
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFree-atom-like d states beyond the dilute limit of single-atom alloys†
Andrew S.
Rosen
 abc,
Sudarshan
Vijay
ac and
Kristin A.
Persson
*ad
abc,
Sudarshan
Vijay
ac and
Kristin A.
Persson
*ad
aDepartment of Materials Science and Engineering, University of California, Berkeley, Berkeley, California 94720, USA
bMiller Institute for Basic Research in Science, University of California, Berkeley, Berkeley, California 94720, USA
cMaterials Science Division, Lawrence Berkeley National Laboratory, Berkeley, California 94720, USA
dMolecular Foundry, Lawrence Berkeley National Laboratory, Berkeley, California 94720, USA. E-mail: kristinpersson@berkeley.edu
First published on 19th January 2023
Abstract
Through a data-mining and high-throughput density functional theory approach, we identify a diverse range of metallic compounds that are predicted to have transition metals with “free-atom-like” d states that are highly localized in terms of their energetic distribution. Design principles that favor the formation of localized d states are uncovered, among which we note that site isolation is often necessary but that the dilute limit, as in most single-atom alloys, is not a pre-requisite. Additionally, the majority of localized d state transition metals identified from the computational screening study exhibit partial anionic character due to charge transfer from neighboring metal species. Using CO as a representative probe molecule, we show that localized d states for Rh, Ir, Pd, and Pt tend to reduce the binding strength of CO compared to their pure elemental analogues, whereas this does not occur as consistently for the Cu binding sites. These trends are rationalized through the d-band model, which suggests that the significantly reduced d-band width results in an increased orthogonalization energy penalty upon CO chemisorption. With the multitude of inorganic solids that are predicted to have highly localized d states, the results of the screening study are likely to result in new avenues for heterogeneous catalyst design from an electronic structure perspective.
Catalysts play a central role in the efficient production of chemicals, fuels, and materials across virtually all sectors of industry. In addition to improving existing chemical processes, the discovery of novel catalysts is crucial for addressing major societal challenges involving clean energy and sustainability. With the function of a given catalyst intricately tied to its atomic-scale structure, there has been a surge of interest in single-site heterogeneous catalysts, which are solid-state materials with well-defined and uniform active sites.1,2 Especially in tandem with theoretical methods and recent advances in data science,3–5 studies of such catalysts have made it easier to discover structure–reactivity relationships and ultimately design new materials tailored for a given chemical transformation of interest.
Single-atom alloys (SAAs)—solid-state materials composed of a catalytically active “guest” metal embedded on the surface of a typically less-reactive “host” metal—are one particularly interesting class of single-site heterogeneous catalyst that has received significant attention in recent years.6–12 The density of active sites is purposefully kept low in most SAAs to create atomically dispersed binding sites and to minimize the likelihood of surface aggregation, with monolayer coverages of a few hundredths being common.13 In addition to their ability to break scaling relationships by decoupling the binding sites of reactants and intermediates,6,14–16 some SAAs are known to exhibit unique electronic structure properties that can greatly influence their catalytic activity.7,11,17 As noted by Thirumalai and Kitchin,18 certain combinations of guest and host metals in SAAs (particularly those with Ag or Au as the host) can lead to significant contraction of the guest atom's d-band that is reminiscent of a free metal atom or that of a molecular metal complex. This finding was later supported experimentally in work by Greiner et al. who used in situ photoemission spectroscopy to show that a AgCu SAA with 0.3 at% Cu exhibits Cu d states that are “free-atom-like” in character.19 Recently, several studies have suggested that these free-atom-like d states can drastically alter the reactivity of the guest metal sites in SAAs,18–22 such as by reducing the activation energy for methanol reforming,19 enabling the chemisorption of methane,21 and tailoring selectivity during the activation of crotonaldehyde.22
To date, “free-atom-like” d states have primarily been discussed within the context of SAAs, but this begs the question: is the dilute limit of alloying a pre-requisite for d states that are highly localized in terms of their energetic distribution? If not, what other materials can exhibit this unique electronic structure behavior, and how might it effect their reactivity? To address these questions, we turn to high-throughput density functional theory (DFT) calculations and data-driven approaches built upon the Materials Project: an open-access database of predicted physicochemical properties for approximately 150![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 materials.23,24 Of particular relevance to the present work, the Materials Project contains DFT-optimized structures and corresponding electronic density of states (DOS) for most known inorganic compounds as well as many hypothetical structures.
000 materials.23,24 Of particular relevance to the present work, the Materials Project contains DFT-optimized structures and corresponding electronic density of states (DOS) for most known inorganic compounds as well as many hypothetical structures.
Inspired by the AgCu SAA known to have localized d states,19 we first carried out a simple query of the Ag–Cu compounds on the Materials Project to test whether the dilute limit of alloying is necessary for this electronic structure phenomenon. As shown in Fig. 1a, we found that the hypothetical bulk compound Ag3Cu (mp-984351) exhibits an extremely narrow Cu d-band that is clearly distinct from the comparatively broad d-band of elemental Cu (mp-30) as well as most other Cu-containing metals, such as Pd3Cu (mp-1184119). In fact, the Cu d-projected DOS of bulk Ag3Cu is nearly the same as that reported for the Ag255Cu SAA19 despite a significant difference in Cu density and incorporation of the Cu sites throughout the crystal lattice rather than just on the surface.
