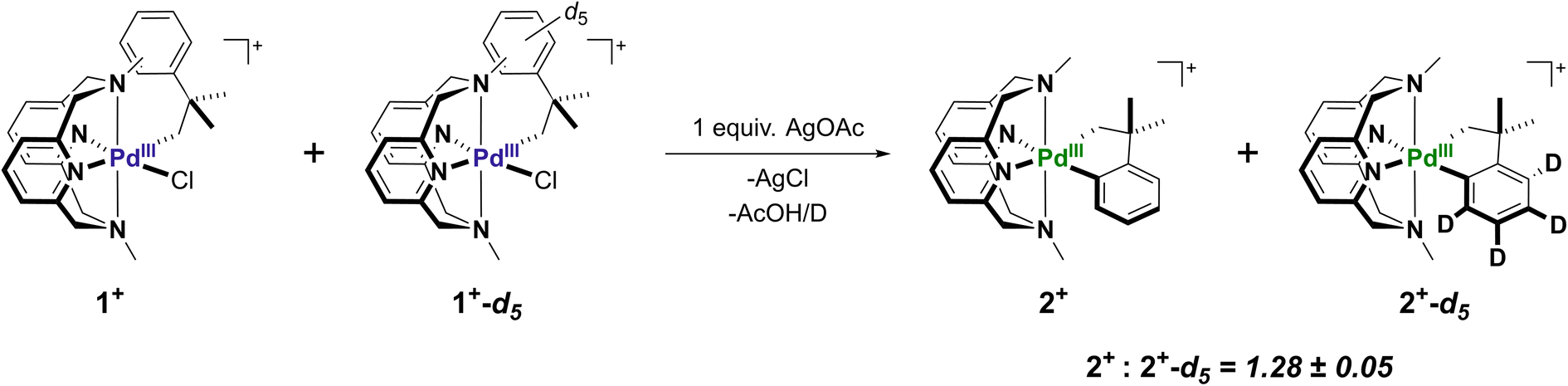DOI:
10.1039/D3SC00034F
(Edge Article)
Chem. Sci., 2023,
14, 3800-3808
C–H bond activation via concerted metalation–deprotonation at a palladium(III) center†
Received
3rd January 2023
, Accepted 12th March 2023
First published on 13th March 2023
Abstract
Herein we report the direct observation of C–H bond activation at an isolated mononuclear Pd(III) center. The oxidation of the Pd(II) complex (MeN4)PdII(neophyl)Cl (neophyl = –CH2C(CH3)2Ph; MeN4 = N,N′-dimethyl-2,11-diaza[3.3](2,6)pyridinophane) using the mild oxidant ferrocenium hexafluorophosphate (FcPF6) yields the stable Pd(III) complex [(MeN4)PdIII(neophyl)Cl]PF6. Upon the addition of an acetate source, [(MeN4)PdIII(neophyl)Cl]PF6 undergoes Csp2–H bond activation to yield the cyclometalated product [(MeN4)PdIII(cycloneophyl)]PF6. This metalacycle can be independently prepared, allowing for a complete characterization of both the starting and final Pd(III) complexes. The C–H activation step can be monitored directly by EPR and UV-Vis spectroscopies, and kinetic isotope effect (KIE) studies suggest that either a pre-association step such as an agostic interaction may be rate limiting, or that the C–H activation is partially rate-limiting in conjunction with ligand rearrangement. Density functional theory calculations support that the reaction proceeds through a κ3 ligand coordination and that the flexible ligand structure is important for this transformation. Overall, this study represents the first example of discrete C–H bond activation occurring at a Pd(III) center through a concerted metalation–deprotonation mechanism, akin to that observed for Pd(II) and Pd(IV) centers.
Introduction
In recent decades, transformations utilizing C–H bond functionalization involving high-valent PdIII and PdIV intermediates have been proposed with increasing frequency.1–18 Therefore, it has become critical to gain a better understanding of the reactivity of these high oxidation states using stable, isolable model compounds. However, most mechanistic studies involving isolated PdIII/PdIV species have focused on reductive elimination reactivity.19–29 Sanford et al. reported the first fully characterized model systems for C–H activation at PdIV, supported by the 4,4′-di-tert-butyl-2,2′-bipyridine ligand in 2011, with another example supported by a tris(2-pyridyl)methane ligand being reported in 2013.30,31 These reactions proceed under mild conditions, with site selectivity complementary to analogous transformations at PdII. Of note, many proposals of C–H activation invoking high-valent palladium intermediates proceed first by C–H activation followed by oxidation.32–34 However, these mechanistic investigations are often unclear as to whether it is possible for oxidation to precede the C–H activation step. Specifically, C–H activation from the PdIII oxidation state is rarely proposed, even in cases where evidence of high-valent PdIII intermediates is provided.35–37 Although C–H activation has been well characterized and studied at both PdII and PdIV,2,9,38 C–H activation at PdIII has not been investigated to date, with no concrete evidence existing for the feasibility of this process. Our group has previously explored the stabilization and organometallic reactivity of high-valent Pd centers by utilizing RN4 pyridinophane ligands (RN4 = N,N′-di-alkyl-2,11-diaza[3.3](2,6)pyridinophane).39–45 As such, we wanted to explore these ligands for the potential study of C–H activation at PdIII. Herein, we describe in detail the characterization of a distinct C–H activation step at PdIII promoted by acetate as an exogenous base, with mechanistic proposals supported by experiments and DFT calculations. We hope such fundamental study might enable more rational incorporation of high-valent Pd-mediated C–H activation into catalytic processes.
Results and discussion
Synthesis and characterization of (MeN4)PdIII complexes
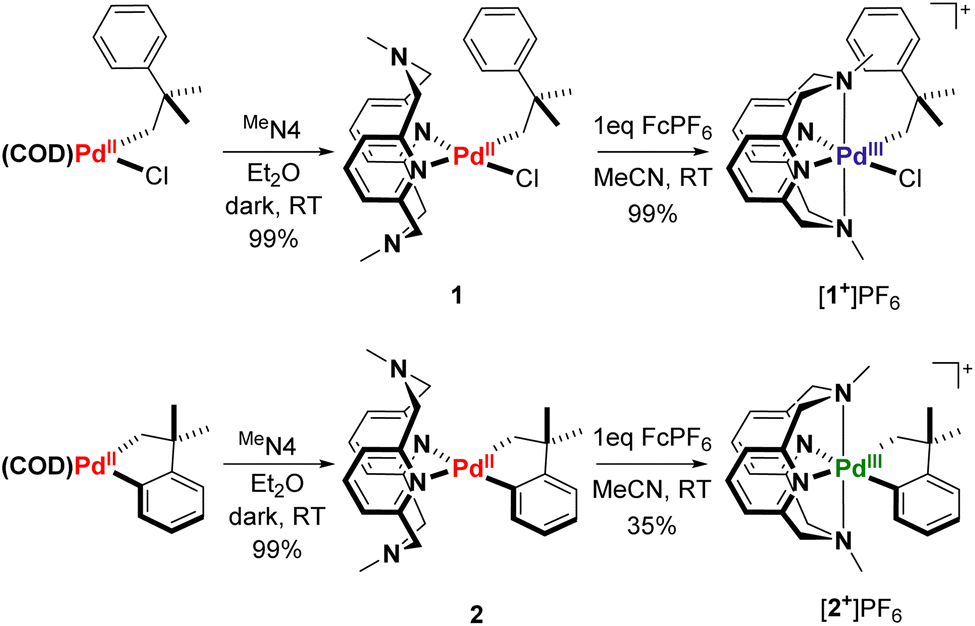
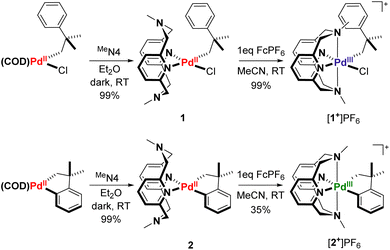
In order to study a potential C–H activation step at the PdIII oxidation state, we envisioned taking an analogous PdII system with a well-characterized C–H activation process and oxidizing it to PdIII by employing ligands known to stabilize high-valent Pd centers. The (COD)PdII(neophyl)Cl complex, a common precursor for the preparation of organometallic palladium compounds,46 is known to readily undergo cyclometalation in the presence of base to form (COD)PdII(cycloneophyl) (cycloneophyl = –CH2C(CH3)2C6H4–).47 Displacement of COD with a ligand capable of stabilizing PdIII, such as RN4-type pyridinophane ligands, could allow for high-valent Pd reactivity studies. Indeed, ligand substitution of (COD)PdII(neophyl)Cl and (COD)PdII(cycloneophyl) with the MeN4 pyridinophane ligand48 generated the PdII complexes (MeN4)PdII(neophyl)Cl, 1, and (MeN4)PdII(cycloneophyl), 2. Subsequent one electron oxidation with FcPF6 resulted in the isolation of both the [(MeN4)PdIII(neophyl)Cl]PF6 ([1+]PF6) and [(MeN4)PdIII(cycloneophyl)]PF6 ([2+]PF6) complexes, respectively (Scheme 1).
