
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAn easy-to-perform evaluation of steric properties of Lewis acids†
Ludwig
Zapf
ab,
Melanie
Riethmann
ab,
Steffen A.
Föhrenbacher
a,
Maik
Finze
 *ab and
Udo
Radius
*a
*ab and
Udo
Radius
*a
aInstitute of Inorganic Chemistry, Julius-Maximilians-Universität Würzburg, Am Hubland, 97074 Würzburg, Germany. E-mail: u.radius@uni-wuerzburg.de; maik.finze@uni-wuerzburg.de; Web: https://www.ak-radius.de Web: https://go.uniwue.de/finze-group
bInstitute for Sustainable Chemistry & Catalysis with Boron (ICB), Julius-Maximilians-Universität Würzburg, Am Hubland, 97074 Würzburg, Germany
First published on 6th February 2023
Abstract
Steric and electronic effects play a very important role in chemistry, as these effects influence the shape and reactivity of molecules. Herein, an easy-to-perform approach to assess and quantify steric properties of Lewis acids with differently substituted Lewis acidic centers is reported. This model applies the concept of the percent buried volume (%VBur) to fluoride adducts of Lewis acids, as many fluoride adducts are crystallographically characterized and are frequently calculated to judge fluoride ion affinities (FIAs). Thus, data such as cartesian coordinates are often easily available. A list of 240 Lewis acids together with topographic steric maps and cartesian coordinates of an oriented molecule suitable for the SambVca 2.1 web application is provided, together with different FIA values taken from the literature. Diagrams of %VBur as a scale for steric demand vs. FIA as a scale for Lewis acidity provide valuable information about stereo-electronic properties of Lewis acids and an excellent evaluation of steric and electronic features of the Lewis acid under consideration. Furthermore, a novel LAB-Rep model (Lewis acid/base repulsion model) is introduced, which judges steric repulsion in Lewis acid/base pairs and helps to predict if an arbitrary pair of Lewis acid and Lewis base can form an adduct with respect to their steric properties. The reliability of this model was evaluated in four selected case studies, which demonstrate the versatility of this model. For this purpose, a user-friendly Excel spreadsheet was developed and is provided in the ESI, which works with listed buried volumes of Lewis acids %VBur_LA and of Lewis bases %VBur_LB, and no results from experimental crystal structures or quantum chemical calculations are necessary to evaluate steric repulsion in these Lewis acid/base pairs.
Introduction
About a century ago, Gilbert N. Lewis reported Lewis acids and characterized these compounds as electron pair acceptors.1 Since then, the synthesis of new Lewis acids and the investigation of their properties have become an intensely studied area of research.2 It was shown that Lewis acids are efficient catalysts for various transformations,3 and that sterically hindered Lewis acids are part of frustrated Lewis pairs (FLPs),4 which are capable of activating small molecules such as H2, CO, CO2, and many more. The addition of a suitable Lewis acid to a transition metal complex often leads to catalytic active species, and thus Lewis acids are often used as co-catalysts for the activation of transition metal complexes.5,6 Research on Lewis acids is often closely linked to the formation of weakly coordinating anions (WCAs),7,8 as they are converted into WCAs upon reaction with a suitable metal complex precursor. For example, the reaction of metal fluoride complexes such as [(Dipp2Im)CuF] (Dipp2Im = 1,3- bis(2,6-diisopropylphenyl)imidazolin-2-ylidene) with tris(pentafluoroethyl)difluorophosphorane (C2F5)3PF2 gave the dimeric cationic copper(I) complex [{(Dipp2Im)Cu}2]2+ stabilized by the WCA [(C2F5)3PF3]− (FAP).9 In addition, the application of a transition metal complex in combination with a Lewis acid can enable tandem or bifunctional catalysis, in which two catalytic processes are combined; one of these processes is catalyzed by the transition metal complex and the other by the Lewis acid.10A common feature of many Lewis acids is that they consist of a central atom that is surrounded by electronegative elements or substituents. Among the easiest and most widely used Lewis acids are group 13 and 15 molecules such as BF3, AlCl3, GaCl3, PF5, AsF5, and SbF5. Since trivalent boron(III) compounds have a vacant pz orbital at boron, these molecules can be regarded as prime examples for Lewis acids.2–11 Consequently, much effort has been made in the synthesis of boron-based Lewis acids.3,11–13 Formal substitution of fluorine in BF3 by strong electron withdrawing groups allows the tuning of both steric and electronic properties of a Lewis acid.3,11–14 Different experimental and theoretical methods serve as measures for Lewis acceptor properties and thus for Lewis acidity.11 As an experimental scale of Lewis acidity the Gutmann–Beckett method is often used, which relies on 31P NMR shifts of Et3PO adducts of the Lewis acid under investigation.15 According to Pearson's HSAB concept (hard and soft acids and bases)16 the “hard” Lewis base Et3PO should readily form adducts with “hard” Lewis acids such as BF3. Thus, to extend the Gutmann–Beckett method towards “soft” Lewis acids, Lichtenberg et al. recently proposed the application of Me3PS and Me3PSe for adduct formation and a proper assessment of “soft” Lewis acids.17 Another scale for Lewis acidity was suggested by Childs and co-workers, which relies on 1H NMR chemical shifts of crotonaldehyde adducts (and other aldehydes) of Lewis acids.18 An evaluation based on IR spectroscopy, that relies on the C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N stretching frequency of acetonitrile CH3CN19 and its deuterated homologue CD3CN,20 which increases with increasing strength of the Lewis acid upon adduct formation, was established.2a Baumgartner, Caputo and co-workers recently introduced a Lewis acidity scale based on the bathochromic shifts of the emission wavelengths of adducts of several Lewis acids and fluorescent dithienophosphole oxides,21 and Ofial et al. developed a Lewis acidity scale for several triarylboranes based on the equilibrium constants of several N-, O-, P-, and S-donor Lewis acid/base adducts.22
N stretching frequency of acetonitrile CH3CN19 and its deuterated homologue CD3CN,20 which increases with increasing strength of the Lewis acid upon adduct formation, was established.2a Baumgartner, Caputo and co-workers recently introduced a Lewis acidity scale based on the bathochromic shifts of the emission wavelengths of adducts of several Lewis acids and fluorescent dithienophosphole oxides,21 and Ofial et al. developed a Lewis acidity scale for several triarylboranes based on the equilibrium constants of several N-, O-, P-, and S-donor Lewis acid/base adducts.22
The most wide-spread measure to evaluate the strength of a Lewis acid nowadays is probably fluoride ion affinity (FIA), which is defined as the negative reaction enthalpy of the addition of a fluoride ion to a Lewis acid in the gas phase.23 Since a naked fluoride ion is difficult to approach by means of quantum chemical calculations, FIAs are typically calculated via isodesmic reactions, which use experimentally determined FIAs (e.g. of carbonyl fluoride OCF2) or FIAs calculated on a high level of theory (e.g. of Me3Si–F) as anchor points.23,24 This concept has been extended to ion affinities with respect to other anions such as chloride, hydride, and methide or alkoxide and also neutral Lewis bases such as water or ammonia and others, in part to also account for the differences between “hard” and “soft” Lewis acids.14,24 Thus, a variety of Lewis acidity scales is available and easily applicable. A great advantage of calculated affinities is that, in contrast to Lewis acidities derived from experimental data, they can also be obtained for hypothetical or not yet isolated Lewis acids. Thus, quantum chemical calculations typically serve as a starting point for the synthesis of Lewis acids with tailored properties.
Besides electronic effects, steric effects play an important role in chemistry, in general. Steric interactions influence the shape and the reactivity of molecules such as Lewis acids and Lewis bases. As steric effects have a decisive impact on properties and reactivity, several methods to assess steric properties have been developed.25 In theory, the steric contributions to the activation free energy were typically analyzed and classified into potential and kinetic energy factors.26 For experimentalists, the introduction of the cone angle to quantify the steric properties of phosphines by Tolman was a game-changer, as this method provided an easy-to-perform evaluation of steric properties of ligands in organometallic chemistry.27 Since then, many approaches for the quantification of steric effects of phosphines and other Lewis bases have been suggested.28,29 However, to date there has been no comprehensive study dealing with steric effects of differently substituted Lewis acidic centers.
