
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCobalt-catalyzed enantioselective desymmetrizing reductive cyclization of alkynyl cyclodiketones†
Ren-Xiao
Liang
 *a,
Heng-Wei
Tang
a,
Jia-Liang
Liu
a,
Jian-Feng
Xu
a,
Ling-Jia
Chen
a and
Yi-Xia
Jia
*ab
*a,
Heng-Wei
Tang
a,
Jia-Liang
Liu
a,
Jian-Feng
Xu
a,
Ling-Jia
Chen
a and
Yi-Xia
Jia
*ab
aCollege of Chemical Engineering, State Key Laboratory Breeding Base of Green-Chemical Synthesis Technology, Zhejiang University of Technology, Chaowang Road 18#, Hangzhou 310014, China. E-mail: liangrx@zjut.edu.cn; yxjia@zjut.edu.cn
bState Key Laboratory of Organometallic Chemistry, Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, Shanghai 200032, P. R. China
First published on 15th May 2023
Abstract
A highly enantioselective cobalt-catalyzed desymmetrizing reductive cyclization of alkynyl cyclodiketones has been developed. Under mild reaction conditions by employing HBpin as a reducing agent and ferrocene-based PHOX as a chiral ligand, a series of polycyclic tertiary allylic alcohols bearing contiguous quaternary stereocenters are achieved in moderate to excellent yields with excellent enantioselectivities (up to 99%). Broad substrate scope and high functional group compatibility are observed in this reaction. A CoH-catalyzed pathway involving alkyne hydrocobaltation followed by nucleophilic addition to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond is proposed. Synthetic transformations of the product are conducted to demonstrate the practical utilities of this reaction.
O bond is proposed. Synthetic transformations of the product are conducted to demonstrate the practical utilities of this reaction.
Introduction
Cyclic allylic alcohols are important building blocks for organic synthesis and unique structural motifs of natural products and biologically active molecules.1 The reductive cyclization of alkynyl aldehydes or ketones provides an efficient, straightforward, and atom-economic access to such structural units.2 A number of transition metal catalysts have been employed in this cyclization reaction by merging with suitable hydride-donating reagents, such as hydrogen, organosilanes, alcohol, and others.3 As a result, the corresponding enantioselective variants were well established in the presence of Rh-,3a,b,r Ni-,3i–n and Pd-3o based chiral catalysts (Scheme 1a). Despite these important advances, the documented reports are predominantly limited to linear aldehydes or ketones and only one single stereogenic carbon center was formed in these transformations. To the best of our knowledge, the reaction of alkynyl cyclic ketones has remained unexplored probably due to the lower reactivity caused by the increasing steric hindrance and the lack of efficient chiral catalysts,4 although it would efficiently assemble chiral polycyclic molecules and build two chiral carbocenters in one step (Scheme 1b).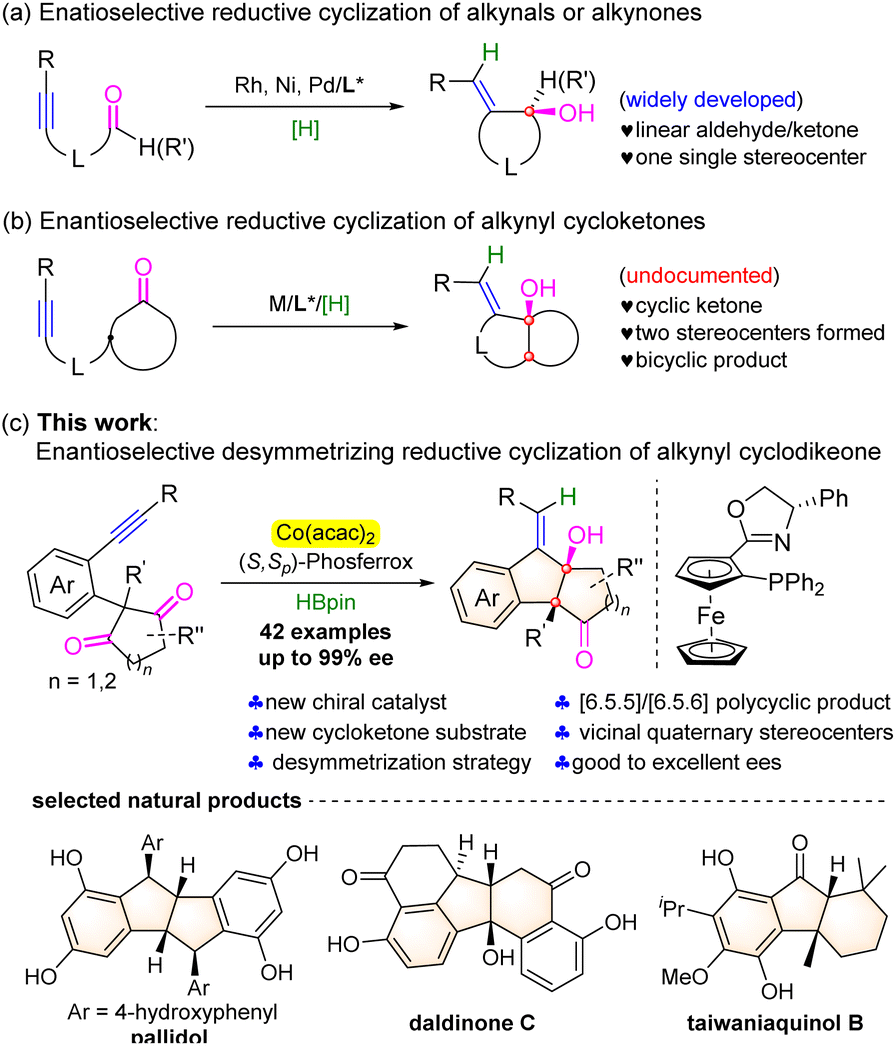 | ||
| Scheme 1 Transition-metal-catalyzed enantioselective reductive couplings of alkynals and alkynones. | ||
