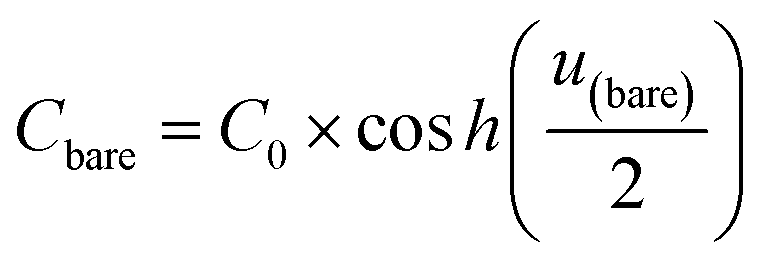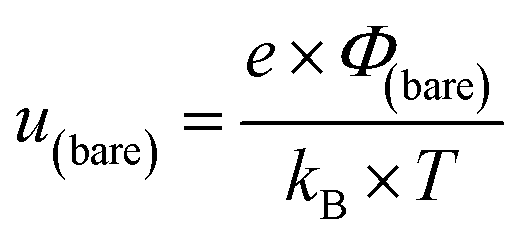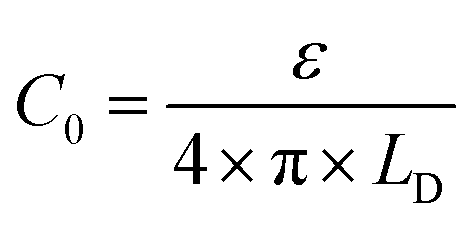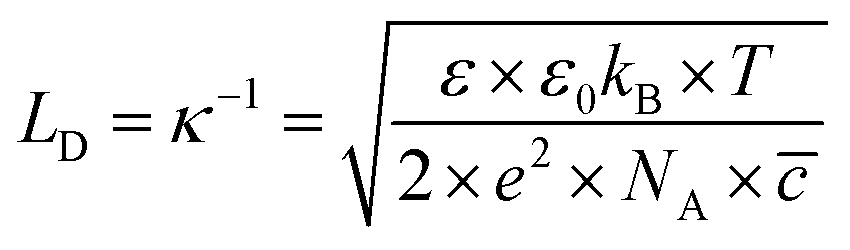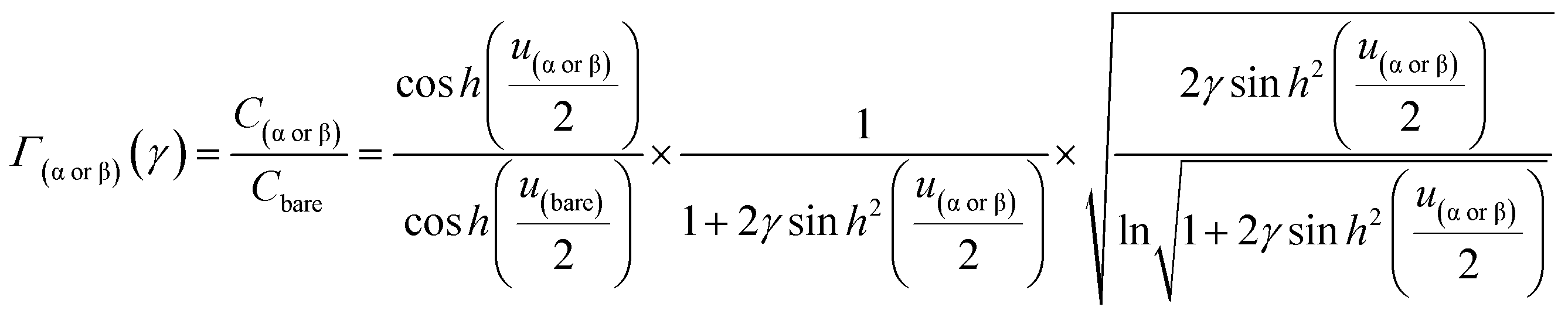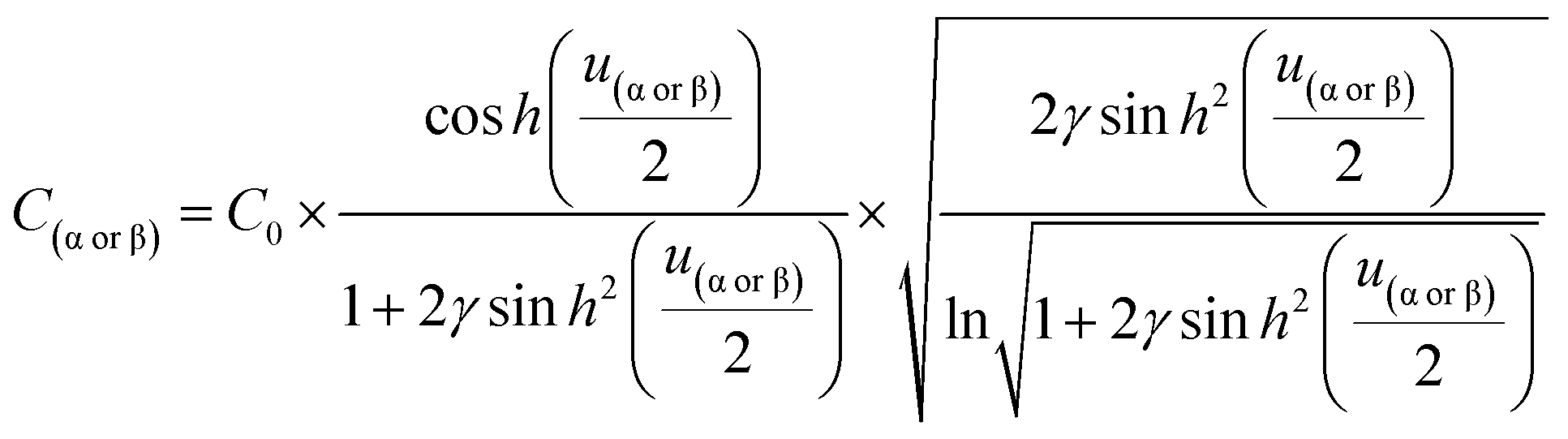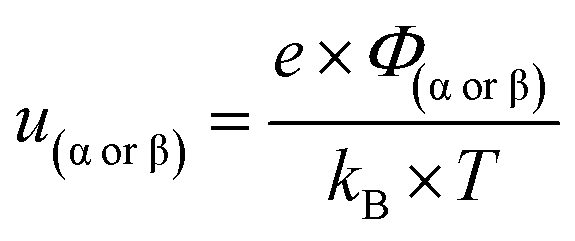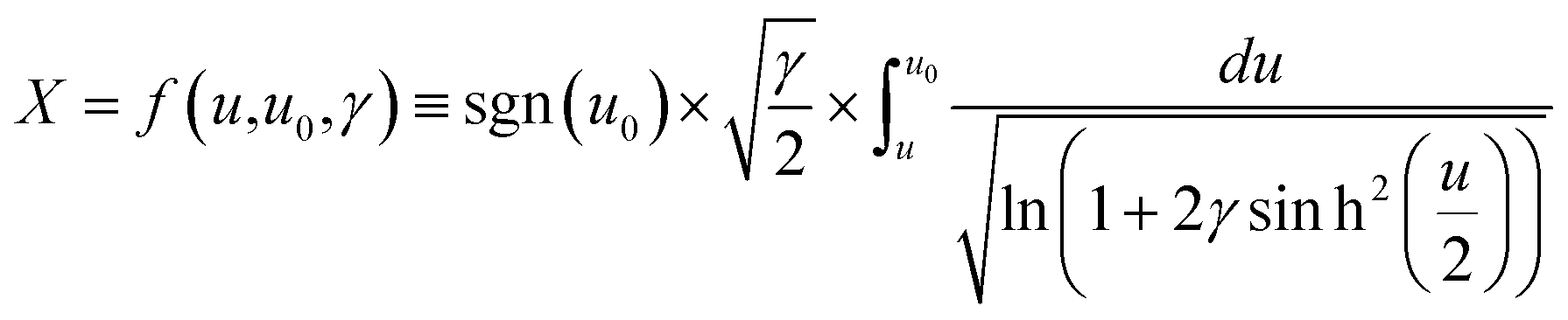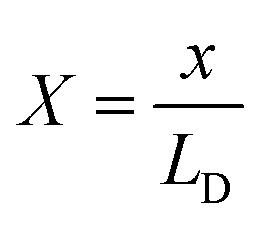DOI:
10.1039/D3SC01692G
(Edge Article)
Chem. Sci., 2023,
14, 6383-6392
Unprecedented energy storage in metal–organic complexes via constitutional isomerism†
Received
2nd April 2023
, Accepted 17th May 2023
First published on 17th May 2023
Abstract
The essence of any electrochemical system is engraved in its electrical double layer (EDL), and we report its unprecedented reorganization by the structural isomerism of molecules, with a direct consequence on their energy storage capability. Electrochemical and spectroscopic analyses in combination with computational and modelling studies demonstrate that an attractive field-effect due to the molecule's structural-isomerism, in contrast to a repulsive field-effect, spatially screens the ion–ion coulombic repulsions in the EDL and reconfigures the local density of anions. In a laboratory-level prototype supercapacitor, those with β-structural isomerism exhibit nearly 6-times elevated energy storage compared to the state-of-the-art electrodes, by delivering ∼535 F g−1 at 1 A g−1 while maintaining high performance metrics even at a rate as high as 50 A g−1. The elucidation of the decisive role of structural isomerism in reconfiguring the electrified interface represents a major step forward in understanding the electrodics of molecular platforms.
