling structure by an activator.18,19 The initiated products are later deactivated by a competing reaction, leading to disassembly and recovery of the starting non-interacting building blocks.20,21 Through delicate design, temporary sol–gel transition processes have been realized, regulated by methylating agents,22,23 glucose,24 enzymatic reaction,25–27 and other chemical fuels.28,29 Recently, CRNs have also been used to control coacervate assembly with applications in drug delivery, nanoreactors and protocell simulation.30–33 These have typically been dispersed nano- and micro-scale assemblies and there are no studies on CRN-regulated coacervate assembly for macroscopic gels.
In the current work, we start from a Michael addition-based CRN to create chemical signal responsive coacervate hydrogels. Specifically, allyl acetates (electrophile) act as an activator which can react with tertiary amines, yielding a cationic quaternary ammonium species. This species can subsequently react with a competing nucleophile (deactivator), re-forming the initial tertiary amine, completing the reaction cycle.34 For example, we selected a pyridine-based polymer as the poly(tertiary amine), whose ionization could be reversibly regulated with the electrophiles (to ionize the polyamine) and competing nucleophiles (amines or thiols, to deionize the charged polyamine). Accordingly, the hydrophobicity and charge state of the pyridine-based polymer could be controlled.35 With this strategy, we have controlled the assembly of hydrophobic surfactant micelles and complex coacervate-core micelles (C3Ms) as well as the degree of swelling in cross-linked gels.35,36
At the base of the current work is the ABA triblock copolymer PVP1, which has pyridine groups incorporated into the A block that can be charged by treatment with an electrophile – methyl-2-(acetoxymethyl)acrylate (ME, activator). The copolymer can then revert to a neutral state after addition of a nucleophile – pyrrolidine (P, a secondary amine, deactivator). Importantly, the positively charged A blocks can interact with a polyanion, spontaneously forming a network of coacervate domains interconnected by the hydrophilic B block chains. Above a threshold polymer concentration, network formation leads to gelation. Addition of P can then revert the positively charged pyridinium units to their initial neutral state, destroying the gel. Gel properties like mechanical strength and gelation time are controllable by means of the amount of activator (ME). We then investigated the self-healing and injectability of these coacervate gels, which demonstrated their potential use as injectable materials. Moreover, our coacervate gels show selective nucleophile-responsiveness and therefore exhibit excellent degradability in cell culture media-based solutions or gels, acting as simulated interstitial fluid or tissue. We propose that these properties make CRN controlled coacervate hydrogels promising candidates for therapeutic biomedical materials.
Design concept of chemical signal regulated coacervate hydrogels
Inspired by reported polymer architectures,37 where the designed ABA triblock copolymer usually requires each block in moderate length for adequate interaction sites and moderate water-solubility, we synthesized the ABA triblock copolymer PVP1 (Scheme 1A) by a two-step RAFT polymerization using a symmetrical RAFT agent. A flexible water-soluble DMA (N,N-dimethylacrylamide) ‘B’ block of 410 units was prepared, followed by symmetrical extension with a random mixture 17 DMA and 41 VP (4-vinyl pyridine) units on either side as the ‘A’ blocks (further details and characterization were shown in Fig. S1–S2, S4–S5, S7 and Tables S1, S3†). Herein, DMA units in the ‘A’ blocks serve to weaken hydrophobic interactions and suppress gel formation in the neutral state. The pendent polyvinyl-pyridine units can be activated to quaternary pyridinium groups by ME, forming the cationic copolymer PVP1+ can interact with anionic polymer PAMPS236 (poly(2-acrylamido-2-methylpropane sulfonic acid sodium salt)), forming coacervate domains.36 When the polymers are of sufficient concentration, these domains are linked through the hydrophilic ‘B’ blocks, forming a coacervate gel network. Upon reaction with P, PVP1+ returns to its neutral state (PVP1), thus the electrostatic interactions disappear and the gel disassociates to a solution (Scheme 1B).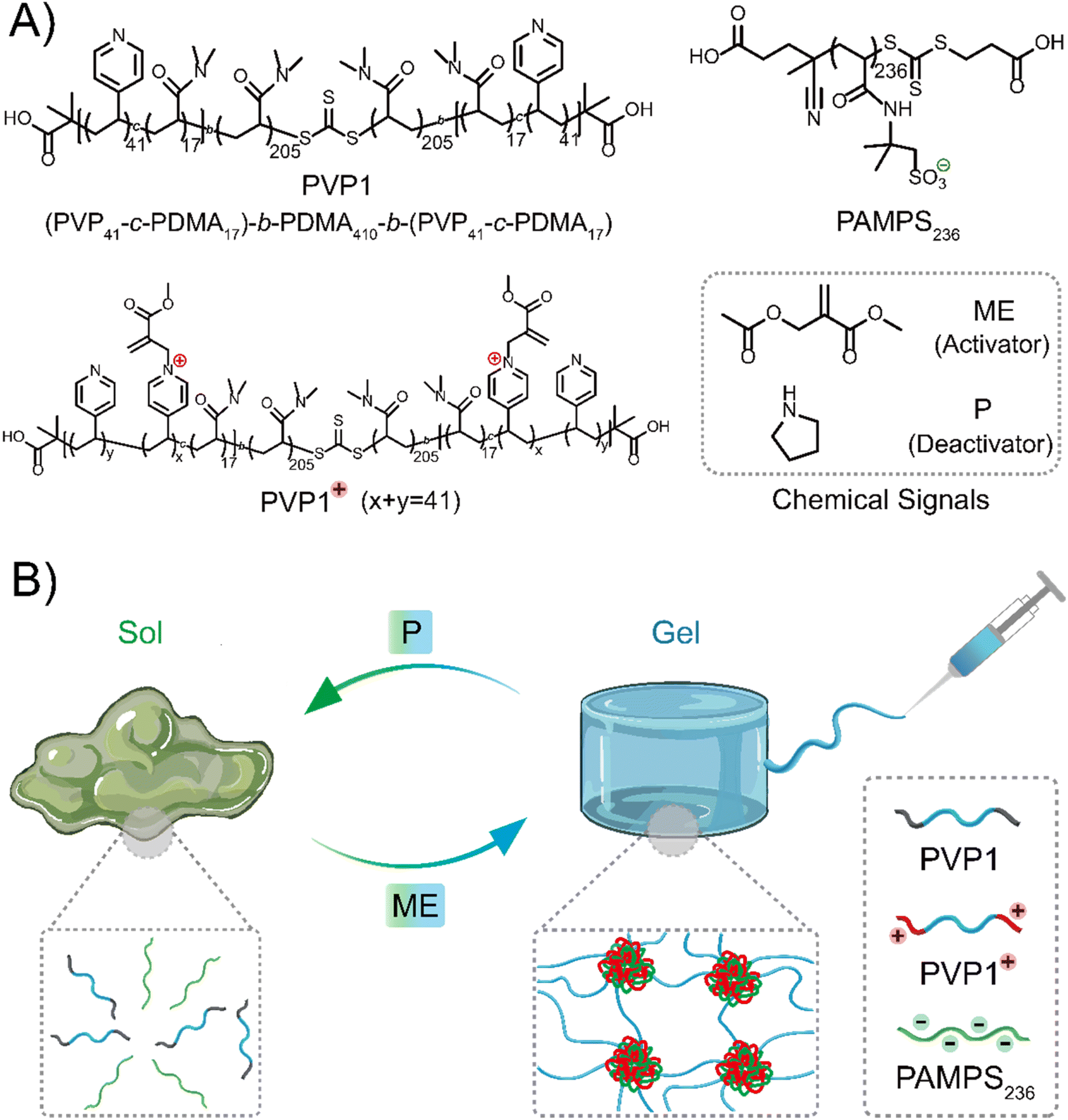 | ||
| Scheme 1 (A) Chemical structures of polymers and reagents involved. (B) Scheme of the responsive coacervate hydrogel formation regulated by chemical signals: ME (activator, an electrophile) activates the pyridine groups (on the polymer chain) from neutral to positive charged state, which can interact with anionic polymer PAMPS236, leading to a coacervate gel. With addition of P (deactivator, a nucleophile), the charged pyridine groups are deactivated, leading to a gel to solution transition. | ||
Results and discussion
Chemical reaction process and assembly mechanism
We began by studying the (de)ionization processes of our system in solution state based on NMR tests (Fig. S8†). Since the pKa of PVP1 is 4.1 (Fig. S9†), we performed the experiments with 0.5 wt% PVP1 in phosphate buffer (100 mM, pH 7.4), where the polymer is in a neutral state. After adding 1.0 eq. ME into the PVP1 solution at t = 0 h, we noticed that the conversion (from PVP1 to PVP1+) increased rapidly to approximately 75% in the first 3 hours and reached to its peak (78%) at t = 5 h (see Fig. 1A and B). Next, we added 1.0 eq. P into the solution at t = 30 h, where we observed a dramatic recovery of PVP1 from PVP1+ over the following 2 hours (from 78% to 20%). After a further 3 hours, the PVP1+ contents stabilized at 10%. From the NMR data for the reaction VP + ME ⇄ VP+ + acetate with an equilibrium conversion as 78%, the Keq for the reaction can be calculated as [VP+][acetate]/[VP][ME] = 0.78 × 0.78/0.22 × 0.22 = 13. This indicates that when an equivalent amount of ME is used (ME/VP = 1), 22% of the pyridine groups remain uncharged. With this Keq, increasing the amount of ME will lead to an increase in the ratio of charged over uncharged pyridines, thereby regulating the strength of polyelectrolyte complexation. This provides the possibility to modulate the material states by adjusting polymer concentration and quaternization degree. | ||
