
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMismatched covalent and noncovalent templating leads to large coiled coil-templated macrocycles†
Kyla J.
Stingley‡
,
Benjamin A.
Carpenter‡
,
Kelsey M.
Kean‡
and
Marcey L.
Waters
 *
*
Department of Chemistry, University of North Carolina at Chapel Hill, CB 3290, Chapel Hill, NC 27599, USA. E-mail: mlwaters@email.unc.edu
First published on 3rd April 2023
Abstract
Herein we describe the use of dynamic combinatorial chemistry to self-assemble complex coiled coil motifs. We amide-coupled a series of peptides designed to form homodimeric coiled coils with 3,5-dithiobenzoic acid (B) at the N-terminus and then allowed each B-peptide to undergo disulfide exchange. In the absence of peptide, monomer B forms cyclic trimers and tetramers, and thus we expected that addition of the peptide to monomer B would shift the equilibrium towards the tetramer to maximize coiled coil formation. Unexpectedly, we found that internal templation of the B-peptide through coiled coil formation shifts the equilibrium towards larger macrocycles up to 13 B-peptide subunits, with a preference for 4, 7, and 10-membered macrocycles. These macrocyclic assemblies display greater helicity and thermal stability relative to intermolecular coiled coil homodimer controls. The preference for large macrocycles is driven by the strength of the coiled coil, as increasing the coiled coil affinity increases the fraction of larger macrocycles. This system represents a new approach towards the development of complex peptide and protein assemblies.
Introduction
Proteins form a complex array of self-assembled structures, ranging from discrete structures such as homo- or heterodimers to extended assemblies such as the protein filaments, keratin, myosin, and others. De novo protein design has provided insight into the factors that drive protein assemblies, including discrete α-helical coiled coil1–8 and β-sheet assemblies.9–11 Higher order supramolecular assemblies of peptides that are driven by covalent linkage, metal templation, and noncovalent assembly, which result in the formation of complex 1-, 2- and 3D assemblies with interesting materials properties, have also been investigated.6,8,12–26The coupling of dynamic combinatorial chemistry (DCC)27 with noncovalent peptide assembly is complementary to de novo design as it allows for the discovery of new, often unexpected assemblies and properties. DCC utilizes building blocks that can reversibly react with one another via covalent bond formation to produce a dynamic combinatorial library (DCL) whose speciation is controlled by thermodynamic stability.27 The introduction of peptide components to a DCC system couples covalent bond formation with noncovalent assembly to create new and often unanticipated structures. We and others have previously reported using this approach to create novel structures.28–41 Among these, globular-like assemblies have been discovered by coupling β-turns,28,29,31 and nucleobases, with DCC while fibril formation has also been observed with short unstructured peptides and β-strand-templated DCLs via nonspecific hydrophobic interactions and β-sheet assembly, respectively.32–37 However, the coupling of DCC with peptides that are designed to form discrete quaternary structures has only been minimally explored.38,39,42 To this end, we sought to investigate how the coupling of a DCC building block to a peptide that favors a discrete quaternary structure influences the balance between formation of covalent bonds between DCC monomers and noncovalent interactions between peptides to give biomimetic peptide assemblies. Herein, we describe the coupling of a series of peptides, which are designed to form homodimeric α-helical coiled coils, with a DCC building block that favors cyclic trimer and tetramer formation via reversible covalent bond formation (Fig. 1a). When combined, these two mismatched components, with competing driving forces to form noncovalent dimers versus covalent trimers and tetramers, create a new class of oligomeric macrocyclic peptide assemblies (Fig. 1). Since the coiled coil sequences favor dimerization and the covalent subunit prefers trimeric and tetrameric species, it would be reasonable to expect both factors to shift the equilibrium speciation toward the tetrameric species due to the creation of two coiled coils (Fig. 1d). However, we find the interplay between the covalent and noncovalent interactions also gives rise to significant amounts of larger cyclic oligomers, overcoming the inherent entropic preference for smaller macrocycles (Fig. 1e). These assemblies represent a middle ground between uncontrolled aggregates and defined protein assemblies, expanding the scope of peptidic complexity of other reported systems.12,33 We find that the shift in equilibrium to larger macrocycles is directly linked to the stability of the coiled coil and that specific macrocyclic ring sizes are favored over others. These findings demonstrate the interplay of effective molarity, binding energy, and degeneracy that give rise to emergent behavior that mimics the factors that contribute to complex biological assemblies.