During our studies on Lewis acids and Lewis acid/base adducts over the last few years9,14,30–32 we became interested in the stereo-electronic properties of Lewis acids, which specifically include the steric properties of Lewis acids and their influence on the formation of Lewis acid/base adducts. Herein we present an easy-to-apply model we developed over the last few years to estimate the steric properties of Lewis acids without accounting for electronic factors or different electronic interaction models. As there is currently no easy approach available to characterize steric properties of differently substituted Lewis acids, we addressed this problem and developed a model to access and quantify steric properties of Lewis acids, which also allows some prediction as to whether Lewis acid/base adduct formation can happen or not just by considering steric properties of the reactants (i.e., waiving electronic factors). This model is based on the well-established concept of the percent buried volume (%VBur) developed by Cavallo and co-workers for ligands in coordination chemistry,29 and we applied this concept to different fluoride ion adducts of Lewis acids for cataloging steric properties of a large number (240) of different fluoride adducts of Lewis acids of group 13, 14, and 15 elements using low level DFT (def2-SV(P)/BP86) optimized geometries. Furthermore, we developed a simple repulsion model which predicts if Lewis acid/base adduct formation is, with respect to sterics, possible or not within seconds, just considering the steric demand of Lewis acids and bases. The capability of this LAB-Rep (Lewis acid/base repulsion) model is demonstrated using several examples.
Results and discussion
Evaluation of the steric demand of Lewis acids via the percent buried volume (%VBur) model
It has been demonstrated in the past that the model of the percent buried volume (%VBur) as developed by Cavallo and co-workers is a versatile descriptor of steric properties of NHCs, phosphines, and related ligands in transition metal chemistry.29 For transition metal complexes, the buried volume serves as a measure of the space occupied by a ligand in the first coordination sphere of a metal center. The calculation requires a definition of the metal center, to which the ligand is coordinated at a certain distance d. Then, a sphere of radius R, which is centered at the metal atom is created and the volume the ligand captures is assigned to the buried volume VBur of this sphere. The buried volume VBur already indicates the volume of the coordination sphere, which is occupied by the ligand, but typically the percentage of the volume buried by the ligand with respect to the volume of the total sphere (%VBur) leads to a meaningful result, which can be compared. However, the results obtained depend on the M–L distance d and the sphere radius R. So, both parameters should be chosen wisely and must be compared for any values given.This concept can be easily adopted to Lewis acid/base adducts, for example to fluoride ion adducts of Lewis acids. If the fluorine atom of an anionic fluoride ion adduct [LA–F]− of the corresponding Lewis acid (LA) is used as an anchor point of the system, %VBur can be estimated. This is shown in Fig. 1 for two examples of Lewis acid/base adducts of a fluoride ion with BPh3 and (C2F5)3PF2. It should be emphasized that any anchor point (chloride, methyl, hydride etc. adducts) can be chosen since electronic factors and hard and soft properties play no role in an exclusively steric model, as long as this anchor point is consistent. We opt here to evaluate the steric properties of fluoride ion adducts [LA–F]− since these geometries are often available from DFT calculations of fluoride ion affinities. In addition, fluoride adducts are also often experimentally accessible via the reaction of a Lewis acid and a fluoride ion source. The model simply uses either calculated (geometry refined) or experimentally determined structures and the fluorine atom of the fluoride adducts [LA–F]− is placed at the center of the sphere, which is then processed using the user-friendly SambVca 2.1 web application.29f,g SambVca 2.1 was used to determine %VBur for several Lewis acids for a radius R = 3.50 Å. The latter radius is typically used for the determination of %VBur of transition metal ligands. Thus, we chose the same radius R for the assessment of the steric demand of Lewis acids because all atoms that pose steric pressure to the center of consideration typically lie within this radius. For example, for the anion [F–AlPh3]− the aluminum atom, the ipso carbon atom and the ortho-C–H units pointing at the fluoride are located within R = 3.50 Å (for an illustration of the lighter homologue [F–BPh3]− see Fig. 1, left side). Table 1 provides a list of %VBur of selected group 13 Lewis acids obtained from DFT optimized geometries, Table 2 a list for selected group 14 and Table 3 a list of group 15 Lewis acids. A full list of all 240 Lewis acids considered herein together with cartesian coordinates of the oriented molecules suitable for the SambVca 2.1 web application, as well as different FIA values taken from the literature, is provided in Table S1 of the ESI.† In addition, topographic steric maps29f,g of the Lewis acids BF3, B(CN)3, BCl3, BPh3, B(C6F5)3, and B(C6Cl5)3 in their fluoride adducts [BF4]−, [(NC)3BF]−, [Cl3BF]−, [Ph3BF]−, [(C6F5)3BF]−, and [(C6Cl5)3BF]− (R = 3.50 Å) are provided in Fig. 2, and the full set of topographic steric maps of all Lewis acids studied is provided in Fig. S1 of the ESI.†
 | ||
| Fig. 1 Illustration of %VBur discussed herein, exemplified by the Lewis acid/base adducts of a fluoride ion with BPh3 (left) and (C2F5)3PF2 (right); R = 3.50 Å; d(B–F) = 1.40 Å; d(P–F) = 1.67 Å. | ||
| LA | %VBur | LA | %VBur | LA | %VBur |
|---|---|---|---|---|---|
| a ArF = 3,5-(CF3)2C6H3. | |||||
| BH3 | 27.3 | AlH3 | 24.8 | GaH3 | 24.7 |
