
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFine-tuning the sequential drug release of nano-formulated mutual prodrugs dictates the combination effects†
Haiping
Zhong
a,
Xingwei
Li
a,
Na
Yu
a,
Xi
Zhang
a,
Jingqing
Mu
a,
Tao
Liu
a,
Bo
Yuan
bc,
Xiaoyong
Yuan
bcd and
Shutao
Guo
 *a
*a
aKey Laboratory of Functional Polymer Materials of Ministry of Education, State Key Laboratory of Medicinal Chemical Biology, Frontiers Science Center for New Organic Matter, College of Chemistry, Nankai University, Tianjin, 300071, China. E-mail: stguo@nankai.edu.cn
bTianjin Key Laboratory of Ophthalmology and Visual Science, Tianjin Eye Institute, Tianjin Eye Hospital, Tianjin, 300020, China
cSchool of Medicine, Nankai University, Tianjin, 300071, China
dClinical College of Ophthalmology, Tianjin Medical University, Tianjin, 300052, China
First published on 17th March 2023
Abstract
The maintenance of robust ratiometric loading of dual therapeutic agents and fine-tuning release kinetics for consistent in vitro and in vivo optimization of combination effects is vital for discovering new anticancer drug combinations and remains challenging. Smart nanomedicine strategies have been investigated for this purpose, but most of the reported strategies focus either on ratiometric delivery or on unimodal sequential release of the two different agents, which hampers effective optimization of combination effects. Herein we report a sequential drug release system based on nanoformulated mutual prodrugs constructed by the formation of ketal linkages with different acid sensitivities, thus enabling the acid-triggered release of two anticancer drugs, paclitaxel and gemcitabine, in various sequences. We found that in several cell lines, the sequence of drug release substantially affected the combination effects; specifically, in A549 cells, time-staggered release profiles showed enhanced synergistic effects relative to those of a simultaneous release profile. Moreover, in vivo assessment of the antitumor efficacy of the nanoformulations in A549 xenograft models indicated that the best therapeutic effects were obtained with time-staggered release profiles, which was consistent with the in vitro results. Our strategy for precisely controlled sequential drug release can be expected to facilitate the screening of optimal drug combinations and maximize combination effects both in vitro and in vivo.
Introduction
Drug combinations have successfully been used to improve therapeutic efficacy, overcome multidrug resistance, and reduce systemic toxicity,1,2 but the complexities of signaling pathways and the genetic diversity of cancer require precisely defined combinatorial regimens.3 Combinations of drugs can act synergistically, additively, or antagonistically, depending on the dosage, drug ratio, and administration schedule.4 Discovering synergistic drug combinations by in vitro screening of large libraries of drug combinations is a powerful approach for identifying effective combinatorial therapies, and genomic screening using CRISPR technology has recently been exploited for high-throughput in vitro evaluation of synergistic drug combinations for cancer treatment.5 However, the variation in the pharmacokinetics of different drugs makes it very difficult to deliver drugs to the targeted tissue at predefined ratios and in the desired sequence using the current regimens, which hampers optimization of combination effects. Compared to in vitro screening of multidrug treatments, in vivo screening has been much less thoroughly investigated. In a recent study, Gray's group used an implantable microdevice to identify optimal combination effects upon administration of multiple drugs at different sites in tumors to investigate the response of tumor cells to various treatment regimens.6 Although this microdevice allows for in vivo screening for effective drug combinations, the release profiles of the drugs were not explored, and the need for implantation limits the device's utility for certain types of tumors.Nanotechnology has enabled the systemic delivery of multiple drugs with various properties to maximize the efficacy of combination therapies.7–9 For example, Vyxeos, a liposomal formulation co-loaded with cytarabine and daunorubicin at a fixed ratio of 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, enables co-delivery of both drugs to kill cancer cells; in a phase III clinical trial among patients with acute myeloid leukemia, Vyxeos extended patient survival by 6–10 months compared to standard treatments.10 However, formulations such as this one focus mainly the co-loading of the drugs into the carrier without considering their controlled release. The order in which drugs are presented to cancer cells has been shown to play an important role in combination effects. In a representative study, Yaffe's group investigated the influence of the sequence of application of the epidermal growth factor receptor inhibitor erlotinib and the topoisomerase inhibitor doxorubicin on the response of cancer cells and found that pretreatment with erlotinib significantly enhanced the cells' response to doxorubicin. This result emphasizes the importance of time-staggered application of combination treatments for enhancing anticancer effects.11 Current strategies for controlling drug release sequence rely on adjustment of the administration schedule,12 passive diffusion of two drugs from carriers,13 or the release of each drug by means of various stimuli.14–18 For example, Morton et al. used a liposomal delivery platform for time-staggered release of erlotinib and doxorubicin on the basis of the differences in their hydrophilicities.19 The existing strategies focus mainly on a single release profile, whereby one drug is released prior to the other drug; and achieving well-defined drug release in different sequences is challenging. Moreover, loading of drugs with different properties is difficult to control with delivery systems such as liposomes or micelles, and consistency between in vitro and in vivo drug loading and release profiles is poor. The interactions between drugs in living systems are complex, and the development of a delivery system that allows robust ratiometric loading of multiple therapeutic agents and fine-tuning of their release kinetics both in vitro and in vivo can be expected to facilitate optimization of formulations for precision cancer therapy and is therefore urgently needed.
:1, enables co-delivery of both drugs to kill cancer cells; in a phase III clinical trial among patients with acute myeloid leukemia, Vyxeos extended patient survival by 6–10 months compared to standard treatments.10 However, formulations such as this one focus mainly the co-loading of the drugs into the carrier without considering their controlled release. The order in which drugs are presented to cancer cells has been shown to play an important role in combination effects. In a representative study, Yaffe's group investigated the influence of the sequence of application of the epidermal growth factor receptor inhibitor erlotinib and the topoisomerase inhibitor doxorubicin on the response of cancer cells and found that pretreatment with erlotinib significantly enhanced the cells' response to doxorubicin. This result emphasizes the importance of time-staggered application of combination treatments for enhancing anticancer effects.11 Current strategies for controlling drug release sequence rely on adjustment of the administration schedule,12 passive diffusion of two drugs from carriers,13 or the release of each drug by means of various stimuli.14–18 For example, Morton et al. used a liposomal delivery platform for time-staggered release of erlotinib and doxorubicin on the basis of the differences in their hydrophilicities.19 The existing strategies focus mainly on a single release profile, whereby one drug is released prior to the other drug; and achieving well-defined drug release in different sequences is challenging. Moreover, loading of drugs with different properties is difficult to control with delivery systems such as liposomes or micelles, and consistency between in vitro and in vivo drug loading and release profiles is poor. The interactions between drugs in living systems are complex, and the development of a delivery system that allows robust ratiometric loading of multiple therapeutic agents and fine-tuning of their release kinetics both in vitro and in vivo can be expected to facilitate optimization of formulations for precision cancer therapy and is therefore urgently needed.