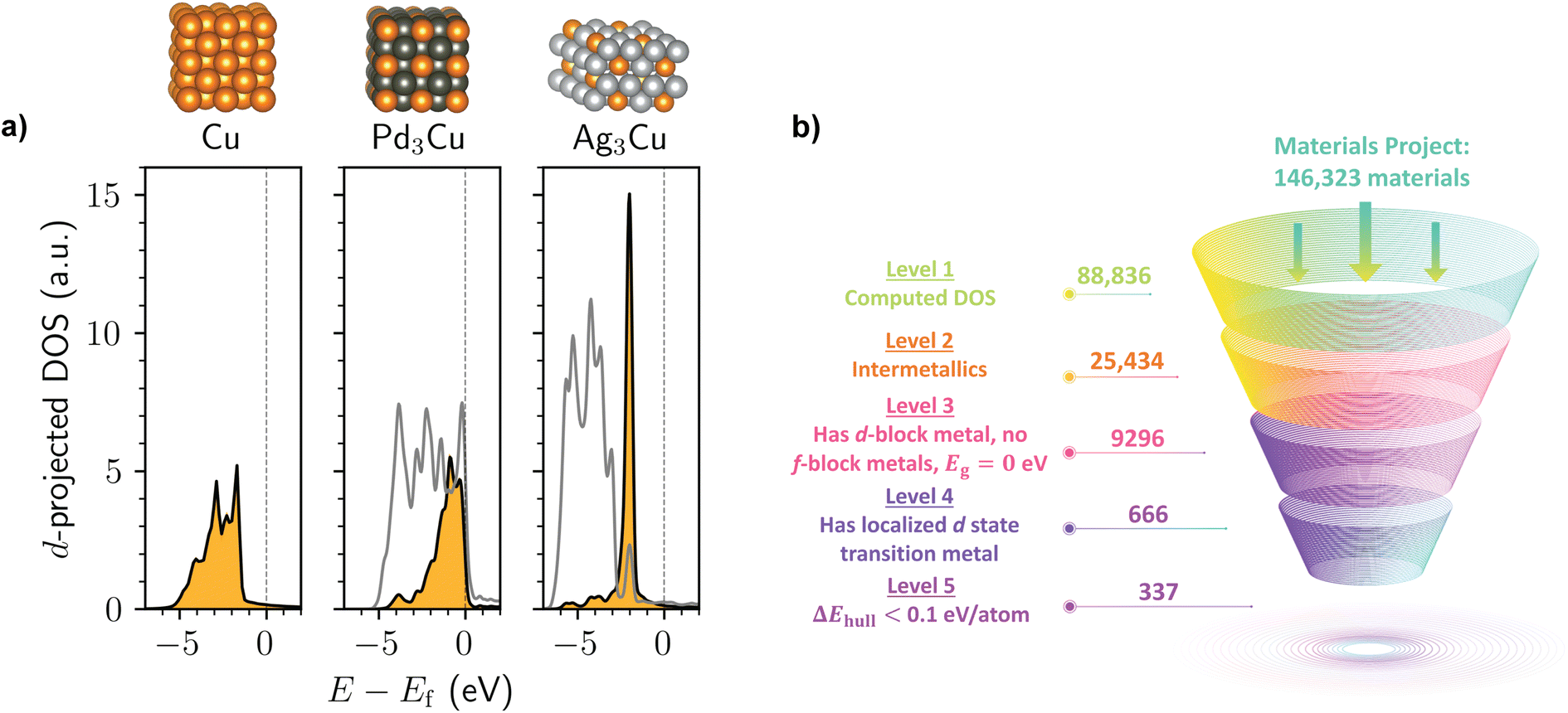 | ||
| Fig. 1 (a) Calculated d-projected DOS for bulk structures of Cu (mp-30), Pd3Cu (mp-1184119), and Ag3Cu (mp-984351), as obtained from the Materials Project. The orange filled DOS is the Cu d-band, and the gray unfilled DOS is the Pd or Ag d-band. The DOS values are normalized by the number of Cu atoms in the unit cell for ease-of-comparison. Energies, E, are shown with respect to the Fermi level, Ef. Atom color code: Cu (orange), Pd (dark gray), Ag (silver). (b) Summary of the computational screening process to identify intermetallics with localized d state transition metals on the Materials Project. | ||
With the Ag3Cu proof-of-concept suggesting that localized d states are neither restricted to the highly dilute limit of SAAs nor confined to surface sites, we sought to explore the broader chemical space of intermetallic compounds and identify more general structure–property–reactivity relationships. Intermetallics, which contain well-defined and uniform local coordination environments, have received significant attention in recent years for a variety of catalytic reactions and exhibit substantial chemical diversity.25–27 As demonstrated by Wegener et al.,28 intermetallics can also serve as appealing alternatives to SAAs, as the density of active sites can be made significantly higher, and their structural uniformity makes it possible to more precisely tailor their geometric and electronic properties.29
In a more data-driven mode, we screened the Materials Project for both known and hypothetical intermetallic compounds with transition metals that exhibit localized d states based on their computed DOS (defined here as having a Gaussian fit with an R2 ≥ 0.9 and standard deviation less than 0.3 eV; refer to the ESI† for additional details). We note that some of the intermetallics on the Materials Project are better classified as alloys with the crystallographic disorder removed. Additionally, some of the metals in the investigated intermetallic compounds may be immiscible in the bulk state, as can be expected for the Ag3Cu proof-of-concept.30 Nonetheless, our primary goal is to investigate the chemical space of metallic compounds beyond SAAs in the dilute limit to better understand the nature of localized d states.
As shown in Fig. 1b, several hundred intermetallics on the Materials Project are predicted to exhibit localized d-state transition metals. This suggests that—although the phenomenon is relatively uncommon amongst all intermetallic structures—it is certainly not restricted to Ag3Cu. To filter out structures that are likely to be highly unstable,31 we analyzed the thermodynamic phase diagrams for each material and selected the subset of intermetallics that are within 0.1 eV per atom of the convex hull. This process resulted in over 300 unique intermetallics with localized d states from the starting dataset of ∼25000 intermetallic compounds. We note that if a much stricter 0.01 eV per atom tolerance for the energy above hull were used as suggested in prior work,32 211 structures still remain at the end of the screening process.
By investigating the structures obtained from the screening process, several clear principles emerge for the design of intermetallics with localized d states. As shown in Fig. 2a, the median interatomic distance between transition metals with localized d states in the Materials Project dataset is only 4.6 Å. As a result, having metal species in the dilute limit—as is commonly the case with SAAs where this phenomenon has previously been reported—is not a pre-requisite. Nonetheless, some degree of site isolation is found for nearly all the examples identified in this work, even if the relevant metals are separated by only two coordination spheres. The importance of site isolation is expected based on prior work showing that two- and three-atom ensembles of dopant metals in SAAs can introduce hybridization and splitting of the dopant metal's d-band.33
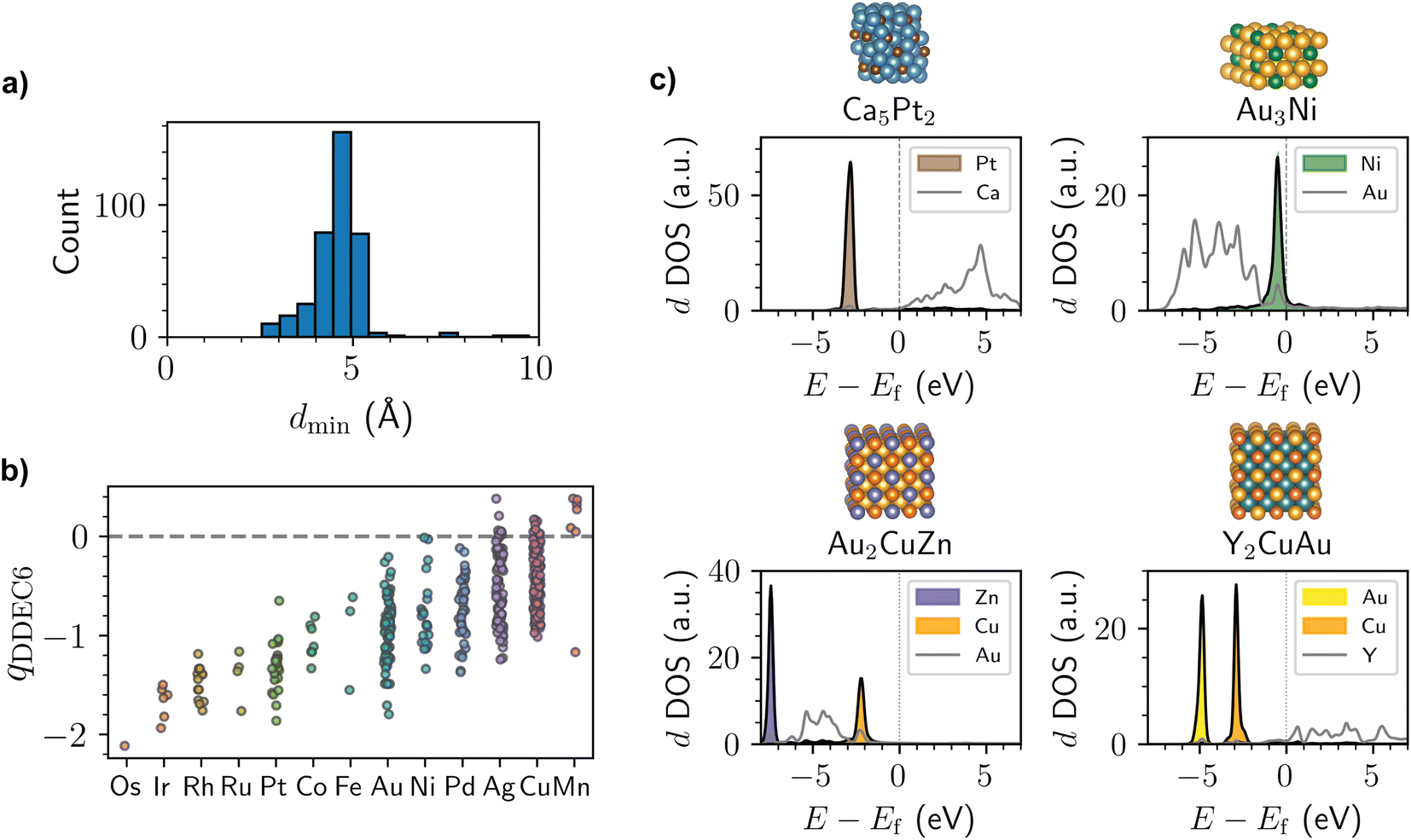 | ||
| Fig. 2 (a) Histogram of the minimum distance, dmin, between transition metals with localized d states for the intermetallics identified in the screening process. (b) Distribution of DDEC6 (ref. 34–36) partial atomic charges, qDDEC6, for the transition metals with localized d states identified in the screening process. Partial charges are averaged across transition metals of the same identity within a given structure. The points are jittered horizontally for ease-of-visualization. (c) Structure and calculated d-projected DOS for bulk Ca5Pt2 (mp-1103570), bulk Au3Ni (mp-976806), bulk Au2CuZn (mp-864623), and bulk Y2CuAu (mp-1187723). Energies, E, are shown with respect to the Fermi level, Ef. Atom color code: Ca (turquoise), Pt (brown), Au (yellow), Ni (green), Cu (orange), Zn (light purple), Y (teal). | ||