| BF3 | 33.3 | AlF3 | 29.6 | GaF3 | 28.9 |
| BCl3 | 40.9 | AlCl3 | 34.5 | GaCl3 | 33.3 |
| BBr3 | 43.0 | AlBr3 | 35.6 | GaBr3 | 34.4 |
| BI3 | 45.5 | AlI3 | 37.2 | GaI3 | 36.0 |
| B(CN)3 | 38.9 | Al(CN)3 | 33.4 | Ga(CN)3 | 32.7 |
|
B(CCH)3
|
39.3 |
Al(CCH)3
|
33.5 |
Ga(CCH)3
|
32.8 |
| B(CH3)3 | 40.7 | Al(CH3)3 | 34.1 | Ga(CH3)3 | 33.4 |
| B(CF3)3 | 50.8 | Al(CF3)3 | 40.5 | Ga(CF3)3 | 39.7 |
| B(C2F5)3 | 63.0 | Al(C2F5)3 | 47.5 | Ga(C2F5)3 | 46.9 |
| BPh3 | 53.1 | AlPh3 | 44.2 | GaPh3 | 42.7 |
| BArF3 | 53.7 | AlArF3 | 45.5 | GaArF3 | 45.1 |
| B(C6F5)3 | 58.9 | Al(C6F5)3 | 47.4 | Ga(C6F5)3 | 46.7 |
| B(C6Cl5)3 | 70.2 | Al(C6Cl5)3 | 59.0 | Ga(C6Cl5)3 | 58.1 |
| LA | Anion | %VBur | LA | Anion | %VBur | LA | Anion | %VBur |
|---|---|---|---|---|---|---|---|---|
| SiF4 | [SiF5]− | 37.9 | GeF4 | [GeF5]− | 37.2 | SnF4 | [SnF5]− | 36.3 |
| SiCl4 | [Cl4SiF]− (eq) | 44.6 | GeCl4 | [Cl4GeF]− (eq) | 42.6 | SnCl4 | [Cl4SnF]− (eq) | 39.7 |
| [Cl4SiF]− (ax) | 46.0 | [Cl4GeF]− (ax) | 44.2 | [Cl4SnF]− (ax) | 41.8 | |||
| SiBr4 | [Br4SiF]− (eq) | 46.7 | GeBr4 | [Br4GeF]− (eq) | 44.4 | SnBr4 | [Br4SnF]− (eq) | 41.1 |
| [Br4SiF]− (ax) | 48.3 | [Br4GeF]− (ax) | 46.3 | [Br4SnF]− (ax) | 43.5 | |||
| SiI4 | [I4SiF]− (eq) | 49.4 | GeI4 | [I4GeF]− (eq) | 46.9 | SnI4 | [I4SnF]− (eq) | 43.1 |
| [I4SiF]− (ax) | 51.3 | [I4GeF]− (ax) | 49.1 | [I4SnF]− (ax) | 45.9 | |||
| Si(CN)4 | [(NC)4SiF]− (eq) | 43.2 | Ge(CN)4 | [(NC)4GeF]− (eq) | 41.7 | Sn(CN)4 | [(NC)4SnF]− (eq) | 39.3 |
| [(NC)4SiF]− (ax) | 44.2 | [(NC)4GeF]− (ax) | 42.7 | [(NC)4SnF]− (ax) | 40.8 | |||
| Si(CF3)4 | [(CF3)4SiF]− (eq) | 54.6 | Ge(CF3)4 | [(CF3)4GeF]− (eq) | 51.8 | Sn(CF3)4 | [(CF3)4SnF]− (eq) | 47.1 |
| [(CF3)4SiF]− (ax) | 57.4 | [(CF3)4GeF]− (ax) | 54.6 | [(CF3)4SnF]− (ax) | 50.0 | |||
| Si(C6F5)4 | [(C6F5)4SiF]− (eq) | 63.9 | Ge(C6F5)4 | [(C6F5)4GeF]− (eq) | 61.8 | Sn(C6F5)4 | [(C6F5)4SnF]− (eq) | 58.7 |
| [(C6F5)4SiF]− (ax) | 65.4 | [(C6F5)4GeF]− (ax) | 61.9 | [(C6F5)4SnF]− (ax) | 56.6 |
| LA | Anion | %VBur | LA | Anion | %VBur | LA | Anion | %VBur |
|---|---|---|---|---|---|---|---|---|
| PF3 | [PF4]− | 29.6 | AsF3 | [AsF4]− | 19.2 | |||
| PCl3 | [Cl3PF]− | 42.0 | AsCl3 | [Cl3AsF]− | 34.1 | |||
| PBr3 | [Br3PF]− | 45.0 | AsBr3 | [Br3AsF]− | 34.4 | |||
| PI3 | [I3PF]− | 45.9 | AsI3 | [I3AsF]− | 34.5 | |||
| P(CF3)3 | [(CF3)3PF]− | 46.4 | As(CF3)3 | [(CF3)3AsF]− | 35.3 | |||
| P(C2F5)3 | [(C2F5)3PF]− | 51.6 | As(C2F5)3 | [(C2F5)3AsF]− | 43.3 | |||
| PF5 | [PF6]− | 38.4 | AsF5 | [AsF6]− | 35.6 | SbF5 | [SbF6]− | 37.4 |
| PCl5 | [Cl5PF]− | 49.6 | AsCl5 | [Cl5AsF]− | 45.1 | SbCl5 | [Cl5SbF]− | 45.0 |
| P(CN)5 | [(NC)5PF]− | 48.0 | As(CN)5 | [(NC)5AsF]− | 44.4 | Sb(CN)5 | [(NC)5SbF]− | 43.4 |
| P(CF3)5 | [(CF3)5PF]− | 63.9 | As(CF3)5 | [(CF3)5AsF]− | 59.1 | Sb(CF3)5 | [(CF3)5SbF]− | 55.4 |
| P(C6F5)5 | [(C6F5)5PF]− | 77.8 | As(C6F5)5 | [(C6F5)5AsF]− | 73.5 | Sb(C6F5)5 | [(C6F5)SbF]− | 69.5 |
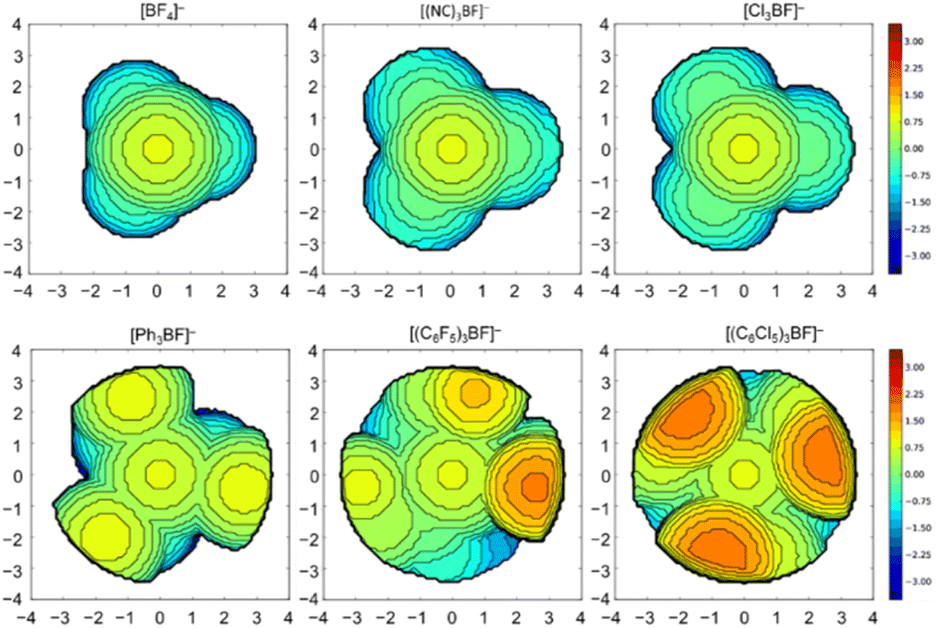 | ||
| Fig. 2 Topographic steric maps of the Lewis acids BF3, B(CN)3, BCl3, BPh3, B(C6F5)3, and B(C6Cl5)3 in their fluoride adducts [BF4]−, [(NC)3BF]− (MFB), [Cl3BF]−, [Ph3BF]−, [(C6F5)3BF]−, and [(C6Cl5)3BF]− (d(B–F) = 1.40 Å and R = 3.50 Å). The isocontour scheme is given in Å, red and blue zones indicate the more- and less-hindered zones with respect of the origin. | ||
As intuitively expected, the smallest %VBur of the boranes studied was obtained for BH3 (27.3%). Substitution of hydrogen by halogen led to an increase of %VBur: BH3 (27.3 %VBur) < BF3 (33.3 %VBur) < BCl3 (40.9 %VBur) < BBr3 (43.0 %VBur) < BI3 (45.5 %VBur). The steric demand of boron Lewis acids with linear substituents such as B(CN)3 (38.9 %VBur) and B(CCH)3 (39.3 %VBur) is similar to that of BCl3 (40.9 %VBur). The substituents CH3, CF3, and C2F5 are bulkier: B(CH3)3 (40.7 %VBur) < B(CF3)3 (50.8 %VBur) < B(C2F5)3 (63.0 %VBur); the latter imposes more steric bulk than C6H5, C6F5, and 3,5-(CF3)2C6H3: BPh3 (53.1 %VBur) < BArF3 (ArF = 3,5-(CF3)2C6H3; 53.7 %VBur) < B(C6F5)3 (58.9 %VBur). The extraordinary steric protection of the pentafluoroethyl substituent –C2F5 has been demonstrated experimentally, earlier.9,30a,33 Substitution of hydrogen in the meta-position of BPh3 with CF3 groups has only little influence on the bulkiness of the Lewis acid (cf. 53.1 %VBur for BPh3vs. 53.7 %VBur for BArF3). In B(C6Cl5)3 the high steric demand of the C6Cl5 group results in an increase of %VBur to 70.2 % VBur. ortho-Substituted boranes exceed these values and the bulkiest boron Lewis acid considered in this study is B{2,4,6-(CF3)3C6H2}3 (85.9 %VBur; see Table S1 in the ESI†). However, B{2,4,6-(CF3)3C6H2}3 and the related borane B{2,6-(CF3)2C6H3}3 are unknown. Examples for synthetically accessible sterically highly demanding Lewis acids are B{OC(CF3)3}3 (FIA = 423 kJ mol−1)34 and B(1,2-C2B10H11)3 (FIA = 605 kJ mol−1)35 with a buried volume of 71.9 %VBur. For aluminum, the Lewis acids Al{OC(CF3)3}3 (FIA = 543 kJ mol−1; 56.8 %VBur) and36a Al{N(C6F5)2}3 (FIA = 555 kJ mol−1; 68.1 %VBur),36b and the recently reported Al(OTeF5)3 (FIA = 591 kJ mol−1; 51.5 %VBur)36c provide excellent combinations of steric bulk and high Lewis-acidity (see also the ESI, Fig. S5†).
A chart of %VBurvs. Lewis-acidic atoms of all Lewis acids considered shows that there is a wide range of %VBur covered by the different Lewis acids and that any steric demand between 30 %VBur and 75 %VBur can be realized easily (Fig. S2 of the ESI†). Thus, easy tuning of %VBur is possible by proper choice of (i) the central atom and (ii) the substituents. Lewis acids of aluminum and gallium typically exhibit smaller % VBur than related boron-based Lewis acids, which reflects the larger E–X distances of E = Al, Ga compared to that of E = B. The values of the buried volume range from 29.6 %VBur (AlF3) to 37.2 %VBur (AlI3) for aluminum halides and from 28.9 %VBur (GaF3) to 36.0 %VBur (GaI3) for gallium halides. All %VBur values decrease in the order B ≫ Al > Ga.