 |
| | Scheme 1 Synthesis of PdIII complexes 1+ and 2+. | |
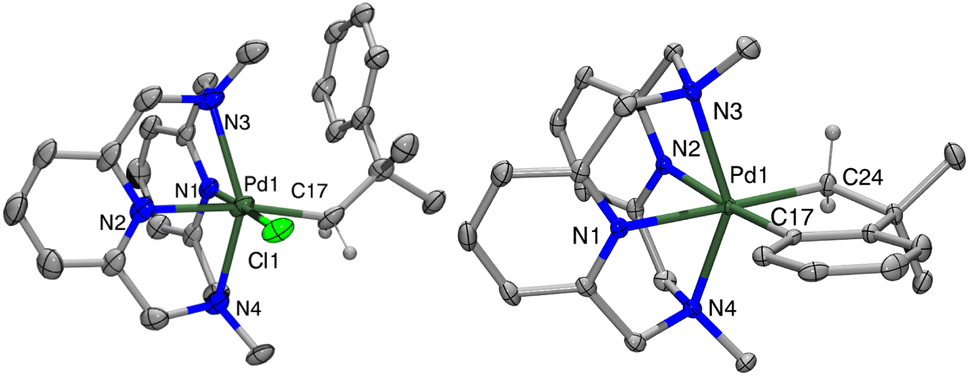
Compounds [1+]PF6 and [2+]PF6 were characterized by X-ray crystallography, with both structures showing distorted octahedral geometries due to the constrained interaction between the axial tertiary nitrogen atoms and the Pd center (Fig. 1). The axial Pd–amine bonds are significantly longer than the equatorial Pd–pyridine bonds by ∼0.2 Å, with the Pd1–N3 interaction in 1+ being even longer, possibly due to steric interactions with the neophyl ligand. 1+ also shows a larger desymmetrization of the equatorial Pd–pyridine bond distances vs. those in 2+, likely due to the stronger trans influence of the organometallic ligand relative to the chloride. The cyclic voltammetry (CV) data for 1 show redox waves for the PdII/III couple, corresponding to different conformers of the MeN4 ligand in solution at room temperature as established previously,42 with events occurring at −360 mV, 0 mV, and 376 mV vs. Fc0/+ in MeCN, and a higher PdIII/IV redox wave at 640 mV. By comparison, complex 2 shows a single feature for PdII/III occurring at −400 mV, and a PdIII/IV redox wave at −260 mV vs. Fc0/+ (Fig. S24 and S25†).
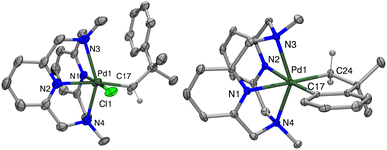 |
| | Fig. 1 ORTEP representation (50% probability ellipsoids) of 1+ (left) and 2+ (right). Counterions omitted for clarity. Selected bond distances (Å) 1+: Pd1–N1 2.061(5), Pd1–N2 2.158(5), Pd1–N3 2.394(5), Pd1–N4 2.301(5), Pd1–C17 2.076(6), Pd1–Cl1 2.367(2); 2+: Pd1–N1 2.156(2), Pd1–N2 2.152(2), Pd1–N3 2.350(2), Pd1–N4 2.383(3), Pd1–C17 1.994(3), Pd1–C24 2.045(3). | |
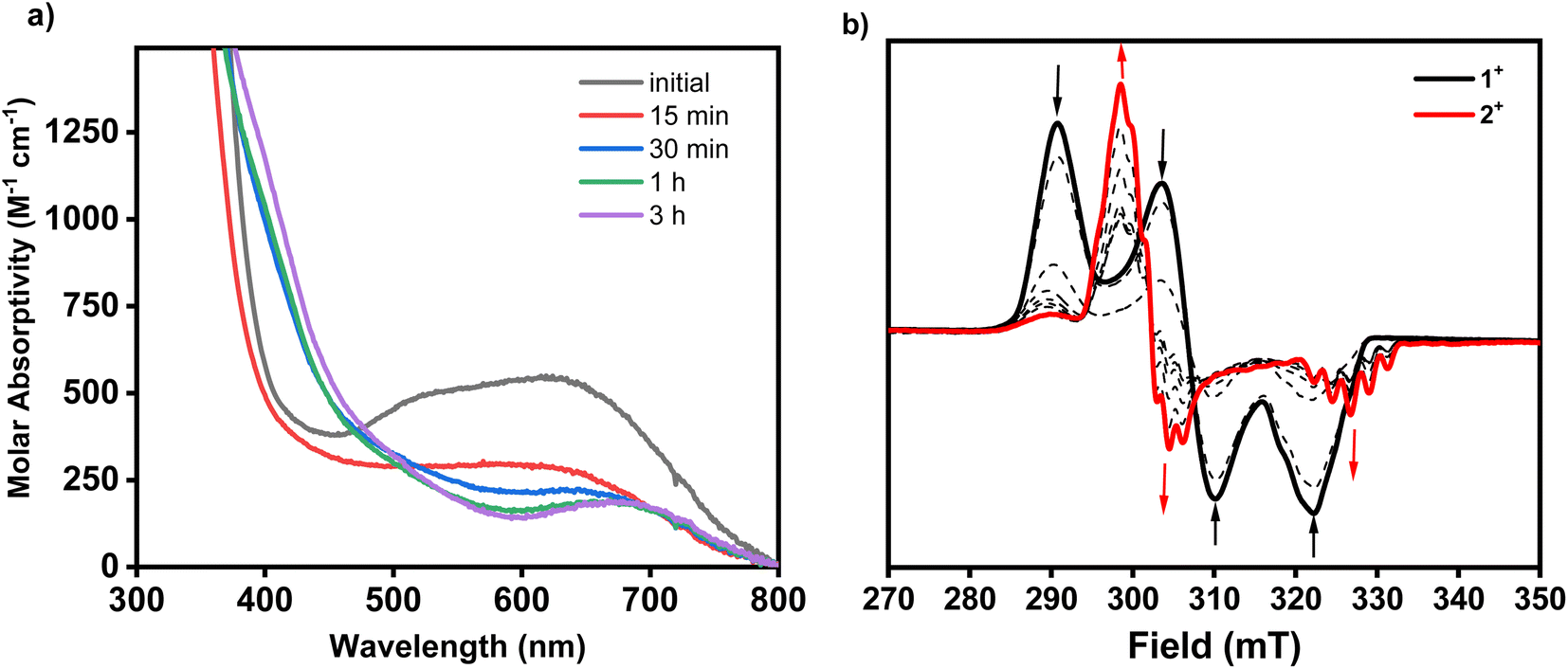
The electronic differences between 1+ and 2+ can also be seen in their EPR spectra. The frozen spectra of 1+ in 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 butyronitrile:acetonitrile shows a rhombic EPR signal with a weakly resolved quintet in the gz region arising from the two axial amine nitrogens (Fig. 2). In contrast, 2+ shows a pseudo-axial signal with coupling from two nitrogens in the gx, gy, and gz directions. The shift from a rhombic spectrum to an axial spectrum after C–H activation and cyclization is consistent with substitution of the chloride in 1+ with a second organometallic interaction from the aryl carbon atom, as well as the changes in bond lengths between the two compounds observed by X-ray crystallography. The UV-Vis spectrum of 1+ shows a strong absorption peak at 625 nm, which is tentatively assigned to a ligand to metal charge transfer (LMCT).39,40,49 The UV-Vis spectrum of complex 2+ reveals a red shift in the LMCT relative to 1+, with an absorption peak at 675 nm (Fig. 2). A similar red shift going from an organometallic ligand plus a halide ligand to two organometallic ligands has been observed previously in the UV-Vis spectra of [(MeN4)PdIIIMeCl]+ and [(MeN4)PdIIIMe2]+.40 Overall, these characteristic differences in the EPR and UV-Vis spectra of complexes 1+ and 2+ allow for the study of the C–H activation process by in situ reaction monitoring by EPR and UV-Vis spectroscopy.