The majority of intermetallics with localized d states also have non-negligible partial atomic charges (Fig. 2b), although we note that the degree of charge transfer is rather sensitive to the charge partitioning scheme used (Fig. S2 and S3†). The presence of partial anionic character for many of the localized d states is reminiscent of prior experiments on the AgCu SAA, which was shown to have Cu sites with anionic character based on X-ray photoelectron spectroscopy and near edge X-ray absorption fine structure measurements.19 While not necessarily associated with localized d states, charge transfer has recently been reported for several other SAAs in the literature as well.37 With this in mind, intermetallics and alloys with localized d states may be worthwhile materials to consider if catalysts with anionic metals are desired. For instance, prior work has shown that single-atom catalysts with anionic metal centers are promising for several electrocatalytic reduction reactions.38
A few representative intermetallics identified from the screening process are shown in Fig. 2c. Most of the elements with localized d states are late transition metals (Fig. S4†). In addition, the elements with localized d states are often surrounded by group 1–3 metals (Fig. S5 and S6†), as is the case for Na3Pd (mp-1186067), Ca5Pt2 (mp-1103570), and Sr7Ag3 (mp-1106092) with localized Pd, Pt, and Ag d states, respectively. That said, there are also numerous examples of localized d state metals surrounded by late transition metals, as is the case for Ag3Cu (mp-984351) and Au3Ni (mp-976806) with localized Cu and Ni d states, respectively. Notably, the presence of localized d states is not restricted to binary intermetallics, and many examples of ternary compounds exist with this electronic structure behavior. This includes ternary compounds where two different metal elements have localized d states, such as Y2CuAu (mp-1187723), Au2CuZn (mp-864623), and Pd2CuSn (mp-1184057) with localized Cu/Au, Cu/Zn, and Cu/Sn d states, respectively. Given the substantial diversity of intermetallic compounds, the presence of localized d states in these materials opens up a wider range of compositions that can be considered as compared to SAAs where the dopant metal is in the dilute limit. Additionally, the presence of two different localized d states in the ternary compounds may make it possible to design multi-functional catalysts that can be tailored for specific reactions of interest, similar to recent work on dual-atom catalysts.39
Drawing from crystal structures that appeared several times in the screening process, we took cubic Na3X (Fm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m space group), orthorhombic Y3X (Pnma space group), hexagonal Ag3X (P63/mmc space group), and hexagonal Au3X (P63/mmc space group) and varied the identity of metal X to be any of the group 7–11 metals to identify periodic trends. The d-projected DOS are shown for the 3d metals in Fig. 3, with the 4d and 5d metals shown in Fig. S7–S10.† Although not all compositions yield highly localized d states, there exists a general trend that elements with higher group numbers tend to decrease the width of the d-band while shifting the d-band center to lower energies with respect to the Fermi level (Fig. 3 and S7–S10†), similar to that recently reported for SAAs.22 We note that the d-band trends are different for intermetallics with magnetic d states, such as Ag3X (X = Mn, Fe, Co), which have asymmetric spin-channels. Carrying out a spin-unpolarized calculation for these compounds restores the periodic trend of decreasing d-band center with increasing group number (Fig. S11†). The width of the minority atom's d-band also tends to be narrower for the 3d metals than the 4d and 5d metals (Fig. S7–S10†), likely due to the larger spatial extent of the valence orbitals in the latter, which leads to increased orbital overlap and broadens the d-band.
m space group), orthorhombic Y3X (Pnma space group), hexagonal Ag3X (P63/mmc space group), and hexagonal Au3X (P63/mmc space group) and varied the identity of metal X to be any of the group 7–11 metals to identify periodic trends. The d-projected DOS are shown for the 3d metals in Fig. 3, with the 4d and 5d metals shown in Fig. S7–S10.† Although not all compositions yield highly localized d states, there exists a general trend that elements with higher group numbers tend to decrease the width of the d-band while shifting the d-band center to lower energies with respect to the Fermi level (Fig. 3 and S7–S10†), similar to that recently reported for SAAs.22 We note that the d-band trends are different for intermetallics with magnetic d states, such as Ag3X (X = Mn, Fe, Co), which have asymmetric spin-channels. Carrying out a spin-unpolarized calculation for these compounds restores the periodic trend of decreasing d-band center with increasing group number (Fig. S11†). The width of the minority atom's d-band also tends to be narrower for the 3d metals than the 4d and 5d metals (Fig. S7–S10†), likely due to the larger spatial extent of the valence orbitals in the latter, which leads to increased orbital overlap and broadens the d-band.