Even more valuable than solely considering steric aspects are diagrams that correlate %VBur and fluoride ion affinity (FIA) as a scale for the Lewis acidity of the compound. In the 1970s, Tolman demonstrated that the IR frequency of the A1 stretch in Ni(CO)3L, where L was a monodentate phosphorus donor ligand of interest, was a useful probe to quantify the electron-donating ability of the ligand L,37 nowadays known as the Tolman electronic parameters TEPs. Charts of the Tolman electronic parameter with the Tolman cone angle as the quantification of the steric demand for many phosphines usually serve the organometallic community as a basis for phosphine ligand choice. Recently, the method was applied to the determination of the electronic and steric properties of various NHC donor ligands.38 We believe that diagrams which correlate the steric properties via %VBur and the Lewis acidity via fluoride ion affinity (FIA) can serve in a similar way for a proper choice of Lewis acids. Such correlation diagrams combine steric and electronic features of the Lewis acid and a plot of %VBurversus FIA of all Lewis acids considered herein is provided in Fig. S3 in the ESI.† This chart shows that a wide range of combinations of %VBur (30–75%) and FIA (200–600 kJ mol−1) is available by choosing a central atom and its substituents. In the ESI,† separate diagrams are also provided for boron (Fig. S4†), aluminum (Fig. S5†), and gallium (Fig. S6†).
A comparison of the stereo-electronic properties of boron, aluminum, and gallium halides is shown in Fig. 3. This chart reveals the dispersion in the stereo-electronic features of boron-based Lewis acids, which span a range of 12.2 %VBur and 100 kJ mol−1 in FIA,39 compared to those of aluminum- (7.6 %VBur and 28 kJ mol−1) and gallium-based (7.1 %VBur and 20 kJ mol−1) Lewis acids. The wider range of %VBur and FIAs accessible with boron-based Lewis acids, in general (see Fig. S4 to S6 of the ESI†), is mainly due to shorter E–X distances between boron and its substituents compared to Al and Ga. These relatively short B–X distances compared to Al–X and Ga–X distances are accompanied by a pronounced impact not only on the steric situation but also on the electronic features of the central boron atom compared to aluminum or gallium.40
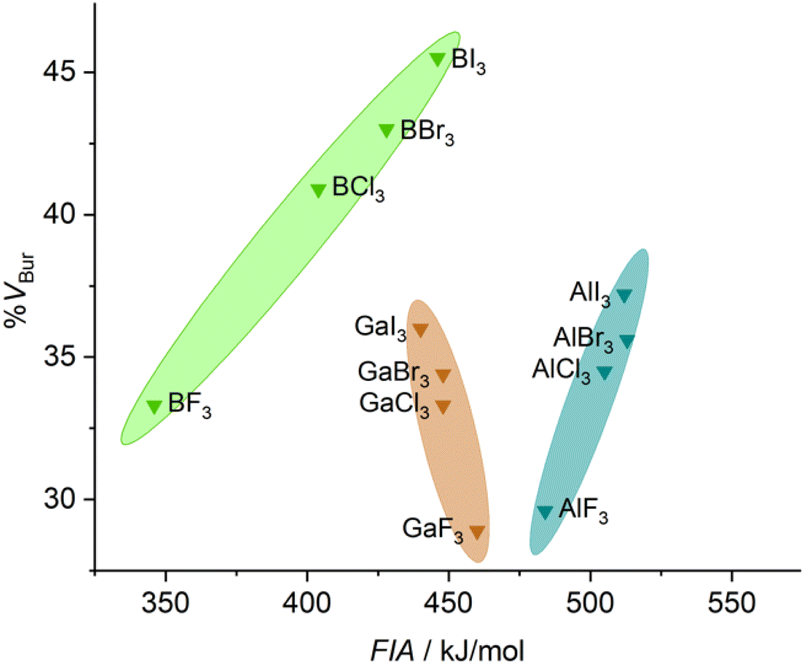 | ||
| Fig. 3 Comparison of the stereo-electronic properties of group 13 halides (FIAs were taken from the literature39). | ||
For the evaluation of group 14 Lewis acids EX4 (Fig. 4) we considered two isomers formed with the Lewis base (LB), e.g., the fluoride ion, either in equatorial or in axial position (see Scheme 1).
 | ||
| Fig. 4 Comparison of the stereo-electronic properties of homoleptic group 14 (top) and group 15 (bottom) halides (FIAs were taken from the literature39). | ||
 | ||
| Scheme 1 Stereoisomers of Lewis acid/base adducts of a Lewis base LB with a group 14 Lewis acid EX4. | ||
A selection of %VBur values of group 14 and group 15 Lewis acids (LAs) obtained from geometry optimized fluoride ion adducts [LA–F]− is provided in Tables 2 and 3. First of all, even the small Lewis acids EF4 (E = Si, 37.9 %VBur; Ge, 37.2 %VBur; Sn, 36.3 %VBur) are sterically more demanding than their group 13 and group 15 counterparts EF3 (E = Al, 29.6 %VBur; Ga, 28.9 %VBur; P, 29.6 %VBur; As 19.2 %VBur), which is mostly a consequence of the additional fourth substituent. Thus, the increase in steric demand is even more pronounced for group 15 Lewis acids EF5 (E = P, 38.4 %VBur; As, 35.6 %VBur; Sb, 37.4 %VBur; Table 3). In the case of group 14 Lewis acids, the Lewis base bonded in the axial position usually experiences a larger EX4 group than the Lewis base coordinated in the equatorial position; cf. SiCl4 (eq: 44.6 %VBur; ax: 46.0 %VBur) or Si(CF3)4 (eq: 54.6 %VBur; ax: 57.4 %VBur), which is in line with the general expectation that the equatorial positions of trigonal bipyramids are less sterically demanding.
For group 13–15 element halides FIA increases for the lighter elements such as B, Al, Si, and P with the heavier and larger halogen, i.e. EFn < ECln < EBrn < EIn (E = B, Al, Si, P; n = 3 {B, Al, P}, 4 {Si}, 5 {P}), whereas for the heavier elements Ge, Sn, As, and Sb the opposite trend occurs, i.e. FIA increases on going to the lighter and sterically less demanding halogen, EFn > ECln > EBrn > EIn (E = Ge, Sn, As, and Sb; n = 4 {Ge, Sn}, 5 {Sb}). The halides of Ga and As are borderline cases, as the dispersion of the FIA values of Ga(III) halides is small (FIA{GaF3} = 460 kJ mol−1vs. FIA{GaI3} = 440 kJ mol−1; a larger FIA value for the fluoride) and for As the FIAs are in opposite direction for the As(III) halides (FIA{AsF3} = 244 kJ mol−1vs. FIA{AsBr3} = 286 kJ mol−1; a smaller FIA value for the fluoride) and the As(V) halides (FIA{AsF5} = 439 kJ mol−1vs. FIA{AsBr5} = 393 kJ mol−1; a larger FIA value for the fluoride).
Since the Lewis acidities of B, Al, and Si halides EX3 and EX4 increase with the less electronegative, larger halogen substituents, “cigars” with a positive slope result in the diagrams shown in Fig. 3 and 4. In contrast, “cigars” with a negative slope are observed for Ge and Sn halides EX4 with the higher, less electronegative halogen substituents because the Lewis acidity decreases on going to the heavier halogens. The FIA calculated for the gallium halides GaX3 are almost invariant to changes in the halogen substituent and thus differ only in their steric demand.
In addition, we examined the percent buried volume of PF5 and the related pentafluoroethyl(fluoro)phosphoranes (C2F5)nPF5−n. Due to the higher coordination number of the phosphorus(V) derivatives, the %VBur of PF5 (38.4 %VBur) is higher than those of AlF3 (29.6 %VBur), SiF4 (37.9 %VBur), and PF3 (29.6 %VBur). The evaluation of %VBur of unsymmetrical Lewis acids such as the phosphoranes (C2F5)nPF5−n is easily resolved using the model described herein. The evaluation of %VBur can be performed for the different isomers that are either experimentally observed or considered in a theoretical study. The assessment of the different isomers of the pentafluoroethyl(fluoro)phosphate anions in Fig. 5 demonstrates the strong influence of isomers on the steric demand via the %VBur, in general. Typically, the lowest value for %VBur is the most reasonable. A detailed discussion on the steric aspects of the pentafluoroethyl(fluoro)phosphoranes (C2F5)nPF5−n and the corresponding pentafluoroethyl(fluoro)phosphate anions [(C2F5)nPF6−n]− depicted in Fig. 5 can be found in the ESI.†
 | ||
| Fig. 5 Pentafluoroethyl(fluoro)phosphoranes (C2F5)nPF5−n and their %VBur values. | ||
LAB-Rep: an empirical model for the evaluation of Lewis acid/base adduct formation
With the percent buried volumes %VBur of various Lewis acids in hand, we developed an empirical model (LAB-Rep model; Lewis Acid/Base Repulsion model) to predict whether an arbitrary pair of Lewis acid and Lewis base may form an adduct based on the steric properties of the Lewis acid and Lewis base. This model just requires the principal shape of the Lewis acid and Lewis base and their buried volumes %VBur_LA and %VBur_LB, respectively, to provide a prediction if Lewis acid/base complexes can form for steric reasons. It should be emphasized here that this estimation is executed at no cost (computational time, etc.) and an Excel sheet (see also Fig. 9) for this purpose that requires minimum input data is provided in the ESI.† The shape of the components can be derived from experimental data, from quantum chemical calculations, from models prepared with Chem3D41 or similar programs or even simply by chemical intuition.For the evaluation of whether an acid/base adduct can be formed or not, both entities, the Lewis acid and Lewis base, were projected into a single sphere S with radius R (see Fig. 6).