| Fig. 1 (A) Scheme of chemical reaction network (CRN) for reversible cationization of pyridine groups. Specifically, ME can induce the ionization of tertiary amines via a nucleophilic substitution reaction, yielding the cationic amines and acetate. The starting neutral tertiary amines can then be regenerated by addition of a competing nucleophile P followed by the production of waste. (B) Reversible formation of PVP1+ following sequential 1.0 eq. additions of ME and P to PVP1 (0.5 wt% PVP1, in 100 mM pH 7.4 phosphate buffer). The reaction cycle was also studied by dynamic light scattering for 2-cycles of 1.0 eq. ME and P sequential additions followed with (C) scatter count and (D) Z-average diameter (0.1 wt% PVP1 and 1.0 eq. PAMPS236, in 100 mM pH 7.4 phosphate buffer). | ||
After NMR experiments demonstrated signal responsive control over the ionization state of PVP1, we next investigated whether this induced polycation could be used to trigger coacervate formation with an anionic polymer. For this we synthesized an AMPS (2-acrylamido-2-methylpropane sulfonic acid sodium salt) polymer of approximately 236 units (PAMPS236, for synthesis details see Fig. S3, S6 and Tables S2, S3†). We studied their interactions via dynamic light scattering (DLS) at a relatively low PVP1 concentration (0.1 wt% PVP1, in 100 mM pH 7.4 phosphate buffer) in the presence of 1.0 eq. PAMPS236 (meaning the ionic molarity for AMPS and VP are equal). Fig. 1C and D show that, although the neutral PVP1 cannot interact with anionic PAMPS236, it can still form micellar structures by itself in an aqueous environment based on hydrophobic interactions (the presence of PAMPS236 did not influence the result, see Fig. S11A and B†). Thus, the initial system had a high scatter count and large Z-average size (15 Mcps, 175 nm). Charging the pyridine groups with 1.0 eq. ME led to disassociation of hydrophobic micelles. As a result, in first 2 hours, both the scatter count and size decreased. Interestingly, this was followed by an increase in both parameters for the next 11 hours, reaching stable values around 12 Mcps, 150 nm. This implies that once sufficient ionization of PVP1+ is achieved, its coacervate interactions with PAMPS236 start to dominate, leading to the formation of coacervate micellar structures (for the dilute 0.1 wt% PVP1 case). Then, 1.0 eq. P was added to neutralize the charged pyridine groups, triggering a reduction in the scatter count and size of our system due to dissociation of the coacervate micelles. Interestingly, the scatter count and corresponding particle size stabilized here (1.5 Mcps and 50 nm) and did not return to the values observed before ME addition. This implies that the hydrophobic association is not reversible in our system, probably because of a small amount of residual PVP1+. A second cycle was then conducted which demonstrated the reversibility of the system, with 1.0 eq. ME triggering the appearance of coacervate micelles and 1.0 eq. P destroying them. Since there was no hydrophobic micelles disassembly process during the second cycle, the time for reaching the maximum count rate (∼7 hours) was much shorter than in the first cycle (∼13 hours). Promisingly, the peak values for light scatter intensity and particle size (13 Mcps, 145 nm) were approximately the same as in the first cycle.
To further study why the hydrophobic micelles did not form again, we repeated the experiment without PAMPS236. At the start of the process, again hydrophobic micelles of similar size and scatter count as the system with PAMPS236 were observed. Addition of 1.0 eq. ME similarly decreased these values, indicating micelle disassociation (Fig. S11A and B†). After this point, further additions of P were unable to recover the hydrophobic micelles, even when treated with 3.0 eq. excess of P (Fig. S12A and B†).
Based on these experiments, we speculate that the incomplete recovery of PVP1 from PVP1+ (as indicated in NMR experiments) leads to weaker hydrophobicity of recovered PVP1, blocking the re-formation of hydrophobic micelles. At the same time, the presence of a small amount of residual cationic pyridine groups after P addition appears to be insufficient to allow coacervate formation, opening the door to repeated transitions between coacervate states.
Coacervate hydrogels formation controlled by chemical signals