Herein, we present a novel nanoformulation that can sequentially release two anticancer drugs, paclitaxel (PTX) and gemcitabine (Gem), at a fixed delivery ratio and in different sequences determined by ketal linkages with tunable hydrolysis rates (Scheme 1). PTX and Gem are widely used and are often administered together in the clinic.20,21 It has been suggested that their administration sequence affects their combination effects.12 Thus, in this proof-of-concept study, we conjugated PTX and Gem to form mutual ![[P with combining low line]](https://www.rsc.org/images/entities/char_0050_0332.gif) TX–
TX–![[G with combining low line]](https://www.rsc.org/images/entities/char_0047_0332.gif) em prodrugs (PGs), which were then formulated into nanoparticles (designated PG NPs). The hydroxyl groups of PTX and Gem were linked via asymmetric ketal bonds with different distances from electron-withdrawing ester bonds. In this way, three release sequence profiles were realized: Gem released faster than PTX, Gem released slower than PTX, or Gem and PTX released simultaneously. Ketal bonds are sensitive to mildly acidic conditions but are stable under normal physiological conditions, which guaranteed the in vivo stability of the PG NPs during circulation. The cytotoxicities and synergistic effects of the formulations in various cancer cell lines depended on the release profile. More important, the combination effects of the formulations in a single cell line also depended on the release profile. Moreover, an in vivo A549 tumor model showed varied response rates to the PG NPs that were correlated with the varied synergistic effects observed the in vitro cytotoxicity experiments. Our nanoformulation enables the fine-tuned delivery of two drugs and thus provides the opportunity to optimize the sequential release of therapeutic agents and maximize synergistic effects in both in vitro and in vivo studies. More important, our platform will inspire the exploitation of novel prodrug technologies, in vitro screening of drug combinations, and nanotechnology for developing more efficacious combinations.
em prodrugs (PGs), which were then formulated into nanoparticles (designated PG NPs). The hydroxyl groups of PTX and Gem were linked via asymmetric ketal bonds with different distances from electron-withdrawing ester bonds. In this way, three release sequence profiles were realized: Gem released faster than PTX, Gem released slower than PTX, or Gem and PTX released simultaneously. Ketal bonds are sensitive to mildly acidic conditions but are stable under normal physiological conditions, which guaranteed the in vivo stability of the PG NPs during circulation. The cytotoxicities and synergistic effects of the formulations in various cancer cell lines depended on the release profile. More important, the combination effects of the formulations in a single cell line also depended on the release profile. Moreover, an in vivo A549 tumor model showed varied response rates to the PG NPs that were correlated with the varied synergistic effects observed the in vitro cytotoxicity experiments. Our nanoformulation enables the fine-tuned delivery of two drugs and thus provides the opportunity to optimize the sequential release of therapeutic agents and maximize synergistic effects in both in vitro and in vivo studies. More important, our platform will inspire the exploitation of novel prodrug technologies, in vitro screening of drug combinations, and nanotechnology for developing more efficacious combinations.
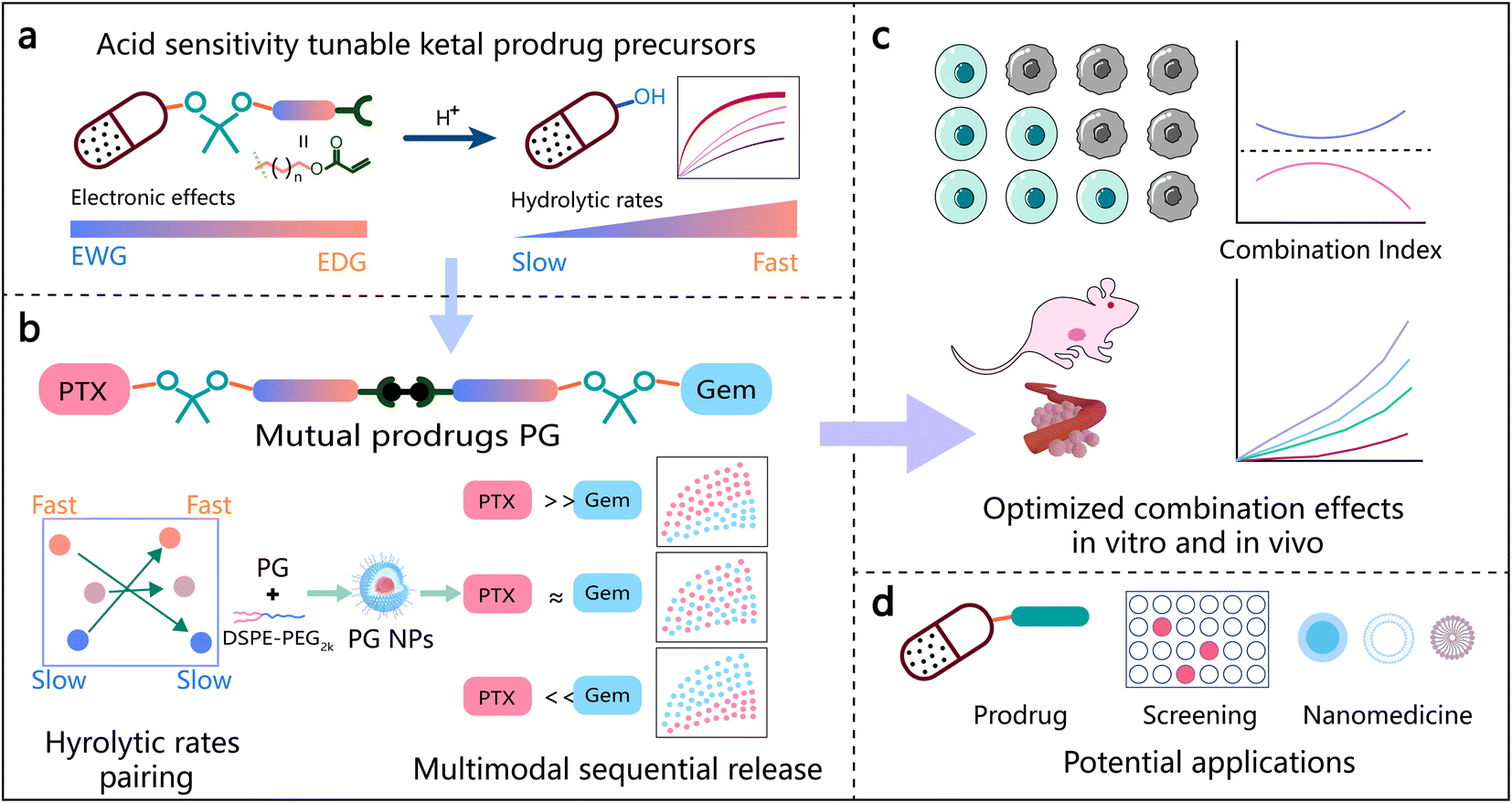 | ||
| Scheme 1 Design and synergistic antitumor effects of mutual ketal prodrug precursors and their nanoformulations that allow predefined sequential release of paclitaxel (PTX) and gemcitabine (Gem). (a) The substituent groups of ketal prodrug precursors influence the hydrolytic rates of drugs. EWG: electron-withdrawing groups; EDG: electron-donating groups. (b) Pairing of different prodrug precursors to construct mutual prodrugs with different release sequences of two drugs. (c) Sequential release of PTX and Gem influences the combination effects both in vitro and in vivo. (d) Potential applications of this sequential release delivery platform. | ||
Results and discussion
Rational design of mutual ketal prodrugs
We reasoned that a mutual prodrug approach involving tunable stimulus-responsive linkers would allow for ratiometric, sequential release of therapeutic molecules. The mutual prodrug approach, which would allow us to maintain a fixed drug ratio, has been used to enhance therapeutic efficacy, but controlling the release profiles of different drugs by means of this approach has rarely been achieved.22 Recently, ketals have emerged as promising linkers for prodrugs.23,24 Because ketal bonds are sensitive to acidic environments but are stable under mildly basic conditions, they are ideal linkers for acid-responsive drug release. What is more, the acid sensitivity of ketal bonds can readily be tuned by adjusting the substituents adjacent to the ketal bond, which offers the possibility for constructing prodrugs with various release profiles.25 Thus, in this proof-of-concept study, we used an acetone-based ketal linkage to construct acid-sensitive mutual prodrugs.The hydrolysis rate of ketals is significantly influenced by the electronic effects of substituents and will be decreased by electron-withdrawing groups and increased by electron-donating groups.26 Given acrylate group can easily react with dithiols via Michael addition to afford asymmetric mutual prodrugs, we chose acrylate compounds to function as electron-withdrawing groups to construct ketal prodrug precursors with various hydrolysis rates (Scheme 1a). We varied the distance between the ester bond and the ketal moiety by changing the number of saturated carbon atoms between them. Then the prodrug precursors with different hydrolytic rates were paired to form mutual prodrugs through conjugation with dithiols. By this way, three modes of release sequences can be achieved (Scheme 1b).