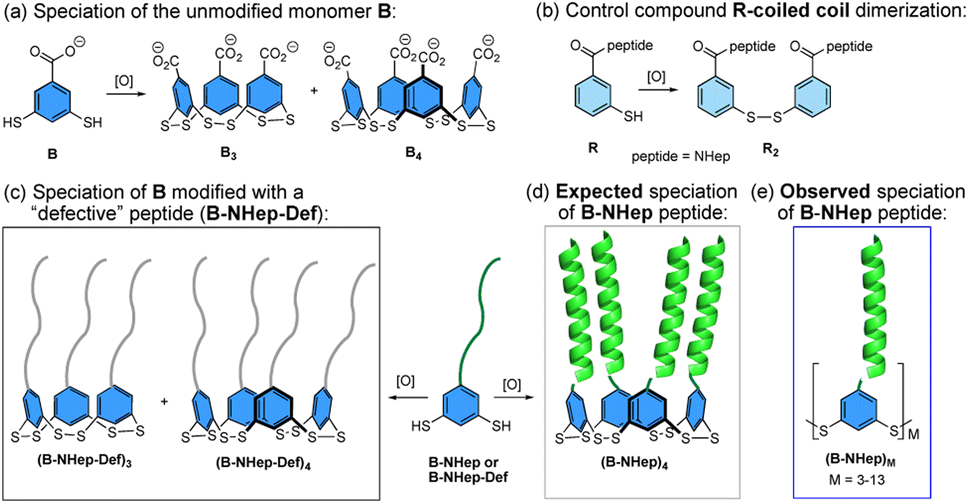 | ||
| Fig. 1 Cartoon depiction of the (a) speciation of unmodified monomer B in a dynamic combinatorial library (DCL). (b) Structure and dimerization of control monomer R. (c) Speciation of B-NHep-Def monomers; (d) and (e) Expected and Observed speciation of the DCL of B-NHep libraries. | ||
Results
System design
The monomer design couples a DCC building block with a peptide designed to form a homodimeric coiled coil with the expectation that speciation of the library and formation of the coiled coil would be energetically coupled, resulting in novel protein-like assemblies (Fig. 1). We chose to use 3,5-dithiobenzoic acid, B, as the DCC building block capable of forming reversible covalent bonds.40 This building block has been utilized previously in studies where peptides have been coupled to the B subunit via amide coupling.33,36,40 The two thiol groups of B allow for disulfide formation in atmospheric oxygen, and equilibrium is subsequently reached via thiol–disulfide exchange. Libraries were generated by adding desired monomer in 50 mM borate buffer (pH 8.4) and equilibrating at room temperature for at least 7 days before analysis. Each library was prepared at 150 μM total monomer concentration unless otherwise noted; the concentration of monomer stocks was determined via absorbance (Fig. S1†). Under similar conditions, libraries of unmodified B have been shown to almost exclusively favor trimeric and tetrameric macrocycles (Fig. 1a), as larger rings are entropically less favorable.36,40 As a control, we also investigated an analog of B, 3-thiobenzoic acid, dubbed monomer R, which only contains a single thiol (Fig. 1b). Thus, libraries of R will only form dimeric species and function as a control for the influence of covalent bond formation on effective molarity and coiled coil formation.Homodimeric coiled coil peptide sequences were chosen based on previously described sequence design from the Woolfson group.2–5,7 Each peptide consists of heptad repeats with hydrophobic residues at the a and d position to create the dimerization interface (Fig. 2a). The placement of Ile (I) at the a positions and Leu (L) at the d positions, in addition to a single Asn (N) residue at one a position, has been shown to specify parallel dimerization due to “knobs-into-holes” packing of the hydrophobic interface coupled with buried hydrogen bonding of the Asn residues between two peptides.2–4 Placing Arg (R) at the e position and Glu (E) at the g position favors homodimerization through complementary electrostatic interactions.3,4 The remaining b, c, and f positions, which do not participate in these noncovalent interactions, were respectively populated with Ala (A) residues to improve helicity, Glu to increase water solubility, or Trp (W) for concentration determination. A Gly (G) spacer on the N-terminus was incorporated between the DCC building block B and the peptide. Structural analysis of distances between the a positions at the N-termini of a coiled coil (7.5 Å) as well as computational analysis of the distance between the carbonyl carbon position in the B4 macrocycles (ranging from 5.9–10.5 Å, depending on the conformation) suggests that a single Gly spacer is sufficient to allow coiled coil formation within the macrocycles, although not all conformations orient the peptides in the same direction (Fig. 2b–e).