:1 butyronitrile:acetonitrile shows a rhombic EPR signal with a weakly resolved quintet in the gz region arising from the two axial amine nitrogens (Fig. 2). In contrast, 2+ shows a pseudo-axial signal with coupling from two nitrogens in the gx, gy, and gz directions. The shift from a rhombic spectrum to an axial spectrum after C–H activation and cyclization is consistent with substitution of the chloride in 1+ with a second organometallic interaction from the aryl carbon atom, as well as the changes in bond lengths between the two compounds observed by X-ray crystallography. The UV-Vis spectrum of 1+ shows a strong absorption peak at 625 nm, which is tentatively assigned to a ligand to metal charge transfer (LMCT).39,40,49 The UV-Vis spectrum of complex 2+ reveals a red shift in the LMCT relative to 1+, with an absorption peak at 675 nm (Fig. 2). A similar red shift going from an organometallic ligand plus a halide ligand to two organometallic ligands has been observed previously in the UV-Vis spectra of [(MeN4)PdIIIMeCl]+ and [(MeN4)PdIIIMe2]+.40 Overall, these characteristic differences in the EPR and UV-Vis spectra of complexes 1+ and 2+ allow for the study of the C–H activation process by in situ reaction monitoring by EPR and UV-Vis spectroscopy.
 |
| | Fig. 2 (a) EPR spectrum of 1+ in 3:1 PrCN:MeCN at 77 K and the simulated spectrum using the mentioned parameters. (b) EPR spectrum of 2+ in 3:1 PrCN:MeCN at 77 K and the simulated spectrum using the mentioned parameters. (c) UV-Vis spectrum of 1+ in MeCN. The molar absorptivity of each band is shown in parentheses. (d) UV-Vis spectrum of 2+ in MeCN. The molar absorptivity of each of the UV-Vis bands are shown in parentheses. | |
Study of C–H activation at [(MeN4)PdIII(neophyl)Cl]+
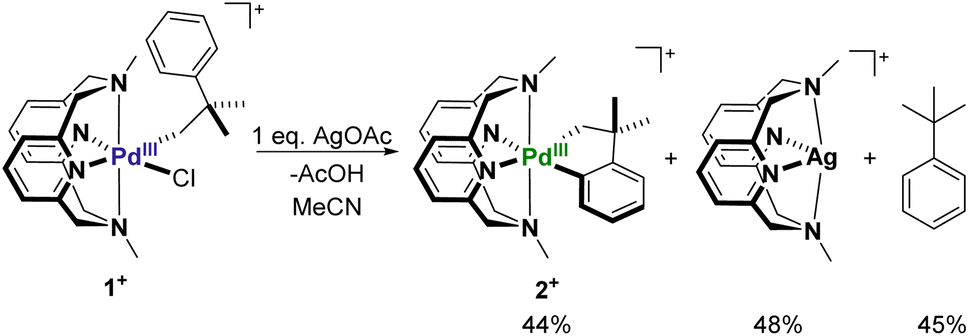
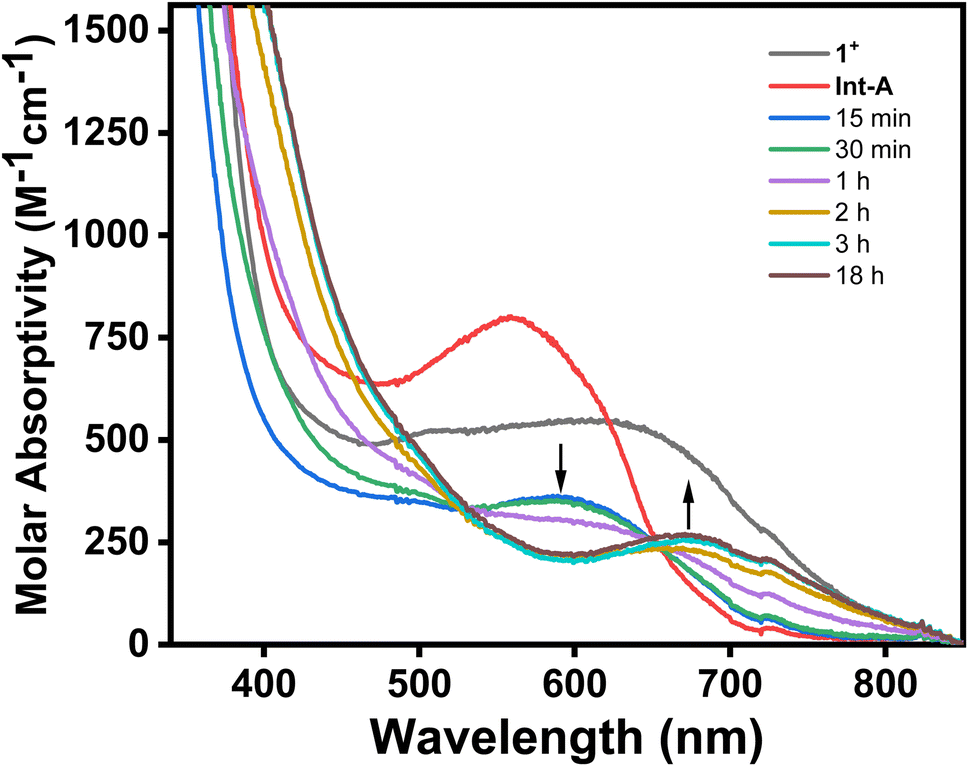
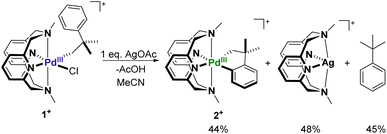
The treatment of 1+ with 1 equiv. of AgOAc in MeCN at room temperature readily results in the formation of the palladacycle 2+, which can be tracked by UV-Vis spectroscopy (Fig. 3a). The spectral feature at 625 nm corresponding to 1+ readily disappears with concomitant formation of 2+, as seen by the growth of the characteristic 675 nm band. The generation of 2+ occurs steadily, with maximum formation of the product after 3 hours, representing a 44% yield determined by molar absorptivity. Additionally, since the reaction occurs at the paramagnetic PdIII oxidation state, the C–H activation process could also be tracked by EPR spectroscopy (Fig. 3b). In a similar fashion to the UV-Vis experiment, the addition of 1 equiv. of AgOAc to 1 in MeCN resulted in the conversion to 1+ in 45% yield by EPR, without observation of other paramagnetic side products. In order to identify possible explanations for the low conversion, 1H NMR experiments were performed on 1 in order to analyze the diamagnetic products (Fig. S20†). Adding up to 2 equiv. of AgOAc does not result in the detection of MeN4PdIV(cycloneophyl), which was independently prepared, ruling out PdIV formation as the main contributing factor to the decreased yield. The 1H NMR does, however, show tert-butylbenzene in 45% yield and shifted 1H NMR resonances for the MeN4 ligand in 48% yield, which was also confirmed to be detection of the diamagnetic ion [(MeN4)Ag]+ by ESI-MS. Altogether, these results suggest that protodemetalation of the neophyl ligand and subsequent decomplexation PdII are the main contributing factors towards the decreased yield of the C–H activated product. Protodemetalation may arise from the formation of 1 equiv. of acetic acid during the C–H activation process, which could then protodemetalate one additional equiv. of 1+, producing tert-butyl benzene and complex decomposition. This is further supported by the fact that cyclometalation only proceeds in approximately 50% yield (Scheme 2). An alternative mechanism was considered that includes the formation of a PdIII-di(neophyl) species, as observed for the Ni analogs,50 however this was ruled out since no di-neophyl coupled product was observed by NMR or MS, as well as given the very slow kinetics or alkyl ligand exchange that we have observed previously for (RN4)PdIII complexes.39–45 Combined with the independent synthesis and characterization of 1+ and 2+, these UV-Vis and EPR spectroscopic results strongly suggests an acetate-assisted C–H activation/cyclometalation event occurring at the PdIII center, which represents, to the best of our knowledge, the first example of such a process.