We next sought to use such coacervate assembly to construct macroscopic hydrogels and study their chemical signal-triggered sol–gel transitions. Herein, 5 wt% PVP1 and 1.0 eq. PAMPS236 were dissolved in 100 mM pH 7.4 phosphate buffer. Then, the solution was mixed with 1.0 eq. ME, and a gel formed in 30 min (Fig. 2A, for detailed gel preparation see ESI†). With addition of 1.0 eq. P, the obtained gel can revert to a solution in 5 min with similar rheological properties as the starting solution (Fig. 2A and B). To clearly illustrate the (de)generation of the coacervate gel, we conducted time sweep rheological measurements. As shown in Fig. 2B, at the beginning the storage modulus (G′) of the polymer solution was much lower than loss modulus (G′′), indicating a solution state. After treatment with 1.0 eq. ME, both G′ and G′′ increased rapidly. Within 30 min, G′ surpassed G′′, suggesting the gelation of polymer solutions. After further 60 min, G′ stabilized at about 1000 Pa, showing a stable gel state. Then, 1.0 eq. P was added to neutralize the charged pyridine groups, which should lead to disassociation of the coacervation domains in the gel. After 3 min shaking, the gel converted back to solution (G′′ > G′) (shaking does not induce sol–gel transitions in this system, see Fig. S13†). To demonstrate the reversibility of the chemical signal-induced coacervate gel, we completed two more cycles with sequential additions of 1.0 eq. ME and 1.0 eq. P. Note that the gelation time for cycles 2 and 3 were about 15 min (much shorter than the gelation time (∼30 min) of cycle 1), which is in good agreement with our DLS test results. Moreover, this sol–gel cycle can bear at least 8 cycles, otherwise the materials will keep as a solution state, possibly due to the accumulation of reagents waste.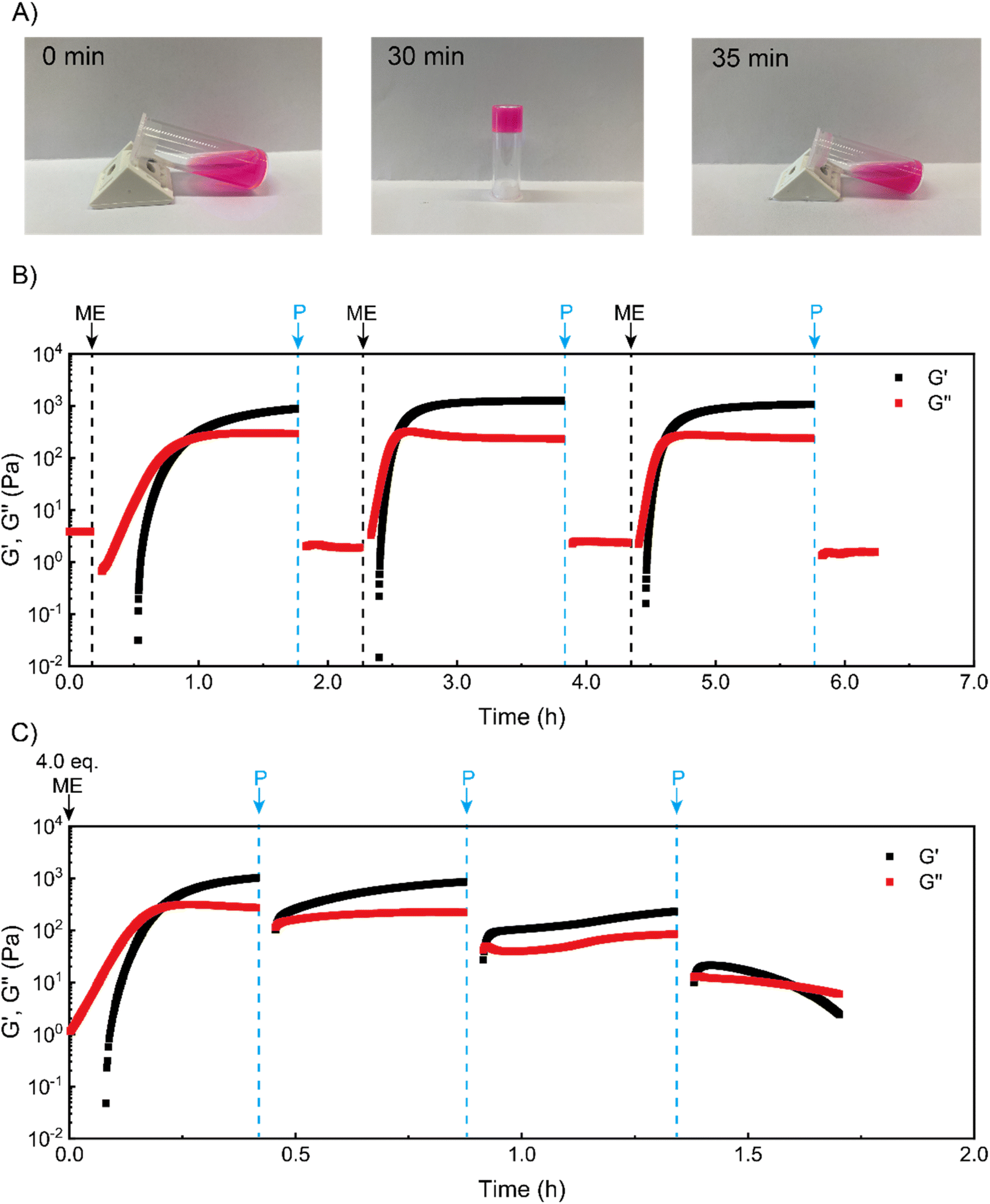 | ||
| Fig. 2 Chemical signal-programmed sol–gel transition process. (A) Photographs showing the gelation of PVP1 solution (in the presence of 1.0 eq. PAMPS236) driven by 1.0 eq. ME and the disassembly of coacervate gels induced by 1.0 eq. P. Rhodamine B was added for coloring (1 μM). Mixing the PVP1 solution with 1.0 eq. ME at 0 min led to gelation after 30 min. After adding 1.0 eq. P, the formed coacervate gel converted to the solution state in 5 min. (B) Time sweep rheological tests of the ME-driven gelation and P-induced disassembly process of the coacervate gels (3 cycles). (C) Time sweep rheological tests monitoring autonomous recovery of coacervate gels (triggered by 4.0 eq. ME) with re-additions of 1.0 eq. P. For the time sweep oscillatory tests, the strain (γ) and frequency (ω) were set as 5% and 10 rad s−1, respectively. All samples: 5 wt% PVP1 and 1.0 eq. PAMPS236 (overall polymer content: 6.8 wt%), in 100 mM pH 7.4 phosphate buffer. | ||
As a further test, we tried to achieve an ‘autonomous recovery’ by preparing a gel with excess ME (4.0 eq.) and using successive 1.0 eq. additions of P to transiently modulate gel properties. We observed that the gelation of polymer solutions (triggered by 4.0 eq. ME) quickly happened in 10 min (see Fig. 2C), which is much faster than the 1.0 eq. ME-triggered gelation. It seems plausible that a larger amount of ME leads to a higher ionization rate for PVP1, resulting in faster gelation. After the storage modulus stabilized around 1000 Pa, 1.0 eq. P was added to weaken the gel (as there is still some excess ME inside, 1.0 eq. P cannot fully convert the gel to a solution). Immediately after adding P, the moduli dropped sharply, but they then recovered to the starting value in minutes. Apparently, the added P initially removes (part of) the charges, but reaction with the remaining excess of ME restores these charges nearly entirely. In the second recovery cycle, an additional 1.0 more eq. P was placed onto the gel surface. Although this time there was a recovery process for gel mechanical strength, the maximum attained storage modulus (∼300 Pa) was lower than the first cycle (∼900 Pa). Furthermore, we noticed that after the third P addition, a solution formed that was unable to recover to a gel. We believe this is due to insufficient amount of remaining ME to achieve the required ionization on PVP1 for coacervate gel formation.
Overall, the experiments illustrate how sol–gel transitions can be achieved by modulating coacervate formation through reaction with an activator electrophile (ME) and a deactivator secondary amine (P). We found that the ‘forward’ reaction to PVP1+ is slower than the reverse reaction. In addition, with excess ME in the solution, a non-monotonic autonomous recovery, namely an initial drop in stiffness followed by a recovery, can be realized.
Effects of chemical signals on the mechanical performance of coacervate hydrogels and diagram of material states
Next, we characterized the impact of different ME equivalents on the mechanical properties and gelation times of the coacervate gels. For this purpose, we first prepared the polymer solutions (5 wt% PVP1 and 1.0 eq. PAMPS236, in 100 mM pH 7.4 phosphate buffer) and activated them with different ME equivalents. The gelation time and mechanical strength of gels were determined by oscillatory rheological measurements (Fig. S14†). The results shown in Fig. 3A suggest that the maximum storage modulus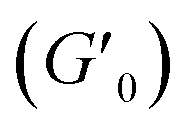 of coacervate gels increased from ∼25 to ∼1000 Pa with increasing ME equivalents (from 0.5 to 1.0 eq.). Clearly, a larger ratio of ME to pyridine groups leads to more charged pyridine groups, leading to stronger coacervation interactions (higher
of coacervate gels increased from ∼25 to ∼1000 Pa with increasing ME equivalents (from 0.5 to 1.0 eq.). Clearly, a larger ratio of ME to pyridine groups leads to more charged pyridine groups, leading to stronger coacervation interactions (higher 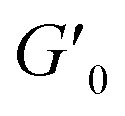 ). However, further increasing the ME equivalents beyond 1.0 eq. does not unlimitedly lead to enhanced mechanical properties such as G′ (Fig. S14†), likely due to reaching a plateau in pyridinium formation on the copolymers. Moreover, the gelation time (first time point where G′ > G′′) can also be regulated by varying ME equivalents. As shown in Fig. 3B, we found that the gelation time decreased from ∼1.5 h to ∼0.3 h with increasing ME equivalents (from 0.5 to 1.5 eq.). This indicates that higher ME concentration increases the ionization reaction rate and therefore shortens the gelation time of polymer solutions.