1. Introduction
Organometallic systems such as metal-phthalocyanines (MPcs) and metal-porphyrins (MPs), have long been explored as electrocatalysts for an assortment of electrochemical applications and, quite recently, as promising energy storage platforms in electrochemical capacitors due to their chemical and thermal stability and tuneable optoelectronic properties.1–7 The wide range of possibilities that exists as a result of refining their chemistry, by changing the central metal ion and by secondary sphere (N4-macrocyclic ligand) functionalization, results in a host of opportunities for targeting complex electrochemical transformations in fuel cells, batteries, and electrochemical sensors and for electro-organic synthesis.2,3,7–11 In electrocatalysis with MPcs and MPs, their central metal ion is known to dictate the reaction mechanism, while the ligand is perceived to control the electrochemical kinetics chiefly via certain electronic factors.7,12–14 With regards to their energy storage capabilities, most research has been focused on improving their charge storage by forming composites with various carbon-derived substrates.4,15–19 Only a fraction of these studies have also suggested enhancing their surface-limited charge storage through a functionalization of the N4-macrocyclic ligand.4,16 The insights and investigations concerning their electrocatalysis and energy storage capabilities have indeed gone a long way toward improving overall efficiency with regards to specific targets, yet the factors commanding the chemistry of the electrified interface remain largely unknown since little attention has been paid to the structure of their electrical double layer (EDL).20–23 Even a minute alteration in the EDL structure can impact the energy stored in electrochemical capacitors and can lead to a dramatic increase in the electrochemical kinetics.20,23–25 Given the fundamental and technological relevance of organometallic architectures, comprehending the factors determining their EDL structure is of undeniable importance, and yet progress is severely hampered by the critical knowledge gap surrounding this issue, particularly at the molecular level.
We show that a ligand's structural isomerism via attractive and repulsive non-covalent interactions contributes to anomalous reorganization of the ionic charge assembly within the EDL. This study establishes that non-covalent interactions play a subtle yet decisive role in reorganizing a molecule's EDL structure, thereby opening new avenues for the design of molecular platforms favoring improved high-energy and high-power supercapacitors. The correlation between the EDL structure and noncovalent interactions in organometallic complexes enriches their fundamental electrochemistry, which in turn is expected to contribute to new generation organometallic systems to target challenging electrochemical transformations.
2. Results and discussion
2.1. Structural isomerism induced interfacial charge assembly
Since intricate molecular interactions govern all domains of chemistry and biochemistry, deciphering the role that non-covalent interactions play in deciding a molecule's EDL structure is crucial as it goes a long way toward comprehending the chemistry of the corresponding electrified interface. In order to bridge the current knowledge gap and to deepen scientific understanding of what defines the EDL structure in organometallic molecules, we experimented with activating and deactivating non-covalent interactions in molecular systems via structural isomerism of the molecule. The structures of the molecular scaffolds used in this investigation are shown in Fig. S1a of the ESI.† They are denoted as α- and β-tetra-amino-substituted metal (metal ions can be based on Co, Zn, Ni, Cu, Fe, etc.) phthalocyanines (denoted as α-TAMPc and β-TAMPc where M = Co, Zn, Ni, Cu, Fe, etc.). These two molecules differ only with respect to the position of the secondary sphere substituent, and they are thus structural isomers. It should also be noted that they are explored mainly in terms of electrocatalysis.12–14 These structural isomers were synthesized and characterized as per the literature (refer to pages S6–S11 of the ESI and Fig. S2–S4† for more details).26,27
As per the molecular structure provided in Fig. S1a,† owing to the proximity of the –NH2 group to iminic nitrogen of the phthalocyanine ring in the α structural isomer, an attractive field effect is expected due to the possible H-bonding interaction between the groups (ESI, Scheme S1†). This should lead to the presence of intramolecular hydrogen bonding in the α isomer. On the other hand, the relatively large spatial separation of H-bonding groups in the β structural isomer should restrict any such attractive field effects in the molecule (ESI, Scheme S1†). In order to strengthen the claim that the structural isomer triggers non-covalent interactions in the studied molecules, we initially turned to density functional theory (DFT) calculations of the α and β isomers of amino-substituted cobalt phthalocyanines (TACoPcs), as shown in Fig. 1 and S5.† The main conclusion drawn from the DFT calculations is that the presence of intramolecular H-bonding in the α isomer in turn restricts the extent of protonation of the N atoms. On the other hand, the absence of such non-covalent interactions in the β isomer allows the majority of its N atoms to be protonated (refer to pages S13–S16 of ESI, Fig. S5, Table S1† for more details). As we show below, these have important consequences in determining the EDL's structure and electrochemical capacity.
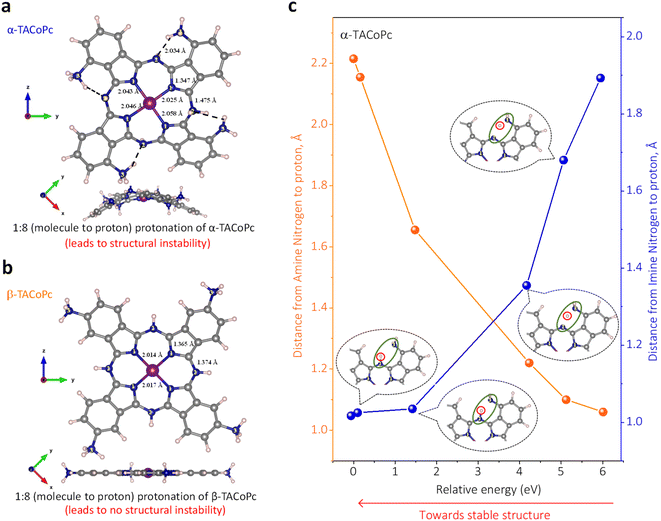 |