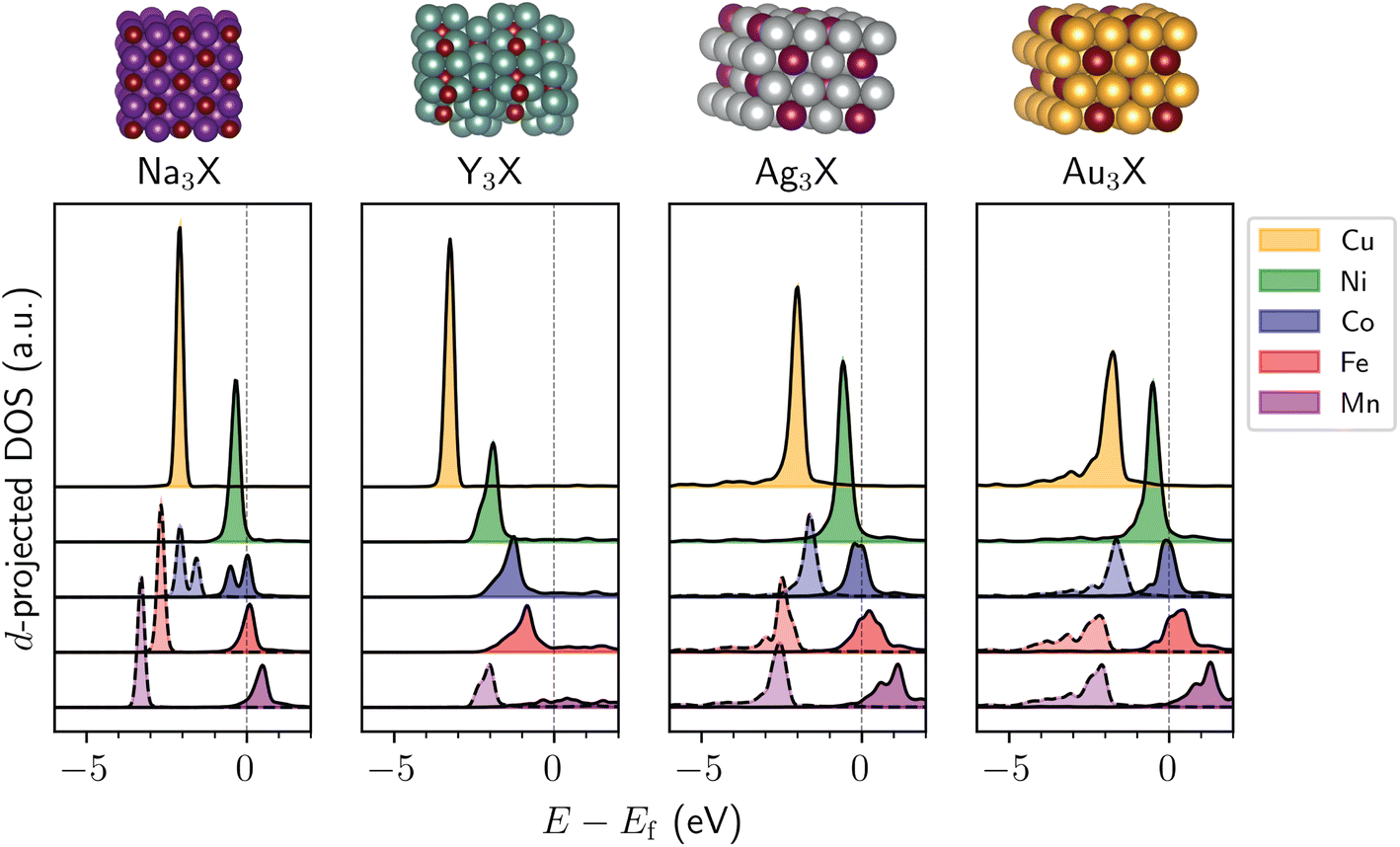 | ||
| Fig. 3 d-Projected DOS for atom X (X = Mn, Fe, Co, Ni, Cu) in tetragonal Na3X, orthorhombic Y3X, hexagonal Ag3X, and hexagonal Au3X. In the case of magnetic structures, the dashed and solid lines distinguish between the spin-up and spin-down channels. Energies, E, are shown with respect to the Fermi level, Ef. Atom color code: X (maroon), Na (dark purple), Y (turquoise), Ag (gray), Au (yellow). | ||
To investigate the effects of localized d states on adsorption trends, we used a high-throughput workflow40 to explore CO adsorption on various surfaces of intermetallics identified from the Materials Project screening process, focusing on the following metal binding sites: Rh, Ir, Pd, Pt, and Cu. These metals were selected because they are commonly invoked as the active site in various SAAs,7 and CO was chosen as it is a common probe molecule and reaction intermediate. Before discussing the adsorption trends, we first note that the localized d states found in the bulk are often still present on the surface. Consistent with prior work,41,42 we find that the d bands tend to be narrower for the surface sites than their bulk counterparts due to the reduced coordination numbers of the former (Fig. S12a†); nonetheless, there exist a few outliers where this is not the case, such as for the Cu binding sites of Pd2CuM (M = Al, Zn, Ga, In), which were subsequently excluded from the CO adsorption analysis. Additionally, the localized d-band centers are generally shifted towards higher energies for the surface sites compared to their bulk analogues (Fig. S12b†).
Fig. 4 shows the distribution of DFT-computed adsorption energies for CO binding atop of the metal sites with localized d states as compared to the same process on elemental reference surfaces. Based on the results in Fig. 4, CO is predicted to bind weaker on the surface of most intermetallic compounds with localized Rh, Ir, Pd, or Pt d states compared to their elemental counterparts (this trend does not hold for Cu, which we will explain below). The reduced binding strength of CO for many of the materials studied here is particularly beneficial for catalyst design, as it suggests that CO poisoning can likely be substantially reduced for many intermetallics that have localized d states. A reduction in CO poisoning has also been known to occur for several SAAs (and Pt–Fe intermetallics28) when compared to elemental reference structures,43,44 although the aforementioned materials were not studied within the context of localized d states. Previous studies on metallic surfaces have suggested that narrowing of the d-band—albeit often not to the degree shown here—is typically associated with stronger CO chemisorption.45,46 However, narrowing of the d-band due to perturbative effects like strain and reduced coordination numbers is often associated with a simultaneous increase in the d-band center,46 which does not generally hold across the varied intermetallic compounds studied in this work (Fig. S13†) since there is no requirement to maintain a constant band filling. As a brief aside, despite the less exothermic adsorption energy of CO on the Rh, Ir, Pd, and Pt atoms with localized d states compared to their elemental analogues, the bound CO is also more significantly reduced in many cases (Fig. S14†)—likely a consequence of the electron-rich nature for many of the metal binding sites.