Recently, chiral cobalt complexes have become reliable catalysts for the asymmetric enyne hydrofunctionalization/cyclization reactions.5 Ge and co-workers disclosed highly enantioselective hydroborylation- or hydrosilylation/cyclization of 1,6- or 1,7-enynes with HBpin or hydrosilanes, leading to a range of important functionalized chiral heterocyclic compounds.6a–c,f In addition, the asymmetric hydroarylative or hydroacylative cyclization of enynes involving a C–H functionalization process was also achieved to afford the cyclic products in excellent enantioselectivities with high atom economy.6d,e,g–i Inspired by these results, we envisioned that the asymmetric reductive coupling of alkynyl cyclic ketones might be possible under the chiral cobalt catalyst. However, this reaction is indeed very challenging since both the triple bond of alkynes7 and the polar CO bond of ketones8 are prone to be reduced. Noteworthy is that Xia and co-workers disclosed recently an intermolecular enantioselective reductive cross-coupling of alkynes and aldehydes by merging cobalt catalysis with photocatalysis.9 Herein, we communicate an enantioselective desymmetrizing reductive coupling of alkynyl cyclic 1,3-diketones by utilizing the complex of Co(acac)2 and a ferrocene-based chiral phosphine-oxazoline ligand as the catalyst and HBpin as the reducing agent. Through a possible sequential alkyne syn-hydrocobaltation followed by desymmetrizing nucleophilic addition to ketones, a number of optically active polycyclic tertiary allylic alcohols bearing vicinal quaternary stereocenters were obtained in moderate to good yields and good to excellent enantioselectivities. The framework of the resulting molecules constitutes the core structure of natural products, such as pallidol, daldinone C, and taiwaniaquinol B (Scheme 1c).10
Results and discussion
We commenced the study with alkynyl cyclopentane-1,3-dione 1a as the model substrate. The initial test showed that the intramolecular reductive cyclization reaction of 1a proceeded smoothly to afford the desired product 2a in 29% yield with 23% ee in the presence of Co(acac)2 catalyst and chiral ligand (S)-SEGPHOS L1 by using HBpin as the reducing reagent in toluene at room temperature (Table 1, entry 1). Encouraged by this result, the solvent effect was then examined (entries 2–6). The yield and ee were both increased in THF and 1,4-dioxane (entries 2 and 5), while lower yields were observed in Et2O, DME, and DCM (entries 3, 4, and 6). No product was detected when using Et3SiH as the reducing reagent (entry 7). To further improve the yield and ee, other chiral ligands were investigated. Diphosphine ligand (S,S)-QuinoxP* L2 led to 2a in 75% ee and a poor yield of 17%, while (S,S)-Ph-BPE L3 resulted in lower yield and lower ee (entries 8 and 9). Moreover, poor yield and ee were observed for a number of BINOL-based chiral phosphoramidite ligands. Next, we turned our attention to the bidentate P,N-ligand PHOX. The use of (S)-iPr-PHOX L4 afforded 2a in 61% yield and 43% ee (entry 10), while the corresponding (S)-tBu-PHOX L5 or (S)-Ph-PHOX L6 led to poor yields and moderate ee values along with the observation of by-product 3 derived from the direct reduction of the CO bond (entries 11 and 12). Gratifyingly, the ferrocene-based chiral PHOX ligands L7 and L8 significantly improved the enantioselectivities to 80% and 99% with 66% and 85% yields, respectively (entries 13 and 14). In comparison, the analogous Bn-ligand L9 delivered product 2a in poor yield and poor ee (entry 15). Finally, a fast reaction was observed at 50 °C while the yield was diminished to 63% (entry 16). Control experiments demonstrated that the desymmetrizing reductive cyclization reaction could not occur in the absence of Co(acac)2 or HBpin.