| | Fig. 1 Structural isomerism induced proton charge assembly. Stable geometry of eight protonated forms of (a) α-TACoPc and (b) β-TACoPc structural isomers. (c) The correlation plot of the N/H distance between the iminic-nitrogen atom N2[C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N, imine], (a) and the amine-nitrogen atom N3[C–NH2, amine], (a) vs. the relative energy for the α-TACoPc structural isomer. N, imine], (a) and the amine-nitrogen atom N3[C–NH2, amine], (a) vs. the relative energy for the α-TACoPc structural isomer. | |
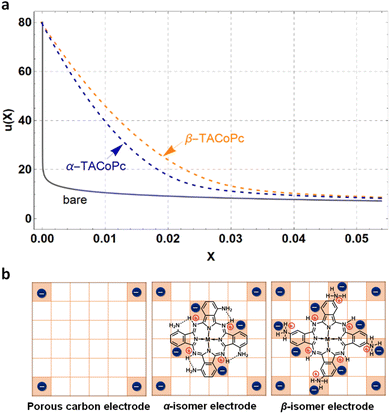
To experimentally validate the theoretical predictions that structural isomerism can trigger non-covalent interactions in the molecule, we performed NMR and X-ray photoelectron spectroscopic (XPS) investigations. NMR analyses were carried out with the corresponding zinc phthalocyanines, as discussed earlier with reference to Fig. S2d and e of the ESI.† 1D and 2D proton NMR spectra of isomeric molecules reveal the existence of intramolecular hydrogen bonding interactions in the α isomer, as shown in Fig. 2a and in ESI, Fig. S6† (refer to pages of the ESI, pages S16, S17 and Fig. S6, S7† for detailed explanation). All these favour the DFT predictions, and suggest that the α isomer has intramolecular hydrogen bonding involving imine nitrogen and amine protons, as shown in Scheme S1.†
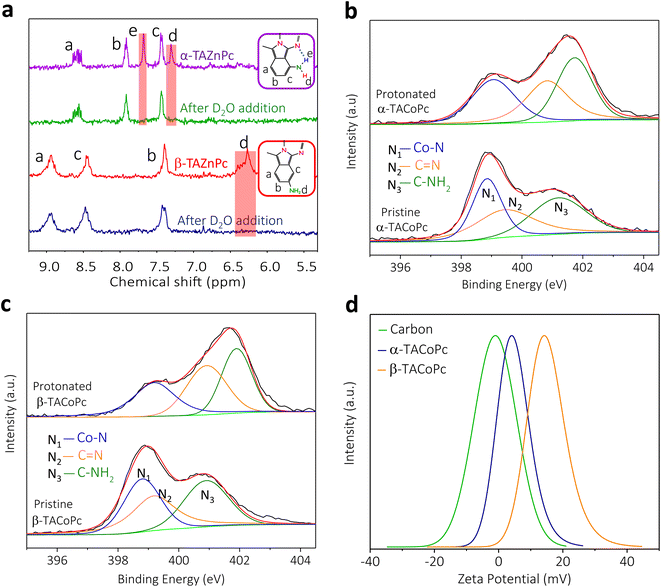 |
| | Fig. 2 Experimental evidence for isomerism directed proton charge assembly. (a) Nuclear magnetic resonance (NMR) spectra of α- and β-TAZnPc structural isomers upon D2O addition. N1s XPS spectra of pristine and protonated forms of (b) α-TACoPc and (c) β-TACoPc structural isomers. (d) Zeta potential profiles of carbon, and α-TACoPc and β-TACoPc structural isomers in a proton-containing medium. | |
As noted earlier based on DFT predictions, this intramolecular hydrogen bonding in the α isomer makes the nitrogen atoms in the molecule less available for protonation (in proton-containing media) as compared to the nitrogen atoms in the β isomer. This claim is strengthened by our N1s XPS analyses, as shown in Fig. 2b and c. The XPS spectra of the α isomer/β isomer reveal three peaks in the N1s regime corresponding to N1 (Co–N), N2 (CN, imine) and N3 (C–NH2, amine) as marked in Fig. 1a, with binding energies (BE) in the following order: N1 < N2 < N3.28,29 The nitrogen atoms, especially N2 and N3 of the pristine α isomer, demonstrated a noticeable shift towards higher binding energy (Fig. S8 and Table S2†) as compared to that of the β isomer, indicating the loss of electron density on nitrogen species in the pristine α isomer, that in turn can be attributed to intramolecular H-bonding interaction. Furthermore, the N1s XPS spectra of both isomers exhibited more of an upshift in their binding energies upon acid treatment than those of the pristine isomers, which was more pronounced in the β-isomer compared to in the α-isomer (Fig. 2b, c and Table S2†). This corroborates the fact that the extent of protonation is significantly higher in the β-isomer as compared to the α isomer, which further strengthens our DFT-based insights. Therefore, the β isomer should possess a higher surface charge density (positive), and consequently a higher electrochemical capacity in proton-containing media which is confirmed by outer sphere redox probe analysis (Fig. S9†) and zeta potential measurements (Fig. 2d). Detailed explanations are provided in page S19 of the ESI and Fig. S9.†
When intramolecular hydrogen bonding is blocked by replacing the amino groups with nitro groups in the secondary sphere (α-TNCoPc and β-TNCoPc structural isomers); please refer to reference numbers 32 and 33, the outer-sphere redox probe responses were almost the same (Fig. S9b and d),† pointing to similar molecular charge densities. Substantiating this, zeta potential measurements of the structural isomers of nitro molecules were almost the same in proton-containing media, as shown in Fig. S10.† This is further validated by DFT calculations of structural isomers of nitro molecules (α- and β-TNCoPc), which did not reveal any disparity in the extent of their protonation (Fig. S11†). In previous work, we observed that the regioisomerism of the –NO2 functionality triggers a repulsive field effect in the α-TNCoPc structural isomer, thereby forcing the –NO2 group to cascade away from the plane of the N4 macrocycle.32,33 The data outlined in Fig. S11,† show that these repulsive field effects do not contribute to the surface charge density. DFT predictions in association with electrochemistry, NMR and XPS analyses, as well as zeta potential measurements, strongly suggest that the nature of the substituents along with their structural isomerism governs the surface charge assembly in molecular systems via non-covalent interactions.
2.2. Structural isomerism at electrified interfaces
To investigate how the structural isomerism induced interfacial proton charge assembly contributes to the EDL structure and electrochemical properties, structural isomers of amino molecules dissolved in dimethylformamide (DMF) solutions were then subsequently adsorbed to form a monolayer on a glassy carbon (GC) electrode by exploiting their π–π interactions (see the Experimental section for more details, ESI, page S22).† Quartz crystal microbalance (QCM) studies, performed during the electrode modification with α-TACoPc and β-TACoPc structural isomers, indicate almost equal changes in mass (ESI, Fig. S12†). The surface coverage values signal the adsorption of structural isomers as a monolayer with an almost flat orientation on the electrode surface (please refer to pages S22–S24 of ESI, Fig. S12 and Table S3†).