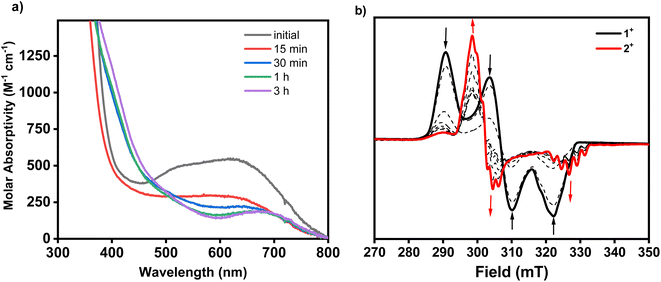 |
| | Fig. 3 (a) UV-Vis spectra of reaction mixture of 1+ and 1 equiv. AgOAc in MeCN at room temperature. The precipitated AgCl was filtered prior to UV-Vis measurement. (b) EPR spectra of the reaction mixture of 1+ and 1 equiv. of AgOAc in MeCN at room temperature at over a 280 minute period (time points: 0 min, 1 min, 5 min, 10 min, 15 min, 20 min, 30 min, 50 min, 110 min, and 280 min). Aliquots were diluted 4-fold with butyronitrile prior to EPR measurement at 77 K. | |
 |
| | Scheme 2 C–H activation at 1+ in the presence of AgOAc. | |
To further understand the exact process by which C–H activation occurs, complex 1+ was treated with either Ag+ or OAc− under similar reaction conditions. Upon mixing 1+ with 1 equiv. of AgBF4 in MeCN at room temperature, a new blue PdIII species was observed along with a grey precipitate (Fig. 4). The UV-Vis of this new PdIII compound showed a new absorption feature at 560 nm (Fig. S13†), and also displayed a new distinct rhombic EPR signal with g values of 2.212, 2.109, and 2.019 (Fig. S14†). Given that the species displays a rhombic signal similar to that of 1+, and that the grey precipitate in the reaction suggests AgCl is produced, this species is assigned to the acetonitrile adduct, [(MeN4)PdIII(neophyl)(MeCN)]2+ (Int-A), generated upon halide abstraction by Ag+ and subsequent binding of a MeCN molecule. Attempts to generate palladacycle 2+ in a non-coordinating solvent (DCM) with AgOAc were unsuccessful, only decomposition of the starting material into an unidentified product being observed (Fig. S18†). In addition, other carboxylate sources such as Et4NOAc or Bu4NOAc were also used in the absence of silver to promote the C–H activation of complex 1+ under similar reaction conditions. Using 1 equiv. of either Et4NOAc·4H2O or Bu4NOAc results in the formation of 2+ in 36% and 41% yield, respectively, with significantly longer reaction times required to reach the maximum yield (20–26 hours, Table S2†). By comparison, the addition of 1 equiv. AgOPiv resulted in formation of 2+ in a similar yield (37%) over 6 hours (Table S2†). While this is significantly faster than the silver-free acetate sources, it is still relatively slower than AgOAc, likely to due to the highly congested coordination geometry of the Pd center in 1+ that precludes coordination of the pivalate anion. In addition, attempts to generate palladacycle 2+ using 2,6-lutidine as a non-nucleophilic base from either 1+ or Int-A were unsuccessful (Fig. S16 and S17†). These results strongly suggest that acetate is the key promoter for the PdIII mediated C–H activation.51 Using more equivalents of the acetate source led to a considerable shortening of the reaction time, however, the maximum yield decreased as a consequence. The addition of 5 equiv. of Bu4NOAc to 1+ resulted in rapid change in the UV-Vis spectrum, without formation of 2+, perhaps due to ligand exchange between acetate and MeN4 to generate a new species (Fig. 4). With all of this in mind, the role of silver in the reaction appears to be facilitating ligand exchange by extracting the chloride and allowing the acetate to bind to the open coordination site, stabilized by the coordinating solvent. To verify this, a sequential addition experiment was performed, by which 1+ was first reacted with 1 equiv. of AgBF4 for 20 minutes, followed by 1 equiv. of Bu4NOAc after the AgCl precipitate was removed by filtration. Addition of acetate to the in situ generated Int-A results in the instantaneous formation of a new species, as observed by both UV-Vis and EPR, which is assumed to be the acetate adduct [(MeN4)PdIII(neophyl)(OAc)]+ (Int-B). This compound exhibits a rhombic EPR signal with a weaker superhyperfine coupling to the two axial nitrogen atoms compared to 1+ or Int-A (17 G vs. 21.5 G or 23 G, respectively, Fig. S39†), and the Int-A and Int-B intermediate species were also detected in situ by ESI-MS, supporting their assignment (Fig. S30 and S31†). Moreover, the addition of the acetate after complete removal of the chloride ligand did not decrease the time necessary for the formation of 2+, with the reaction still requiring 3 hours to reach completion, as observed by UV-Vis spectroscopy. This suggests that halide abstraction is unlikely to be the rate determining step during the C–H activation process. Finally, the presence of a coordinated solvent is likely needed to complete the coordination environment of the high-valent Pd center and preclude any adventitious side-reactions, as the productive C–H activation does not proceed in a non-coordinating solvent (Fig. S18†).
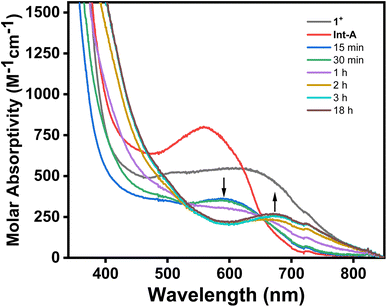 |
| | Fig. 4 UV-Vis spectrum of 1+ after sequential addition of 1 equiv. of AgBF4 followed by 1 equiv. of Bu4NOAc in MeCN. 1+ (black line); 1+ after addition of AgBF4 to generate Int-A (red line); time points after addition of 1 equiv. of NBu4OAc to the in situ generated Int-A. | |
Mechanistic and kinetic isotope effect studies
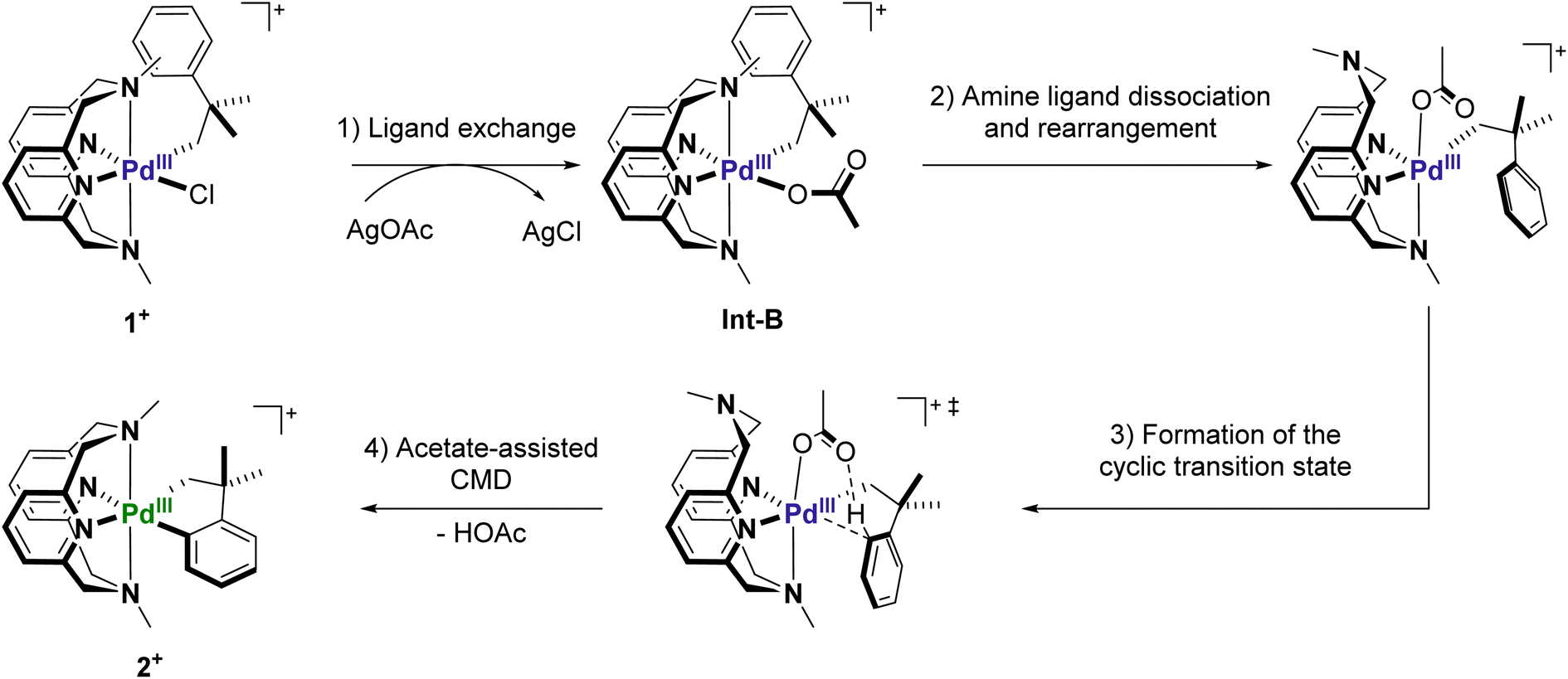
It is well established that carboxylate additives play an important role in facilitating C–H activation processes in PdII-mediated reactions.9,52,53 Numerous experimental and computational studies suggest that the carboxylate group acts as an internal base to deprotonate C–H bonds through a cyclic transition state, a process known as concerted metalation–deprotonation (CMD).54,55 Sanford et al. reported the first known example of acetate-assisted C–H activation at high-valent PdIV, and we consider a similar process is likely occurring at PdIII.31 We hypothesize that the observed C–H activation reaction involves the following steps: (1) chloride-to-acetate ligand exchange facilitated by silver ions; (2) axial amine dissociation and subsequent rearrangement to form either a κ2 or κ3-MeN4 coordinated complex with an axially-oriented acetate; (3) formation of the 5-membered transition state to facilitate C–H activation; (4) cyclometalation and loss of one equiv. of acetic acid to generate the final palladacycle (Scheme 3).