|
|
|||||
|---|---|---|---|---|---|
| Entry | L* | Solvent | 2a | 3 | |
| Yieldb/% | eec/% | Yieldb/% | |||
| a Reaction conditions: 1a (0.2 mmol), Co(acac)2 (3 mol%), L* (4 mol%), and HBpin (1.5 equiv.) in the solvent (1.0 mL) at room temperature for 48 h. b Isolated yields. c Determined by chiral HPLC. d Et3SiH instead of HBpin. e At 50 °C for 24 h. | |||||
| 1 | L1 | Toluene | 29 | 23 | — |
| 2 | L1 | THF | 43 | 57 | — |
| 3 | L1 | Et2O | 26 | 20 | — |
| 4 | L1 | DME | 20 | 41 | — |
| 5 | L1 | 1,4-Dioxane | 53 | 51 | — |
| 6 | L1 | DCM | <10 | nd | — |
| 7d | L1 | 1,4-Dioxane | nd | — | — |
| 8 | L2 | 1,4-Dioxane | 17 | 75 | — |
| 9 | L3 | 1,4-Dioxane | 31 | 34 | — |
| 10 | L4 | 1,4-Dioxane | 61 | 43 | — |
| 11 | L5 | 1,4-Dioxane | 11 | 53 | 21 |
| 12 | L6 | 1,4-Dioxane | 12 | 54 | 18 |
| 13 | L7 | 1,4-Dioxane | 66 | 80 | — |
| 14 | L8 | 1,4-Dioxane | 85 | 99 | — |
| 15 | L9 | 1,4-Dioxane | 14 | 18 | 28 |
| 16e | L8 | 1,4-Dioxane | 63 | 99 | — |
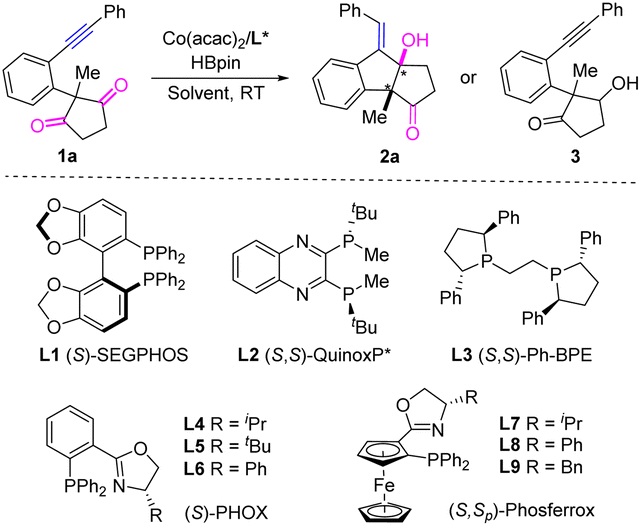
With the optimal conditions in hand, we then investigated the substrate scope of this cobalt-catalyzed reductive cyclization reaction. Excellent enantioselectivities with moderate to good yields were generally observed in the reactions. As shown in Scheme 2, a 1.0 mmol-scale reaction of 1a led to product 2a in a slightly lower yield (81%) and 99% ee. The substrates bearing different aryl groups linked to the alkyne moiety were first examined. Various substituents attached to the para-, meta-, and ortho-positions of the benzene ring, either electron-donating groups (methyl: 2b, 2g, and 2k; methoxy: 2c and 2l) or electron-withdrawing groups (chloride: 2d and 2h; fluoride: 2e and 2i; methoxycarbonyl: 2f) were well tolerated to afford the corresponding products in 52–89% yields and 93–99% ees. No steric effect was observed for these substituents. The reaction of a 3,5-dimethoxy-substrate led to product 2j in 79% yield and 99% ee. Moreover, a 2-naphthyl substituted substrate reacts smoothly to furnish product 2m in 74% yield and 95% ee. The thiophenyl group is also compatible in the reaction and the corresponding product 2n was achieved in 51% yield and 98% ee. The absolute configuration of 2n was determined to be (3aR,8aR,E) based on X-ray crystallographic analysis.11
 | ||
| Scheme 2 Substrate scope. Reaction conditions: 1 (0.2 mmol), Co(acac)2 (3 mol%), L8 (4 mol%), and HBpin (1.5 equiv.) in 1,4-dioxane (1.0 mL) at rt. a In THF. | ||
In addition, a series of linear alkyl and heteroalkyl substituents linked to the alkyne moiety were investigated. As shown in Scheme 2, the reactions of methyl, benzyl, and n-octyl substrates afforded the corresponding products 2o, 2p, and 2w in moderate to good yields with 94–98% ees. High compatibility of the heteroalkyl groups was observed. A number of functional substituents, including phenoxyl (2q), benzyloxyl (2s), silyloxyl (2t), chloride (2u), free hydroxyl (2v), ester (2x), and imide (2y), were all well tolerated to furnish the desired products in 59–83% yields and 90–98% ees. The reaction of a diethoxymethyl-substrate also led to product 2r in 85% yield and 89% ee. Moreover, the substituent effect of R2 attached to the linker benzene ring was also evaluated. The methyl (2z), trifluoromethyl (2aa), methoxycarbonyl (2ab), and difluoro (2ac) groups were introduced into the substrates and the corresponding products were afforded in moderate to good yields (65–83%) and excellent enantioselectivities (90–99%).