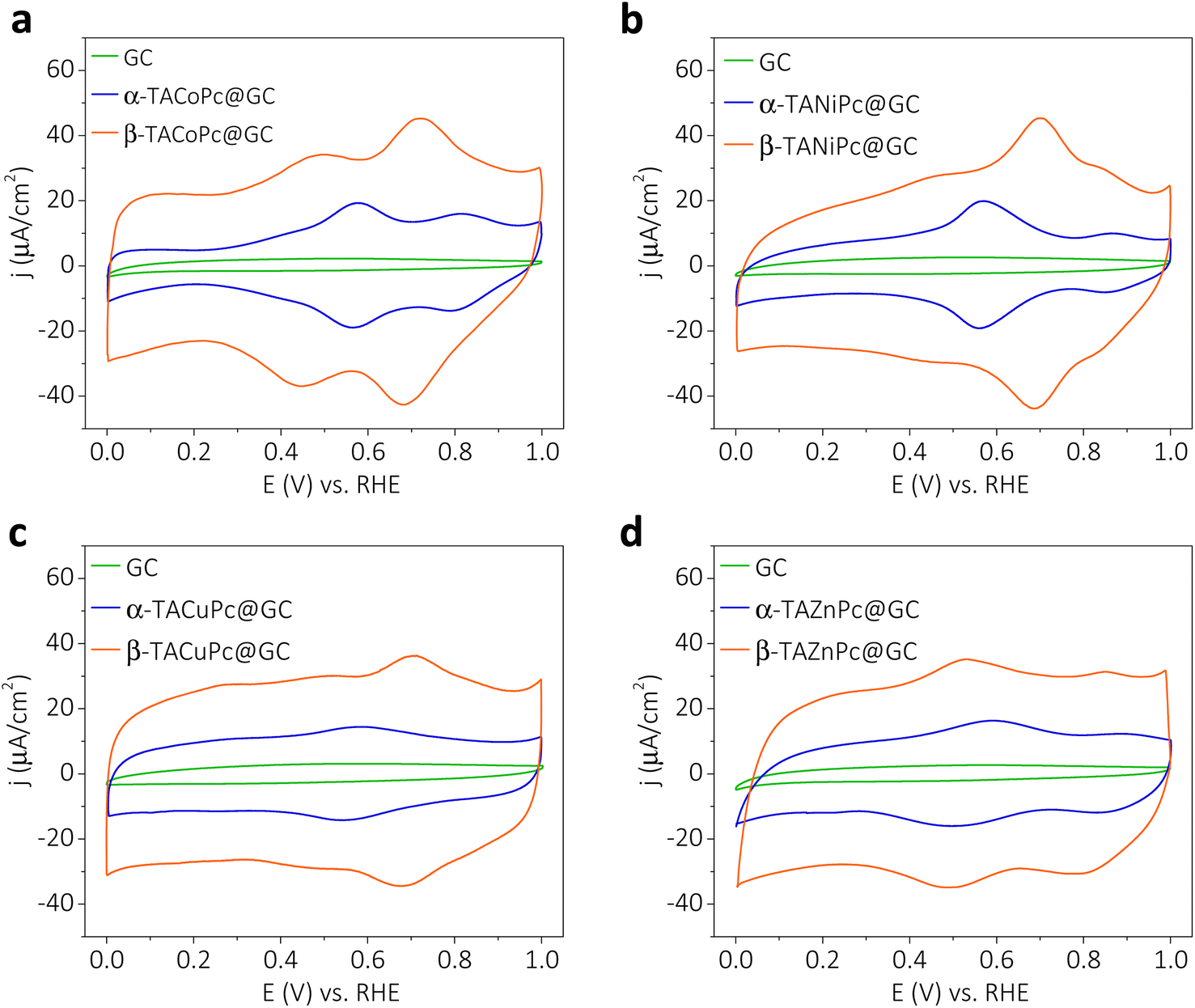
Cyclic voltammograms of GC electrodes, modified with a monolayer of α-TACoPc and β-TACoPc structural isomers in a 0.5 M H2SO4 solution, show distinct redox features corresponding to central cobalt ions and a phthalocyanine (Pc) ring (Fig. 3a). In the literature, the redox peaks at lower and higher positive potentials are assigned to cobalt ions and a Pc ring, respectively (eqn (1) and (2)).30,31,34 The lower positive potential peak originates from the redox transition in the central metal ion (eqn (1)) and the higher positive potential redox pair corresponds to the redox transition in the phthalocyanine (Pc) ring (eqn (2)). The most striking feature, and one that is evident in the voltammograms of α and β isomeric molecules, is their variation in electrochemical capacity, which is notably higher in the case of the β isomer as opposed to the α isomer (Fig. 3a). It should be noted that these distinct differences in electrochemical capacity are observed for those electrode surfaces modified with a monolayer of structural isomers, meaning that they are intrinsic to the nature of the molecules. In order to verify the contribution of pseudocapacitance to the overall capacitance, we have extracted the faradaic contributions by integrating the charge under the redox peaks in Fig. 3. The data as shown in Fig. S13† show that the faradaic contributions are nearly 0.297 mF cm−2 and 0.312 mF cm−2 for the α isomer and β isomer, respectively, suggesting that the contribution of pseudocapacitance to the overall capacitance is almost similar in magnitude in both the structural isomers. To get further insight into the diffusion and surface-controlled contributions, ‘b’ values were extracted from the logarithmic plot of current vs. scan rate, as shown in Fig. S14.† The value of b came out to be close to 1 at most of the potentials. This suggests that the contribution to the total capacitance from the diffusion-controlled process is marginal. Furthermore, we quantified the capacitive contributions by analyzing the cyclic voltammograms at various sweep rates, Fig. S15 shows that the surface capacitive contribution dominates the charge storage especially in the EDLC region, and its contribution to the overall capacitance scales up with the scan rate (please refer pages 26 and 27 of the ESI† for more details). When the central metal ion in TACoPc was changed to Cu, Ni and Zn; all of them maintained this marked contrast in their electrochemical capacity, displaying a more dominant capacitive contribution in the β isomer over the corresponding α isomer (Fig. 3b–d). Interestingly, even the TAZnPc molecule with a non-electrochemically active central metal ion revealed a distinctly higher capacitive contribution in the β isomer than in the α isomer, suggesting that the origin of this intriguing behavior is mainly due to structural isomerism of the ligand. It is worth noting that the redox wave observed in MPcs at lower potentials is due in part to a contribution from imine-like groups within the phthalocyanine structure,4 as shown in eqn (3), and we attribute this to the lower potential redox wave in TAZnPc.
| | | Co2+TAPc2− ⇌ Co3+TAPc2− + e− | (1) |
| | | Co3+TAPc2− ⇌ Co3+TAPc1− + e− | (2) |
| | ![[double bond splayed left]](https://www.rsc.org/images/entities/char_e009.gif) CN– + 2e− + 2H+ ⇌ CH–NH– CN– + 2e− + 2H+ ⇌ CH–NH– | (3) |
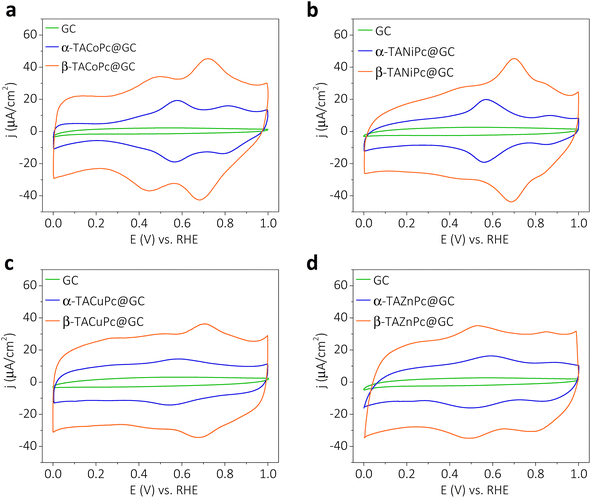 |
| | Fig. 3 Experimental evidence for ligand geometry-directed surface charge. Structural isomerism vs. energy storage: voltammograms for (a) α-TACoPc and β-TACoPc structural isomers, (b) α-TANiPc and β-TANiPc structural isomers, (c) α-TACuPc and β-TACuPc structural isomers and (d) α-TAZnPc and β-TAZnPc structural isomers when they are adsorbed as a monolayer on a glassy carbon electrode surface. Measurements in (a–d) were performed at a scan rate of 20 mV s−1 in N2 saturated 0.5 M H2SO4. | |
Charge–discharge characteristics of the structural isomers are further investigated by cyclic voltammetry and galvanostatic charge–discharge measurements (Fig. S16 and S17, Table S4†). The data reveal that the areal capacitances are more than 2 times higher in the case of the β isomer than in that of the α isomer when they are adsorbed as a monolayer on a glassy carbon electrode surface. Nevertheless, when the –NH2 group in the secondary sphere of the phthalocyanine ligand is replaced by a –NO2 group, the disparity in the isomeric molecule's electrochemical capacity disappears (Fig. S18†). Therefore, as highlighted in the previous section, repulsive field effects do not contribute to the double-layer structure. The fact that the electrochemical capacity is independent of the central metal ion, yet is strongly correlated to the nature as well as isomerism of the secondary sphere functionality (Fig. 3 and S17, Table S4†), suggests that the double-layer structure is governed by the type of non-covalent interactions. A modulation in the double-layer structure is observed when an attractive field effect (by H-bonding in the –NH2-substituted molecule) is in action; however, no such modulation is apparent when a repulsive field effect (by electron cloud repulsion in the –NO2-substituted molecule) is triggered. We note that in the field of molecular charge storage and electrocatalysis, the role of structural isomerism, and thus, the implications of non-covalent interactions, are often neglected; however, our results reveal that this issue certainly deserves consideration if the maximum potential of the molecule is to be exploited for electrochemical applications.