). However, further increasing the ME equivalents beyond 1.0 eq. does not unlimitedly lead to enhanced mechanical properties such as G′ (Fig. S14†), likely due to reaching a plateau in pyridinium formation on the copolymers. Moreover, the gelation time (first time point where G′ > G′′) can also be regulated by varying ME equivalents. As shown in Fig. 3B, we found that the gelation time decreased from ∼1.5 h to ∼0.3 h with increasing ME equivalents (from 0.5 to 1.5 eq.). This indicates that higher ME concentration increases the ionization reaction rate and therefore shortens the gelation time of polymer solutions.
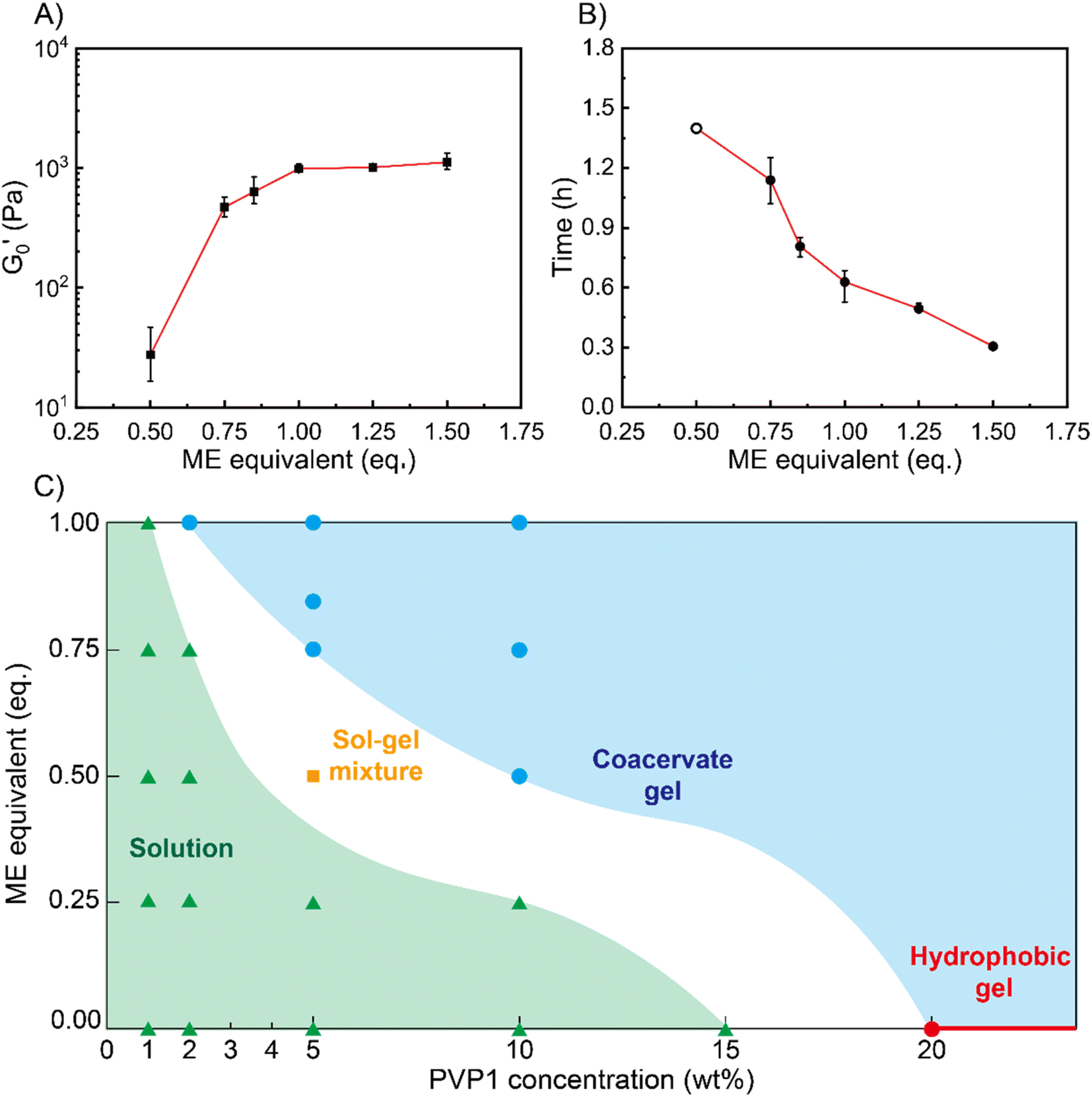 | ||
Fig. 3 Effect of ME equivalents on coacervate gelation process. (A) Maximum storage modulus 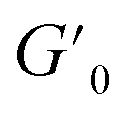 and (B) gelation time of the coacervate hydrogels as a function of ME equivalents. Error bars in (A) and (B) are calculated from three independent measurements. All samples: 5 wt% PVP1 and 1.0 eq. PAMPS236 (overall polymer content: 6.8 wt%), in 100 mM pH 7.4 phosphate buffer. Note that a sol–gel mixture (5 wt% PVP1, 0.5 eq. ME) naturally gives variable results. In three measurements, we found one gel-like curve (from which we got one crossover point/gelation time, the open circle) and two solution-like curves. (C) Diagram of material states by varying both PVP1 concentrations and ME equivalents. The diagram is a compilation of the results from rheological measurements and visual observation (red circle and line – hydrophobic micelle gel state; blue circles and area – coacervate gel state; orange square – sol–gel mixture state; white area – potential sol–gel intermediate state; and green triangles and area – solution state). For 1 wt%, 2 wt%, 5 wt% and 10 wt% samples (overall polymer content: 1.4 wt%, 2.7 wt%, 6.8 wt% and 13.6 wt%): PVP1 and 1.0 eq. PAMPS236, in 100 mM pH 7.4 phosphate buffer; for 15 wt% and 20 wt% samples: PVP1, in 100 mM pH 7.4 phosphate buffer. and (B) gelation time of the coacervate hydrogels as a function of ME equivalents. Error bars in (A) and (B) are calculated from three independent measurements. All samples: 5 wt% PVP1 and 1.0 eq. PAMPS236 (overall polymer content: 6.8 wt%), in 100 mM pH 7.4 phosphate buffer. Note that a sol–gel mixture (5 wt% PVP1, 0.5 eq. ME) naturally gives variable results. In three measurements, we found one gel-like curve (from which we got one crossover point/gelation time, the open circle) and two solution-like curves. (C) Diagram of material states by varying both PVP1 concentrations and ME equivalents. The diagram is a compilation of the results from rheological measurements and visual observation (red circle and line – hydrophobic micelle gel state; blue circles and area – coacervate gel state; orange square – sol–gel mixture state; white area – potential sol–gel intermediate state; and green triangles and area – solution state). For 1 wt%, 2 wt%, 5 wt% and 10 wt% samples (overall polymer content: 1.4 wt%, 2.7 wt%, 6.8 wt% and 13.6 wt%): PVP1 and 1.0 eq. PAMPS236, in 100 mM pH 7.4 phosphate buffer; for 15 wt% and 20 wt% samples: PVP1, in 100 mM pH 7.4 phosphate buffer. | ||
To show how chemical signals may regulate these coacervate materials, we investigated the relationship between PVP1 concentrations, ME equivalents and material states (see Fig. 3C). We firstly prepared polymer solutions of different concentrations (1 wt%, 2 wt%, 5 wt%, 10 wt% PVP1 with 1.0 eq. PAMPS236, 15 wt% and 20 wt% PVP1) and initiated their gelation by addition of different ME equivalents. All samples were characterized by the vial inversion method and frequency sweep rheology experiments to identify the material states (solution or gel) (see Fig. S15 and S16†). We found that 20 wt% PVP1 can form a hydrophobic micelle gel by itself. In agreement with earlier dilute solution DLS analysis, these hydrophobic micelle gels showed poor reversibility, even after we attempted recovery using excess P (3.0 eq.) (see Fig. S11C, S12C and D†). The samples with ≤1 wt% PVP1 never formed a gel with ME (even with >1.0 eq. ME, see Fig. S15A and S16†). Samples with a PVP1 concentration between 1 wt% and 10 wt% were able to form coacervate gels, with less ME required for gel formation as polymer concentration increased (1.0 eq. ME for 2 wt% PVP1 and 0.5 eq. ME for 10 wt% PVP1). Interestingly, for 5 wt% PVP1 treated with 0.5 eq. ME (in the presence of 1.0 eq. PAMPS236), we observed the intermediate case of a sol–gel mixture. It seems likely that such intermediate states can also occur at other combinations of PVP1 concentrations and ME equivalents, just below a threshold.