Synthesis of ketal prodrug precursors with various hydrolysis rates
Using our well-established ketal prodrug synthetic chemistry,23 we first synthesized a set of ketal prodrug precursors (Fig. 1, S1–S19, Schemes S1 and S3†) and then investigated their hydrolysis rates in buffer containing 40% acetonitrile (Fig. S35†). Specifically, Gem derivatives bearing alkyl chains of various lengths (n = 2, 3, 4, 6) between the ketal bond and the electron-withdrawing ester group were prepared: the resulting prodrug precursors are designated G-2 (n = 2), G-3 (n = 3), G-4 (n = 4), and G-6 (n = 6), respectively (Fig. 1a). Their t1/2 s at pH 5.0 ranged from 9.2 to 96.3 min and increased with decreasing chain length (Fig. 1c). Three PTX ketal prodrug precursors were prepared for pairing Gem precursors (Fig. 1b). The 2′-OH of PTX was conjugated with 6-hydroxyhexyl prop-2-enoate to form P2′-6, which had a much longer t1/2 at pH 5.0 (57.3 min) than the corresponding Gem precursor, G-6. To increase the hydrolysis rates of the PTX prodrug precursors, we then conjugated the 7-OH of PTX with 6-hydroxyhexyl prop-2-enoate to form ketal prodrug precursor P7-6 and with the isopropenyl ether of 4-hydroxycyclohexyl acrylate to form P7-C6. As shown in Fig. 1c, the t1/2 s of P7-6 and P7-C6 were 37 and 10.2 min, similar to the values for G-3 (30.1 min) and G-6 (9.2 min), respectively. The different hydrolysis rates of these prodrug precursors suggested that a mutual prodrug system for sequential drug release could be constructed by choosing ketal prodrug precursors with an appropriate combination of hydrolysis rates. | ||
| Fig. 1 Construction of PTX and Gem precursors with various hydrolytic rates. (a) Ketal prodrug precursors of Gem. (b) Ketal prodrug precursors of PTX. Various carbon chain lengths were used to adjust the distance between ketal bonds and electron-withdrawing acrylates. (c) Half-lives (t1/2 s) of ketal prodrug precursors at acidic buffer conditions (pH = 5.0) based on pseudo-first-order kinetics, determined by HPLC (n = 4). | ||
Preparation of mutual ketal prodrug NPs with adjustable release sequence profiles
With the goal of achieving sequential release of the two drugs, we began by conjugating P7-6 with G-6, G-3, and G-2 to form the corresponding mutual PTX-Gem prodrugs (PGs) because the t1/2 of P7-6 was intermediate between those of the three Gem precursors. The resulting mutual prodrugs are designated PG1, PG2, and PG3, respectively (Table 1). The acrylate group of the above-described precursors was used to conjugate them to 1,2-ethanedithiol by means of a Michael addition to form mutual ketal prodrugs (Schemes S4, S5 and Fig. S20–S27†). Because Gem is hydrophilic and PTX is hydrophobic, we anticipated that these amphiphilic PGs would self-assemble into stable NPs.27 However, after several attempts, we were unable to obtain the desired nanoformulations by using the prodrugs alone. Because we suspected that the hydrophilic–hydrophobic balance of the prodrugs was unsuitable for self-assembly, we added the amphiphilic phospholipid 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy(polyethylene glycol)-2000] (DSPE-PEG2k) to facilitate the formation of stable NPs (Fig. 2a). To maintain a relatively high drug content, we added only small amounts of DSPE-PEG2k (10 wt% relative to PG). Liquid 1H NMR has been utilized to analyze the core shell structures of amphiphilic self-assemblies,28 thus we applied this technology to study PG1 NPs in D2O, and the results suggested that hydrophilic Gem was distributed at the surface of the NPs. We observed a peak at 3.73 ppm, which was attributed to Gem; and the peaks at 3.46–3.70 ppm were attributed to the PEG segments (Fig. 2b). The theoretical integration ratio of the Gem and PEG segments is 1:10, and the observed ratio was close to this value. In addition, because the hydrophobic PTX was embedded in the core of the NPs, no PTX peaks were observed in the liquid 1H NMR spectrum.
| PG1 (PsGf) NPsa | PG2 (PiGi) NPsa | PG3 NPs | PG4 (PfGs) NPsa | |
|---|---|---|---|---|
| a The subscripts “f”, “s”, and “i” refer to “fast”, “slow”, and “intermediate” release, respectively. b The value of t1/2 s were calculated according to pseudo-first-order kinetics. c Calculated from the sum of the weights of Gem and PTX (determined by HPLC) relative to the total weight of the NPs. d Sizes, PDI and ζ potential were determined by dynamic light scattering (DLS, mean ± SD, n = 3). Sizes were determined by intensity values. e Determined from transmission electron microscopy (TEM) images by measuring the sizes of particles in different fields. PG3 NPs were not further used thus were not characterized by TEM. | ||||
| PTX precursor | P7-6 | P7-6 | P7-6 | P7-C6 |
| Gem precursor | G-6 | G-3 | G-2 | G-2 |
| t 1/2 of PTX at pH 5.0b (min) | 206.9 | 154.0 | 176.2 | 62.5 |
| t 1/2 of Gem at pH 5.0b (min) | 64.0 | 169.1 | 150.3 | 181.7 |
| Drug contentc (wt%) | 47.5 | 48.9 | 49.3 | 47.8 |
| Size by DLSd (nm) | 110.2 ± 5.7 | 138.9 ± 4.7 | 130.3 ± 2.1 | 111.3 ± 5.6 |
| PDId | 0.11 ± 0.02 | 0.14 ± 0.02 | 0.13 ± 0.02 | 0.13 ± 0.03 |
| ζ potentiald | −14.4 ± 1.2 | −2.4 ± 0.2 | −6.4 ± 0.7 | −15.1 ± 1.1 |
| Size by TEMe | 104.9 ± 20.0 | 93.6 ± 16.5 | — | 88.8 ± 14.5 |
 | ||
| Fig. 2 Preparation and characterization of PG NPs. (a) Illustration of PG NP formation by nanoprecipitation of PG prodrug and DSPE-PEG2k. (b) Liquid 1H NMR spectrum of PG1 NPs in D2O indicates the hydrophilic PEG segments and Gem are distributed on the surface of PG NPs. (c) Hydrolysis kinetics of PG NPs at pH 5.0 (n = 4). The comsumption of PG prodrugs and the release of Gem and PTX were monitored by HPLC. (d) Transmission electron microscopy images of PG NPs obtained by means of negative staining (scale bar: 100 nm), along with average sizes measured from different scopes. | ||
To examine the drug release profiles of the PG NPs, PG degradation and Gem and PTX release in pH 5.0 or 7.4 buffer were monitored by HPLC (Fig. 2c, S36, and S37†). Acid-triggered hydrolysis of the PG NPs obeyed pseudo-first-order kinetics. The t1/2 s of Gem and PTX were calculated accordingly. As expected, Gem was released faster than PTX from the PG1 NPs (the corresponding t1/2 s were 64.0 and 206.9 min), and Gem and PTX were released simultaneously from the PG2 NPs (169.1 and 154.0 min, respectively) (Table 1). However, the results for the PG3 NPs were unexpected: Gem was released almost simultaneously with PTX rather than slower than PTX, as would be predicted from their t1/2 s (150.3 and 176.2 min). Notably, the rates for PG NP hydrolysis in the pH 5.0 and 7.4 buffers were all slower than the rates for the corresponding prodrug precursors in buffers containing 40% acetonitrile. Because the PTX was embedded in the hydrophobic core of the PG NPs, we deduced that PTX release from the PG3 NPs was slowed by the delayed exposure of ketal bonds in the hydrophobic cores to protons.