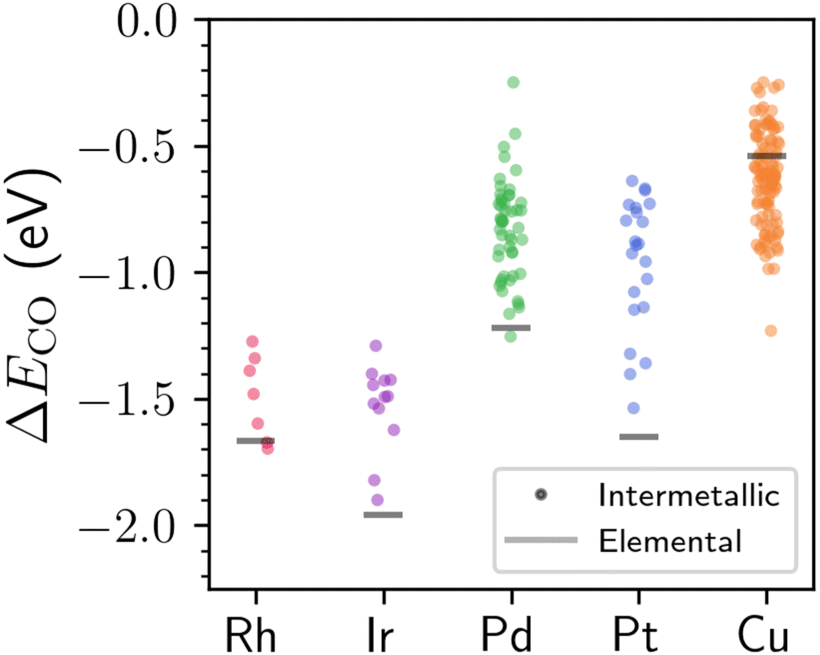 | ||
| Fig. 4 Adsorption energies, ΔECO, for CO on the atop site of a given metal with localized d states. Each point represents the CO adsorption process on a material with a different composition and/or surface facet. The points are jittered horizontally for ease of visualization. The horizontal lines represent the average CO adsorption energy at the atop site for the (100), (110), and (111) surfaces of the corresponding ground-state, elemental reference structure. | ||
Initially, we considered whether the position of the d-band center could explain the difference in the DFT-computed CO adsorption energies, as might be expected from the Hammer–Nørskov model47,48 and prior work.41,49–51 However, as shown in Fig. S15,† the d-band center only loosely captures the CO adsorption energy trends across the intermetallics with localized d states, especially when comparing binding sites of the same metal element. The same is true for various descriptors for the upper edge of the d-band,52,53 as shown in Fig. S16.† The relatively poor correlation is likely due to the wide range of chemical compositions considered in the present study, which differs from the perturbative effects that are typically invoked when using the d-band center as a metric for reactivity trends.17
To better understand variations in the CO adsorption energy between the metal binding sites with localized d states and their elemental analogues, we parameterized an effective chemisorption model introduced by Vijay et al.,54 which is built upon the d-band model55 with a modified term from the Newns–Anderson model of adsorption.56,57 As discussed further in the ESI† (with the model parameters and model fit in Table S1 and Fig. S1,† respectively), the modified Newns–Anderson model makes it possible to split the chemisorption energy, ΔEchemi(εd,wd), into a sum of two components given by
| ΔEchemi(εd,wd) = Ehyb(εd,wd) + Eortho(εd,wd) | (1) |
By varying the d-band width (wd) and d-band center (εd) independently and continuously in the effective chemisorption model, it becomes possible to determine their general effect on the CO adsorption energy. As shown in Fig. 5, increasing the d-band center for a fixed d-band width (i.e. moving up the y-axis for a given x-value) decreases the orthogonalization energy penalty (Eortho) and increases the hybridization energy (Ehyb), which synergistically strengthen the binding of CO. In contrast, decreasing the d-band width while keeping the d-band center fixed (i.e. moving left on the x-axis for a given y-value) increases Eortho while Ehyb remains relatively similar, thereby weakening the adsorption of CO.
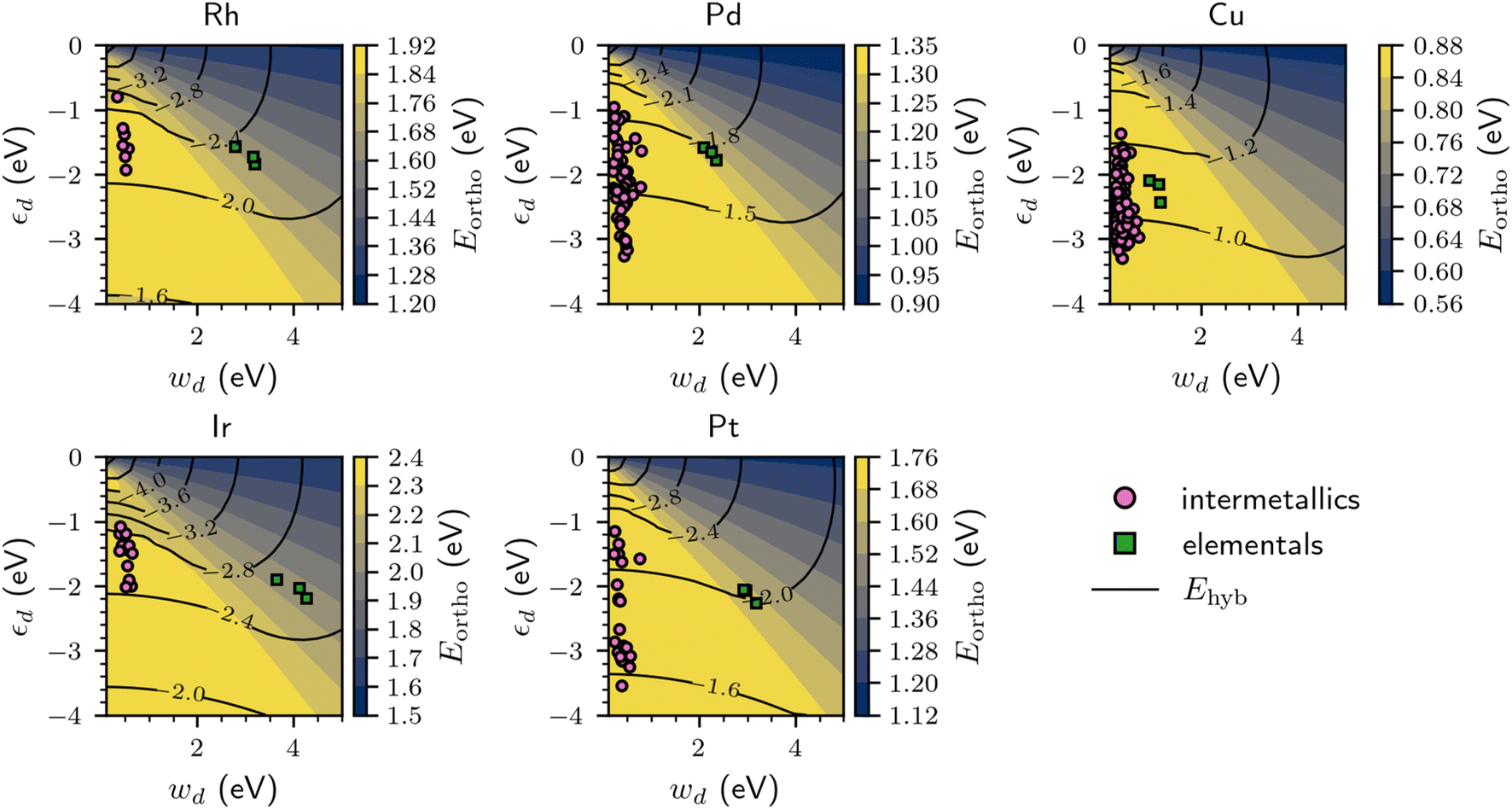 | ||
| Fig. 5 Orthogonalization energy penalty, Eortho (colormap), and hybridization energy, Ehyb (contour lines), when freely varying the d-band center, εd, and d-band width, wd, for different materials considered in this study. The points are from semi-elliptical fits to the d-projected DOS of the metal binding sites on the surface of intermetallics (pink circles) and corresponding transition metals (green squares). | ||
The variation of Ehyb and Eortho with changing εd and wd can be used to rationalize the trends in the DFT-computed CO adsorption energies previously shown in Fig. 4. By the very nature of the computational screening process, the intermetallics with Rh, Ir, Pd, and Pt binding sites have a significantly narrower d-band compared to their corresponding elemental surfaces. The decreased wd for the intermetallics with localized d states often results in a significant energetic penalty due to an increased Eortho, which makes the overall CO chemisorption process less exothermic. We propose that the increased orthogonalization energy penalty is an essential component for explaining why localized d states tend to weaken the binding of CO for most of the intermetallics with Rh, Ir, Pd, or Pt binding sites compared to their elemental analogues. While necessary to consider, the orthogonalization energy penalty is not always the primary distinguishing factor; for instance, many of the intermetallics with localized Pd d states have a reduced d-band center compared to elemental Pd, which decreases Ehyb and further weakens the CO adsorption process. We note that designing a catalyst with localized Rh, Ir, Pd, and Pt d states with stronger CO binding than their elemental counterparts is possible if the d-band center is sufficiently close to the Fermi level, although in practice this may be challenging to achieve based on the dataset considered in this work.