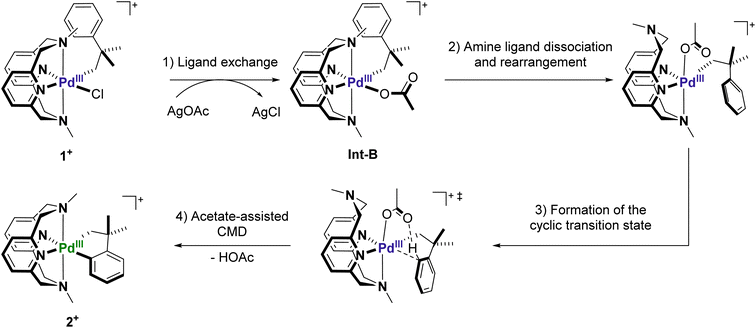 |
| | Scheme 3 Potential mechanism of C–H activation at 1+ in the presence of AgOAc. | |
UV-Vis experiments show that the ligand exchange steps are not rate limiting due to an unaffected reaction time starting from either Int-A or Int-B. However, the ligand rearrangement and C–H activation steps could not be probed by either UV-Vis or EPR due to a lack of other observable intermediates. To further study the proposed mechanism, the fully aryl-deuterated neophyl complex 1+-d5, as well as the corresponding 2+-d4 complex were independently prepared, and kinetic isotope effect (KIE) experiments via in situ ESI-MS were performed to analyze the C–H activation step (Fig. S21 and S22†) A 1:1 mixture of 1+ and 1+-d5 were combined with 1 equiv. of AgOAc in MeCN, and the reaction mixture was analyzed by ESI-MS over time (Fig. S23–S29†). Over the monitored time course of the reaction (5–50 min), the ESI-MS peak intensity ratio of the cyclometalated products 2+:2+-d4 remained constant at ∼1.3 (Fig. S21†). In addition, the consumption of the starting material correlated well with the formation of cyclometalated product, as the ESI-MS peak intensity ratio of the starting material 1+-d5:1+ was also ∼1.3. Overall, a small kinetic isotopic effect (KIE = kH/kD = 1.28 ± 0.05) was obtained. This experimentally determined KIE value suggested two possible scenarios. Firstly, the C–H activation step is partially rate-limiting, in conjunction with a form of MeN4 ligand isomerization. It has been demonstrated previously in N-based ligand-supported Pt-neophyl complexes that KIE values may heavily depend on ligand conformational flexibility.56 Alternatively, there could be a rate-limiting pre-association step between the Pd center and the α carbon, either via an agostic interaction through the C–H bond, or formation of a σ-complex. Previous examples of C–H activation systems employing a pre-activation intermediates in which the C–H bond is weakened by interaction with the electrophilic metal center are known to give KIE values between 1.1 and 1.4,57–59 which is in agreement with our measured value (Scheme 4).
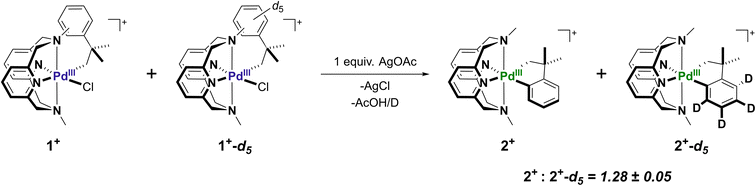 |
| | Scheme 4 Kinetic isotope effect on C–H activation from a mixture of 1+ and 1+-d5 with AgOAc determined by ESI-MS. | |
Computational details
DFT calculations were used to address two main aspects of the proposed reaction coordinate: (1) to probe if C–H activation was involved the rate determining step; (2) to evaluate whether this process occurs via a κ3 or κ2 conformation of the MeN4 ligand, where one or both of the axial amine nitrogens are no longer bound to the palladium center, respectively. It is necessary to analyze these ligand configurations as proposed transition states, since an acetate-assisted CMD mechanism necessitates a cyclic transition state that can only be achieved if the acetate is positioned perpendicular to the C–H bond to be activated.51,60 Energy profiles for the conversion of Int-B to 2+ were constructed for each of the mentioned ligand configurations (Fig. S47†). Comparing the two ligand geometries, the κ2 pathway shows a significantly larger energy penalty for the dissociation of both axial amine arms than the κ3 pathway (+22.0 kcal mol−1vs. +5.3 kcal mol−1). Additionally, attempts to optimize the κ2 geometry containing the axially oriented acetate resulted in a reorientation of the acetate back to the equatorial position. These details suggest that the κ2 pathway is too energetically unfavorable to support this transformation and indicates that the flexible denticity of the MeN4 ligand is essential for promoting this transformation at a PdIII center. However, the calculated KIE for the C–H activation step was found to be 5.7, significantly higher than the experimentally observed value by ESI-MS. This deviation from the experimental value provides further evidence that the C–H activation step is not the sole contributor to the observed experimental KIE. We have also considered rearrangement of the neophyl ligand to the axial position instead of the acetate, however, the calculated energy of that intermediate was large relative to the intermediate with the axial acetate (Fig. S47†). In addition, this migration would require a complete rearrangement of the MeN4 ligand to reach the final cyclometalated product, which would most likely require a high-energy intermediate or transition state. Finally, the possibility of C–H activation occurring in an axial position, trans to the coordinated tert-butyl amine group, followed by aryl migration to the equatorial position and concomitant recoordination of the axial amine donor cannot be completely excluded.