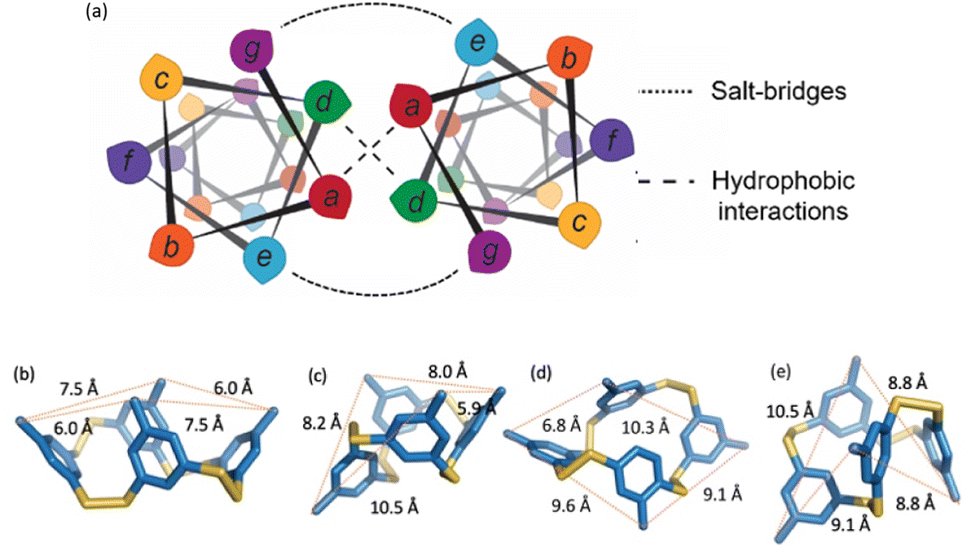 | ||
| Fig. 2 (a) Helical wheel diagram depicting the coiled coil interface and location of each amino acid position in the heptad. (b–e) Computational models of possible conformations of B4 showing distances between neighboring carbonyl carbons. (b) “All up”, corresponding to the orientation in Fig. 1; (c), “1 down”; (d) “1,2-down”; (e) “1,3-alternating”. | ||
Previous reports have shown that de novo coiled coil peptides exhibit length dependent affinity; 4-heptad peptides exhibit low nanomolar affinity, 3-heptad peptides exhibit low micromolar affinity, and so on.2,3,43 Thus, we utilized a series of peptide sequences coupled to monomer B that range from 1.5-heptad to 3-heptad in length, referred to as B-NHep where N is the number of heptads, to systematically vary the strength of the coiled coil (Table 1, Fig. S2–S5†). In doing so, we were able to vary the relative energetic contribution of the coiled coil formation versus the covalent DCC linkage on the speciation preference of the library. Acetyl-capped peptides (Ac-NHep) were used as controls to evaluate the extent of folding in the absence of covalent interactions from the DCC building blocks (Fig. S6–S9†). The 2.5 and 3-heptad peptide were also coupled to building block R to determine the impact of a single covalent linker on folding (Fig. S10 and S11†). Lastly, we synthesized a set of defective peptides (Ac-NHep-Def and B-NHep-Def) that cannot form a coiled coil (Fig. S12–S15†) by replacing the Leu residues at the d positions of each heptad with Gly (G; Table 1). Below we describe the characterization of the speciation of these DCLs by mass spectrometry and SDS-PAGE electrophoresis, as well as the characterization of folding and stability by circular dichroism (CD) spectroscopy.