 | ||
| Fig. 6 Illustration of %VBur exemplified for a Lewis acid (left) and a Lewis base (right) and projection of the compounds into a single sphere (middle); R = 3.50 Å. | ||
However, since the real distance D of a potential acid/base adduct, i.e., the distance between the donor atom of the Lewis base and the acceptor atom of the Lewis acid, is typically shorter than 2d as shown in Fig. 6 (D < 2d) a correction volume Vcorr must be applied. Adding up its percentage share %Vcorr of the sphere S with the buried volumes of the Lewis acid %VBur_LA and the Lewis base %VBur_LB provides a prediction whether a stable acid/base adduct can be formed, or not, by the number of %VBur_all (eqn (1), Fig. 7).
 | ||
| Fig. 7 Illustration of eqn (1) with %VBur_LA, %VBur_LB, and %Vcorr. | ||
When %VBur_all significantly exceeds 100% (>110%), the steric repulsion is too large to allow formation of a stable adduct. In the case that %VBur_all is significantly smaller than 100% (<90%), the steric repulsion is small enough to enable adduct formation. Buried volumes in between 90 and 110% are indicative of weakly interacting Lewis acids and bases, for example with unusually long bonds between the donor and the acceptor atoms, which often lead to equilibria of the free acid and the free base and the respective Lewis acid/base adduct (vide infra).
| %VBur_all = %VBur_LA + %VBur_LB + %Vcorr | (1) |
For a proper evaluation, the different shapes of different Lewis acids have to be taken into account. Thus, three different prototype shapes for the respective Lewis acid and Lewis base were considered, leading to five designs for the correction volume Vcorr (Fig. 8, for a mathematical description see the ESI†). Prototype I comprises cone-shaped molecules, BMe3 as an example for a Lewis acid and PMe3 as an example for a Lewis base. When two prototype I molecules are combined into a Lewis acid/base pair, Vcorr is approximated using the volume of a segment Vseg of sphere S (design 1). Prototype II conflates fan-like and more bulky molecules such as NHCs or PF5. For the combination of two prototype II molecules Vcorr is approximated using the volume of a biconvex lens Vbcl (design 2). Design 3 is suitable for the combination of a prototype I molecule and a prototype II molecule, e.g. BMe3 and a NHC, with Vcorr being a convex lens Vcl (design 3). Prototype III molecules are sterically overcrowded and thus have a % VBur of more than 50%. Examination of a combination of a prototype I and a prototype III molecule leads to an approximation of Vcorr as a half-segment Vhseg (design 4). For the pair of a prototype II and a prototype III molecule, the volume of a half-biconvex lens Vhbcl (design 5) should be applied as correction volume (Fig. 8). To take into account that prototype III molecules extend into the hemisphere of the potential adduct partner when their %Vbur exceeds 50%, an empirical correction was introduced for the correction volumes Vcorr of design 4Vhseg and design 5Vhbcl (for details including the mathematical description of the five designs see the ESI†). The distance h between the different types of correction volumes was estimated to be d/2 (Fig. 8). This estimation gives very accurate results, as outlined below. In principle, the combination of a prototype III Lewis base with a prototype III Lewis acid is possible. However, a combination of two molecules with %VBur of more than 50% usually results in a repulsive, and thus nonbonding interaction.
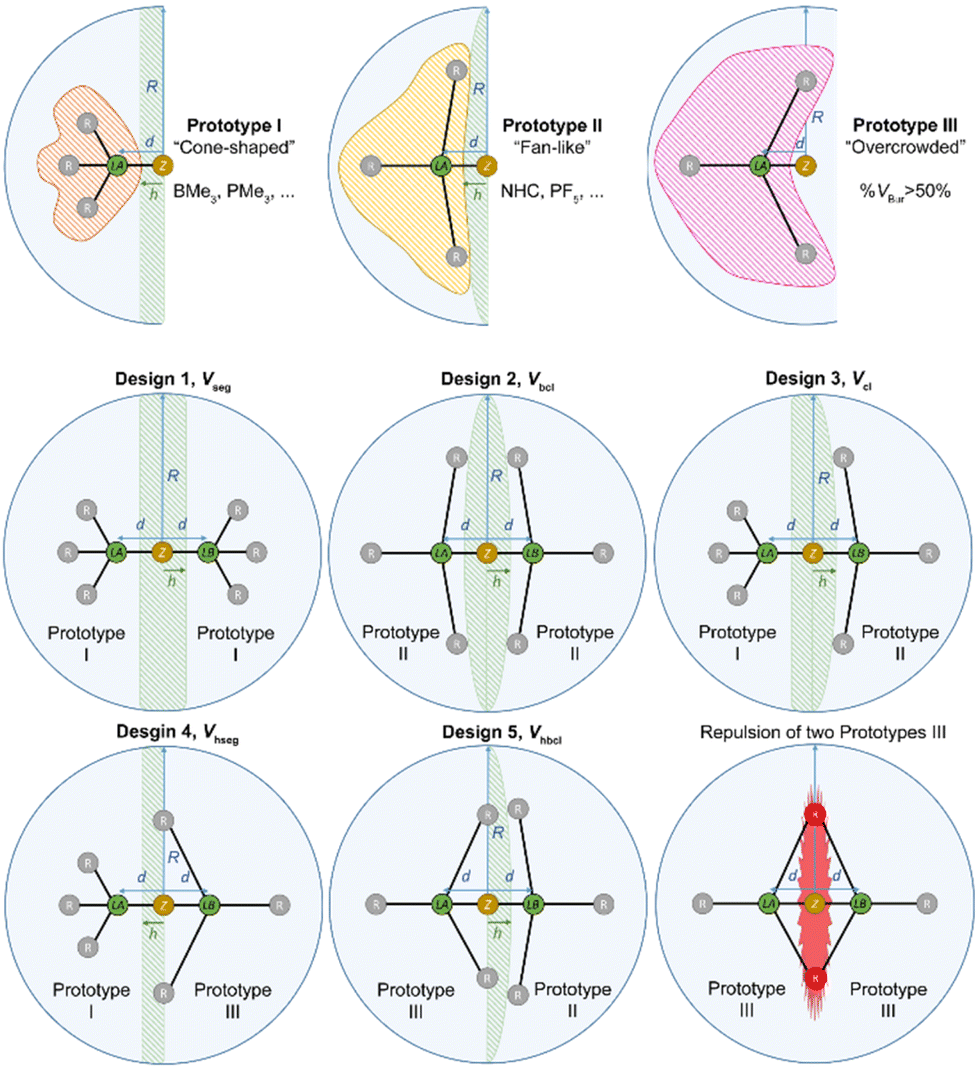 | ||
| Fig. 8 Illustration of the five designs for the correction volume Vcorr. | ||
We would like to point out that the choice of the prototype for the Lewis acid and base requires some intuition and the only necessary inputs for the LAB-Rep model are the buried volumes and the d values of the Lewis acids and bases, which can be derived from tabulated values (Table S1 of the ESI†).