Aerobic oxidation of 1
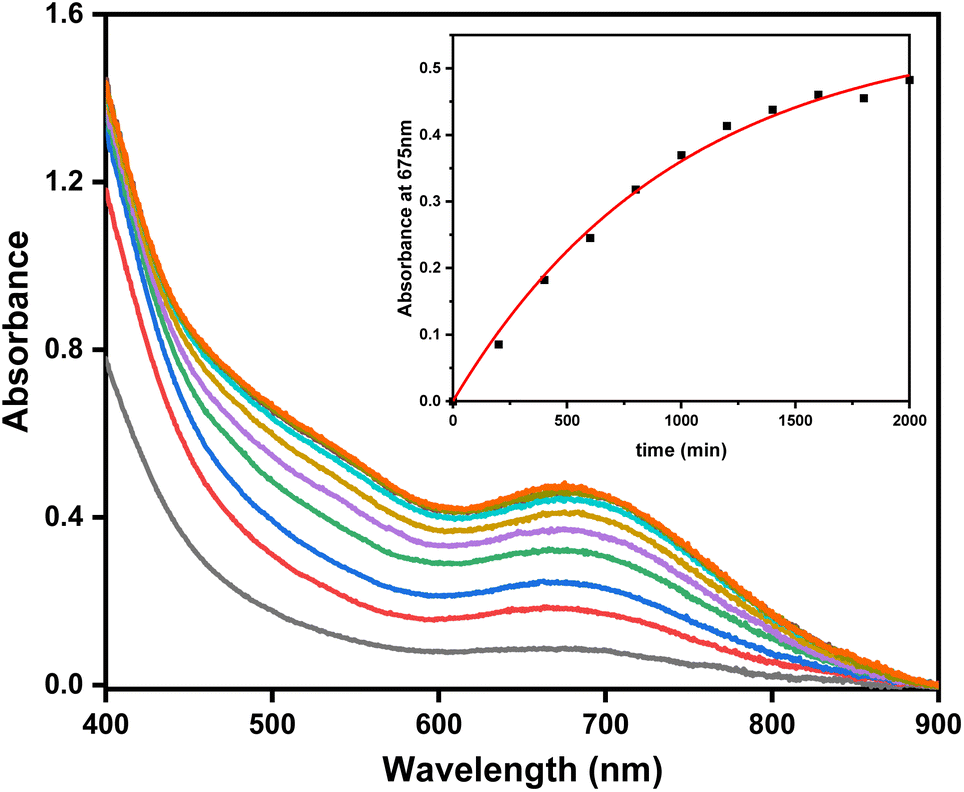
The reaction between organometallic Pd centers and O2, as well as their role in oxidative catalysis, has been extensively studied.1,61,62 Additionally, the previously (MeN4)PdII organometallic complexes reported by our group have demonstrated extremely low PdII/PdIII oxidation potentials, which led to the generation of high-valent PdIII and PdIV intermediates in the presence of O2.40,43,44 In order to determine how 1 would react with O2, aerobic reactivity experiments were performed. Remarkably, subjecting 1 to a O2-saturated solution of 5% H2O in MeCN resulted in the formation of 2+ in 68% yield, as observed by UV-Vis (Fig. 5). The identity of 2+ was confirmed by EPR and ESI-MS spectroscopy (Fig. S41†), and thus we tentatively propose that this tandem aerobic oxidation/C–H activation steps could proceed through a high-valent Pd intermediate.41,43,45,63 Other oxygen-mediated C–H bond activations at Pd centers have been shown previously by Goldberg64 and Vilar,65 and while detailed mechanistic studies are needed to decipher this oxidatively-induced C–H activation process, these exciting initial results point to the potential inclusion of both aerobic oxidation and C–H activation steps in novel Pd-mediated catalytic transformations.
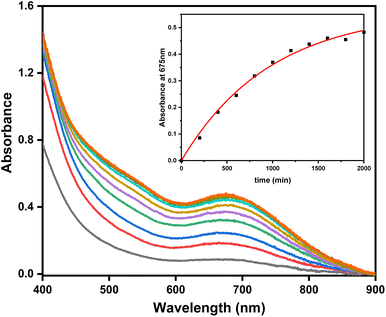 |
| | Fig. 5 Formation of 2+ during aerobic oxidation of complex 1 (1.84 mM) in O2 saturated 5% H2O/MeCN at room temperature, followed by UV-Vis spectroscopy (Δt = 200 min, tmax = 2000 min). Inset: plot of the concentration of 2+ over time, monitored at 675 nm, and the first-order fit (red line, k = 0.00103 min−1, R2 = 0.995). | |
Conclusion
Herein, we report the synthesis of a model complex for the study of C–H activation at a mononuclear PdIII center, (MeN4)PdIII(neophyl)Cl+ (MeN4 = N,N′-dimethyl-2,11-diaza[3.3](2,6)pyridinophane), which undergoes a concerted metalation–deprotonation (CMD) step resulting in the formation of the cyclometalated PdIII complex (MeN4)PdIII(cycloneophyl)+. To the best of our knowledge, this result represents the first example of a fully characterized C–H activation process at a PdIII center. Both the starting complex 1+ and the C–H activated product 2+ were independently synthesized and characterized, allowing for unambiguous reaction monitoring by UV-Vis, EPR, and ESI-MS. Mechanistic studies were performed in order to understand the role of the acetate source, as well as identification of the possible rate determining step(s). Sequential addition of AgBF4 followed by an acetate source reveals that ligand displacement of the chloride with acetate, followed by isomerization leading to a CMD process is the likely reaction mechanism. This process is analogous to previously reported transformations at both PdII and PdIV centers. The obtained KIE value of 1.28 for this transformation could be the result of either a partially rate-limiting C–H activation step, or a rate-limiting pre-activation step such as the formation of an agostic interaction or σ complex between the Pd and the C atom of the C–H bond to be activated. It is important to note that this system is limited to the use of an exogenous base to facilitate C–H activation, where intramolecular processes are known to be significantly easier to perform. However, inclusion of an exogenous base benefits from the simplicity of an additive over sophisticated ligand framework synthesis.15,34 Moreover, the addition of O2 to the PdII complex 1 generates the PdIII C–H activated metallocycle 2+ directly, which sets the stage for future studies into the mechanism of C–H bond activation processes mediated by aerobic oxidation.
Data availability
Crystallographic data for compounds 1, 2, [1+](ClO4), [2+](ClO4), and [22+](ClO4)2 have been deposited at the CCDC under the identifier numbers 2129670–2129674.
Author contributions
L. M. M. directed and secured the funding for this project. B. S. B. and F. T. contributed to the characterization of the transition metal complexes and the reactivity studies. B. S. B. performed control experiments and characterization of Int-A and Int-B. F. T. performed the KIE studies and aerobic oxidation reactivity studies. D. Y. B. and L. M. M. contributed the DFT study and analysis. B. S. B. prepared the initial version of the manuscript and all authors have read and provided comments on the manuscript and the ESI.†
Conflicts of interest
The authors declare no competing financial interest.
Acknowledgements
This work was supported by the National Science Foundation (CHE-2102544). We would also like to thank the Department of Chemistry at the University of Illinois at Urbana-Champaign for all the support, and Dr Nigam Rath for his assistance with X-ray structural determination.
References
- D. Wang, A. B. Weinstein, P. B. White and S. S. Stahl, Ligand-Promoted Palladium-Catalyzed Aerobic Oxidation Reactions, Chem. Rev., 2018, 118(5), 2636–2679 CrossRef CAS PubMed.
- J. He, M. Wasa, K. S. L. Chan, Q. Shao and J.-Q. Yu, Palladium-Catalyzed Transformations of Alkyl C–H Bonds, Chem. Rev., 2017, 117(13), 8754–8786 CrossRef CAS PubMed.
- A. R. Dick, K. L. Hull and M. S. Sanford, A highly selective catalytic method for the oxidative functionalization of C-H bonds, J. Am. Chem. Soc., 2004, 126(8), 2300–2301 CrossRef CAS PubMed.
- L. V. Desai, K. L. Hull and M. S. Sanford, Palladium-catalyzed oxygenation of unactivated sp(3) C-H bonds, J. Am. Chem. Soc., 2004, 126(31), 9542–9543 CrossRef CAS PubMed.
- R. Giri, X. Chen and J. Q. Yu, Palladium-catalyzed asymmetric iodination of unactivated C-H bonds under mild conditions, Angew. Chem., Int. Ed., 2005, 44(14), 2112–2115 CrossRef CAS PubMed.
- R. Giri, J. Liang, J. G. Lei, J. J. Li, D. H. Wang, X. Chen, I. C. Naggar, C. Y. Guo, B. M. Foxman and J. Q. Yu, Pd-catalyzed stereoselective oxidation of methyl groups by inexpensive oxidants under mild conditions: a dual role for carboxylic anhydrides in catalytic C-H bond oxidation, Angew. Chem., Int. Ed., 2005, 44(45), 7420–7424 CrossRef CAS PubMed.
- J. A. Jordan-Hore, C. C. C. Johansson, M. Gulias, E. M. Beck and M. J. Gaunt, Oxidative Pd(II)-Catalyzed C-H Bond Amination to Carbazole at Ambient Temperature, J. Am. Chem. Soc., 2008, 130(48), 16184–16186 CrossRef CAS PubMed.