As previously shown in Fig. 4, this trend of weakened CO adsorption for the intermetallics with localized d states does not hold in general for the Cu binding sites. This can also be explained with the chemisorption model in Fig. 5, as the difference in the d-band width between elemental Cu and the intermetallics with localized Cu d states is small compared that of Rh, Ir, Pd, and Pt, which results in relatively similar Eortho values for the Cu-containing intermetallics and the elemental Cu surfaces. As such, the main factor that dictates that difference in CO adsorption energies for the Cu elementals and intermetallics with localized Cu d states is primarily Ehyb (via a change in the d-band center), which varies substantially across the dataset considered in this work.
To summarize, we have shown that localized (or “free-atom-like”) d states are not restricted to the dilute limit typically associated with single-atom alloys, although some degree of site isolation is generally present. A diverse range of metallic compounds can exhibit transition metals with localized d states based on our screening of the Materials Project, including ternary compounds that have two different localized d states in the crystal structure. The majority of inorganic solids with localized d states also exhibit charge transfer between the metal centers, similar to what was reported experimentally for the AgCu SAA.19 With the large dataset of localized d states identified in this work, we identified several structure–reactivity relationships using CO as the probe molecule. Based on the high-throughput DFT calculations, we predict that localized Rh, Ir, Pd, and Pt d states tend to result in weaker CO adsorption than their elemental analogues, whereas this is not generally true for Cu. We attribute the weaker CO adsorption in the former to an increased orthogonalization energy penalty with narrower d-band widths, which is less pronounced when comparing the Cu-containing compounds. The weaker CO adsorption with narrowed d-band widths mimics the resistance to CO poisoning observed for SAAs43,44 without needing to dilute the density of active sites on the surface. Collectively, with the multitude of inorganic materials that are predicted to have highly localized d states and the direct influence that their electronic structure has on chemical reactivity, new avenues in heterogeneous catalyst design60,61 are likely to become accessible.60,61
Data availability
The data on the Materials Project can be found at https://materialsproject.org. A summary of the tabulated VASP data is stored as a JSON file at the following DOI: http://10.5281/zenodo.7098518. The VASP output files are stored at the following DOI: http://10.17172/NOMAD/2022.11.25-2. The code underlying the effective chemisorption model can be found at the following DOI: http://10.5281/zenodo.7358098.Author contributions
A. S. R. designed the project, carried out and analyzed the DFT calculations, and wrote the original draft of the manuscript. S. V. constructed the effective chemisorption model and interpreted the results. K. A. P. supervised the project. All authors contributed to review and editing of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
A. S. R. acknowledges support via a Miller Research Fellowship from the Miller Institute for Basic Research in Science, University of California, Berkeley. K. A. P. acknowledges support by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences, Materials Sciences and Engineering Division under Contract No. DE-AC02-05-CH11231 (Materials Project program KC23MP). The authors acknowledge computing support from the National Energy Research Scientific Computing Center (NERSC), a U.S. Department of Energy Office of Science User Facility located at Lawrence Berkeley National Laboratory, operated under Contract No. DE-AC02-05CH11231 using NERSC award ERCAP0020225.References
- J. M. Thomas, R. Raja and D. W. Lewis, Single-Site Heterogeneous Catalysts, Angew. Chem., Int. Ed., 2005, 44(40), 6456–6482, DOI:10.1002/anie.200462473
.
- J. D. A. Pelletier and J.-M. Basset, Catalysis by Design: Well-Defined Single-Site Heterogeneous Catalysts, Acc. Chem. Res., 2016, 49(4), 664–677, DOI:10.1021/acs.accounts.5b00518
- J. K. Nørskov, T. Bligaard, J. Rossmeisl and C. H. Christensen, Towards the Computational Design of Solid Catalysts, Nat. Chem., 2009, 1(1), 37–46, DOI:10.1038/nchem.121
- B. W. J. Chen, L. Xu and M. Mavrikakis, Computational Methods in Heterogeneous Catalysis, Chem. Rev., 2021, 121(2), 1007–1048, DOI:10.1021/acs.chemrev.0c01060
- J. A. Esterhuizen, B. R. Goldsmith and S. Linic, Interpretable Machine Learning for Knowledge Generation in Heterogeneous Catalysis, Nat. Catal., 2022, 5(3), 175–184, DOI:10.1038/s41929-022-00744-z
- M. T. Darby, M. Stamatakis, A. Michaelides, E. Sykes and H. Charles, Lonely Atoms with Special Gifts: Breaking Linear Scaling Relationships in Heterogeneous Catalysis with Single-Atom Alloys, J. Phys. Chem. Lett., 2018, 9(18), 5636–5646, DOI:10.1021/acs.jpclett.8b01888
- T. Zhang, A. G. Walsh, J. Yu and P. Zhang, Single-Atom Alloy Catalysts: Structural Analysis, Electronic Properties and Catalytic Activities, Chem. Soc. Rev., 2020, 50(1), 569–588, 10.1039/d0cs00844c
- E. C. H. Sykes and P. Christopher, Recent Advances in Single-Atom Catalysts and Single-Atom Alloys: Opportunities for Exploring the Uncharted Phase Space in-Between, Curr. Opin. Chem. Eng., 2020, 29, 67–73, DOI:10.1016/j.coche.2020.06.004
- R. T. Hannagan, G. Giannakakis, M. Flytzani-Stephanopoulos and E. C. H. Sykes, Single-Atom Alloy Catalysis, Chem. Rev., 2020, 120(21), 12044–12088, DOI:10.1021/acs.chemrev.0c00078