To examine the reliability of the LAB-Rep model and to demonstrate its feasibility, some examples for the application of this model are presented in the following, which also include some selected borderline cases where a prediction whether an acid/base adduct is formed or not is less obvious. The application of the LAB-Rep model is easy and can be performed by using the Excel spreadsheet as provided in the ESI† (see also Fig. 9). This spreadsheet provides a prediction according to the LAB-Rep model for arbitrary Lewis acid/base pairs without the preselection of the prototype of Lewis acid and Lewis base, respectively, but calculates %VBur_all for all five different designs, which makes an assessment of the possible formation of a stable Lewis acid/base adduct even more convenient. All data needed for input are the values for %VBur and the distance d used for the determination of %VBur for the Lewis acid and the Lewis base (see Fig. 9).
 | ||
| Fig. 9 Example for the input and output for the application of the LAB-Rep model by using the Excel spreadsheet provided in the ESI:† the Lewis acid/base pair 2,6-lutidine and BMe3. The input requires the values for %VBur and the distance d used for the determination of %VBur for the Lewis acid (red circles) and the Lewis base (blue circles), i.e. 40.7 %VBur and 1.47 Å for BMe3 (red circles) and 28.6 %VBur and 2.11 Å for 2,6-lutidine (blue circles). The Excel spreadsheet calculates %VBur_all for all five different designs, and the convex lens design 3 is the obvious choice here. The corresponding total buried volume %VBur_all was calculated for BMe3/2,6-lutidine to be 97.9% (red square) for design 3. | ||
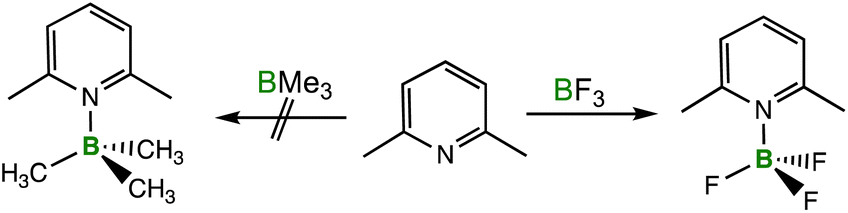 | ||
| Scheme 2 Reaction of 2,6-lutidine with BMe3 and BF3. | ||
By applying the LAB-Rep model, it was found that the convex lens design 3 is the obvious choice for the correction volume Vcorr since BF3 and BMe3 are cone-shaped Lewis acids while 2,6-lutidine can be considered fan-like with the methyl groups in ortho position to the Lewis basic nitrogen. The correction volume of design 3 %Vcl was calculated using eqn (S10) and (S11) (see the ESI†) and values for d are 1.42 Å for BF3, 1.47 Å for BMe3, and 2.11 Å for 2,6-lutidine to give a %Vcl of 28.5% for BF3/2,6-lutidine and 28.6% for BMe3/2,6-lutidine, respectively. The d values have been derived from the calculated structures of the fluoroborate anions [BF4]− and [BMe3F]− and from the 2,6-lutidine nickel tricarbonyl complex [Ni(CO)3(2,6-lutidine)] as outlined in the ESI.† The Excel spreadsheet as provided in the ESI† and illustrated in Fig. 9 for the input and output of the calculations performed on the Lewis acid/base pair 2,6-lutidine and BMe3 was applied. This spreadsheet only requires the values for %VBur and the distance d used for the determination of %VBur for the Lewis acid and the Lewis base as the input. With the buried volumes of BF3 (33.3 %VBur, d = 1.42 Å), BMe3 (40.7 %VBur, d = 1.47 Å), and 2,6-lutidine (28.6 %VBur, d = 2.11 Å), the corresponding total buried volumes %VBur_all were calculated according to eqn (1) (Fig. 7). In the case of BF3/2,6-lutidine, the %VBur_all of 89.7% is significantly smaller than 100%, and therefore the formation of an acid/base-adduct is predicted in accordance with the experiment. For BMe3/2,6-lutidine %VBur_all was estimated to be 97.9%, which is within the limit of our model (90–110%).
In order to check whether the different d values have a significant influence on the results of the LAB-Rep model, the calculations for %Vbur and %Vcl were performed with an equilibrium B–N distance of 1.72 Å, which was derived for BF3/2,6-lutidine by quantum chemical calculations (details in the ESI†). This resulted in a %VBur_all of 91.8% for BF3/2,6-lutidine and 97.7% for BMe3/2,6-lutidine. Thus, the different d values applied do not alter the assessment of the possibility of the formation of a Lewis acid/base adduct. This in turn demonstrates the stability of the LAB-Rep model and its practical value.
Similarly, Stephan and co-worker expanded the combination of Lewis bases capable of FLP chemistry with tris(pentafluorophenyl)borane to the sterically encumbered NHC IDipp (= 1,3-bis(2,5-diisopropyl-phenyl)imidazolin-2-ylidene). This system shows effective FLP reactivity including H–H bond cleavage to yield imidazolium borates and amine N–H bond cleavage to afford aminoborate salts, although the molecular structure of the adduct (C6F5)3B–IDipp was reported.46 By calculation of this system with the LAB-Rep model, using the typical input for B(C6F5)3 (58.9 %VBur, d = 1.46 Å) and IDipp (36.8 %VBur, d = 1.96 Å), %VBur_all was found to be 108.0% for (C6F5)3B/IDipp, which is again at the upper end of the range (110%), where equilibria are expected and weakly bound adducts may form.
The Lewis acid/base combination BPh3 and PPh3 is a closely related example that was investigated by Wittig et al. already in the 1950s. These authors reported that with triphenylborane and triphenylphosphine, 1,2-dehydrobenzene forms an o-phenylene-bridged zwitterionic phosphonium-borate instead of a simple Lewis acid/base adduct between BPh3 and PPh3.47a Later on, Horner and Haufe reported that an adduct Ph3B–PPh3 was formed from the reaction of [{Ph3P}2Hg]2+ and sodium tetraphenylborate.47b Adduct formation of Ph3B–PPh3 from Ph3B and PPh3 was reported at the same time in the patent literature, but no spectroscopic data is available for this adduct.47c Triphenylborane is sterically less demanding than tris(pentafluorophenyl)borane, which leads to a smaller %VBur_all of 105.9% for design 4 using buried volumes of BPh3 (53.1 %VBur, d = 1.46 Å) and PPh3 (31.1 %VBur, d = 2.25 Å). Thus, adduct formation should be possible according to the LAB-Rep model, but the adduct formed should experience steric pressure due to steric repulsion of the Lewis acid BPh3 and the Lewis base PPh3.
To probe this prediction, we reacted BPh3 and PPh3 to yield the Lewis acid/base adduct Ph3B–PPh31 in almost quantitative yield (96%). The reaction was carried out in THF, and product formation and isolation were enhanced by immediate precipitation of the product. Adduct formation was evidenced from solid state NMR spectroscopy, as resonances were detected in the region of four-coordinate boron at δ = −1.7 ppm in the 11B{1H} NMR RSHE/MAS NMR solid state spectrum (see Fig. S27 of the ESI†) and at δ = 4.6 ppm in the 31P{1H} CP/MAS solid state NMR spectrum (see Fig. S28 of the ESI†). Crystals of 1 suitable for X-ray diffraction were obtained by recrystallizing the compound in THF. The adduct Ph3B–PPh31 (Fig. 10) crystallizes in the hexagonal space group P63cm. Adduct 1 is heavily disordered, and thus the crystallographic result serves as mere evidence for the connectivity in the solid state, but a discussion of the metric data would be arbitrary. In solution, only BPh3 (δ = 65.8 ppm in the 11B{1H}-NMR spectrum, see Fig. S23 of the ESI†) and PPh3 (δ = −5.0 ppm in the 31P{1H}-NMR spectrum, see Fig. S24 of the ESI†) can be observed via NMR spectroscopy at room temperature, but not the adduct PPh3–BPh3. Adduct formation in solution takes place at temperatures below approximately −20 °C (see Fig. S25 of the ESI†). Thus, Ph3B–PPh31 can be formed and exists in the solid state, the melting point of the solid is 213 °C, but easily decomposes upon dissolution into the Lewis acidic and Lewis basic components.
 | ||
| Fig. 10 Molecular structure of Ph3B–PPh31. Thermal ellipsoids correspond to 25% probability except for the H atoms that are depicted with arbitrary radii; disorder is omitted for clarity. | ||
 | ||
| Scheme 3 Reactivity of NHCs with (C2F5)3PF2.30a | ||
The reactivity of (C2F5)3PF2 with different sterically demanding bases makes its reactions an ideal case study for the LAB-Rep model. In addition, the closely related but sterically less encumbered Lewis acid PF5 is ideally suited as it forms stable adducts with all bases studied. In Table 4 the applied correction volumes %Vcorr and %VBur, and the calculated %VBur_all are listed for different combinations of (C2F5)3PF2 and PF5 with selected NHCs and phosphines. For PF5, adduct formation is predicted for most bases as %VBur_all is at the lower end (>90%) of the range indicative of equilibria or weakly bound adducts. The calculated %VBur_all for the different Lewis acid/base adducts with (C2F5)3PF2 as the Lewis acid nicely mirrors the experimental findings. Sterically encumbered Lewis bases do not result in stable adducts while sterically less demanding bases lead to %VBur_all which do not exclude adduct formation, which is again in agreement with the experimental data. Furthermore, both experimentally observed isomers of adducts between (C2F5)3PF2 and PMe3 (mer and fac) give similar %VBur_all. For example, (C2F5)3PF2 (67.7 %VBur, d = 1.69 Å) reacted with IMe (27.4 %VBur, d = 2.01 Å) to yield the crystallographically characterized adduct (C2F5)3PF2·IMe (%VBur_all = 102.3), whereas IDipp (36.5%VBur, d = 1.96 Å; %VBur_all = 111.0) revealed in the presence of (C2F5)3PF2 FLP reactivity and slowly converted in the absence of substrates to the adduct of the abnormal NHC aIDipp (31.8 %VBur, d = 1.98 Å; %VBur_all = 106.4; see Table 4).