- J. Zhang, E. Khaskin, N. P. Anderson, P. Y. Zavalij and A. N. Vedernikov, Catalytic aerobic oxidation of substituted 8-methylquinolines in PdII-2,6-pyridinedicarboxylic acid systems, Chem. Commun., 2008,(31), 3625–3627 RSC.
- T. W. Lyons and M. S. Sanford, Palladium-Catalyzed Ligand-Directed C-H Functionalization Reactions, Chem. Rev., 2010, 110(2), 1147–1169 CrossRef CAS PubMed.
- R. Y. Guo, J. L. Portscheller, V. W. Day and H. C. Malinakova, An allylpalladium(IV) intermediate in the synthesis of highly substituted benzoxepines and benzopyrans via reactions of stable pallada(II)cycles with allyl bromides, Organometallics, 2007, 26(15), 3874–3883 CrossRef CAS.
- H. Malinakova, Palladium(IV) Complexes as Intermediates in Catalytic and Stoichiometric Cascade Sequences Providing Complex Carbocycles and Heterocycles, Top. Organomet. Chem., 2011, 503, 85–110 CrossRef PubMed.
- K. L. Hull, E. L. Lanni and M. S. Sanford, Highly Regioselective Catalytic Oxidative Coupling Reactions: Synthetic and Mechanistic Investigations, J. Am. Chem. Soc., 2006, 128(43), 14047–14049 CrossRef CAS PubMed.
- J. J. Topczewski and M. S. Sanford, Carbon-hydrogen (C-H) bond activation at PdIV: a Frontier in C-H functionalization catalysis, Chem. Sci., 2015, 6(1), 70–76 RSC.
- C. F. Rosewall, P. A. Sibbald, D. V. Liskin and F. E. Michael, Palladium-Catalyzed Carboamination of Alkenes Promoted by N-Fluorobenzenesulfonimide via C-H Activation of Arenes, J. Am. Chem. Soc., 2009, 131(27), 9488–9489 CrossRef CAS PubMed.
- Z. Li, Z. Wang, N. Chekshin, S. Q. Qian, J. X. Qiao, P. T. Cheng, K. S. Yeung, W. R. Ewing and J. Q. Yu, A tautomeric ligand enables directed C-H hydroxylation with molecular oxygen, Science, 2021, 372(6549), 1452–1457 CrossRef CAS PubMed.
- P. A. Sibbald, C. F. Rosewall, R. D. Swartz and F. E. Michael, Mechanism of N-Fluorobenzenesulfonimide Promoted Diamination and Carboamination Reactions: Divergent Reactivity of a Pd(IV) Species, J. Am. Chem. Soc., 2009, 131(43), 15945–15951 CrossRef CAS PubMed.
- X. Wang, D. Leow and J.-Q. Yu, Pd(II)-Catalyzed para-Selective C–H Arylation of Monosubstituted Arenes, J. Am. Chem. Soc., 2011, 133(35), 13864–13867 CrossRef CAS PubMed.
- Y.-H. Zhang and J.-Q. Yu, Pd(II)-Catalyzed Hydroxylation of Arenes with 1 atm of O2 or Air, J. Am. Chem. Soc., 2009, 131(41), 14654–14655 CrossRef CAS PubMed.
- D. C. Powers and T. Ritter, Palladium(III) in Synthesis and Catalysis, Top. Organomet. Chem., 2011, 35, 129–156 CrossRef CAS PubMed.
- L.-M. Xu, B.-J. Li, Z. Yang and Z.-J. Shi, Organopalladium(iv) chemistry, Chem. Soc. Rev., 2010, 39(2), 712–733 RSC.
- P. Sehnal, R. J. K. Taylor and I. J. S. Fairlamb, Emergence of Palladium(IV) Chemistry in Synthesis and Catalysis, Chem. Rev., 2010, 110(2), 824–889 CrossRef CAS PubMed.
- N. R. Deprez and M. S. Sanford, Reactions of hypervalent iodine reagents with palladium: mechanisms and applications in organic synthesis, Inorg. Chem., 2007, 46(6), 1924–1935 CrossRef CAS PubMed.
- K. Muniz, High-Oxidation-State Palladium Catalysis: New Reactivity for Organic Synthesis, Angew. Chem., Int. Ed., 2009, 48(50), 9412–9423 CrossRef CAS PubMed.
- A. J. Canty, Organopalladium and platinum chemistry in oxidising milieu as models for organic synthesis involving the higher oxidation states of palladium, J. Chem. Soc., Dalton Trans., 2009,(47), 10409–10417 RSC.
- X. D. Zhao and V. M. Dong, Carbon-Sulfur Reductive Elimination from Palladium(IV) Sulfinate Complexes, Angew. Chem., Int. Ed., 2011, 50(4), 932–934 CrossRef CAS PubMed.
- W. Oloo, P. Y. Zavalij, J. Zhang, E. Khaskin and A. N. Vedernikov, Preparation and C-X Reductive Elimination Reactivity of Monoaryl PdIV-X Complexes in Water (X = OH, OH2, Cl, Br), J. Am. Chem. Soc., 2010, 132(41), 14400–14402 CrossRef CAS PubMed.
- D. C. Powers, D. Benitez, E. Tkatchouk, W. A. Goddard III and T. Ritter, Bimetallic reductive elimination from dinuclear Pd(III) complexes, J. Am. Chem. Soc., 2010, 132(40), 14092–14103 CrossRef CAS PubMed.
- T. Furuya, D. Benitez, E. Tkatchouk, A. E. Strom, P. P. Tang, W. A. Goddard and T. Ritter, Mechanism of C-F Reductive Elimination from Palladium(IV) Fluorides, J. Am. Chem. Soc., 2010, 132(11), 3793–3807 CrossRef CAS PubMed.
- W. Oloo, P. Y. Zavalij, J. Zhang, E. Khaskin and A. N. Vedernikov, Preparation and C-X Reductive Elimination Reactivity of Monoaryl Pd-IV-X Complexes in Water (X = OH, OH2, Cl, Br), J. Am. Chem. Soc., 2010, 132(41), 14400–14402 CrossRef CAS PubMed.
- J. M. Racowski, N. D. Ball and M. S. Sanford, C-H Bond Activation at Palladium(IV) Centers, J. Am. Chem. Soc., 2011, 133(45), 18022–18025 CrossRef CAS PubMed.
- A. Maleckis, J. W. Kampf and M. S. Sanford, A Detailed Study of Acetate-Assisted C–H Activation at Palladium(IV) Centers, J. Am. Chem. Soc., 2013, 135(17), 6618–6625 CrossRef CAS PubMed.
- G. He, G. Lu, Z. W. Guo, P. Liu and G. Chen, Benzazetidine synthesis via palladium-catalysed intramolecular C-H amination, Nat. Chem., 2016, 8(12), 1131–1136 CrossRef CAS.
- D. Saha, P. Das, P. Biswas and J. Guin, Synthesis of Phenolic Compounds via Palladium Catalyzed C-H Functionalization of Arenes, Chem.–Asian J., 2019, 14(24), 4534–4548 CrossRef CAS PubMed.
- Z. Li, H. S. Park, J. X. Qiao, K. S. Yeung and J. Q. Yu, Ligand-Enabled C-H Hydroxylation with Aqueous H2O2 at Room Temperature, J. Am. Chem. Soc., 2022, 144(39), 18109–18116 CrossRef CAS PubMed.
- S. L. Zultanski and S. S. Stahl, Palladium-catalyzed aerobic acetoxylation of benzene using NOx-based redox mediators, J. Organomet. Chem., 2015, 793, 263–268 CrossRef CAS PubMed.
- C. Ma, C. Q. Zhao, Y. Q. Li, L. P. Zhang, X. T. Xu, K. Zhang and T. S. Mei, Palladium-catalyzed C-H activation/C-C cross-coupling reactions via electrochemistry, Chem. Commun., 2017, 53(90), 12189–12192 RSC.
- H. G. Xia, Y. Y. An, X. C. Zeng and J. Wu, Palladium-catalyzed direct sulfonylation of C-H bonds with the insertion of sulfur dioxide, Chem. Commun., 2017, 53(93), 12548–12551 RSC.