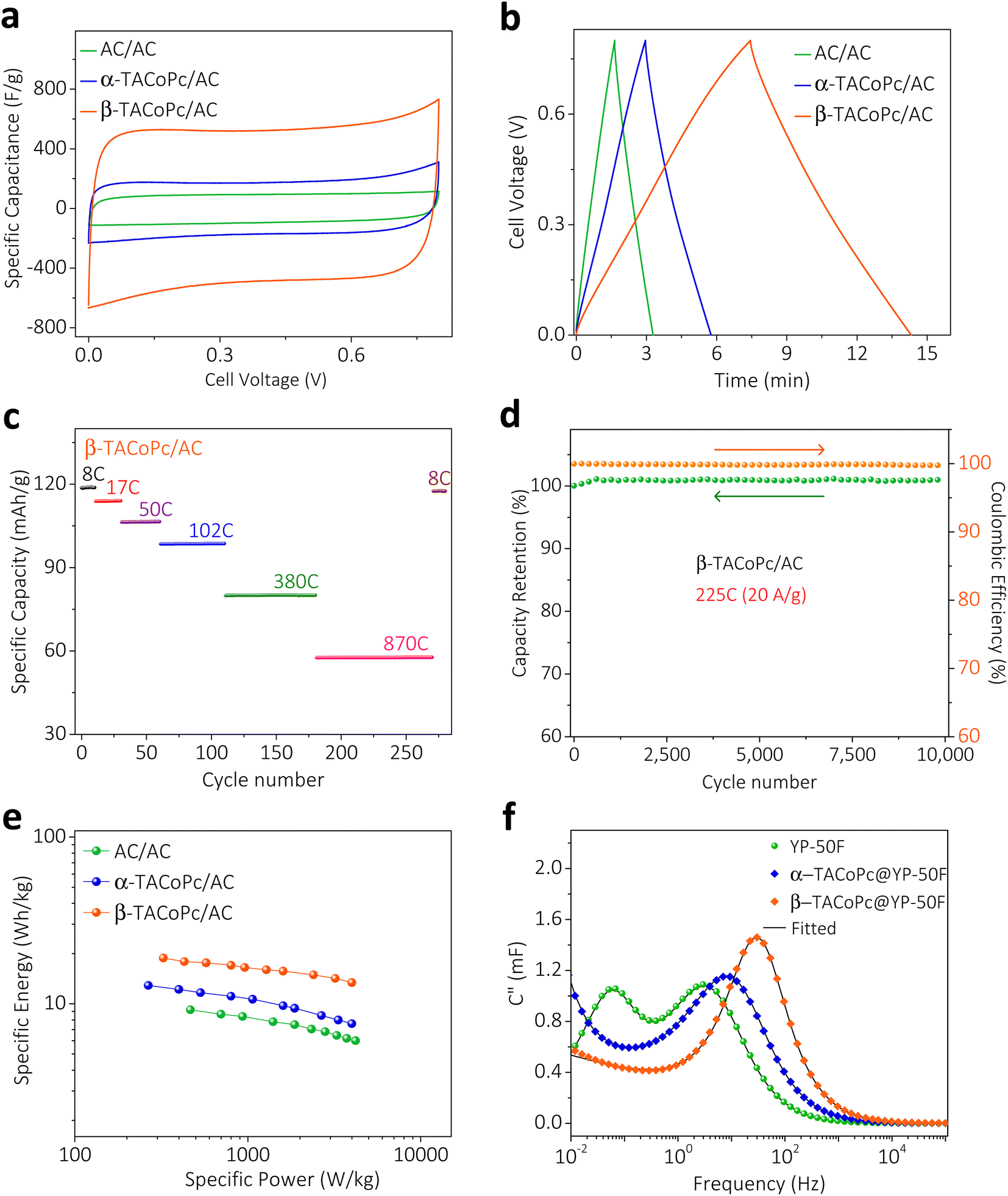
To take advantage of these peculiar structural characteristics for practical charge storage applications, we have made different weight ratios of isomeric phthalocyanine molecules on porous carbon material (YP-50F) (see Section 9 for more details, ESI, pages S30–S32, Fig. S19–S21†).35,36 These kinds of modified electrodes demonstrated similar characteristics as monolayer modified electrodes, with a dominant electrochemical capacity in β-TACoPc@YP-50F as compared to α-TACoPc@YP-50F (Fig. 3, S16 and Table S4 of the ESI).† The performance was optimized as detailed in pages S33 and S34 of the ESI (Fig. S22 and Table S5).† The gravimetric capacitance of β-TACoPc@YP-50F is found to be ∼730 F g−1, as compared to ∼360 F g−1 for α-TACoPc@YP-50F, which is more than double the capacitance observed for the latter. Rate capability by cyclic voltammetry and galvanostatic charge–discharge measurements further reinforces the superior charge storage capability of β-TACoPc@YP-50F as compared to α-TACoPc@YP-50F (Fig. S23†). Therefore, as discussed earlier in the section concerning monolayer modified electrodes (Fig. 3 and S16, S17†), the presence of H-bonding interactions in –NH2-substituted phthalocyanines contributes significantly to their double-layer structure. As a whole, the results presented here indicate that β-TACoPc@YP-50F has the potential to fulfil the role of a practical electrode material for charge storage, as opposed to α-TACoPc@YP-50F. To show that these results remain valid with respect to an operational supercapacitor device, we carried out studies with a realistic two-electrode device architecture in an asymmetric configuration. We assembled two-electrode cells using α/β-TACoPc@YP-50F and activated carbon (AC) as the positive and negative electrodes, respectively. The two-electrode cell device architecture is shown in Fig. S24,† and the fabrication procedure is discussed in page S38. Cyclic voltammetry and galvanostatic charge–discharge measurements, shown in Fig. 4a and b, using the two-electrode cell setup yield similar results to that of the three-electrode measurements with a monolayer and composite electrodes (Fig. 3 and discussed in the context of the ESI, Fig. S16, S17, S23, Tables S4 and S6†). Similarly, in the two-electrode device setup, the performance of the β-TACoPc-based device is higher than that of the α-TACoPc one. The specific capacity of β-TACoPc/AC is found to be ∼119 mA h g−1, which is more than double that observed with α-TACoPc/AC (∼48 mA h g−1), as shown in Fig. S25a, b and Table S7.† Since the β-TACoPc/AC device consistently outperforms the α-TACoPc/AC one, we carried out further rate capability and cycling tests with β-TACoPc/AC alone. Rate capability results via cyclic voltammetry and galvanostatic charge–discharge measurements further reinforce the excellent charge storage capability of β-TACoPc/AC (Fig. S25c–e and Table S8†). The rate capability tests at different C-rates using β-TACoPc/AC show good stability and, even at an extraordinarily high rate of 870C (50 A g−1), the cell displays a capacity of over 58 mA h g−1 and is able to return to lower rates of 8C (1 A g−1), exhibiting a capacity of 119 mA h g−1 without a loss in performance (Fig. 4c and Table S8†). Furthermore, cycling stability even at high rates of 225C (20 A g−1) indicates a nearly 100% coulombic efficiency, with negligible loss in capacity over 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cycles (Fig. 4d and S25f†). The Ragone plot, shown in Fig. 4e and Table S8,† further reinforces the fact that the structural isomerism of the amine functionality in the secondary sphere profoundly influences the charge storage, as well as enhances the energy density of the molecules without compromising their power capability. As a whole, the results discussed above serve to demonstrate the effect of structural isomerism on the charge storage mechanism to pave the way for commercial high energy density supercapacitors, without forfeiting power capability.
000 cycles (Fig. 4d and S25f†). The Ragone plot, shown in Fig. 4e and Table S8,† further reinforces the fact that the structural isomerism of the amine functionality in the secondary sphere profoundly influences the charge storage, as well as enhances the energy density of the molecules without compromising their power capability. As a whole, the results discussed above serve to demonstrate the effect of structural isomerism on the charge storage mechanism to pave the way for commercial high energy density supercapacitors, without forfeiting power capability.