- R. Réocreux and M. Stamatakis, One Decade of Computational Studies on Single-Atom Alloys: Is In Silico Design within Reach?, Acc. Chem. Res., 2022, 55(1), 87–97, DOI:10.1021/acs.accounts.1c00611
- Z. Chen and P. Zhang, Electronic Structure of Single-Atom Alloys and Its Impact on The Catalytic Activities, ACS Omega, 2022, 7(2), 1585–1594, DOI:10.1021/acsomega.1c06067
- J. D. Lee, J. B. Miller, A. V. Shneidman, L. Sun, J. F. Weaver, J. Aizenberg, J. Biener, J. A. Boscoboinik, A. C. Foucher, A. I. Frenkel, J. E. S. van der Hoeven, B. Kozinsky, N. Marcella, M. M. Montemore, H. T. Ngan, C. R. O'Connor, C. J. Owen, D. J. Stacchiola, E. A. Stach, R. J. Madix, P. Sautet and C. M. Friend, Dilute Alloys Based on Au, Ag, or Cu for Efficient Catalysis: From Synthesis to Active Sites, Chem. Rev., 2022, 122(9), 8758–8808, DOI:10.1021/acs.chemrev.1c00967
- G. Giannakakis, M. Flytzani-Stephanopoulos and E. C. H. Sykes, Single-Atom Alloys as a Reductionist Approach to the Rational Design of Heterogeneous Catalysts, Acc. Chem. Res., 2019, 52(1), 237–247, DOI:10.1021/acs.accounts.8b00490
- J. Pérez-Ramírez and N. López, Strategies to Break Linear Scaling Relationships, Nat. Catal., 2019, 2(11), 971–976, DOI:10.1038/s41929-019-0376-6
- M. T. Darby, R. Réocreux, E. Sykes, H. Charles, A. Michaelides and M. Stamatakis, Elucidating the Stability and Reactivity of Surface Intermediates on Single-Atom Alloy Catalysts, ACS Catal., 2018, 8(6), 5038–5050, DOI:10.1021/acscatal.8b00881
- C. F. Nwaokorie and M. M. Montemore, Alloy Catalyst Design beyond the Volcano Plot by Breaking Scaling Relations, J. Phys. Chem. C, 2022, 126(8), 3993–3999, DOI:10.1021/acs.jpcc.1c10484
-
G. O. Kayode, Z. Zhang and M. M. Montemore, Linking Electronic Structure to Adsorption Energies: Metal Surfaces and Single-Atom Catalyst, in Catalysis, ed. J. Spivey, Y.-F. Han and D. Shekhawat, 2022, vol. 34, pp. 17–55 Search PubMed
- H. Thirumalai and J. R. Kitchin, Investigating the Reactivity of Single Atom Alloys Using Density Functional Theory, Top. Catal., 2018, 61(5–6), 462–474, DOI:10.1007/s11244-018-0899-0
- M. T. Greiner, T. E. Jones, S. Beeg, L. Zwiener, M. Scherzer, F. Girgsdies, S. Piccinin, M. Armbrüster, A. Knop-Gericke and R. Schlögl, Free-Atom-like d States in Single-Atom Alloy Catalysts, Nat. Chem., 2018, 10(10), 1008–1015, DOI:10.1038/s41557-018-0125-5
- N. Zhang, F. Chen and L. Guo, Catalytic Activity of Palladium-Doped Silver Dilute Nanoalloys for Formate Oxidation from a Theoretical Perspective, Phys. Chem. Chem. Phys., 2019, 21(40), 22598–22610, 10.1039/c9cp04530a
- V. Fung, G. Hu and B. Sumpter, Electronic Band Contraction Induced Low Temperature Methane Activation on Metal Alloys, J. Mater. Chem. A, 2020, 8(12), 6057–6066, 10.1039/d0ta00375a
- T. D. Spivey and A. Holewinski, Selective Interactions between Free-Atom-like d-States in Single-Atom Alloy Catalysts and Near-Frontier Molecular Orbitals, J. Am. Chem. Soc., 2021, 143(31), 11897–11902, DOI:10.1021/jacs.1c04234
- A. Jain, G. Hautier, C. J. Moore, S. P. Ong, C. C. Fischer, T. Mueller, K. A. Persson and G. Ceder, A High-Throughput Infrastructure for Density Functional Theory Calculations, Comput. Mater. Sci., 2011, 50(8), 2295–2310, DOI:10.1016/j.commatsci.2011.02.023
- A. Jain, S. P. Ong, G. Hautier, W. Chen, W. D. Richards, S. Dacek, S. Cholia, D. Gunter, D. Skinner, G. Ceder and K. A. Persson, Commentary: The Materials Project: A Materials Genome Approach to Accelerating Materials Innovation, APL Mater., 2013, 1(1), 011002, DOI:10.1063/1.4812323
- M. Armbrüster, R. Schlögl and Y. Grin, Intermetallic Compounds in Heterogeneous Catalysis—a Quickly Developing Field, Sci. Technol. Adv. Mater., 2014, 15(3), 034803, DOI:10.1088/1468-6996/15/3/034803
- M. Krajčí and J. Hafner, Intermetallic Compounds as Selective Heterogeneous Catalysts: Insights from DFT, ChemCatChem, 2015, 8(1), 34–48, DOI:10.1002/cctc.201500733
- Y. Yang and M. Wei, Intermetallic Compound Catalysts: Synthetic Scheme, Structure Characterization and Catalytic Application, J. Mater. Chem. A, 2019, 8(5), 2207–2221, 10.1039/c9ta09448b
- E. C. Wegener, B. C. Bukowski, D. Yang, Z. Wu, A. J. Kropf, W. N. Delgass, J. Greeley, G. Zhang and J. T. Miller, Intermetallic Compounds as an Alternative to Single-Atom Alloy Catalysts: Geometric and Electronic Structures from Advanced X-Ray Spectroscopies and Computational Studies, ChemCatChem, 2020, 12(5), 1325–1333, DOI:10.1002/cctc.201901869
- A. P. Tsai, S. Kameoka, K. Nozawa, M. Shimoda and Y. Ishii, Intermetallic: A Pseudoelement for Catalysis, Acc. Chem. Res., 2017, 50(12), 2879–2885, DOI:10.1021/acs.accounts.7b00476
- V. Elofsson, G. A. Almyras, B. Lü, R. D. Boyd and K. Sarakinos, Atomic Arrangement in Immiscible Ag–Cu Alloys Synthesized Far-from-Equilibrium, Acta Mater., 2016, 110, 114–121, DOI:10.1016/j.actamat.2016.03.023
- C. J. Bartel, Review of Computational Approaches to Predict the Thermodynamic Stability of Inorganic Solids, J. Mater. Sci., 2022, 1–24, DOI:10.1007/s10853-022-06915-4
- S. Curtarolo, D. Morgan and G. Ceder, Accuracy of Ab Initio Methods in Predicting the Crystal Structures of Metals:
A Review of 80 Binary Alloys, CALPHAD, 2005, 29(3), 163–211, DOI:10.1016/j.calphad.2005.01.002
- A. P. Monasterial, C. A. Hinderks, S. Viriyavaree and M. M. Montemore, When More Is Less: Nonmonotonic Trends in Adsorption on Clusters in Alloy Surfaces, J. Chem. Phys., 2020, 153(11), 111102, DOI:10.1063/5.0022076
- T. A. Manz and N. G. Limas, Introducing DDEC6 Atomic Population Analysis: Part 1. Charge Partitioning Theory and Methodology, RSC Adv., 2016, 6(53), 47771–47801, 10.1039/c6ra04656h
- N. G. Limas and T. A. Manz, Introducing DDEC6 Atomic Population Analysis: Part 2. Computed Results for a Wide Range of Periodic and Nonperiodic Materials, RSC Adv., 2016, 6(51), 45727–45747, 10.1039/c6ra05507a