For example, B2pin2 (45.1 %VBur, d = 1.44 Å) as the most common and very well established diboron(4) compound in organic and inorganic syntheses was reacted with the NHC IiPr (28.8 %VBur, d = 1.99 Å) and the formation of the mono-NHC adduct B2pin2·IiPr (%VBur_all = 101.3; design 3; Vcl) was observed (see Scheme 4).31d This stable adduct was isolated and characterized including by single-crystal X-ray diffraction. However, during our work on the defluoroborylation of fluoroaromatics using [Ni(NHC)2] complexes as catalysts,47 we recognized that formation of this adduct leads to degradation of the nickel catalyst. This side reaction can be suppressed if an NHC is used which cannot react with the boron source, and application of the LAB-Rep model pointed to IMes (35.9 %VBur, d = 1.97 Å) as a likely NHC ligand that should not react with B2pin2 (45.1 %VBur, d = 1.44 Å) to yield an adduct B2pin2·IMes (%VBur_all = 108.2; design 3; VCl). The use of [Ni(IMes)2] then paved the way for successful defluoroborylation catalysis.48 In contrast, for the ethyl glycol ether B2eg2 (40.5 %VBur, d = 1.43 Å) adduct formation to yield B2eg2·IMes (%VBur_all = 103.5; design 3; Vcl) was observed and the adduct was structurally characterized.31d
 | ||
| Scheme 4 Reaction of B2pin2 with IiPr to yield the mono-NHC adduct B2pin2·IiPr. | ||
| LA | LB | Isomer | V corr | d(LA)/Å | d(LB)/Å | %Vcorr | %VLA | %VLB | %VBur_all |
|---|---|---|---|---|---|---|---|---|---|
| PF5 | IMe | — | bcl | 1.65 | 2.01 | 20.1 | 38.4 | 27.4 | 85.9 |
| PF5 | IiPr | — | bcl | 1.65 | 1.99 | 21.0 | 38.4 | 28.8 | 89.7 |
| PF5 | ItBu | — | bcl | 1.65 | 2.06 | 20.3 | 38.4 | 37.9 | 96.6 |
| PF5 | aItBu | — | bcl | 1.65 | 2.01 | 20.1 | 38.4 | 28.8 | 87.3 |
| PF5 | IDipp | — | bcl | 1.65 | 1.96 | 19.8 | 38.4 | 36.5 | 94.7 |
| PF5 | aIDipp | — | bcl | 1.65 | 1.98 | 19.9 | 38.4 | 31.8 | 90.1 |
| PF2(C2F5)3 | IMe | mer–trans | hbcl | 1.69 | 2.01 | 16.0 | 67.7 | 27.4 | 102.3 |
| PF2(C2F5)3 | IiPr | mer–trans | hbcl | 1.69 | 1.99 | 15.8 | 67.7 | 28.8 | 103.5 |
| PF2(C2F5)3 | ItBu | mer–trans | hbcl | 1.69 | 2.06 | 16.4 | 67.7 | 37.9 | 113.1 |
| PF2(C2F5)3 | aItBu | mer–trans | hbcl | 1.69 | 2.01 | 16.0 | 67.7 | 28.8 | 103.7 |
| PF2(C2F5)3 | IDipp | mer–trans | hbcl | 1.69 | 1.96 | 15.6 | 67.7 | 36.5 | 111.0 |
| PF2(C2F5)3 | aIDipp | mer–trans | hbcl | 1.69 | 1.98 | 15.8 | 67.7 | 31.8 | 106.4 |
| PF5 | PMe3 | — | cl | 1.65 | 2.21 | 30.8 | 38.4 | 24.1 | 93.3 |
| PF2(C2F5)3 | PMe3 | mer–cis | hseg | 1.67 | 2.21 | 18.2 | 59.3 | 24.1 | 101.6 |
| fac | 1.67 | 20.5 | 54.8 | 99.4 | |||||
| PF5 | PPh3 | — | cl | 1.65 | 2.25 | 31.1 | 38.4 | 31.1 | 100.6 |
| PF2(C2F5)3 | PPh3 | mer–cis | hseg | 1.67 | 2.25 | 18.7 | 59.3 | 31.1 | 109.1 |
| fac | 1.67 | 20.9 | 54.8 | 106.8 |
Conclusions
Steric and electronic effects are decisive parameters in chemistry which determine the shape and the reactivity of molecules. For Lewis acids and Lewis bases, different models have been developed in the last few decades to scale their acid/base strengths in a rather easy way; the most prominent are probably fluoride ion affinity (FIA) for Lewis acids and the Tolman parameter for Lewis bases. Both can be derived from experiments, but in practice they are most often evaluated by simple, low-cost (DFT) calculations. Steric effects of Lewis bases are nowadays quantified by judging the percent buried volume (%VBur), which can be easily performed thanks to the SambVca 2.1 web application. It has been demonstrated over the last decade that %VBur is a versatile and reliable descriptor of steric properties of different ligands such as NHCs and phosphines. As there is currently no easy approach to dealing with steric effects of differently substituted Lewis acidic centers, we introduce herein the first general approach to easily access and quantify steric properties of Lewis acids. In addition, based on this approach an easy-to-use model for the prediction of whether a specific Lewis acid/base adduct may be formed considering steric effects was developed. This model implies the application of the concept of the percent buried volume (%VBur) to fluoride ion adducts of Lewis acids. In principle, this model can be extended to other adducts, but fluoride adducts were chosen as those are frequently calculated to judge fluoride ion affinities, and thus data such as cartesian coordinates are easily available. Furthermore, many fluoride adducts have been characterized crystallographically, providing additional access to the coordinates required.We applied this model to a large number (240) of different fluoride adducts of Lewis acids of group 13, 14, and 15 elements using low level DFT (def2-SV(P)/BP86) optimized geometries and report their percent buried volume as well as their topographic steric maps. This evaluation does not require the fluoride atom of the anion fluoride adduct [LA–F]− located in a fixed distance to the Lewis acidic center of the LA d(LA–F), as the values obtained for d(LA–F) of a certain Lewis-acidic center (e.g., of boron) vary only little for the different systems and these small differences translate into only minor differences of %VBur. Note also that these distances [LA–F]− can be set to any value wanted within the user-friendly SambVca 2.1 web application if this is required. A chart of %VBurvs. Lewis-acidic main group element of all Lewis acids considered in this study revealed that there is a wide range of %VBur covered by known Lewis acids and that any steric demand between %VBur = 30 and %VBur = 75 can be realized easily by the choice of the proper element, coordination number and substituent. Very valuable are charts which correlate %VBur and FIA (fluoride ion affinity) as a scale for the Lewis acidity of the compound, which combine steric and electronic features of the Lewis acid under consideration and provide valuable information about stereo-electronic properties of the Lewis acid. As there is no general correlation between Lewis acidity and steric demand, both factors have to be addressed. Thus, the present model presents a highly valuable tool for synthetic and materials chemists.
With these data in hand, we introduce the novel LAB-Rep (Lewis Acid/Base Repulsion) model, which judges steric repulsion in Lewis acid/base pairs and helps to predict if an arbitrary pair of Lewis acid and Lewis base can form an adduct with respect to their steric properties. The reliability of this model is demonstrated by four selected case studies, which show the versatility of our model. Using the listed buried volumes of Lewis acids %VBur_LA and of Lewis bases %VBur_LB it has to be emphasized that no crystal structure or quantum chemical calculation is required to evaluate steric repulsion in Lewis acid/base pairs. A user-friendly Excel spreadsheet is provided in the ESI† to this publication, which can be used for this purpose.