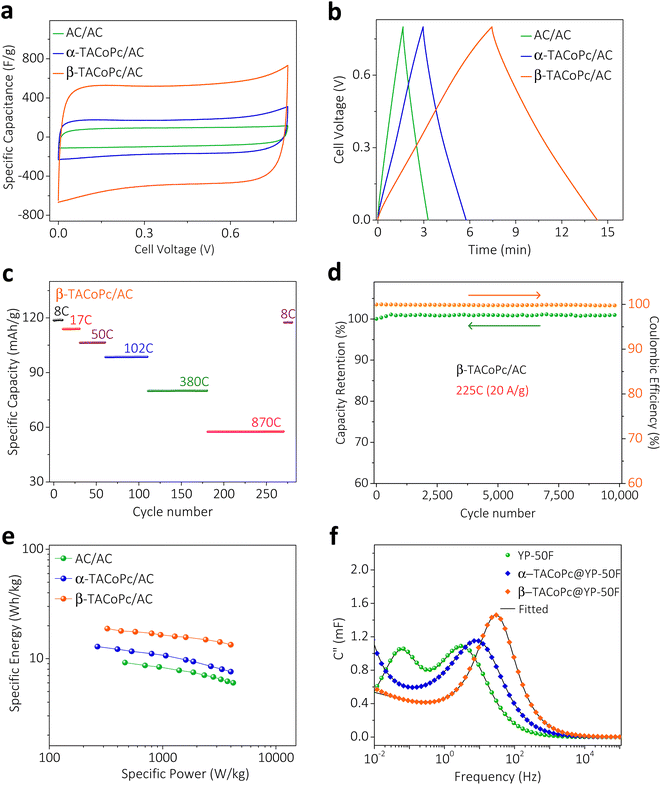 |
| | Fig. 4 Structural isomerism at electrified interfaces for a high energy supercapacitor. Characterization of a two-electrode split test cell assembled with α/β-TACoPc (+) electrodes and activated carbon (−) electrodes. (a) Cyclic voltammetry and (b) galvanostatic charge–discharge of the device. (c) Capacity retention of the β-TACoPc/AC cell at sequentially higher rates. After GCD cycling up to an extremely high rate of 870C, the rate can be lowered back to 8C while maintaining the original capacity. (d) Capacity retention and coulombic efficiency of the β-TACoPc/AC device over 10000 cycles at a rate of 225C (20 A g−1). (e) Ragone plots for the two-electrode split test cell. (f) Plot of the imaginary part of capacitance (C′′) vs. frequency at OCV vs. RHE, acquired in the frequency range of 100 kHz to 5 mHz with a 10 mV (peak to peak) AC excitation signal. Measurements in (a–e) were performed using a 0.5 M H2SO4 aqueous electrolyte with an α/β-TACoPc electrode mass loading of 3.25 mg cm−2. | |
In order to gain further insight into the capacitive behavior of the α and β isomers, comparative electrochemical impedance spectroscopy (EIS) experiments were carried out with the composite electrodes in a three-electrode configuration (Fig. S26, please refer to page S42 of the ESI† for details). The Bode plot (log of frequency vs. phase angle, Fig. S26b†) reveals that resistance to capacitive switching takes place at a higher frequency for β-TACoPc@YP-50F than for α-TACoPc@YP-50F, which is indicative of the superior supercapacitor characteristics of the former over the latter. The “knee” or “onset” frequency (fo) reflecting the power capability of a supercapacitor, was extracted from the frequency corresponding to the maximum of the imaginary part of the capacitance (C′′) in the C′′ versus frequency plot (Fig. 4f).4,37,38C′′ signifies the boundary between purely capacitive and purely resistive behavior (Fig. 4f).4,38,39 The fo decreases in the following order: β-TACoPc@YP-50F (32 Hz) ≫ α-TACoPc@YP-50F (9 Hz) ≫ YP50F (3 Hz). Accordingly, the higher fo value of β-TACoPc@YP-50F confirms its higher power capability as compared to α-TACoPc@YP-50F and YP-50F, and this is consistent with the comparative CV and charge–discharge data shown in ESI, Fig. S23.† Furthermore, the relaxation time ((τ0 = 1/fo),40–42 Table S9†) which reflects the minimum time required to fully discharge the capacitor was shorter for the β isomer as compared to the α isomer, (Table S10†), pointing to the superior power capability of the former over the latter. The measured self-discharge rates and leakage currents of the supercapacitors suggest remarkably lower self-discharge rates and leak currents in the β isomer as compared to the α isomer (Fig. S27 and Table S10†). The above results clearly demonstrate that structural isomerism in conjunction with the nature of the functionality profoundly influences the double-layer structure via non-covalent interactions, and that these characteristics tend to combine synergistically to promote rapid charging, high capacitance, and long-term stability (additional data and explanation are provided in page 44 of the ESI and Fig. S28†).
2.3. Model for structural isomerism driven electrochemical double-layer capacity
The objective of the following section is to analyze the capacity values obtained in the experimental part, as shown in Tables S4 and S6 of the ESI.†Table 1 summarizes the values obtained according to the modifications.
Table 1 The capacity extracted from cyclic voltammetry and galvanostatic charge–discharge. Charge storage by bare, α-TACoPc-modified and β-TACoPc-modified flat and porous electrodes
| Electrodes |
Flat electrode |
Porous electrode |
| From cyclic voltammetry Csp (mF cm−2) |
From galvanostatic charge–discharge Csp (mF cm−2) |
From cyclic voltammetry Csp (F g−1) |
From galvanostatic charge–discharge Csp (F g−1) |
| Bare |
0.068 |
0.083 |
180 |
217 |
| α-TACoPc |
0.553 |
0.645 |
323 |
359 |
| β-TACoPc |
1.410 |
1.670 |
675 |
729 |
Kornyshev et al. proposed a simple explanation with the concept of a superionic state in a metallic nanopore caused by the screening effect of ion–ion pair interactions in a nanogap, thereby enabling more ions to be adsorbed.43 In fact, when an anion approaches the positive electrode, it undergoes two opposing forces: an electrostatic attractive force with the positively charged electrode, and an electrostatic repulsive force between the anions. In our case, a third force of electrostatic attraction is added by the protonated (i.e., positively charged) amine on the anions, decreasing the steric effect, that in turn served to increase the local density of the anions.
Introducing α and β isomeric molecules onto the electrode surface can increase ion compactness, Γα or β(γ), resulting from this new attractive force. Scheme S1,† illustrates this change in local anion density according to the isomerism. The capacitance of the electrodes with surface modification, C(α or β), modulated by a compactness parameter can be denoted as:
| | | Cα or β = Cbare × Γα or β(γ), | (4) |
| | 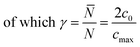 | (5) |
where
Cbare is the capacitance of the bare electrode,
γ is the compactness parameter, defined as the total number of cations and anions in the bulk (
![[N with combining macron]](https://www.rsc.org/images/entities/i_char_004e_0304.gif)
) compared to the total number of sites available for them (
N).
c0 is the charge concentration of cations and anions
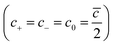
, and
cmax is the maximum ion concentration in the bulk, respectively.
In this formalism, the bare electrode capacitance can be defined by using the Gouy–Chapman equation as:
| | 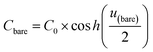 | (6) |
| | 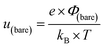 | (7) |
| | 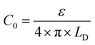 | (8) |
| | 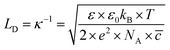 | (9) |
From eqn (6)–(9), we consider the following variables: ε is the dielectric constant, ε0 is the free-space permittivity (8.854 × 10−12 F m−1), LD is the Debye length (m), e is the electron charge (1.602 × 10−19 C), Φ is the potential (in V), T is the temperature (293.15 K), kB is the Boltzmann constant (1.381 × 10−23 J K−1) and NA is the Avogadro constant (6.022 × 1023 mol−1), while the compactness can be expressed according to Kornyshev and was validated by Levin et al.:44
| | 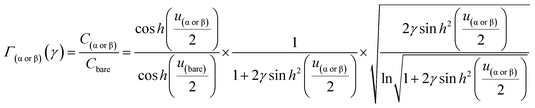 | (10) |
| | 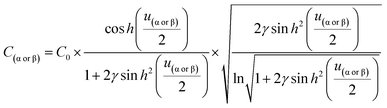 | (11) |
| | 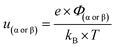 | (12) |
From eqn (10), Γα(γ) and Γβ(γ)—ratio of the experimental capacitance associated with the α (or β) isomeric molecule to the capacitance of the bare electrode (see ESI, Table S11†).
To extract the value of the γ coefficient, it was essential to determine the potential dependence on the distance from the electrode, u(X), to establish the conditions close to the electrode surface. Confirming modelling precision, we thus reproduced Kornyshev's model44 by using Mathematica tool based on the Wolfram language (Fig. S29 and S30†) with eqn (11)–(14), which can then be used reliably to determine the value of γ:
| | 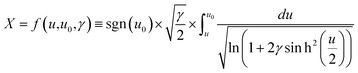 | (13) |
| | 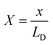 | (14) |
where
x and
X are the distances from the electrode in meter and dimensionless form, respectively. At the electrode surface, the potential is equivalent to
u(
X0). However, the potential is varied with
X (Fig. S29
†) and
γ parameters as per Kornyshev's study. Hence,
Γ(α or β) (
eqn (10)) is a function of both
u(
X) and
γ. So, the determination
γ coefficient from the experimental data could only be obtained by solving for both (
Γ(α or β) value) and
u(
X)
u(
X) simultaneously. Now with
X (dimensionless distance) equivalent to the thickness of the α (or β) molecular layer (
x ≈ 0.3 nm), the
![[c with combining macron]](https://www.rsc.org/images/entities/i_char_0063_0304.gif)
= 0.5 M ion concentration in bulk,
ε = 80.1 (water at 293.15 K),
LD can be obtained using
eqn (9), which is found to be ∼13.627 nm. Hence, the thickness
X = 0.022 is the point where
u(
X) will be determined for different electrodes.