Variation of the cyclic diketone moiety was also conducted. The reaction of the 2-ethyl 1,3-cyclopentanedione substrate led to 2ad in 74% yield and 98% ee. Excellent ees with moderate to excellent yields were also obtained for the polycyclic [6.5.5.6] products 2ae–2aj that bear additional fused benzene on the ring of cyclopentanedione. Furthermore, the reactions of the six-membered 1,3-cyclohexanedione-derived substrates also proceeded smoothly to afford the corresponding products 2ak–2an, although their ee values (73–87%) were relatively lower than those for 1,3-cyclopentanedione substrates. The substrate bearing an additional CH2 group between benzene and cyclodiketone is also suitable for the desymmetrizing reductive cyclization reaction, affording the desired product 2ao in 23% yield and 73% ee by using L8 as the chiral ligand in THF. It is noteworthy that the present chiral cobalt catalyst is inefficient in the intermolecular reductive coupling reaction between alkynes and ketones/aldehydes9 and the intramolecular reductive cyclization reactions of acyclic alkynyl monoaldehydes or monoketones.3
To gain insight into the reaction mechanism, control experiments were conducted (Scheme 3). As shown in eqn (1) and (2), the reactions of 1a employing either the Co(acac)2/dppbz complex or the preformed CoH(dppbz)2 as the catalyst6a could lead to rac-2a in 37% and 30% yields, which might imply that CoH was the active catalyst for this reductive cyclization reaction.12
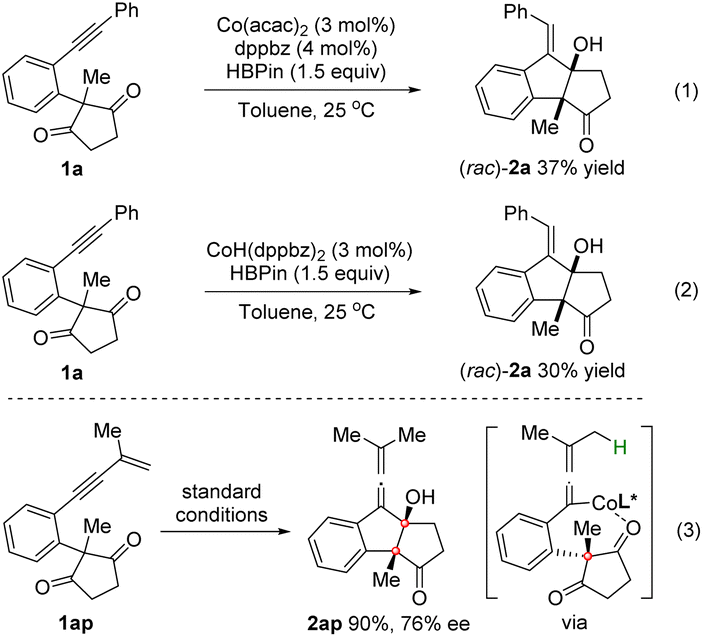 | ||
| Scheme 3 Control experiments. | ||
Moreover, the reaction of enynone substrate 1ap under the standard conditions afforded an allenylic alcohol product 2ap in 90% yield with 76% ee, in which an allenyl-Co intermediate generated by enyne hydrocobaltation was likely involved (eqn (3)). Based on these results, a CoH-catalyzed reaction pathway was proposed. As depicted in Scheme 4, the active CoH species was initially formed by the reaction of Co(acac)2 with HBpin. Vinyl-Co intermediate A was then generated via alkyne hydrocobaltation. Subsequent desymmetrizing nucleophilic addition to the CO bond followed by transmetalation of the resulting cobalt alkoxide B with HBpin affords intermediate C, which furnishes the product after hydrolysis. The active CoH catalyst is simultaneously regenerated to complete the catalytic cycle.