|
Characterization of speciation by LC-MS and MALDI-TOF MS
To assess the influence of each peptide on speciation of the DCL, we utilized LC-MS analysis to compare speciation of the B-NHep libraries to the parent B monomer alone. The parent compound B exhibits two peaks representing B3 and B4 with B3 as the major species, as has been observed previously (Fig. S16†).36,40 Similarly the single-thiol monomer R behaved as expected, forming exclusively R2 (Fig. S17†). Because the peptide sequences favor coiled coil dimer formation in a length-dependent manner, we expected that the appended peptide would shift the library toward tetramer, allowing for the formation of two coiled coils per macrocycle, and that the shift towards B4 would correlate with increasing peptide length. Analysis of the B-1.5Hep library indicates the formation of B3 and B4 with B3 as the major species (Fig. S18†), similar to B alone. This suggests that the 1.5Hep peptide is too short to form significantly stabilizing coiled coils that would perturb the equilibrium, even when in a covalent complex. However, while B-2Hep, B-2.5Hep, and B-3Hep also form B3 and B4 with a shift towards B4 as expected, additional broad peaks that did not ionize well are also observed (Fig. S19–S21†). Attempts to optimize the chromatography did not improve the peak resolution, and the species giving rise to these peaks did not ionize well by ESI-MS. In contrast, the defective B-peptide conjugates, B-2Hep-Def and B-2.5Hep-Def form only B3 and B4, similar to B-1.5Hep and B alone (Fig. S22 and S23†), suggesting that the broad peaks are not solely due to peptide length but correlate with the ability to form a coiled coil. Further, R-2.5Hep and R-2.5Hep-Def libraries showed only the expected peaks for covalent dimers, suggesting that the unidentified species are not noncovalent aggregates (Fig. S24 and S25†). Based on these data and the size of some species reported in related systems,32,35 these unidentifiable masses were hypothesized to be large oligomers or assemblies.To further characterize the speciation of the libraries, we analyzed DCLs of B-1.5Hep-3Hep and B-2.5Hep-Def by MALDI-TOF mass spectrometry using at least 20-fold higher concentration libraries to observe higher mass species (3–8 mM, Fig. S26–S30†). We also utilized a relatively high laser intensity to observe higher mass species, sacrificing resolution, such that the isotopic envelopes were not resolved.17 Analysis of the peak maxima, representing the isotope average mass, demonstrate a difference of a single B-NHep unit between each peak (Tables S2–S6†). This analysis indicates that B-2Hep, B-2.5Hep, and B-3Hep all show masses up to at least B11, indicating that large oligomers are formed in these DCLs. By contrast, B-2.5Hep-Def only shows species up to B7, suggesting the importance of coiled coil formation in forming higher mass species. As only B3 and B4 were observed by LC-MS in the B-2.5Hep-Def library (Fig. S23†), the presence of some amount of B5–B7 in the MALDI data may be due to the much higher concentrations of these experiments. While analysis through MALDI provided evidence for the formation of higher order oligomers, it did not provide reliable information regarding relative abundance. Thus, we turned to gel electrophoresis to gain further insight into the speciation of these libraries.