To obtain a release profile that was opposite that of the PG1 NPs, we further conjugated P7-C6, which had faster release kinetics, with G-2 to form a prodrug designated PG4 (Table 1, Fig. S28 and S29†); and then PG4 NPs were prepared in the manner described for the other PG NPs. The hydrolytic profile of PG4 NPs at pH 5.0 was studied as well, and Gem release was found to be slower than PTX release (the t1/2 s were 181.7 and 62.5 min, respectively) (Fig. 2c and Table 1). Overall, HPLC confirmed that Gem and PTX were released at different rates from these PG NPs. Thus, we were able to generate NPs of mutual ketal prodrugs with three different sequences for Gem and PTX release by fine-tuning the ketal prodrug precursors and appropriately combining the precursors; specifically, two types of NPs showed time-staggered Gem and PTX release profiles (Gem then PTX or PTX then Gem) and one type released the two drugs simultaneously.
All the PG NPs were similar in size (110–140 nm determined by intensities from dynamic light scattering) and showed narrow size distributions with small polydispersity index values, as determined by dynamic light scattering and transmission electron microscopy (Fig. 2d, S38† and Table 1). The sizes and polydispersity index values remained almost unchanged upon storage for up to 5 days at 4 °C, indicating the good stability of these NPs (Fig. S38d†). Moreover, because of the small amounts of other ingredients added to the NP formulations, they all exhibited drug contents close to 50 wt%, calculated from the sum of the weights of Gem and PTX (as determined by HPLC after complete release of the native drugs from the PG NPs) relative to the total weight of the NPs (Table 1).
To better reveal the different release profiles, these three types of PG NPs are hereafter referred to as PsGf NPs (PG1 NPs), PiGi NPs (PG2 NPs) and PfGs NPs (PG4 NPs), where the subscripts “f”, “s”, and “i” refer to “fast”, “slow”, and “intermediate” release, respectively.
Drug release profile of PG NPs under cell culture conditions
We investigated the drug release profiles of PG NPs under in vitro cell culture conditions. After incubation of PG NPs with A549 human lung adenocarcinoma cells for 6 h, the microtubules were stained with α-tubulin red. The PfGs NPs showed a faster intracellular PTX release profile than the other two PG formulations and inhibited microtubule depolymerization to a greater extent (Fig. 3a). Gem has a relative short plasma circulation time, therefore it is not feasible to detect the release of Gem under cell culture conditions.29 To track Gem release, we used the nucleoside analog 5-ethynyl-2′-deoxyuridine (EdU) to replace Gem, and we generated PE prodrugs to mimic the PG prodrugs. Like Gem, EdU can be incorporated into DNA, and the alkyl group of EdU can undergo a click reaction with a fluorescent azide dye, allowing us to visually detect nucleotide analog incorporation into the nuclei of cells (Fig. S39a†).30 Similar to Gem precursors, we generated EdU precursors E-2, E-3, and E-6, respectively (Scheme S2 and Fig. S30–S32†). To prove the various release rates of Gem from PG NPs, we intended to synthesized PE1 (from P7-6 and E-6), PE2 (from P7-6 and E-3), and PE4 (from P7-C6 and E-2) to mimic the corresponding Gem precursors. However, the synthesis of PE4 failed after several trials. Thus, less-than-ideal alternative, PE1 and PE2 were selected to investigate the different release rates of EdU in cell culture conditions and designated as PEf and PEs (Fig. S33, S34 and S39b†). PEf and PEs prodrugs were synthesized and formulated as PE NPs in the same manner as described for the PG NPs. | ||
| Fig. 3 Varied drug release rates of PTX/EdU from PG NPs/PE NPs under cell culture conditions. (a) Inhibition of microtubule depolymerization by PG NPs in A549 cells. Blue color: nucleus stained by DAPI; red color: microtubule stained by α-tubulin red. Lower right panel shows fluorescence intensity profile crossing a 10 μm length. The differences in shrinkage degree of microtubules indicate the difference in release rates from PG NPs. (b) Distribution of 5-ethynyl-2′-deoxyuridine (EdU) in A2780 cells after incubation with EdU or PE NPs. The green fluorescence of azide-488 overlapping with the blue-stained cell nuclei forms a cyan color, indicating EdU incorporation into the nuclei. The green fluorescence spread to the cytoplasm after treatment with the PEs NPs indicates the rate of EdU release was slower from these PEs NPs. | ||
Then A2780 human ovarian adenocarcinoma cells were incubated with the PE NPs for 6 h and subsequently stained with the fluorescent dye azide-488. As shown in Fig. 3b, the distribution of green fluorescence correlated with the rates of ketal bond hydrolysis. The green fluorescence of azide-488 colocalized with the nucleus indicates that the EdU was released from the NPs and integrated to the DNA of the cell, while the green color spread in the cytoplasm suggests the EdU was not efficiently released from the NPs. The green fluorescence was centralized in the area of the nucleus after incubation with free EdU and the PEf NPs, whereas the fluorescence spread to the cytoplasm after treatment with the PEs NPs, indicating the rate of EdU release was slower from these PEs NPs. The difference between the EdU release rates from the PE NPs suggests that the Gem release profile from the PG NPs would be similar under cell culture conditions. Taken together, these results confirm that we could achieve intracellularly sequential release of PTX and Gem by fine-tuning the ketal prodrug structure.
Effect of sequence of Gem and PTX release from PG NPs on combination effects
The administration schedule-dependent cytotoxicities of PTX and Gem have been reported by several research groups,12,31 and the results reveal the important relationship between the administration order of the two drugs and their synergistic effects. In this study, we measured the cytotoxicities of three PG NPs in different cell lines to elucidate the relationship between release sequence and combination effects. Because the PTX/Gem molar ratio was fixed at 1:1 in the PG NPs, we used an equimolar mixture of free PTX and Gem as a control (designated PTX + Gem). Three different cancer cell lines were incubated with PG NPs, free PTX, free Gem, or PTX + Gem (Fig. 4a–c).