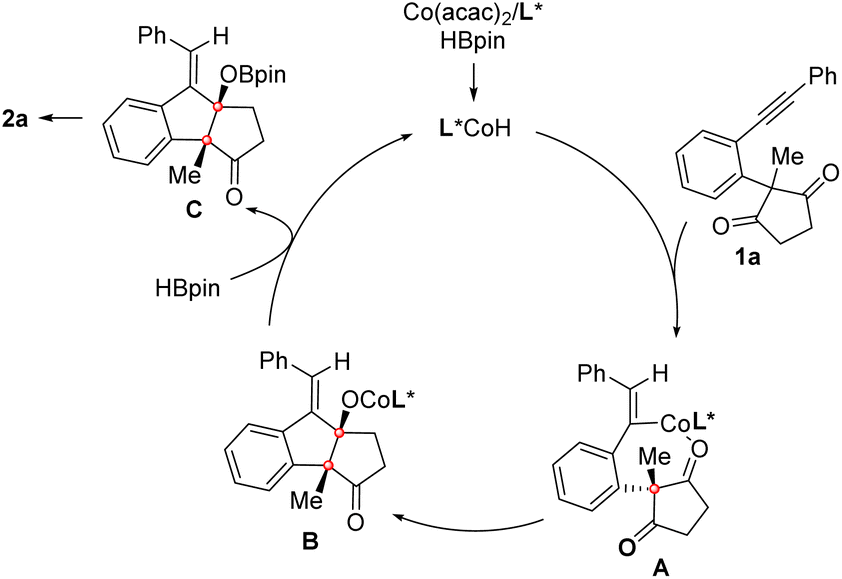 | ||
| Scheme 4 Proposed reaction mechanism. | ||
A number of synthetic transformations of the product were then investigated. The Wittig reaction of 2a with Ph3PMeBr was first tested in the presence of tBuOK as a base. The target exocyclic olefin 4 was obtained in excellent yield, while the enantiomeric excess was irregularly eroded. We assumed that a retro-Aldol reaction of the β-hydroxyl ketone 2a followed by aldol reaction might occur under the basic conditions, which would cause racemization of 2a and consequently lead to a decreased ee value of 4 (Scheme 5). Indeed, the erosion of the ee value was observed when treating 2a with tBuOK.
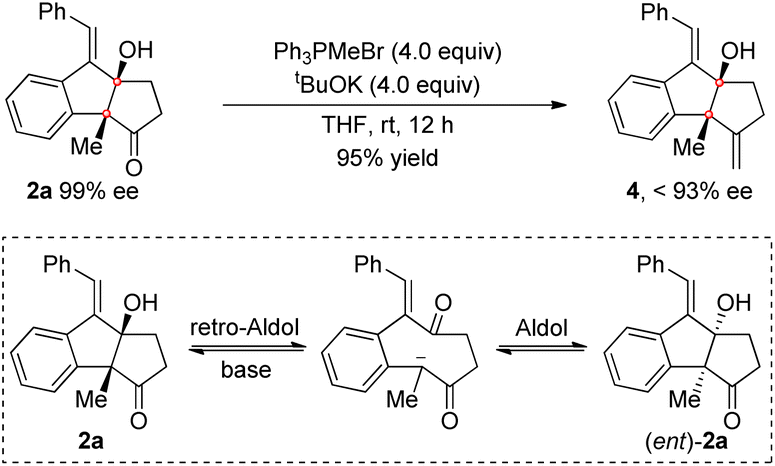 | ||
| Scheme 5 ee erosion in the Wittig reaction of 2a. | ||
As shown in Scheme 6, a MOM-protected compound 5 was then prepared to be used as the starting material for the Wittig reaction. As expected, the corresponding product 6 was obtained in 90% yield with 99% ee. Compound 6 was further converted to cyclopropane 7 in 58% yield and 99% ee through deprotection followed by a Simmons–Smith reaction. Meanwhile, the condensation of 2a with TsNHNH2 led to hydrozone 8, which further coupled with PhB(OH)2 to afford compound 9 (ref. 13) bearing three contiguous stereocenters in 53% yield, 99% ee, and > 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 dr.
:1 dr.
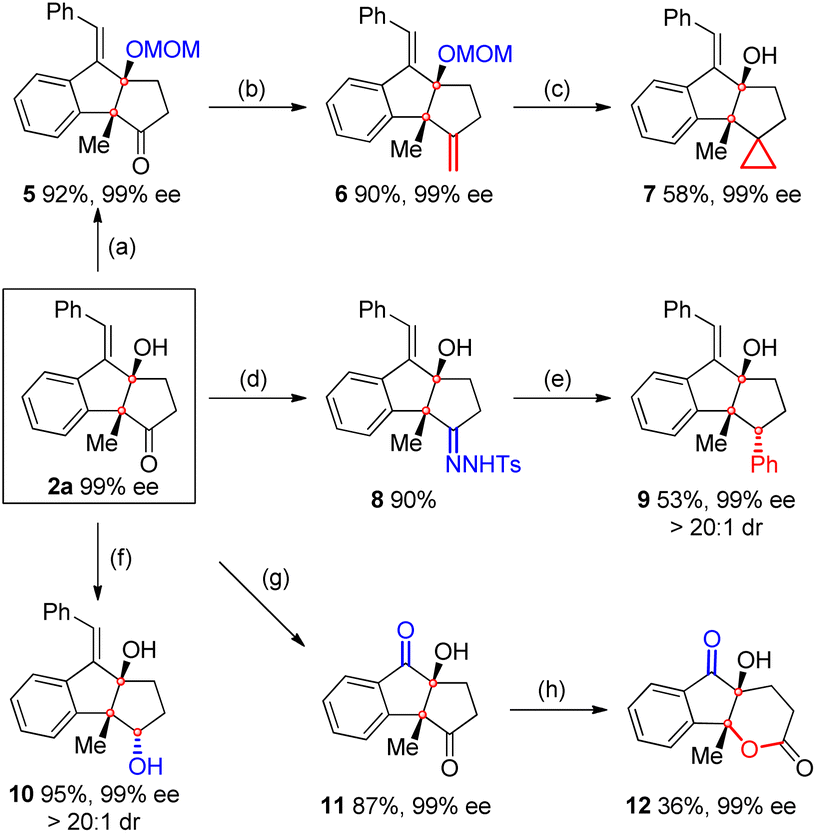 | ||
| Scheme 6 Synthetic transformations. reaction conditions: (a) 2a (0.2 mmol), CH3OCH2Br (2.8 equiv.), and DIPEA (4.4 equiv.) in DCM (4 mL) at 80 °C for 12 h. (b) 5 (0.2 mmol), PPh3MeBr (4.0 equiv.), and tBuOK (4.0 equiv.) in THF (4 mL) at rt for 12 h. (c) (i) 6 (0.2 mmol) and HCl (1 N, 10 mL) in CHCl3/MeOH (1/1, 8 mL) at rt for 12 h; (ii) Et2Zn (5.0 equiv., 1.0 M in toluene), CH2I2 (5.0 equiv.) in DCM for 6 h, −10 °C-rt. (d) 2a (0.2 mmol) and TsNHNH2 (1.0 equiv.) in MeOH at rt for 24 h. (e) 8 (0.2 mmol), PhB(OH)2 (1.5 equiv.), and K2CO3 (1.5 equiv.) in 1,4-dioxane at 110 °C for 12 h. (f) 2a (0.2 mmol) and NaBH4 (1.2 equiv.) in MeOH at 0 °C for 10 min. (g) 2a (0.2 mmol) in MeOH/DCM (1/1, 10 mL) was purged with O3 at −78 °C for 5 min, then PPh3 (2.0 equiv.) was added. (h) 11 (0.2 mmol) and m-CPBA (4.0 equiv.) in DCM at 0 °C for 48 h. | ||