Based on these observations, a ‘diagram of material states’ can be constructed (Fig. 3C). To clarify the diagram, datapoints referring to different states were given different colors. In the diagram, the red circle and line represents the material states which can assemble into hydrophobic micelle gels by themselves without any reagents added. The blue circles and area represent the coacervate gel state, where the gelation of polymer solutions can be activated by ME. The green triangles and area represent the liquid state, in which ME cannot trigger the gelation owing to insufficient polymer concentration. For the white area (containing the orange square) between the green and blue areas, we defined it as potential sol–gel intermediate state. Combined, the diagram clearly shows that as polymer concentration increases, the critical degree of PVP1 ionization (or eq. ME added) required to form a gel decreases.
Self-healing and injectability of coacervate gels
We then took the 5 wt% coacervate gels (initiated by 1.0 eq. ME) as a basis to investigate their self-healing properties. Strain sweep rheological tests showed that the gel networks are temporarily disrupted upon application of over 200% strain (see Fig. 4A). In the fixed-frequency measurement (with repeated strain), the gels were subjected to cycles of 5% and 500% strain. At 500% strain, G′ decreased from 800 Pa to 80 Pa and G′′ from 300 Pa to 200 Pa (Fig. 4B). In other words, under a large amplitude oscillatory force, G′′ was higher than G′, indicating the disruption of gel networks and conversion to a viscous fluid. When the strain returned to 5%, G′ and G′′ rapidly recovered to starting value (800 Pa and 300 Pa, respectively), suggesting the recovery of coacervate gels. Moreover, the gels can be subjected to at least 5 cycles without any loss of mechanical strength. In macroscopic self-healing experiments (Fig. 4C), we prepared two gels (one gel was colored by rhodamine B, and another one was non-dyed) and hand-pressed them together into a whole gel. After 5 min, the two separate gels reconnected and could be easily lifted by a tweezer. After 2 further days, the dye diffused into the non-dyed section, demonstrating the initially separate gels have completely self-healed to an integrated gel. These results provide clear evidence that these coacervate gels are capable of self-healing, as expected for gels with reversible, physical bonds.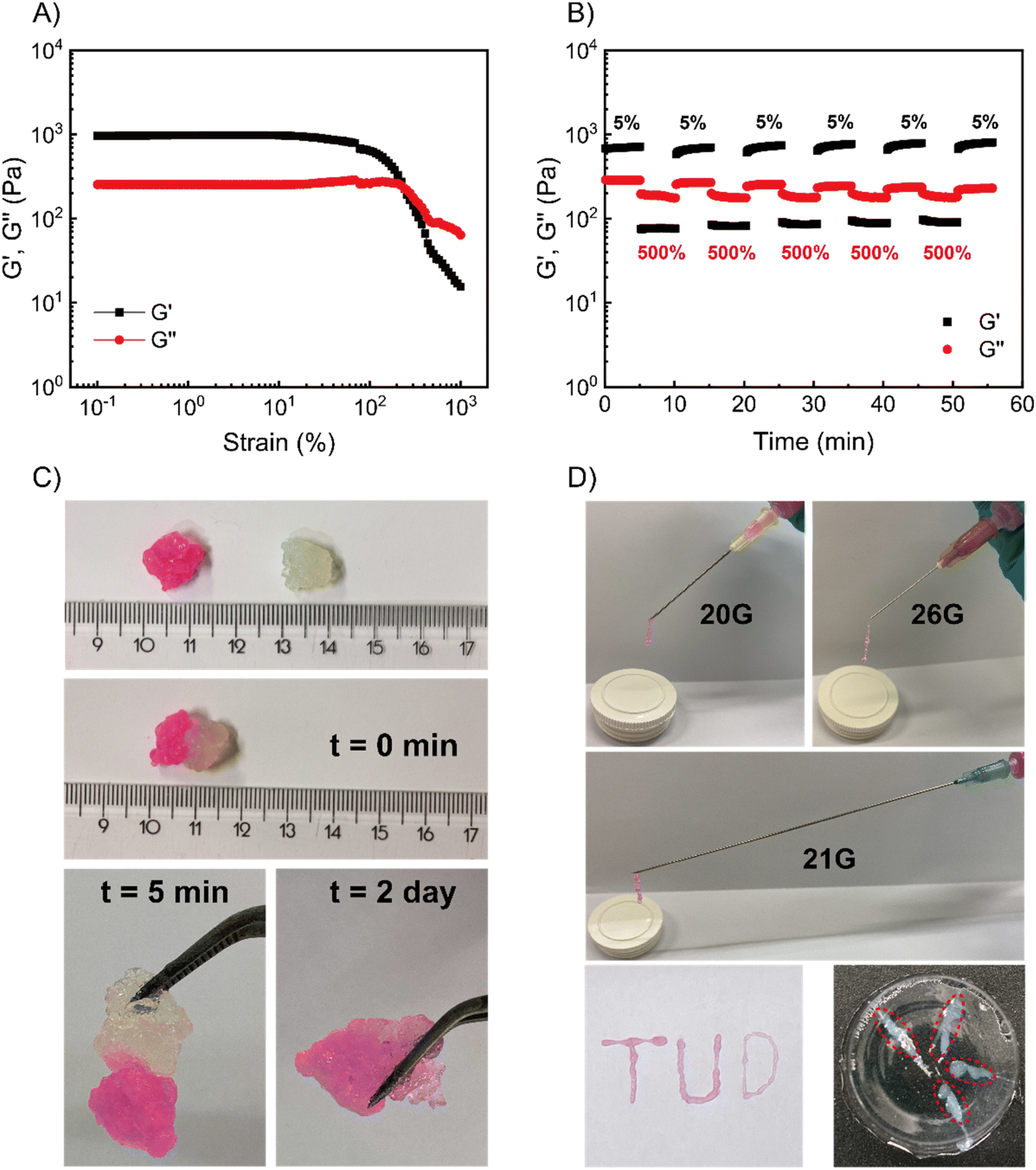 | ||
| Fig. 4 Self-healing and injectability of coacervate gels. (A) Strain sweep and of the coacervate hydrogels. Strain sweep measurements were performed from 0.1% to 1000% at fixed frequency (ω = 10 rad s−1). (B) Fixed-frequency (ω = 10 rad s−1) measurement with repeated strain jumps from 5% to 500% and back. (C) Self-healing of coacervate hydrogels. Two separate gels (one gel was dyed by rhodamine B, another gel was non-dyed) were pressed together. After 5 min, the two gels had become a single gel; after 2 days, the dye diffused from the stained gel to non-stained gel. (D) Coacervate hydrogels injection in air or into a PAAm gel (the coacervate gels can be extruded from 20G, 21G and 26G needles by hand-pressing). The injected gels (which were dyed by rhodamine B) allowed for printing ‘TUD’ letters (‘T’, ‘U’ and ‘D’ were extruded from 20G, 21G and 26G needles, respectively). Also, the coacervate gels (in red circles, non-dyed gels) can be injected into a PAAm gel via a 26G needle. All samples: 5 wt% PVP1 and 1.0 eq. PAMPS236 (overall polymer content: 6.8 wt%), in 100 mM pH 7.4 phosphate buffer (initiated by 1.0 eq. ME). | ||
Since the gels were self-healable and responded quickly to the strain changes, we investigated whether our coacervate gels were also injectable.5,38–40 As shown in Fig. 4D, a rhodamine B dyed gel was prepared and put into a syringe, then we extruded it from needles with various diameters (20G – 0.9 mm, 21G – 0.8 mm and 26G – 0.45 mm). Using hand pressure, these gels could be extruded through all investigated needles; after extruding from the needles (the high shear force was removed), the gel state was recovered instantly. As a proof of injectability, we wrote three letters (‘T’, ‘U’ and ‘D’) through different sized needles (Fig. 4D), a video showing the gel injection can be found as Movie S1.† In future therapeutic applications, such gels would have to injected into various tissues. Thus, we synthesized a polyacrylamide (PAAm) gel as a simulated tissue (the synthesis procedure of the PAAm gel can be found in the ESI†). With a 26G needle, these gels can be easily injected into the simulated tissue (Movie S2†), leading to gel-in-gel hybrid materials.