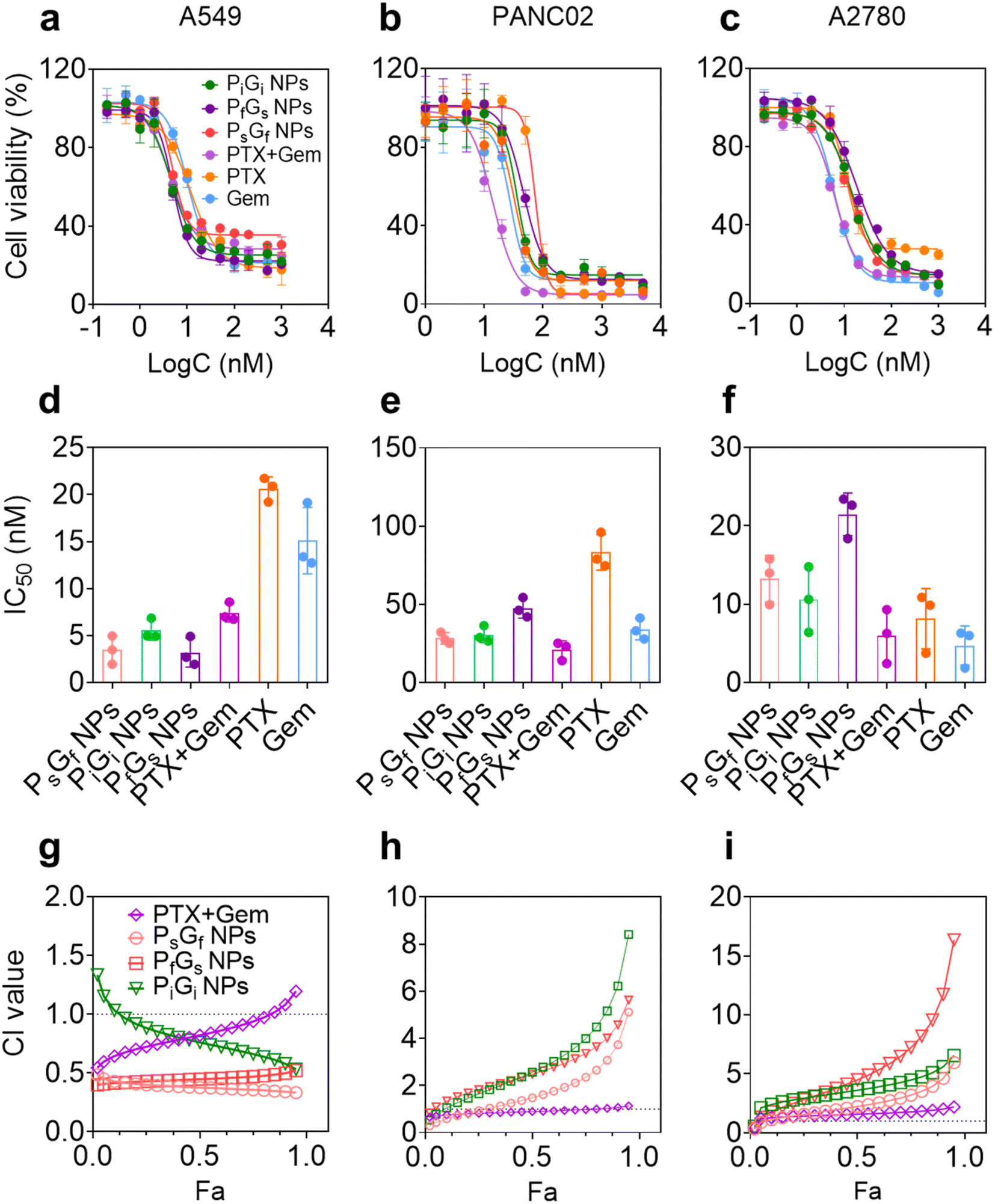 | ||
| Fig. 4 Cytotoxicities and combination effects of PG NPs in (a) A549, (b) PANC02, and (c) A2780 cells. 5 × 103 of the three cell lines seeded in 96 plates were incubated with PG NPs or free drugs for 72 h, and the cell viabilities were determined by CCK-8. Values are mean ± SD (n = 3). (d–f) IC50 values for PG NPs and free drugs determined using the dose–response-inhibition model in GraphPad Prism ver.8.0. Values are mean ± SD (n = 3). (g–i) Combination effects of PG NPs or PTX + Gem were determined from data of cell viabilities. CI values of 0.9–1.1 indicate additivity, values of <0.9 indicate synergism, and values of >1.1 indicate antagonism. Fa means fraction affected. Combination index (CI) values were calculated using CalcuSyn ver.2.0. | ||
The cytotoxicities of the PG NPs to the three cell lines differed (Fig. 4d–f and Table S1†). In A549 cells, PG NPs and PTX + Gem showed lower IC50 values than free PTX and Gem (Fig. 4d). Although the IC50 values of different formulations showed approximate results, the combination effects varied to each other. Combination effects were analyzed by using CalcuSyn 2.0 to calculate combination index (CI) values. Generally, CI values of 0.9–1.1 indicate additivity, values of <0.9 indicate synergism, and values of >1.1 indicate antagonism. As shown in Fig. 4g, PsGf NPs and PfGs NPs showed strong synergies: all the CI values were <0.5 in the range of fractions affected. In contrast, the CI values for the PiGi NP and PTX + Gem groups were similar to one another, being <1.0 in the partial range of fractions affected. Because the molar ratio of the two drugs was fixed at 1:1, the sequence of PTX and Gem release played major role in the combination effects, and the NPs with the time-staggered release profiles showed greater synergism than the NPs with the simultaneous release profile. However, the combination effects observed in the other two cell lines differed from those observed in the A549 cell line. In PANC02 mouse pancreatic adenocarcinoma cells, the IC50 values of all the PG NPs were higher than the value for free Gem (Fig. 4e). As a result, all the calculated CI values were >1.0, indicating antagonistic effects (Fig. 4h). Nevertheless, the CI values of the PfGs NPs were lower than those of the PsGf NPs and PiGi NPs. Similar results were observed in A2780 cells (Fig. 4f and i), and it should be noted that the CI values for the PTX + Gem group also exceeded 1.0, indicating antagonistic effects. Thus, the combination effects exhibited by the PG NPs depended on cell types.