Moreover, the reduction of 2a with NaBH4 delivered diol 10 in 95% yield, 99% ee, and > 20:1 dr. Relative configurations of compounds 9 and 10 were determined by 2D-NOESY analysis. The oxidation of 2a with O3 furnished diketone 11 in 87% yield and 99% ee, which was subsequently converted to a polycyclic lactone 12 in 36% yield and 99% ee through a Baeyer–Villiger reaction.
Conclusions
In summary, we have developed the first example of cobalt-catalyzed enantioselective desymmetrizing reductive cyclization reaction of alkynyl cyclic 1,3-diketones. An array of polycyclic tertiary alcohols containing an exocyclic trisubstituted olefin and contiguous quaternary stereocenters are afforded in moderate to good yields and good to excellent ees (up to 99%). The protocol features high functional group tolerance and mild reaction conditions. A reaction pathway involving alkyne hydrocobaltation followed by desymmetrizing nucleophilic addition to the CO bond is proposed. Synthetic transformations are conducted to convert the product to a range of valuable polycyclic molecules.
Data availability
All experimental and characterization data, as well as NMR spectra are available in the ESI.† Crystallographic data for compound 2n have been deposited in the Cambridge Crystallographic Data Centre (CCDC 2025743).Author contributions
R.-X. L. and Y.-X. J. conceived the idea and wrote the manuscript. Y.-X. J. supervised the project. R.-X. L. and H.-W. T. performed most of the experiments. J.-L. L., J.-F. X and L.-J. C. prepared some of the substrates, and collected and analysed the spectroscopic data. All authors contributed to discussing the results.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful for generous support from the National Natural Science Foundation of China (no. 22071217 and 91956117). This research is also supported by the Natural Science Foundation of Zhejiang Province (no. LY22B020008, LQ22B020011 and LZ23B020006).Notes and references
- (a) A. S. Franklin and L. E. Overman, Chem. Rev., 1996, 96, 505–522 CrossRef CAS PubMed; (b) H.-B. Wang, X.-Y. Wang, L.-P. Liu, G.-W. Qin and T.-G. Kang, Chem. Rev., 2015, 115, 2975–3011 CrossRef CAS PubMed; (c) S. P. Simeonov, J. P. M. Nunes, K. Guerra, V. B. Kurteva and C. A. M. Afonso, Chem. Rev., 2016, 116, 5744–5893 CrossRef CAS PubMed; (d) J. Cossy, S. Arseniyadis and C. Meyer, Metathesis in Natural Product Synthesis: Strategies, Substrates, and Catalysts, Wiley-VCH, Weinheim, Germany, 1st edn, 2010 CrossRef.