- N. G. Limas and T. A. Manz, Introducing DDEC6 Atomic Population Analysis: Part 4. Efficient Parallel Computation of Net Atomic Charges, Atomic Spin Moments, Bond Orders, and More, RSC Adv., 2018, 8(5), 2678–2707, 10.1039/c7ra11829e
- R. Réocreux, E. C. H. Sykes, A. Michaelides and M. Stamatakis, Stick or Spill? Scaling Relationships for the Binding Energies of Adsorbates on Single-Atom Alloy Catalysts, J. Phys. Chem. Lett., 2022, 13, 7314–7319, DOI:10.1021/acs.jpclett.2c01519
- J. Gu, Y. Zhao, S. Lin, J. Huang, C. R. Cabrera, B. G. Sumpter and Z. Chen, Single-Atom Catalysts with Anionic Metal Centers: Promising Electrocatalysts for the Oxygen Reduction Reaction and Beyond, J. Energy Chem., 2021, 63, 285–293, DOI:10.1016/j.jechem.2021.08.004
- W. Zhang, Y. Chao, W. Zhang, J. Zhou, F. Lv, K. Wang, F. Lin, H. Luo, J. Li, M. Tong, E. Wang and S. Guo, Emerging Dual-Atomic-Site Catalysts for Efficient Energy Catalysis, Adv. Mater., 2021, 33(36), 2102576, DOI:10.1002/adma.202102576
- J. H. Montoya and K. A. Persson, A High-Throughput Framework for Determining Adsorption Energies on Solid Surfaces, npj Comput. Mater., 2017, 3(1), 14, DOI:10.1038/s41524-017-0017-z
- B. Hammer, O. H. Nielsen and J. K. Nørskov, Structure Sensitivity in Adsorption: CO Interaction with Stepped and Reconstructed Pt Surfaces, Catal. Lett., 1997, 46(1–2), 31–35, DOI:10.1023/a:1019073208575
- B. Hammer and J. K. Nørskov, Theoretical Surface Science and Catalysis—Calculations and Concepts, Adv. Catal., 2000, 45, 71–129, DOI:10.1016/s0360-0564(02)45013-4
- J. Liu, F. R. Lucci, M. Yang, S. Lee, M. D. Marcinkowski, A. J. Therrien, C. T. Williams, E. C. H. Sykes and M. Flytzani-Stephanopoulos, Tackling CO Poisoning with Single-Atom Alloy Catalysts, J. Am. Chem. Soc., 2016, 138(20), 6396–6399, DOI:10.1021/jacs.6b03339
- M. T. Darby, E. C. H. Sykes, A. Michaelides and M. Stamatakis, Carbon Monoxide Poisoning Resistance and Structural Stability of Single Atom Alloys, Top. Catal., 2018, 61(5), 428–438, DOI:10.1007/s11244-017-0882-1
- A. Ruban, B. Hammer, P. Stoltze, H. L. Skriver and J. K. Nørskov, Surface Electronic Structure and Reactivity of Transition and Noble Metals, J. Mol. Catal. A: Chem., 1997, 115(3), 421–429, DOI:10.1016/s1381-1169(96)00348-2
- J. R. Kitchin, J. K. Nørskov, M. A. Barteau and J. G. Chen, Role of Strain and Ligand Effects in the Modification of the Electronic and Chemical Properties of Bimetallic Surfaces, Phys. Rev. Lett., 2004, 93(15), 156801, DOI:10.1103/physrevlett.93.156801
- B. Hammer and J. K. Nørskov, Electronic Factors Determining the Reactivity of Metal Surfaces, Surf. Sci., 1995, 343(3), 211–220, DOI:10.1016/0039-6028(96)80007-0
- J. K. Nørskov, F. Abild-Pedersen, F. Studt and T. Bligaard, Density Functional Theory in Surface Chemistry and Catalysis, Proc. Natl. Acad. Sci. U. S. A., 2011, 108(3), 937–943, DOI:10.1073/pnas.1006652108
- T. Jiang, D. J. Mowbray, S. Dobrin, H. Falsig, B. Hvolbæk, T. Bligaard and J. K. Nørskov, Trends in CO Oxidation Rates for Metal Nanoparticles and Close-Packed, Stepped, and Kinked Surfaces, J. Phys. Chem. C, 2009, 113(24), 10548–10553, DOI:10.1021/jp811185g
- C. Lu, I. C. Lee, R. I. Masel, A. Wieckowski and C. Rice, Correlations between the Heat of Adsorption and the Position of the Center of the d-band: Differences between Computation and Experiment, J. Phys. Chem., 2002, 106(13), 3084–3091, DOI:10.1021/jp0136359
- H. Abe, H. Yoshikawa, N. Umezawa, Y. Xu, G. Saravanan, G. V. Ramesh, T. Tanabe, R. Kodiyath, S. Ueda, N. Sekido, Y. Yamabe-Mitarai, M. Shimoda, T. Ohno, F. Matsumoto and T. Komatsu, Correlation between the Surface Electronic Structure and CO-Oxidation Activity of Pt Alloys, Phys. Chem. Chem. Phys., 2014, 17(7), 4879–4887, 10.1039/c4cp03406f
- A. Vojvodic, J. K. Nørskov and F. Abild-Pedersen, Electronic Structure Effects in Transition Metal Surface Chemistry, Top. Catal., 2014, 57(1–4), 25–32, DOI:10.1007/s11244-013-0159-2
- H. Xin, A. Vojvodic, J. Voss, J. K. Nørskov and F. Abild-Pedersen, Effects of d-band Shape on the Surface Reactivity of Transition-Metal Alloys, Phys. Rev. B, 2014, 89(11), 115114, DOI:10.1103/physrevb.89.115114
- S. Vijay, G. Kastlunger, K. Chan and J. K. Nørskov, Limits to Scaling Relations between Adsorption Energies?, J. Chem. Phys., 2022, 156(23), 231102, DOI:10.1063/5.0096625
- B. Hammer, Y. Morikawa and J. K. Nørskov, CO Chemisorption at Metal Surfaces and Overlayers, Phys. Rev. Lett., 1996, 76(12), 2141–2144, DOI:10.1103/physrevlett.76.2141
- P. W. Anderson, Localized Magnetic States in Metals, Phys. Rev., 1961, 124(1), 41–53, DOI:10.1103/physrev.124.41
- D. M. Newns, Self-Consistent Model of Hydrogen Chemisorption, Phys. Rev., 1969, 178(3), 1123–1135, DOI:10.1103/physrev.178.1123
- H. Xin and S. Linic, Exceptions to the d-band Model of Chemisorption on Metal Surfaces: The Dominant Role of Repulsion between Adsorbate States and Metal d-States, J. Chem. Phys., 2010, 132(22), 221101, DOI:10.1063/1.3437609
- S. Wang, H. S. Pillai and H. Xin, Bayesian Learning of Chemisorption for Bridging the Complexity of Electronic Descriptors, Nat. Commun., 2020, 11(1), 6132, DOI:10.1038/s41467-020-19524-z
- J. G. Freeze, H. R. Kelly and V. S. Batista, Search for Catalysts by Inverse Design: Artificial Intelligence, Mountain Climbers, and Alchemists, Chem. Rev., 2019, 119(11), 6595–6612, DOI:10.1021/acs.chemrev.8b00759
- F. Göltl, Three Grand Challenges for the Computational Design of Heterogeneous Catalysts, J. Phys. Chem. C, 2022, 126(7), 3305–3313, DOI:10.1021/acs.jpcc.1c10291
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2sc05772g |
| This journal is © The Royal Society of Chemistry 2023 |