Validation of combination effects of PG NPs by means of in vivo antitumor efficacy studies
Encouraged by the synergistic effects of the PG NPs in A549 cells, we evaluated their in vivo combination effects in female nude mice bearing A549 human lung adenocarcinoma xenografts (Fig. 5). A relatively low PTX dose (20 mg kg−1) was chosen to evaluate the therapeutic effects, and the Gem dose was 6.4 mg kg−1. For convenience of administration, a micellar PTX formulation (designated PTX Mic) was used. Three doses of each formulation were intravenously administered mice on days 0, 3, and 6, respectively. As shown in Fig. 5a, a low dose of Gem or PTX Mic failed to effectively suppress tumor growth in the xenografted mice. Simultaneously administered PTX Mic and Gem only moderately inhibited tumor growth. In contrast, all three PG NPs showed enhanced antitumor effects relative to the above-mentioned treatments, although the PiGi NPs, which released the two drugs simultaneously, showed slightly weaker combination effects than the PsGf NPs and PfGs NPs. The weights of tumors resected at the end of treatment also indicated that the PsGf NPs and PfGs NPs showed better tumor growth inhibition than the PiGi NPs (Fig. 5b). Ki67, hematoxylin and eosin (H&E), and terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) immunohistochemical staining confirmed this pattern (Fig. 5c, d and S40†). Notably, although PTX + Gem showed synergism similar to that observed for the PiGi NPs in vitro, the in vivo antitumor efficacy of simultaneously administered PTX Mic + Gem was not as good as that of the PiGi NPs. What is more, the consistent trends of in vitro inhibition of A549 cell proliferation and in vivo suppression of A549 xenografted tumor growth by the three PG NPs demonstrates that the release profile and synergism of the two drugs were maintained both under cell culture conditions and in living systems. We further evaluated the safety of the PG NPs on the basis of body weight changes, blood tests, and histological analysis. The body weights of all the mice slightly increased during treatment (Fig. S41†), and the mice remained healthy. In addition, H&E staining of major organs show no obvious tissue damage or anomalous biochemical indicators in any of the groups (Fig. S42 and S43†).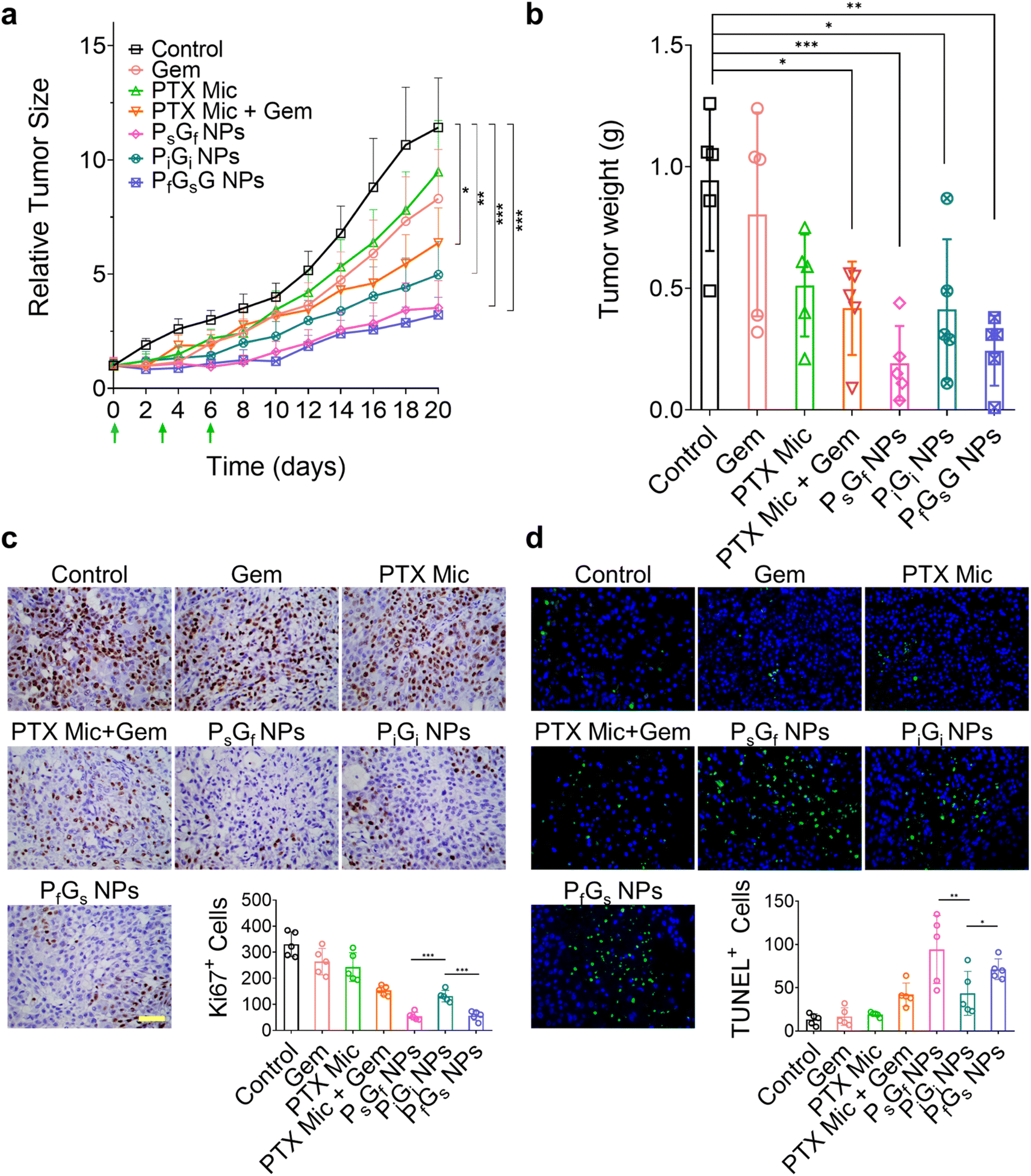 | ||
| Fig. 5 Enhanced antitumor efficacy of PG NPs in A549 human lung adenocarcinoma xenografts. (a) A549 tumor growth in mice treated with various formulations. Relative tumor size was determined by dividing the tumor size on a given day by the starting tumor size. Green arrows indicate the injection of formulations at day 0, day 3, and day 6, respectively. Data are mean ± SD (n = 5). (b) Tumor weights at end of treatment. At day 20, mice were sacrificed and tumor tissues were cut from mice and weighed. (c) Ki67 staining of tumor tissues collected from mice after treatment. The lower right corner graph demonstrates the positive Ki67 positive cells counted from different scopes under microscopy. (d) TUNEL staining of tumor tissues collected from mice after treatment. The lower right corner graph demonstrates the TUNEL positive cells counted from different scopes under microscopy. Blue color: nucleus stained by DAPI; red color: cleaved DNA stained by FITC. Scale bars: 50 μm. The unpaired two-tailed Student's t-test was used for statistical analysis, *P < 0.05, **P < 0.01, and ***P < 0.001. | ||
Conclusion
As understanding of the interactions between drugs and living systems improves, methods for systematically identifying synergistic combinations of multiple therapeutic agents will be urgently needed for precision cancer therapy. The currently available nanotechnological strategies for optimizing combination effects include maintaining predefined drug ratios7 and releasing drugs in a unimodal sequence.19 Although the influences of drug ratio and drug release sequence on combination effects have been widely investigated, and strategies for controlling drug ratios are plentiful, approaches for fine-tuning the release of different drugs are lacking, in that all of the strategies developed to date are limited to a unimodal release profile.We have designed a nanoformulated mutual prodrug release system that allowed us to sequentially release paclitaxel and gemcitabine by tuning the rate of hydrolysis of a ketal linkage. Three different release sequences of the two drugs were accomplished by rationally varying the acid sensitivity of the ketal linkage. The sequence of paclitaxel and gemcitabine release significantly influenced their combination effects, as indicated by the variation of CI values observed for the PG NPs in different cell lines. The enhanced in vivo antitumor efficacy of time-staggered release profile relative to simultaneously released profile in an A549 tumor model further verified the importance of release sequence. Although quantifying each drug in tumors and tissues is essential for co-delivery, the complicated metabolism of Gem in vivo restricts the validation of the spatiotemporal delivery of drugs after administration with fidelity. Overall, our sequential release system could provide new opportunities for investigating the correlations between drug release sequence and combination effects and could facilitate the discovery of optimal formulations for precision cancer therapy. However, our platform does have some limitations. For instance, the ketal linkage can be used only with drugs containing hydroxyl groups, and the drug ratio cannot be readily adjusted. Nevertheless, by varying the drug linkers and valency, libraries of mutual prodrugs and their combinations can be synthesized in parallel for more extensive scope optimization. We expect that our platform will inspire the exploitation of novel prodrug technologies, drug combinations, and nanotechnology for developing more efficacious combinations.