Accelerated degradation of coacervate gels in cell culture media-based environment
One important requirement for injectable materials is whether they can degrade in human tissue or interstitial fluid. Conventional coacervate hydrogels are rendered degradable because of their salt, pH or temperature responsiveness, however the degradation time can be exceedingly long under physiological conditions.9,11,12,39,41 Previous work in our group on related pyridine-based polymeric systems has shown that (in addition to secondary amines) primary amines and thiols can be used to trigger deionization.35,36 Treating our coacervate gels with two representative chemical signals – glycine (Gly, a primary amine) and sodium-3-mercaptopropane-sulfonate (SH-Na, a thiol), we indeed observed gel–sol transitions at a similar rate to that achieved with P (within 5 min). Interestingly, the recovered loss modulus depended on the nucleophilicity of the applied nucleophile,42,43 with stronger nucleophiles giving a lower recovered loss modulus (SH-Na ∼0.3 Pa, P ∼2 Pa, Gly ∼4 Pa) (Fig. S17†). A stronger nucleophile implies a larger reaction free energy, and, hence, a larger conversion of cationic into neutral pyridine groups. This means that after treatment with a stronger nucleophile, a smaller number of physical bonds would have to be broken under flow, observed as solutions with lower viscosity (lower G′′). Since the disassembly of these coacervate gels is responsive to these chemical signals, we anticipate that the degradation for such gels could be accelerated by physiological signals such as amino acids and thiols present in the cytosol or extracellular fluids.We first selected a cell culture medium (Ham's-F12, no phenol red, CM) as the simulated amino acids-rich interstitial fluid, which was anticipated to accelerate gel degradation. 150 mM phosphate buffer (PB) was used to mimic the physiological salt environment without nucleophiles present, isolating the effects of high salt concentration (which is known to degrade non-responsive coacervate gels).6,9,11,44,45 Pure water was applied as a blank culture environment (see Fig. S18 and Table S4†). At 25 °C, the coacervate gels completely disappeared within 4 h when incubated in CM, while those in PB or water did not disassemble even after 24 hours (Fig. 5A). When the degradation temperature was elevated to 37 °C, the coacervate gels in CM degraded in 2 h, again significantly faster than those in PB (24 h) and water (gel fragments still observable after 24 hours, Fig. 5B). These results suggest that the gels are sensitive to nucleophilic biological signals but not to physiological salt or temperature in liquid environments.
 | ||
| Fig. 5 Gel degradation tests in various liquid environments. Degradation of the coacervate gels in water, or 150 mM pH 7.4 phosphate buffer (PB) or cell culture medium (CM) at (A) 25 °C and (B) 37 °C over time. All samples: 5 wt% PVP1 and 1.0 eq. PAMPS236 (overall polymer content: 6.8 wt%), in 100 mM pH 7.4 phosphate buffer (initiated by 1.0 eq. ME). | ||
We then evaluated cell cytotoxicity of these gels by a viability test (CCK-8 assay) on NIH-3T3 mouse fibroblast cells (Fig. S19A†). These tests showed that the ME-activated gels show negligible toxicity at low concentration (gel concentration ≤1000 μg mL−1) but substantial cytotoxicity at high concentration (gel concentration >1000 μg mL−1). To investigate the source of toxicity within the ME-activated gels, we also evaluated the gel components separately. This showed that PAMPS236 and PVP1 are non-toxic even at high concentrations (viability above 80% up to 350 μg mL−1 and above 95% up to 500 μg mL−1 polymer concentration, respectively), while PVP1+ and ME decreased cell viability below 65% at 250 and 10 μg mL−1, respectively (Fig. S19B–E†). To increase biocompatibility, we replaced ME with a more biocompatible electrophile,35,36 for which we have found diethyl(α-acetoxymethyl) vinylphosphonate (DVP) retains ∼80% cell viability at high concentrations (236 μg mL−1, Fig. S20A†). Gels prepared using DVP were found to be significantly less toxic, with cell viability remaining above 85% even at high gel concentrations (10 mg mL−1, Fig. S20B†). Alternatively, the hydrogel may be applied as degradable scaffold or mold in 3D printing, because of its proven extrudability and controllable degradability.46 For example, the coacervate gel could be printed as a mold or scaffold to support the printing of a second material, after which the mold or scaffold can be easily removed by immersing it in an amine–rich medium. Overall, we conclude that this chemical-signal-regulated coacervate hydrogel could be used as injectable material (preferably with DVP as the activator for obtaining a more biocompatible material) or as scaffold in 3D printing.
In future in vivo applications, such gel materials can be introduced in the body by intradermal, subcutaneous or intramuscular injection, which is a reason to examine the gel degradation process in viscoelastic solid environments. Hence, we used swollen PAAm gels as tissue mimics. In brief, PAAm hydrogels were swollen in different media (water, PB and CM) for 18 hours, allowing for solvent exchange (further details shown in ESI and Table S5†). Frequency sweep rheological measurements on these swollen PAAm gels showed that they possess similar viscoelastic properties as many tissues (500 Pa < G′, G′′ < 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 Pa, see Fig. S21†).47 The coacervate gels were then injected into the different swollen PAAm gels and kept at 25 °C or 37 °C, and photos of gel degradation were taken over time (Fig. S22 and S23†).
000 Pa, see Fig. S21†).47 The coacervate gels were then injected into the different swollen PAAm gels and kept at 25 °C or 37 °C, and photos of gel degradation were taken over time (Fig. S22 and S23†).
At 25 °C, the coacervate gels decomposed completely in CM-swollen PAAm gels within 2 h (Fig. 6A). In contrast, the coacervate gels were still observable in PB-swollen PAAm gels even after 48 hours (blue pixels retain 0.6%, Fig. S22 and S24†). At 37 °C, coacervate gels that were injected into a CM-swollen PAAm gel degraded completely within 1 h (Fig. 6B). In PB-swollen PAAm gels the coacervate gels completely disappeared in 36 h (Fig. S23†). However, in water swollen PAAm gels, there was almost no observable degradation within 24 h at 25 °C or 37 °C (blue pixels retain 8.1% and 11.7%, respectively; Fig. S24†), and even after 36 hours the coacervate gel residues can still be observed (blue pixels retain 7.4% and 7.8%, respectively; Fig. S23 and S24†).