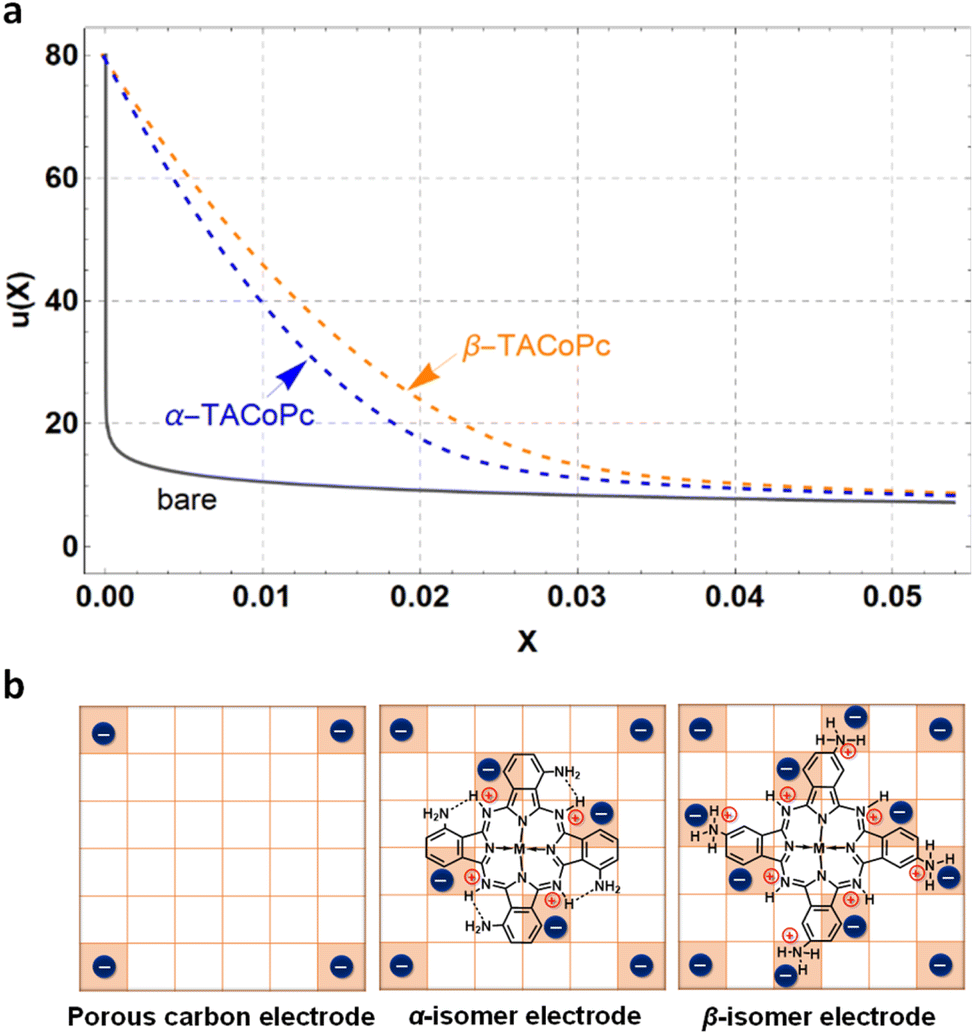
The potential of the bare electrode (γ = 0) at X = 0.022 was determined using eqn (13), which is summarized in ESI, Table S12.† However, to obtain u(X) of the α (or β) isomer molecule modified electrode, eqn (10) and (13) were employed to perform simultaneous calculations, as concluded in ESI, Table S12.† Lastly, the potential profiles of both flat and porous electrodes, with and without α-TACoPc and β-TACoPc coating layers, are outlined in the ESI (Fig. 5a and S31†) suggesting that surface modifications providing the highest capacitances were the result of variations in the compactness parameter, which in turn leads to different potential profiles. Taken together, the probability of an anion positioning itself on the neighbouring square of another anion is almost zero in the case of a bare electrode, since the repulsive effect is too great (Fig. 5b). With an α (or β) isomer molecule, the positive charge of the N-containing functionality causes a screening of the neighbouring anion. This model explains the experimental results demonstrating that structural isomerism can indeed lead to a favorable modulation of the double-layer structure, which thus promises to enhance future electrode design, since this structure is at the crux of electrodics.
 |
| | Fig. 5 Potential distribution profiles and the model for the structural isomerism induced ion charge assembly. (a) Bare (grey bold line), α-TACoPc-modified (blue dashed line) and β-TACoPc-modified (orange dashed line) porous electrodes. (b) Model of the EDL with a porous carbon, and α and β structural isomer electrodes. | |
3. Conclusions
We endeavour to demonstrate the role of structural isomerism of a macrocycle based electrode in reorganizing the EDL structure that in turn fine-tunes its electrochemical capacity via non-covalent interactions. A range of physicochemical analyses, including theoretical investigation, point to the role of intramolecular hydrogen bonding in suppressing the electrochemical capacity of the α-amino structural isomer molecule by largely preventing the protonation of nitrogen atoms in the ligand. The absence of such interactions in the β-amino structural isomer molecule, due to the relatively large spatial separation of H-bonding groups, leaves a large percentage of the nitrogen atoms available for protonation and thereby leads to an enhanced electrochemical capacity. However, nitro substituted structural isomeric molecules that cannot participate in H-bonding interactions predominantly experience a repulsive field effect due to electron cloud repulsion, which contributes only to a geometric rotation of the –NO2 functionality with respect to the N4-macrocyclic plane alone, without affecting the electrochemical capacity. Furthermore, operational two-electrode supercapacitor devices are designed in order to harness this structural isomerism-directed charge-storage mechanism. Those devices containing β-amino structural isomer molecules exhibit a capacity of 535 F g−1 at a 1 A g−1 rate, which is ∼2.5 times higher than that of their α-amino counterparts, and display a remarkably high capacity retention, even at a rate of 50 A g−1 with a leakage current as low as 79 μA/F/V. The above findings serve to demonstrate that non-covalent molecular interactions play a subtle yet decisive role in determining the EDL structure, and that they should thus be taken into consideration when designing new molecular platforms in order take full advantage of their potential for electrochemical applications. We conclude that the correlation between the EDL structure and non-covalent interactions shown herein breaks new ground with respect to designing more effective high-energy and high-power electrochemical capacitors built around organometallic platforms.
Data availability
Experimental data are available from the corresponding author upon reasonable request.
Author contributions
A. R. K. executed extensive characterization studies of isomeric molecules, electrochemical measurements and data analysis, assisted by M. C. D. and Z. M. B. M. A. N. carried out the DFT calculations. C. P. V. performed the XPS spectroscopy measurements. H. M. N. K. and M. O. T. conceived the idea and designed the synthetic methodology for the synthesis of isomeric molecules, synthesized the isomeric molecules and performed their initial physicochemical/electrochemical characterization studies. Theoretical modelling was performed by O. F. along with S. D., J. D. and Y. Z. M. O. T. supervised the work and H. M. N. K. and O. F. contributed to manuscript preparation.
Conflicts of interest
The authors declare no conflict of interest.
Acknowledgements
A. R. K. is grateful for the financial assistance received from UGC-India. The authors thank Prof. Menny Shalom for TEM (transmission electron microscopy), Department of Chemistry, Ben-Gurion University of the Negev, Israel. M. O. T. acknowledges the financial assistance received from DST-SERB (CRG/2020/002549) and DST-WTI (DST/TMD-EWO/WTI/2K19/EWFH/2019/272). O. F. would like to thank the Institute Universitaire de France for their support. O. F., Y. Z. and J. D. appreciate the assistance offered by the College of Food and Biological Engineering, Chengdu University. O. F. and M. O. T. thank the France-India collaboration for supporting the project. O. F. thanks National Research Council of Thailand (NRCT), through the contract numbers N42A650196 and N42A650255.
Notes and references
- Y. Wu, Z. Jiang, Z. Lin, Y. Liang and H. Wang, Nat. Sustain., 2021, 4, 725–730 CrossRef.
- C. J. Kaminsky, S. Weng, J. Wright and Y. Surendranath, Nat. Catal., 2022, 5, 430–442 CrossRef CAS.
- E. J. Kim, P. A. Maughan, E. N. Bassey, R. J. Clément, L. A. Ma, L. C. Duda, D. Sehrawat, R. Younesi, N. Sharma, C. P. Grey and A. R. Armstrong, Adv. Energy Mater., 2022, 12, 2102325 CrossRef CAS.
- A. T. Chidembo, K. I. Ozoemena, B. O. Agboola, V. Gupta, G. G. Wildgoose and R. G. Compton, Energy Environ. Sci., 2010, 3, 228–236 RSC.
- Y. Liang, D. McLaughlin, C. Csoklich, O. Schneider and A. S. Bandarenka, Energy Environ. Sci., 2019, 12, 351–357 RSC.
- N. Mahne, B. Schafzahl, C. Leypold, M. Leypold, S. Grumm, A. Leitgeb, G. A. Strohmeier, M. Wilkening, O. Fontaine, D. Kramer, C. Slugovc, S. M. Borisov and S. A. Freunberger, Nat. Energy, 2017, 2, 1–9 Search PubMed.