Data availability
ESI† is available including detailed experimental methods, 1H NMR and 13C NMR spectra, and other relevant figures related to this article.Author contributions
Shutao Guo and Haiping Zhong designed this study. Haiping Zhong did most parts of this work. Xingwei Li helped to carry out the in vivo anticancer and safety evaluation experiments. Xi Zhang carried out the cytotoxicity experiments. The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (no: 32171386 and 32201157), the Natural Science Foundation of Tianjin of China (no: 21JCZDJC01250), the Tianjin Key Medical Discipline (Specialty) Construction Project (TJYXZDXK-016A), and the China Postdoctoral Science Foundation (no: 2021M690793).References
- A. Wang-Gillam, C.-P. Li, G. Bodoky, A. Dean, Y.-S. Shan, G. Jameson, T. Macarulla, K.-H. Lee, D. Cunningham, J. F. Blanc, R. A. Hubner, C.-F. Chiu, G. Schwartsmann, J. T. Siveke, F. Braiteh, V. Moyo, B. Belanger, N. Dhindsa, E. Bayever, D. D. Von Hoff, L.-T. Chen, C. Adoo, T. Anderson, J. Asselah, A. Azambuja, C. Bampton, C. H. Barrios, T. Bekaii-Saab, M. Bohuslav, D. Chang, J.-S. Chen, Y.-C. Chen, H. J. Choi, I. J. Chung, V. Chung, T. Csoszi, A. Cubillo, L. DeMarco, M. de Wit, T. Dragovich, W. Edenfield, L. E. Fein, F. Franke, M. Fuchs, V. Gonzales-Cruz, A. Gozza, R. H. Fernando, R. Iaffaioli, J. Jakesova, Z. Kahan, M. Karimi, J. S. Kim, E. Korbenfeld, I. Lang, F.-C. Lee, K.-D. Lee, L. Lipton, W. W. Ma, L. Mangel, R. Mena, D. Palmer, S. Pant, J. O. Park, P. Piacentini, U. Pelzer, J. G. Plazas, C. Prasad, K.-M. Rau, J.-L. Raoul, D. Richards, P. Ross, L. Schlittler, M. Smakal, V. Stahalova, C. Sternberg, T. Seufferlein, N. Tebbutt, J. J. Vinholes, R. Wadlow, M. Wenczl and M. Wong, Nanoliposomal irinotecan with fluorouracil and folinic acid in metastatic pancreatic cancer after previous gemcitabine-based therapy (NAPOLI-1): a global, randomised, open-label, phase 3 trial, Lancet, 2016, 387(10018), 545 CrossRef CAS PubMed.
- V. Kunzmann, J. T. Siveke, H. Algül, E. Goekkurt, G. Siegler, U. Martens, D. Waldschmidt, U. Pelzer, M. Fuchs, F. Kullmann, S. Boeck, T. J. Ettrich, S. Held, R. Keller, I. Klein, C.-T. Germer, H. Stein, H. Friess, M. Bahra, R. Jakobs, I. Hartlapp, V. Heinemann, E. Hennes, U. Lindig, T. Geer, M. Stahl, M. Senkal, T. Südhoff, M. Egger, C. Kahl, C. Große-Thie, M. Reiser, S. Mahlmann, P. Fix, H. Schulz, G. Maschmeyer and W. Blau, Nab-paclitaxel plus gemcitabine versus nab-paclitaxel plus gemcitabine followed by FOLFIRINOX induction chemotherapy in locally advanced pancreatic cancer (NEOLAP-AIO-PAK-0113): a multicentre, randomised, phase 2 trial, Lancet Gastroenterol. Hepatol., 2021, 6(2), 128 CrossRef PubMed.
- J. A. Kemp, M. S. Shim, C. Y. Heo and Y. J. Kwon, Combo nanomedicine: Co-delivery of multi-modal therapeutics for efficient, targeted, and safe cancer therapy, Adv. Drug Delivery Rev., 2016, 98, 3 CrossRef CAS PubMed.
- J. Jia, F. Zhu, X. Ma, Z. Cao, Z. W. Cao, Y. Li, Y. X. Li and Y. Z. Chen, Mechanisms of drug combinations: interaction and network perspectives, Nat. Rev. Drug Discovery, 2009, 8(2), 111 CrossRef CAS PubMed.
- K. Han, E. E. Jeng, G. T. Hess, D. W. Morgens, A. Li and M. C. Bassik, Synergistic drug combinations for cancer identified in a CRISPR screen for pairwise genetic interactions, Nat. Biotechnol., 2017, 35(5), 463 CrossRef CAS PubMed.
- Z. Tatarova, D. C. Blumberg, J. E. Korkola, L. M. Heiser, J. L. Muschler, P. J. Schedin, S. W. Ahn, G. B. Mills, L. M. Coussens, O. Jonas and J. W. Gray, A multiplex implantable microdevice assay identifies synergistic combinations of cancer immunotherapies and conventional drugs, Nat. Biotechnol., 2022, 40(12), 1823 CrossRef CAS PubMed.
- F. Yu, Y. Tu, S. Luo, X. Xiao, W. Yao, M. Jiang, X. Jiang, R. Yang and Y. Yuan, Dual-Drug Backboned Polyprodrug with a Predefined Drug Combination for Synergistic Chemotherapy, Nano Lett., 2021, 21(5), 2216 CrossRef CAS PubMed.
- Y. Li, Y. Jiang, Z. Zheng, N. Du, S. Guan, W. Guo, X. Tang, J. Cui, L. Zhang, K. Liu, Q. Yu and Z. Gan, Co-Delivery of Precisely Prescribed Multi-Prodrug Combination by an Engineered Nanocarrier enables Efficient Individualized Cancer Chemotherapy, Adv. Mater., 2022, 34(12), e2110490 CrossRef PubMed.
- X. Xiao, Q. Zong, J. Li and Y. Yuan, Tumor-Selective Cascade-Amplified Dual-Prodrugs Activation for Synergistic Oxidation-Chemotherapy, CCS Chem., 2022, 1, 3878–3888 CrossRef.
- J. E. Lancet, G. L. Uy, J. E. Cortes, L. F. Newell, T. L. Lin, E. K. Ritchie, R. K. Stuart, S. A. Strickland, D. Hogge, S. R. Solomon, R. M. Stone, D. L. Bixby, J. E. Kolitz, G. J. Schiller, M. J. Wieduwilt, D. H. Ryan, A. Hoering, K. Banerjee, M. Chiarella, A. C. Louie and B. C. Medeiros, CPX-351 (cytarabine and daunorubicin) Liposome for Injection Versus Conventional Cytarabine Plus Daunorubicin in Older Patients With Newly Diagnosed Secondary Acute Myeloid Leukemia, J. Clin. Oncol., 2018, 36(26), 2684 CrossRef CAS PubMed.