- For reviews and book, see: (a) J. Montgomery, Acc. Chem. Res., 2000, 33, 467–473 CrossRef CAS PubMed; (b) H.-Y. Jang and M. J. Krische, Acc. Chem. Res., 2004, 37, 653–661 CrossRef CAS PubMed; (c) E. Skucas, M.-Y. Ngai, V. Komanduri and M. J. Krische, Acc. Chem. Res., 2007, 40, 1394–1401 CrossRef CAS PubMed; (d) M. J. Krische, Metal Catalyzed Reductive C–C Bond Formation, Springer Berlin Heidelberg, 2007 CrossRef; (e) R. M. Moslin, K. Miller-Moslinb and T. F. Jamison, Chem. Commun., 2007, 4441–4449 RSC; (f) K. Tanaka and Y. Tajima, Eur. J. Org. Chem., 2012, 3715–3725 CrossRef CAS; (g) E. P. Jackson, H. A. Malik, G. J. Sormunen, R. D. Baxter, P. Liu, H. Wang, A.-R. Shareef and J. Montgomery, Acc. Chem. Res., 2015, 48, 1736–1745 CrossRef CAS PubMed; (h) E. A. Standley, S. Z. Tasker, K. L. Jensen and T. F. Jamison, Acc. Chem. Res., 2015, 48, 1503–1514 CrossRef CAS PubMed; (i) M. Holmes, L. A. Schwartz and M. J. Krische, Chem. Rev., 2018, 118, 6026–6052 CrossRef CAS PubMed; (j) Y.-L. Liu and X.-T. Lin, Adv. Synth. Catal., 2019, 361, 876–918 CrossRef CAS; (k) E. Ortiz, J. Shezaf, Y.-H. Chang and M. J. Krische, ACS Catal., 2022, 12, 8164–8174 CrossRef CAS PubMed.
- For hydrogen: (a) J. U. Rhee and M. J. Krische, J. Am. Chem. Soc., 2006, 128, 10674–10675 CrossRef CAS PubMed; (b) J. U. Rhee, R. A. Jones and M. J. Krische, Synthesis, 2007, 21, 3427–3430 Search PubMed ; For organosilanes: ; (c) I. Ojima, M. Tzamarioudaki and C.-Y. Tsai, J. Am. Chem. Soc., 1994, 116, 3643–3644 CrossRef CAS; (d) W. E. Crowe and M. J. Rachita, J. Am. Chem. Soc., 1995, 117, 6787–6788 CrossRef CAS; (e) X. Q. Tang and J. Montgomery, J. Am. Chem. Soc., 1999, 121, 6098–6099 CrossRef CAS; (f) X.-Q. Tang and J. Montgomery, J. Am. Chem. Soc., 2000, 122, 6950–6954 CrossRef CAS; (g) G. M. Mahandru, G. Liu and J. Montgomery, J. Am. Chem. Soc., 2004, 126, 3698–3699 CrossRef CAS PubMed; (h) B. Knapp-Reed, G. M. Mahandru and J. Montgomery, J. Am. Chem. Soc., 2005, 127, 13156–13157 CrossRef CAS PubMed; (i) M. R. Chaulagain, G. J. Sormunen and J. Montgomery, J. Am. Chem. Soc., 2007, 129, 9568–9569 CrossRef CAS PubMed; (j) W. Fu, M. Nie, A. Wang, Z. Cao and W. Tang, Angew. Chem., Int. Ed., 2015, 54, 2520–2524 CrossRef CAS PubMed; (k) G. Liu, W. Fu, X. Mu, T. Wu, M. Nie, K. Li, X. Xu and W. Tang, Commun. Chem., 2018, 1, 90 CrossRef; (l) X.-W. Zhang, M.-H. Zhu, H.-X. Zeng, Q.-Y. Li and W.-B. Liu, Angew. Chem., Int. Ed., 2021, 60, 27225–27229 CrossRef CAS PubMed; (m) Y. Mu, T. Zhang, Y. Cheng, W. Fu, Z. Wei, W. Chen and G. Liu, Catal. Sci. Technol., 2021, 11, 2306–2315 RSC; (n) W. Chen, Y. Cheng, T. Zhang, Y. Mu, W. Jia and G. Liu, J. Org. Chem., 2021, 86, 5166–5182 CrossRef CAS PubMed ; For alcohols: ; (o) K. Shen, X. Han and X. Lu, Org. Lett., 2013, 15, 1732–1735 CrossRef CAS PubMed ; For others: ; (p) E. Oblinger and J. Montgomery, J. Am. Chem. Soc., 1997, 119, 9065–9066 CrossRef CAS; (q) R. L. Patman, M. R. Chaulagain, V. M. Williams and M. J. Krische, J. Am. Chem. Soc., 2009, 131, 2066–2067 CrossRef CAS PubMed; (r) R. Tanaka, K. Noguchi and K. Tanaka, J. Am. Chem. Soc., 2010, 132, 1238–1239 CrossRef CAS PubMed.
- For a nickel-catalyzed non-asymmetric reaction: N. Saito, Y. Sugimura and Y. Sato, Org. Lett., 2010, 12, 3494–3497 CrossRef CAS PubMed.