-
J. H. Zagal, F. Bedioui and J. P. Dodelet, N4-Macrocyclic Metal Complexes, Springer, New York, 2006, pp. 83–147 Search PubMed.
- H. Zhang, C. Xu, X. Zhan, Y. Yu, K. Zhang, Q. Luo, S. Gao, J. Yang and Y. Xie, Nat. Commun., 2022, 13, 1–11 Search PubMed.
- D. Wright, Q. Lin, D. Berta, T. Földes, A. Wagner, J. Griffiths, C. Readman, E. Rosta, E. Reisner and J. J. Baumberg, Nat. Catal., 2021, 4, 157–163 CrossRef CAS.
- S. Ren, D. Joulié, D. Salvatore, K. Torbensen, M. Wang, M. Robert and C. P. Berlinguette, Science, 2019, 365, 367–369 CrossRef CAS PubMed.
- J. Chen, K. Zou, P. Ding, J. Deng, C. Zha, Y. Hu, X. Zhao, J. Wu, J. Fan and Y. Li, Adv. Mater., 2019, 31, 1–7 Search PubMed.
- J. H. Zagal and M. T. M. Koper, Angew. Chem., Int. Ed., 2016, 55, 14510–14521 CrossRef CAS PubMed.
- J. Masa, K. Ozoemena, W. Schuhmann and J. H. Zagal, J. Porphyrins Phthalocyanines, 2012, 16, 761–784 CrossRef CAS.
- J. H. Zagal, S. Griveau, J. F. Silva, T. Nyokong and F. Bedioui, Coord. Chem. Rev., 2010, 254, 2755–2791 CrossRef CAS.
- M. Wang, H. Shi, P. Zhang, Z. Liao, M. Wang, H. Zhong, F. Schwotzer, A. S. Nia, E. Zschech, S. Zhou, S. Kaskel, R. Dong and X. Feng, Adv. Funct. Mater., 2020, 30, 2002664 CrossRef CAS.
- A. T. Chidembo and K. I. Ozoemena, Electroanalysis, 2010, 22, 2529–2535 CrossRef CAS.
- K. Makgopa, P. M. Ejikeme and K. I. Ozoemena, Electrochim. Acta, 2016, 212, 876–882 CrossRef CAS.
- Y. Lu, Q. Zheng, J. Wu and Y. Yu, Electrochim. Acta, 2018, 265, 594–600 CrossRef CAS.
- N. Taşaltln, C. Taşaltln, H. Aydln, U. Şahintürk and A. G. Gürek, J. Porphyrins Phthalocyanines, 2019, 23, 1616–1621 CrossRef.
- H. Mistry, A. S. Varela, S. Kühl, P. Strasser and B. R. Cuenya, Nat. Rev. Mater., 2016, 1, 16009 CrossRef CAS.
- E. Mourad, L. Coustan, P. Lannelongue, D. Zigah, A. Mehdi, A. Vioux, S. A. Freunberger, F. Favier and O. Fontaine, Nat. Mater., 2016, 16, 446–454 CrossRef PubMed.
- V. R. Stamenkovic, D. Strmcnik, P. P. Lopes and N. M. Markovic, Nat. Mater., 2016, 16, 57–69 CrossRef PubMed.
- J. Chmiola, G. Yushin, Y. Gogotsi, C. Portet, P. Simon and P. L. Taberna, Science, 2006, 313, 1760–1763 CrossRef CAS PubMed.
- P. Sebastián-Pascual, Y. Shao-Horn and M. Escudero-Escribano, Curr. Opin. Electrochem., 2022, 32, 100918 CrossRef.
- Y. Y. Birdja, E. Pérez-Gallent, M. C. Figueiredo, A. J. Göttle, F. Calle-Vallejo and M. T. M. Koper, Nat. Energy, 2019, 4, 732–745 CrossRef CAS.
- J. Alzeer, P. J. C. Roth and N. W. Luedtke, Chem. Commun., 2009, 15, 1970 RSC.
- A. K. Al-lami, N. N. Majeed and A. H. Al-mowali, J. Chem. Mater. Res., 2013, 3, 59–68 Search PubMed.
- V. Y. Aristov, O. V. Molodtsova, V. V. Maslyuk, D. V. Vyalikh, T. Bredow, I. Mertig, A. B. Preobrajenski and M. Knupfer, Org. Electron., 2010, 11, 1461–1468 CrossRef CAS.
- T. Mthethwa, E. Antunes and T. Nyokong, Dalton Trans., 2014, 43, 8230–8240 RSC.
- J. Obirai and T. Nyokong, Electrochim. Acta, 2005, 50, 3296–3304 CrossRef CAS.
- J. Obirai and T. Nyokong, Electrochim. Acta, 2005, 50, 5427–5434 CrossRef CAS.
- A. R. Kottaichamy, S. Begum, M. A. Nazrulla, N. C. Dargily, M. C. Devendrachari, Z. Manzoor Bhat, R. Thimmappa, H. Makri Nimbegondi Kotresh, C. P. Vinod and M. O. Thotiyl, J. Phys. Chem. Lett., 2020, 11, 263–271 CrossRef CAS PubMed.
- A. R. Kottaichamy, S. Begum, M. C. Devendrachari, Z. M. Bhat, R. Thimmappa, H. M. Nimbegondi Kotresh, C. P. Vinod and M. O. Thotiyl, Anal. Chem., 2020, 92, 4541–4547 CrossRef CAS PubMed.
- S. Nyoni and T. Nyokong, Electrochim. Acta, 2014, 136, 240–249 CrossRef CAS.
- Z. Le, F. Liu, P. Nie, X. Li, X. Liu, Z. Bian, G. Chen, H. Bin Wu and Y. Lu, ACS Nano, 2017, 11, 2952–2960 CrossRef CAS PubMed.
- C. Merlet, B. Rotenberg, P. A. Madden, P. L. Taberna, P. Simon, Y. Gogotsi and M. Salanne, Nat. Mater., 2012, 11, 306–310 CrossRef CAS PubMed.
- M. Shrestha, I. Amatya, K. Wang, B. Zheng, Z. Gu and Q. H. Fan, J. Energy Storage, 2017, 13, 206–210 CrossRef.
- T. Purkait, G. Singh, D. Kumar, M. Singh and R. S. Dey, Sci. Rep., 2018, 8, 1–13 CAS.
- K. Sheng, Y. Sun, C. Li, W. Yuan and G. Shi, Sci. Rep., 2012, 2, 3–7 Search PubMed.
- K. Yang, K. Cho and S. Kim, Sci. Rep., 2018, 8, 1–7 Search PubMed.
- P. L. Taberna, P. Simon and J. F. Fauvarque, J. Electrochem. Soc., 2003, 150, A292 CrossRef CAS.
- Y. Yoo, M. S. Kim, J. K. Kim, Y. S. Kim and W. Kim, J. Mater. Chem. A, 2016, 4, 5062–5068 RSC.
- S. Kondrat, N. Georgi, M. V. Fedorov and A. A. Kornyshev, Phys. Chem. Chem. Phys., 2011, 13, 11359–11366 RSC.
- A. A. Kornyshev, J. Phys. Chem. B, 2007, 111, 5545–5557 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Siraprapha
Deebansok
b,
Jie
Deng
a,
Siraprapha
Deebansok
b,
Jie
Deng
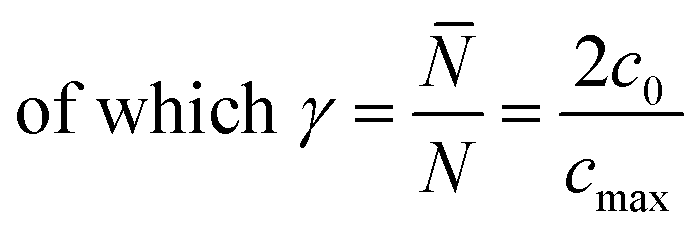
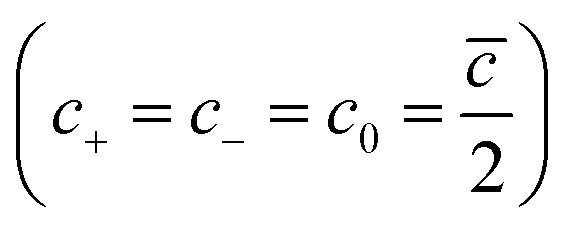 , and cmax is the maximum ion concentration in the bulk, respectively.
, and cmax is the maximum ion concentration in the bulk, respectively.