- M. J. Lee, A. S. Ye, A. K. Gardino, A. M. Heijink, P. K. Sorger, G. MacBeath and M. B. Yaffe, Sequential application of anticancer drugs enhances cell death by rewiring apoptotic signaling networks, Cell, 2012, 149(4), 780 CrossRef CAS PubMed.
- R. Zhang, J. Yang, M. Sima, Y. Zhou and J. Kopecek, Sequential combination therapy of ovarian cancer with degradable N-(2-hydroxypropyl)methacrylamide copolymer paclitaxel and gemcitabine conjugates, Proc. Natl. Acad. Sci. U. S. A., 2014, 111(33), 12181 CrossRef CAS PubMed.
- T. Okuda, K. Tominaga and S. Kidoaki, Time-programmed dual release formulation by multilayered drug-loaded nanofiber meshes, J. Controlled Release, 2010, 143(2), 258 CrossRef CAS PubMed.
- L. Liao, J. Liu, E. C. Dreaden, S. W. Morton, K. E. Shopsowitz, P. T. Hammond and J. A. Johnson, A convergent synthetic platform for single-nanoparticle combination cancer therapy: ratiometric loading and controlled release of cisplatin, doxorubicin, and camptothecin, J. Am. Chem. Soc., 2014, 136(16), 5896 CrossRef CAS PubMed.
- R. K. Pathak and S. Dhar, A Nanoparticle Cocktail: Temporal Release of Predefined Drug Combinations, J. Am. Chem. Soc., 2015, 137(26), 8324 CrossRef CAS PubMed.
- T. Jiang, W. Sun, Q. Zhu, N. A. Burns, S. A. Khan, R. Mo and Z. Gu, Furin-mediated sequential delivery of anticancer cytokine and small-molecule drug shuttled by graphene, Adv. Mater., 2015, 27(6), 1021 CrossRef CAS PubMed.
- J. Huang, Y. Xu, H. Xiao, Z. Xiao, Y. Guo, D. Cheng and X. Shuai, Core-Shell Distinct Nanodrug Showing On-Demand Sequential Drug Release To Act on Multiple Cell Types for Synergistic Anticancer Therapy, ACS Nano, 2019, 13(6), 7036 CrossRef CAS PubMed.
- G. F. Boafo, Y. Shi, Q. Xiao, K. T. Magar, M. Zoulikha, X. Xing, C. Teng, E. Brobbey, X. Li, X. Jiang, X. Wang, Y. Yang, S. Kesse and W. He, Targeted co-delivery of daunorubicin and cytarabine based on the hyaluronic acid prodrug modified liposomes, Chin. Chem. Lett., 2022, 33(10), 4600 CrossRef CAS.
- S. W. Morton, M. J. Lee, Z. J. Deng, E. C. Dreaden, E. Siouve, K. E. Shopsowitz, N. J. Shah, M. B. Yaffe and P. T. Hammond, A nanoparticle-based combination chemotherapy delivery system for enhanced tumor killing by dynamic rewiring of signaling pathways, Sci. Signaling, 2014, 7(325), ra44 CrossRef PubMed.
- D. Goldstein, R. H. El-Maraghi, P. Hammel, V. Heinemann, V. Kunzmann, J. Sastre, W. Scheithauer, S. Siena, J. Tabernero, L. Teixeira, G. Tortora, J. L. Van Laethem, R. Young, D. N. Penenberg, B. Lu, A. Romano and D. D. Von Hoff, nab-Paclitaxel plus gemcitabine for metastatic pancreatic cancer: long-term survival from a phase III trial, J. Natl. Cancer Inst., 2015, 107(2), dju413 CrossRef PubMed.
- K. K. Frese, A. Neesse, N. Cook, T. E. Bapiro, M. P. Lolkema, D. I. Jodrell and D. A. Tuveson, nab-Paclitaxel potentiates gemcitabine activity by reducing cytidine deaminase levels in a mouse model of pancreatic cancer, Cancer Discovery, 2012, 2(3), 260 CrossRef CAS PubMed.
- P. Huang, D. Wang, Y. Su, W. Huang, Y. Zhou, D. Cui, X. Zhu and D. Yan, Combination of small molecule prodrug and nanodrug delivery: amphiphilic drug-drug conjugate for cancer therapy, J. Am. Chem. Soc., 2014, 136(33), 11748 CrossRef CAS PubMed.
- N. Yu, Y. Xu, T. Liu, H. Zhong, Z. Xu, T. Ji, H. Zou, J. Mu, Z. Chen, X. J. Liang, L. Shi, D. S. Kohane and S. Guo, Modular ketal-linked prodrugs and biomaterials enabled by organocatalytic transisopropenylation of alcohols, Nat. Commun., 2021, 12(1), 5532 CrossRef CAS PubMed.
- H. Zhong, J. Mu, Y. Du, Z. Xu, Y. Xu, N. Yu, S. Zhang and S. Guo, Acid-Triggered Release of Native Gemcitabine Conjugated in Polyketal Nanoparticles for Enhanced Anticancer Therapy, Biomacromolecules, 2020, 21(2), 803 CrossRef CAS PubMed.
- B. Liu and S. Thayumanavan, Substituent Effects on the pH Sensitivity of Acetals and Ketals and Their Correlation with Encapsulation Stability in Polymeric Nanogels, J. Am. Chem. Soc., 2017, 139(6), 2306 CrossRef CAS PubMed.
- E. H. Cordes and H. G. Bull, Mechanism and catalysis for hydrolysis of acetals, ketals, and ortho esters, Chem. Rev., 1974, 74(5), 581 CrossRef CAS.
- J. Caron, A. Maksimenko, J. Mougin, P. Couvreur and D. Desmaële, Combined antitumoral therapy with nanoassemblies of bolaform polyisoprenoyl paclitaxel/gemcitabine prodrugs, Polym. Chem., 2014, 5(5), 1662 RSC.
- C. R. Heald, S. Stolnik, K. S. Kujawinski, C. De Matteis, M. C. Garnett, L. Illum, S. S. Davis, S. C. Purkiss, R. J. Barlow and P. R. Gellert, Poly(lactic acid)−Poly(ethylene oxide) (PLA−PEG) Nanoparticles: NMR Studies of the Central Solidlike PLA Core and the Liquid PEG Corona, Langmuir, 2002, 18(9), 3669 CrossRef CAS.
- E. Moysan, G. Bastiat and J. P. Benoit, Gemcitabine versus Modified Gemcitabine: a review of several promising chemical modifications, Mol. Pharm., 2013, 10(2), 430 CrossRef CAS PubMed.
- A. Salic and T. J. Mitchison, A chemical method for fast and sensitive detection of DNA synthesis in vivo, Proc. Natl. Acad. Sci. U. S. A., 2008, 105(7), 2415 CrossRef CAS PubMed.
- Y. Liu, H. Tamam and Y. Yeo, Mixed Liposome Approach for Ratiometric and Sequential Delivery of Paclitaxel and Gemcitabine, AAPS PharmSciTech, 2018, 19(2), 693 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed experimental methods, 1H NMR and 13C NMR spectra, and other relevant figures. See DOI: https://doi.org/10.1039/d3sc00550j |
| This journal is © The Royal Society of Chemistry 2023 |