- X. Chen, K. M. Engle, D. H. Wang and J. Q. Yu, Palladium(II)-Catalyzed C-H Activation/C-C Cross-Coupling Reactions: Versatility and Practicality, Angew. Chem., Int. Ed., 2009, 48(28), 5094–5115 CrossRef CAS PubMed.
- J. R. Khusnutdinova, N. P. Rath and L. M. Mirica, Stable Mononuclear Organometallic Pd(III) Complexes and Their C-C Bond Formation Reactivity, J. Am. Chem. Soc., 2010, 132(21), 7303–7305 CrossRef CAS PubMed.
- F. Tang, F. Qu, J. R. Khusnutdinova, N. P. Rath and L. M. Mirica, Structural and Reactivity Comparison of Analogous Organometallic Pd(III) and Pd(IV) Complexes, Dalton Trans., 2012, 41(46), 14046–14050 RSC.
- J. W. Schultz, N. P. Rath and L. M. Mirica, Improved Oxidative C–C Bond Formation Reactivity of High-Valent Pd Complexes Supported by a Pseudo-Tridentate Ligand, Inorg. Chem., 2020, 59(16), 11782–11792 CrossRef CAS PubMed.
- J. R. Khusnutdinova, N. P. Rath and L. M. Mirica, The Conformational Flexibility of the Tetradentate Ligand tBuN4 is Essential for the Stabilization of (tBuN4)PdIII Complexes, Inorg. Chem., 2014, 53, 13112–13129 CrossRef PubMed.
- F. Tang, Y. Zhang, N. P. Rath and L. M. Mirica, Detection of Pd(III) and Pd(IV) Intermediates during the Aerobic Oxidative C-C Bond Formation from a Pd(II) Dimethyl Complex, Organometallics, 2012, 31(18), 6690–6696 CrossRef CAS.
- F. Z. Tang, S. V. Park, N. P. Rath and L. M. Mirica, Electronic versus steric effects of pyridinophane ligands on Pd(III) complexes, Dalton Trans., 2018, 47(4), 1151–1158 RSC.
- J. R. Khusnutdinova, N. P. Rath and L. M. Mirica, The Aerobic Oxidation of a Pd(II) Dimethyl Complex Leads to Selective Ethane Elimination from a Pd(III) Intermediate, J. Am. Chem. Soc., 2012, 134, 2414–2422 CrossRef CAS PubMed.
- E. Gutiérrez, M. C. Nicasio, M. Paneque, C. Ruiz and V. Salazar, Synthesis and reactivity of new palladium alkyl complexes containing PMe3 ligands: insertion reactions and formation of bis(pyrazolyl)borate derivatives, J. Organomet. Chem., 1997, 549(1–2), 167–176 CrossRef.
- J. Campora, J. A. Lopez, P. Palma, D. del Rio, E. Carmona, P. Valerga, C. Graiff and A. Tiripicchio, Synthesis and Insertion Reactions of the Cyclometalated Palladium–Alkyl Complexes Pd(CH2CMe2-o-C6H4)L2. Observation of a Pentacoordinated Intermediate in the Insertion of SO2, Inorg. Chem., 2001, 40(17), 4116–4126 CrossRef CAS PubMed.
- F. Bottino, M. Di Grazia, P. Finocchiaro, F. R. Fronczek, A. Mamo and S. Pappalardo, Reaction of Tosylamide Monosodium Salt with Bis(halomethyl) Compounds: An Easy Entry to Symmetrical N-tosyl aza macrocycles, J. Org. Chem., 1988, 53(15), 3521–3529 CrossRef CAS.
- J. R. Khusnutdinova, N. P. Rath and L. M. Mirica, Dinuclear Palladium(III) Complexes with a Single Unsupported Bridging Halide Ligand: Reversible Formation from Mononuclear Palladium(II) or Palladium(IV) Precursors, Angew. Chem., Int. Ed., 2011, 50(24), 5532–5536 CrossRef CAS PubMed.
- J. Campora, M. D. Conejo, K. Mereiter, P. Palma, C. Perez, M. L. Reyes and C. Ruiz, Synthesis of dialkyl, diaryl and metallacyclic complexes of Ni and Pd containing pyridine, alpha-diimines and other nitrogen ligands crystal structures of the complexes cis-NiR2py2 (R = benzyl, mesityl), J. Organomet. Chem., 2003, 683(1), 220–239 CrossRef CAS.
- D. L. Davies, S. A. Macgregor and C. L. McMullin, Computational Studies of Carboxylate-Assisted C–H Activation and Functionalization at Group 8–10 Transition Metal Centers, Chem. Rev., 2017, 117(13), 8649–8709 CrossRef CAS PubMed.
- J. Yamaguchi, A. D. Yamaguchi and K. Itami, C-H Bond Functionalization: Emerging Synthetic Tools for Natural Products and Pharmaceuticals, Angew. Chem., Int. Ed., 2012, 51(36), 8960–9009 CrossRef CAS PubMed.
- S. R. Neufeldt and M. S. Sanford, Controlling Site Selectivity in Palladium-Catalyzed C-H Bond Functionalization, Acc. Chem. Res., 2012, 45(6), 936–946 CrossRef CAS PubMed.
- D. L. Davies, S. M. A. Donald and S. A. Macgregor, Computational Study of the Mechanism of Cyclometalation by Palladium Acetate, J. Am. Chem. Soc., 2005, 127(40), 13754–13755 CrossRef CAS PubMed.
- M. Lafrance, S. I. Gorelsky and K. Fagnou, High-Yielding Palladium-Catalyzed Intramolecular Alkane Arylation: Reaction Development and Mechanistic Studies, J. Am. Chem. Soc., 2007, 129(47), 14570–14571 CrossRef CAS PubMed.
- D. C. Griffiths and G. B. Young, Mechanisms of Thermolytic Rearrangement of Dineophylplatinum(II) Complexes via Intramolecular C-H Activation, Organometallics, 1989, 8(4), 875–886 CrossRef CAS.
- M. Gomez-Gallego and M. A. Sierra, Kinetic Isotope Effects in the Study of Organometallic Reaction Mechanisms, Chem. Rev., 2011, 111(8), 4857–4963 CrossRef CAS PubMed.
- R. H. Grubbs and G. W. Coates, α-Agostic interactions and olefin insertion in metallocene polymerization catalysts, Acc. Chem. Res., 1996, 29(2), 85–93 CrossRef CAS.
- M. H. Prosenc, C. Janiak and H. H. Brintzinger, Agostic Assistance to Olefin Insertion in Alkylzirconocene Cations - a Molecular-Orbital Study by the Extended Huckel-Method, Organometallics, 1992, 11(12), 4036–4041 CrossRef CAS.
- D. Lapointe and K. Fagnou, Overview of the Mechanistic Work on the Concerted Metallation–Deprotonation Pathway, Chem. Lett., 2010, 39(11), 1118–1126 CrossRef.
- L. Boisvert and K. I. Goldberg, Reactions of Late Transition Metal Complexes with Molecular Oxygen, Acc. Chem. Res., 2012, 45(6), 899–910 CrossRef CAS PubMed.
- K. M. Gligorich and M. S. Sigman, Recent advancements and challenges of palladium(II)-catalyzed oxidation reactions with molecular oxygen as the sole oxidant, Chem. Commun., 2009,(26), 3854–3867 RSC.
- L. Boisvert, M. C. Denney, H. S. Kloek and K. I. Goldberg, Insertion of Molecular Oxygen into a Palladium(II) Methyl Bond: A Radical Chain Mechanism Involving Palladium(III) Intermediates, J. Am. Chem. Soc., 2009, 131(43), 15802–15814 CrossRef CAS PubMed.
- M. L. Scheuermann, D. W. Boyce, K. A. Grice, W. Kaminsky, S. Stoll, W. B. Tolman, O. Swang and K. I. Goldberg, Oxygen-Promoted C-H Bond Activation at Palladium, Angew. Chem., Int. Ed., 2014, 53(25), 6492–6495 CrossRef CAS PubMed.
- V. Dura-Vila, D. M. P. Mingos, R. Vilar, A. J. P. White and D. J. Williams, Insertion of O-2 into a Pd(I)-Pd(I) dimer and subsequent C-O bond formation by activation of a C-H bond, Chem. Commun., 2000,(16), 1525–1526 RSC.
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Fengzhi
Tang
b,
Dae Young
Bae
a,
Fengzhi
Tang
b,
Dae Young
Bae