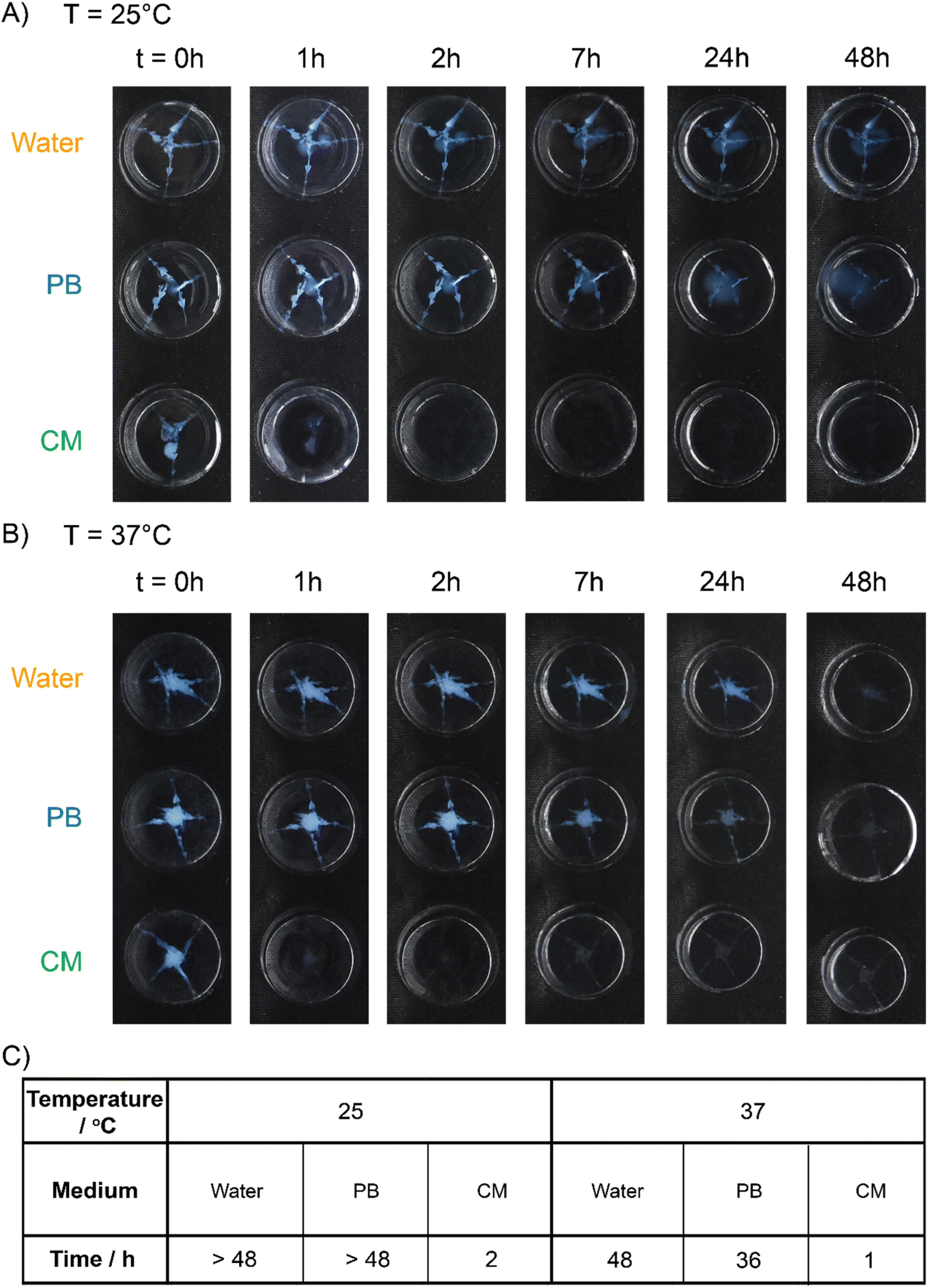 | ||
| Fig. 6 Degradation tests in the swollen PAAm gels. Degradation of the coacervate gels in water, or 150 mM pH 7.4 phosphate buffer (PB) or cell culture media (CM) swollen PAAm gels at (A) 25 °C and (B) 37 °C over time. Red arrows are added to point out the gel residue. (C) Degradation time (time needed for no visible gel remaining or below 0.5% blue pixels remaining, see ESI†) of coacervate gels (in the swollen PAAm gels). All samples: 5 wt% PVP1 and 1.0 eq. PAMPS236 (overall polymer content: 6.8 wt%), in 100 mM pH 7.4 phosphate buffer (initiated by 1.0 eq. ME). | ||
On the basis of these results, we concluded that the degradation of coacervate gels can be highly accelerated in both simulated interstitial fluid and simulated tissue. While relatively insensitive to the non-specific signals of physiological temperature (37 °C) and salt concentration (150 mM), gel degradation is then triggered by specific nucleophilic biochemical signals, enabling a tissue-responsive degradation process while maintaining gel integrity ex vivo.
Conclusions
This work shows the development and potential application of a chemical signal responsive coacervate hydrogel based on a Michael addition-based CRN. Allyl acetate electrophiles can trigger the cationization of pyridine groups located in the A blocks of an ABA triblock copolymer. In the presence of a polyanion, the electrophile addition triggers the formation of coacervate gels. The gels can be disassociated to solutions by reaction with a competing nucleophile, regenerating the starting neutral pyridine-based polymers. Further electrophile additions can recover the gel state, allowing for repeated and reversible sol–gel transitions. Moreover, varying the electrophile dose can tune the mechanical strength and gelation time of the coacervate gels. We further demonstrated promising self-healing properties of these coacervate gels, with application as injectable materials. Importantly, in cell culture media-based (rich in amino acids) environments, gel degradation can be highly accelerated due to the nucleophile-responsiveness of the coacervate gels. Extrapolating these findings, the concept of nucleophile-responsive coacervate gels, being injectable and selectively degradable, shows potential for application as therapeutical injectable materials or degradable scaffold in 3D printing.Data availability
The datasets generated during and/or analysed during the current study are available from the authors on reasonable request.Author contributions
B. W. carried out the experiments and analyzed the results, R. W. L. performed the cell cytotoxicity measurements (DVP) and assisted with NMR analysis. R. W. L. and G. L. provided suggestions on synthesis, NMR and DLS experiments. Y. G. and B. W. performed the cell cytotoxicity measurements (gel, PAMPS236, PVP1, PVP1+ and ME). B. F. provided suggestions on rheology measurements. B. K. provided suggestions on NMR experiments. J. H. assisted with schematic diagram design. B. W. wrote the manuscript. B. W., R. W. L., and R. E. designed the experiments. R. W. L., J. W., M. A. C. S. and R. E. revised the manuscript. All authors commented on the work and the manuscript.Conflicts of interest
The authors declare no financial/commercial conflict of interest.Acknowledgements
Financial support by the China Scholarship Council (B. W., the State Scholarship Fund File No. 202006740035) and the European Research Council (R. E., ERC consolidator grant 726381) is acknowledged. We thank Prof. Stephen Picken (TU Delft) for inspiring discussions on rheological measurements as well as Ruben Boot (TU Delft) for provision and assistance with NIH-3T3 cell line. We thank Jinbo Liu (ECUST) for assistance with photographing.References
- B. P. Purcell, D. Lobb, M. B. Charati, S. M. Dorsey, R. J. Wade, K. N. Zellars, H. Doviak, S. Pettaway, C. B. Logdon, J. A. Shuman, P. D. Freels, J. H. Gorman, R. C. Gorman, F. G. Spinale and J. A. Burdick, Nat. Mater., 2014, 13, 653–661 CrossRef CAS PubMed.
- M. Vazquez-Gonzalez and I. Willner, Angew. Chem., Int. Ed., 2020, 59, 15342–15377 CrossRef CAS PubMed.
- P. Lavrador, M. R. Esteves, V. M. Gaspar and J. F. Mano, Adv. Funct. Mater., 2021, 31, 2005941 CrossRef CAS.
- V. Yesilyurt, M. J. Webber, E. A. Appel, C. Godwin, R. Langer and D. G. Anderson, Adv. Mater., 2016, 28, 86–91 CrossRef CAS PubMed.
- F. Rizzo and N. S. Kehr, Adv. Healthcare Mater., 2021, 10, 2001341 CrossRef CAS PubMed.
- M. Lemmers, J. Sprakel, I. K. Voets, J. van der Gucht and M. A. C. Stuart, Angew. Chem., Int. Ed., 2010, 49, 708–711 CrossRef CAS PubMed.
- J. N. Hunt, K. E. Feldman, N. A. Lynd, J. Deek, L. M. Campos, J. M. Spruell, B. M. Hernandez, E. J. Kramer and C. J. Hawker, Adv. Mater., 2011, 23, 2327–2331 CrossRef CAS PubMed.
- W. D. Wang, L. Xiang, L. Gong, W. H. Hu, W. J. Huang, Y. J. Chen, A. B. Asha, S. Srinivas, L. Y. Chen, R. Narain and H. B. Zeng, Chem. Mater., 2019, 31, 2366–2376 CrossRef CAS.