- (a) M. Jeganmohan and C.-H. Cheng, Chem.–Eur. J., 2008, 14, 10876–10886 CrossRef CAS PubMed; (b) C.-H. Wei, S. Mannathan and C.-H. Cheng, J. Am. Chem. Soc., 2011, 133, 6942–6944 CrossRef CAS PubMed.
- (a) S. Yu, C. Wu and S. Ge, J. Am. Chem. Soc., 2017, 139, 6526–6529 CrossRef CAS PubMed; (b) C. Wang and S. Ge, J. Am. Chem. Soc., 2018, 140, 10687–10690 CrossRef CAS PubMed; (c) C. Wu, J. Liao and S. Ge, Angew. Chem., Int. Ed., 2019, 58, 8882–8886 CrossRef CAS PubMed; (d) A. Whyte, J. Bajohr, A. Torelli and M. Lautens, Angew. Chem., Int. Ed., 2020, 59, 16409–16413 CrossRef CAS PubMed; (e) A. Whyte, A. Torelli, B. Mirabi, L. Prieto, J. F. Rodríguez and M. Lautens, J. Am. Chem. Soc., 2020, 142, 9510–9517 CrossRef CAS PubMed; (f) Y. You and S. Ge, Angew. Chem., Int. Ed., 2021, 60, 12046–12052 CrossRef CAS PubMed; (g) D. Kong, H. Wu, J. Ge, Z. Shen and G. Huang, J. Org. Chem., 2022, 87, 6438–6443 CrossRef CAS PubMed; (h) J. Ge, H. Wu, D. Kong and G. Huang, Org. Chem. Front., 2022, 9, 2368–2374 RSC; (i) K. Maji, P. R. Thorve, P. Rai and B. Maji, Chem. Commun., 2022, 58, 9516–9519 RSC.
- For reviews: (a) H. Wen, G. Liu and Z. Huang, Coord. Chem. Rev., 2019, 386, 138–153 CrossRef CAS; (b) Z. Cheng, J. Guo and Z. Lu, Chem. Commun., 2020, 56, 2229–2239 RSC ; For selected examples: ; (c) J. V. Obligacion, J. M. Neely, A. N. Yazdani, I. Pappas and P. J. Chirik, J. Am. Chem. Soc., 2015, 137, 5855–5858 CrossRef CAS PubMed; (d) Z. Zuo and Z. Huang, Org. Chem. Front., 2016, 3, 434–438 RSC; (e) J. H. Docherty, J. Peng, A. P. Dominey and S. P. Thomas, Nat. Chem., 2017, 9, 595–600 CrossRef CAS PubMed; (f) H. Ben-Daat, C. L. Rock, M. Flores, T. L. Groy, A. C. Bowman and R. J. Trovitch, Chem. Commun., 2017, 53, 7333–7336 RSC; (g) J. Guo, B. Cheng, X. Shen and Z. Lu, J. Am. Chem. Soc., 2017, 139, 15316–15319 CrossRef CAS PubMed.
- For selected examples: (a) Y. Ohtsuka, K. Koyasu, D. Miyazaki, T. Ikeno and T. Yamada, Org. Lett., 2001, 3, 3421–3424 CrossRef CAS PubMed; (b) T. Yamada, T. Nagata, K. D. Sugi, K. Yorozu, T. Ikeno, Y. Ohtsuka, D. Miyazaki and T. Mukaiyama, Chem.–Eur. J., 2003, 9, 4485–4509 CrossRef CAS PubMed; (c) A. Kokura, S. Tanaka, T. Ikeno and T. Yamada, Org. Lett., 2006, 8, 3025–3027 CrossRef CAS PubMed; (d) J. Guo, J. Chen and Z. Lu, Chem. Commun., 2015, 51, 5725–5727 RSC.
- Y.-L. Li, S.-Q. Zhang, J. Chen and J.-B. Xia, J. Am. Chem. Soc., 2021, 143, 7306–7313 CrossRef CAS PubMed.
- (a) Y. Oshima and Y. Ueno, Phytochemist, 1993, 33, 179–182 CrossRef CAS; (b) E. Fillion and D. Fishlock, J. Am. Chem. Soc., 2005, 127, 13144–13145 CrossRef CAS PubMed; (c) W. Gu, H. M. Ge, Y. C. Song, H. Ding, H. L. Zhu, X. A. Zhao and R. X. Tan, J. Nat. Prod., 2007, 70, 114–117 CrossRef CAS PubMed.
- CCDC 2025743 (2n).†.
- J. Chen, J. Guo and Z. Lu, Chin. J. Chem., 2018, 36, 1075–1109 CrossRef CAS.
- J. Barluenga, M. Tomás-Gamasa, F. Aznar and C. Valdés, Nat. Chem., 2009, 1, 494–499 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2025743. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3sc00119a |
| This journal is © The Royal Society of Chemistry 2023 |