Data availability
%VBur values of different Lewis acids and Lewis bases, their topographic steric maps, additional data and spectra, crystallographic data, NMR spectra, details on the DFT calculations and Cartesian coordinates of the DFT optimized structures can be found in the ESI.†Author contributions
U. R., L. Z., M. R., S. A. F., and M. F. conceived the project. M. R. and U. R. performed the quantum chemical calculations and the determination of the %VBur of the Lewis acids. L. Z. developed the LAB-Rep model. U. R., M. F., and L. Z. wrote the manuscript. All authors discussed the results and contributed to the final manuscript.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
L. Z. thanks the Studienstiftung des Deutschen Volkes for generous support. U. R. and M. F. thank the Julius-Maximilians-Universität Würzburg and U. R. the Deutsche Forschungsgemeinschaft (DFG; U. R.: Ra720/13) for financial support.Notes and references
-
(a)
G. N. Lewis, Valence and the Structure of Atoms and Molecules, Chemical Catalog Company, New York, 1923 Search PubMed
; (b) G. N. Lewis, J. Franklin Inst., 1938, 226, 293–313 CrossRef
- See for example:
(a) L. Greb, Chem.–Eur. J., 2018, 24, 17881–17896 CrossRef CAS PubMed
-
(a)
Lewis Acids in Organic Synthesis, ed. H. Yamamoto, Wiley-VCH, Weinheim, 2000 Search PubMed
-
(a) G. C. Welch, R. R. San Juan, J. D. Masuda and D. W. Stephan, Science, 2006, 314, 1124–1126 CrossRef CAS PubMed
- See for example:
(a) A. Andresen, H.-G. Cordes, J. Herwig, W. Kaminsky, A. Merck, R. Mottweiler, J. Pein, H. Sinn and H.-J. Vollmer, Angew. Chem., Int. Ed., 1976, 15, 630–632 CrossRef
- E. Y. X. Chen and T. J. Marks, Chem. Rev., 2000, 100, 1391–1434 CrossRef CAS PubMed
-
(a) W. Beck and K. Suenkel, Chem. Rev., 1988, 88, 1405–1421 CrossRef CAS
-
C. Knapp, Halogenated borates and carborates as weakly coordinating anions, in Comprehensive Inorganic Chemistry II, ed. T. Chivers, 2012, vol. 1, pp. 651–679 Search PubMed
- S. A. Föhrenbacher, M. J. Krahfuss, L. Zapf, A. Friedrich, N. V. Ignat’ev, M. Finze and M. Radius, Chem.–Eur. J., 2021, 27, 3504–3516 CrossRef PubMed
-
(a) D. E. Fogg and E. N. dos Santos, Coord. Chem. Rev., 2004, 248, 2365–2379 CrossRef CAS
- I. B. Sivaev and V. I. Bregadze, Coord. Chem. Rev., 2014, 270–271, 75–88 CrossRef CAS
-
(a) E. Krause and R. Nitsche, Ber. Dtsch. Chem. Ges. A/B, 1922, 55, 1261–1265 CrossRef
-
(a) D. Williams, B. Pleune, J. Kouvetakis, M. D. Williams and R. A. Andersen, J. Am. Chem. Soc., 2000, 122, 7735–7741 CrossRef CAS
-
(a) M. Finze, E. Bernhardt, M. Zähres and H. Willner, Inorg. Chem., 2004, 43, 490–505 CrossRef CAS PubMed
-
(a) U. Mayer, V. Gutmann and W. Gerger, Monatsh. Chem., 1975, 106, 1235–1257 CrossRef CAS
- R. G. Pearson, J. Am. Chem. Soc., 1963, 85, 3533–3539 CrossRef CAS
-
(a) A. Hanft, K. Radacki and C. Lichtenberg, Chem.–Eur. J., 2021, 27, 6230–6239 CrossRef CAS PubMed
- R. F. Childs, D. L. Mulholland and A. Nixon, Can. J. Chem., 1982, 60, 809–812 CrossRef CAS
-
(a) I. R. Beattie and T. Gilso, J. Chem. Soc., 1964, 2292–2295 RSC
-
(a) D. F. Shriver and B. Swanson, Inorg. Chem., 1971, 10, 1354–1365 CrossRef CAS
-
(a) J. R. Gaffen, J. N. Bentley, L. C. Torres, C. Chu, T. Baumgartner and C. B. Caputo, Chem, 2019, 5, 1567–1583 CrossRef CAS
-
(a) M. M. Morgan, A. J. V. Marwitz, W. E. Piers and M. Parvez, Organometallics, 2013, 32, 317–322 CrossRef CAS
-
(a) J. W. Larson and T. B. McMahon, J. Am. Chem. Soc., 1985, 107, 766–773 CrossRef CAS
-
(a) I. Krossing and I. Raabe, Chem.–Eur. J., 2004, 10, 5017–5030 CrossRef CAS PubMed
- For a discussion of the history and different methods to assess steric properties see for example: B. Pinter, T. Fievez, F. M. Bickelhaupt, P. Geerlings and F. De Proft, Phys. Chem. Chem. Phys., 2012, 14, 9846–9854 RSC
-
(a) I. Fernández and F. M. Bickelhaupt, Chem. Soc. Rev., 2014, 43, 4953–4967 RSC
-
(a) C. A. Tolman, J. Am. Chem. Soc., 1970, 92, 2956–2965 CrossRef CAS
-
(a) N. Fey, A. G. Orpen and J. N. Harvey, Coord. Chem. Rev., 2009, 253, 704–722 CrossRef CAS
-
(a) A. C. Hillier, W. J. Sommer, B. S. Yong, J. L. Petersen, L. Cavallo and S. P. Nolan, Organometallics, 2003, 22, 4322–4326 CrossRef CAS
-
(a) S. A. Föhrenbacher, V. Zeh, M. J. Krahfuss, N. Ignat'ev, M. Finze and U. Radius, Eur. J. Inorg. Chem., 2021, 1941–1960 CrossRef
-
(a) S. Pietsch, E. C. Neeve, D. C. Apperley, R. Bertermann, F. Mo, D. Qiu, M. S. Cheung, L. Dang, L. Liu, J. Wang, U. Radius, Z. Lin, C. Kleeberg and T. B. Marder, Chem.–Eur. J., 2015, 21, 7082–7098 CrossRef CAS PubMed
-
(a) H. Schneider, D. Schmidt and U. Radius, Chem.–Eur. J., 2015, 21, 2793–2797 CrossRef CAS PubMed
-
(a) S. Pelzer, B. Neumann, H.-G. Stammler, N. Ignat'ev and B. Hoge, Angew. Chem., Int. Ed., 2016, 55, 6088–6092 CrossRef CAS PubMed
-
(a) D. E. Young, L. R. Anderson and W. B. Fox, Chem. Commun., 1971, 736 RSC
- M. O. Akram, J. R. Tidwell, J. L. Dutton and C. D. Martin, Angew. Chem., Int. Ed., 2022, 61, e202212073 CrossRef CAS PubMed
-
(a) J. F. Kögel, D. A. Sorokin, A. Khvorost, M. Scott, K. Harms, D. Himmel, I. Krossing and J. Sundermeyer, Chem. Sci., 2018, 9, 245–253 RSC
-
(a) W. Strohmeier and F.-J. Müller, Chem. Ber., 1967, 100, 2812–2821 CrossRef CAS
-
(a) D. G. Gusev, Organometallics, 2009, 28, 6458–6461 CrossRef CAS
- We consider here Grebs’s FIA calculations, see ref. 24f and 24g; for the values obtained by Krossing (ref. 24d) see the ESI.†.
-
(a) V. Branchadell, A. Sbai and A. Oliva, J. Phys. Chem., 1995, 99, 6472–6476 CrossRef CAS
- https://perkinelmerinformatics.com/products/research/chemdraw .
-
(a) H. C. Brown, H. I. Schlesinger and S. Z. Cardon, J. Am. Chem. Soc., 1942, 64, 325–329 CrossRef CAS
- A. G. Massey and A. J. Park, J. Organomet. Chem., 1964, 2, 245–250 CrossRef CAS
- H. Jacobsen, H. Berke, S. Döring, G. Kehr, G. Erker, R. Fröhlich and O. Meyer, Organometallics, 1999, 18, 1724–1735 CrossRef CAS
- G. C. Welch, T. Holtrichter-Roessmann and D. W. Stephan, Inorg. Chem., 2008, 47, 1904–1906 CrossRef CAS PubMed
- P. A. Chase and D. W. Stephan, Angew. Chem., Int. Ed., 2008, 47, 7433–7437 CrossRef CAS PubMed
-
(a) G. Wittig and E. Benz, Chem. Ber., 1959, 92, 1999–2013 CrossRef CAS
-
(a) J. Zhou, M. Kuntze-Fechner, R. Bertermann, U. S. D. Paul, J. H. J. Berthel, A. Friedrich, Z. Du, T. B. Marder and U. Radius, J. Am. Chem. Soc., 2016, 138, 5250–5253 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2231981. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3sc00037k |
| This journal is © The Royal Society of Chemistry 2023 |