- D. V. Krogstad, N. A. Lynd, S. H. Choi, J. M. Spruell, C. J. Hawker, E. J. Kramer and M. V. Tirrell, Macromolecules, 2013, 46, 1512–1518 CrossRef CAS.
- M. C. Koetting, J. T. Peters, S. D. Steichen and N. A. Peppas, Mater. Sci. Eng., R, 2015, 93, 1–49 CrossRef PubMed.
- S. Ishii, J. Kaneko and Y. Nagasaki, Macromolecules, 2015, 48, 3088–3094 CrossRef CAS.
- A. L. Z. Lee, Z. X. Voo, W. Chin, R. J. Ono, C. Yang, S. J. Gao, J. L. Hedrick and Y. Y. Yang, ACS Appl. Mater. Interfaces, 2018, 10, 13274–13282 CrossRef CAS PubMed.
- S. A. P. van Rossum, M. Tena-Solsona, J. H. van Esch, R. Eelkema and J. Boekhoven, Chem. Soc. Rev., 2017, 46, 5519–5535 RSC.
- G. Ashkenasy, T. M. Hermans, S. Otto and A. F. Taylor, Chem. Soc. Rev., 2017, 46, 2543–2554 RSC.
- M. Weissenfels, J. Gemen and R. Klajn, Chem, 2021, 7, 23–37 CAS.
- S. Amano, S. Borsley, D. A. Leigh and Z. H. Sun, Nat. Nanotechnol., 2021, 16, 1057–1067 CrossRef CAS PubMed.
- A. Paikar, A. I. Novichkov, A. I. Hanopolskyi, V. A. Smaliak, X. M. Sui, N. Kampf, E. V. Skorb and S. N. Semenov, Adv. Mater., 2022, 34, 2106816 CrossRef CAS PubMed.
- S. Yang, G. Schaeffer, E. Mattia, O. Markovitch, K. Liu, A. S. Hussain, J. Ottele, A. Sood and S. Otto, Angew. Chem., Int. Ed., 2021, 60, 11344–11349 CrossRef CAS PubMed.
- N. Singh, A. Lopez-Acosta, G. J. M. Formon and T. M. Hermans, J. Am. Chem. Soc., 2022, 144, 410–415 CrossRef CAS PubMed.
- H. X. Wang, Y. Y. Wang, B. W. Shen, X. Liu and M. Lee, J. Am. Chem. Soc., 2019, 141, 4182–4185 CrossRef CAS PubMed.
- N. Singh, G. J. M. Formon, S. De Piccoli and T. M. Hermans, Adv. Mater., 2020, 32, 1906834 CrossRef CAS PubMed.
- J. Boekhoven, W. E. Hendriksen, G. J. M. Koper, R. Eelkema and J. H. van Esch, Science, 2015, 349, 1075–1079 CrossRef CAS PubMed.
- B. G. P. van Ravensteijn, W. E. Hendriksen, R. Eelkema, J. H. van Esch and W. K. Kegel, J. Am. Chem. Soc., 2017, 139, 9763–9766 CrossRef CAS PubMed.
- S. H. Yu, S. J. Xian, Z. Ye, I. Pramudya and M. J. Webber, J. Am. Chem. Soc., 2021, 143, 12578–12589 CrossRef CAS PubMed.
- S. Debnath, S. Roy and R. V. Ulijn, J. Am. Chem. Soc., 2013, 135, 16789–16792 CrossRef CAS PubMed.
- T. Heuser, E. Weyandt and A. Walther, Angew. Chem., Int. Ed., 2015, 54, 13258–13262 CrossRef CAS PubMed.
- M. Jain and B. J. Ravoo, Angew. Chem., Int. Ed., 2021, 60, 21062–21068 CrossRef CAS PubMed.
- M. Tena-Solsona, B. Riess, R. K. Grotsch, F. C. Lohrer, C. Wanzke, B. Kasdorf, A. R. Bausch, P. Muller-Buschbaum, O. Lieleg and J. Boekhoven, Nat. Commun., 2017, 8, 15895 CrossRef CAS PubMed.
- N. Singh, B. Lainer, G. J. M. Formon, S. De Piccoli and T. M. Hermans, J. Am. Chem. Soc., 2020, 142, 4083–4087 CrossRef CAS PubMed.
- H. L. Che, J. Z. Zhu, S. D. Song, A. F. Mason, S. P. Cao, I. A. B. Pijpers, L. K. E. A. Abdelmohsen and J. C. M. van Hest, Angew. Chem., Int. Ed., 2019, 58, 13113–13118 CrossRef CAS PubMed.
- C. Donau, F. Spath, M. Sosson, B. A. K. Kriebisch, F. Schnitter, M. Tena-Solsona, H. S. Kang, E. Salibi, M. Sattler, H. Mutschler and J. Boekhoven, Nat. Commun., 2020, 11, 5167 CrossRef CAS PubMed.
- J. Deng and A. Walther, Chem, 2020, 6, 3329–3343 CAS.
- F. Spath, C. Donau, A. M. Bergmann, M. Kranzlein, C. V. Synatschke, B. Rieger and J. Boekhoven, J. Am. Chem. Soc., 2021, 143, 4782–4789 CrossRef PubMed.
- J. M. Zhuang, B. Zhao, X. X. Meng, J. D. Schiffman, S. L. Perry, R. W. Vachet and S. Thayumanavan, Chem. Sci., 2020, 11, 2103–2111 RSC.
- B. Klemm, R. Lewis, I. Piergentili and R. Eelkema, Nat. Commun., 2022, 13, 6242 CrossRef CAS PubMed.
- R. W. Lewis, B. Klemm, M. Macchione and R. Eelkema, Chem. Sci., 2022, 13, 4533–4544 RSC.
- J. M. Kim, T. Y. Heo and S. H. Choi, Macromolecules, 2020, 53, 9234–9243 CrossRef CAS.
- B. W. Fan, K. Zhang, Q. Liu and R. Eelkema, ACS Macro Lett., 2020, 9, 776–780 CrossRef CAS PubMed.
- C. Yang, J. Dawulieti, K. B. Zhang, C. X. Cheng, Y. W. Zhao, H. Z. Hu, M. Li, M. Zhang, L. Chen, K. W. Leong and D. Shao, Adv. Funct. Mater., 2022, 32, 2111698 CrossRef CAS.
- S. D. Li, J. Q. Yu, M. Zhang, Z. Y. Ma, N. Chen, X. P. Li, J. M. Ban, J. N. Xie, Z. N. Chen, J. S. Ma, C. X. Tian, Y. S. Qin, J. Wang, W. C. Gao, L. X. Long, J. Zhao, X. Hou and X. B. Yuan, Adv. Funct. Mater., 2022, 32, 2110617 CrossRef CAS.
- Y. T. Sun, A. L. Wollenberg, T. M. O'Shea, Y. X. Cui, Z. H. Zhou, M. V. Sofroniew and T. J. Deming, J. Am. Chem. Soc., 2017, 139, 15114–15121 CrossRef CAS PubMed.
- F. Brotzel, Y. C. Chu and H. Mayr, J. Org. Chem., 2007, 72, 3679–3688 CrossRef CAS PubMed.
- F. Brotzel and H. Mayr, Org. Biomol. Chem., 2007, 5, 3814–3820 RSC.
- M. Lemmers, I. K. Voets, M. A. C. Stuart and J. van der Gucht, Soft Matter, 2011, 7, 1378–1389 RSC.
- D. V. Krogstad, N. A. Lynd, D. Miyajima, J. Gopez, C. J. Hawker, E. J. Kramer and M. V. Tirrell, Macromolecules, 2014, 47, 8026–8032 CrossRef CAS.
- Z. Y. Huang, G. B. Shao and L. Q. Li, Prog. Mater. Sci., 2023, 131, 101020 CrossRef CAS.
- G. Scionti, M. Moral, M. Toledano, R. Osorio, J. D. G. Duran, M. Alaminos, A. Campos and M. T. Lopez-Lopez, J. Biomed. Mater. Res., Part A, 2014, 102, 2573–2582 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2sc06935k |
| This journal is © The Royal Society of Chemistry 2023 |