Coiled coils shift the equilibrium to higher mass species as observed by SDS-PAGE analysis
To investigate the higher mass species, we used Bis-Tris SDS-PAGE to analyze libraries of each of the B-NHep conjugates (Fig. 3). In addition to its widespread use as a protein and peptide visualization tool, there also is precedence of SDS-PAGE use with complex DCC systems.9,11,41,44 We examined differences in speciation of the B-NHep DCLs containing different length peptides, along with the B-2.5Hep-Def (unable to form coiled coils) and Ac-3Hep (lacking covalent linkages from the DCC monomer) controls. Libraries of each peptide length were set up at 150 μM, the same concentration used for the LC-MS.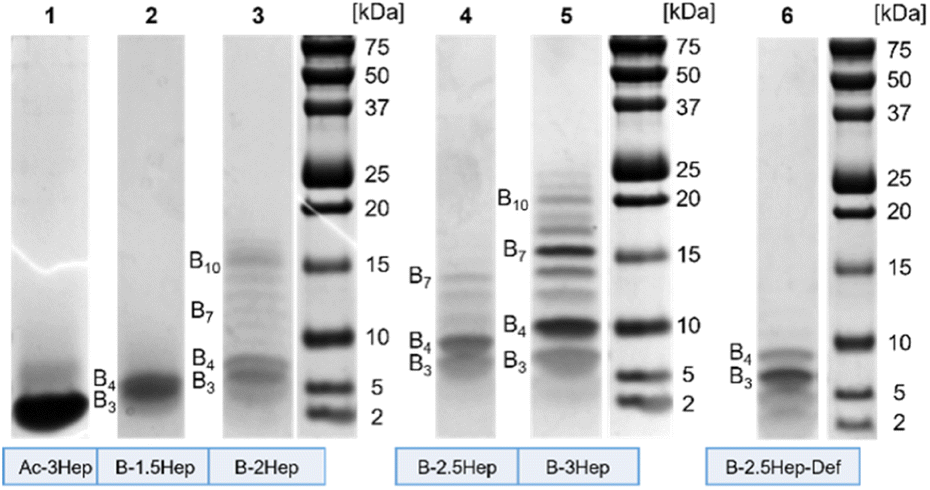 | ||
| Fig. 3 Bis-Tris SDS-PAGE analysis of libraries of monomer B with varying peptide length (B-1.5Hep-3Hep) set up at 150 μM, shown in order of increasing heptad length. Ac-3Hep was also included as a mass reference. Gels were stained with Coomassie Blue for visualization. | ||
As seen in Fig. 3, a number of discrete species were visualized on the gel that were not resolved in the LC-MS traces. Libraries showed a significant number of higher mass species beyond B3 and B4, excepting the B-1.5Hep and B-2.5Hep-Def libraries (Fig. 3, lane 2 and 6). In accordance with the LC-MS trace, the B-1.5Hep libraries exhibit two poorly resolved bands in the gel, corresponding in mass to the B3 and B4 species (Table S7†). This indicates that the 1.5-heptad peptides, which are expected to have the weakest coiled coil interactions, have little impact on speciation of the DCL. The intensity of these bands and their relatively small (1524.6 Da) mass difference makes it difficult to distinguish, but the two separate bands can be seen more clearly at lower concentrations (Fig. S31,† lane 1–3; Fig. S32,† lane 3). Similarly, the B-2.5Hep-Def library also results in bands for only B3 and B4, with a more intense band for B3, in agreement with the results from LC-MS and MALDI analysis. These controls support the importance of coiled coil formation for the emergence of higher mass species.
Samples of B-2Hep, B-2.5Hep, and B-3Hep monomers (Fig. 3, lanes 3, 4, 5, respectively) display multiple bands corresponding to higher mass species in addition to the trimers (B3) and tetramers (B4) favored by unmodified monomer B. Thus, coiled coil formation appears to overcome the entropic restriction to smaller rings. Moreover, different numbers and patterns of bands were observed depending on the peptide length. The B-2Hep library (Fig. 3, lane 3) shows preferential formation of a species around the B10 mass in addition to B3 and B4, while the B-2.5Hep library (Fig. 3, lane 4) favors B4 over B3 and exhibits a significant B7 band. The B-3Hep library (Fig. 3, lane 5) more strongly favors the formation of higher mass species than libraries of other monomers, with a significant preference for B4, B7, and to a lesser extent, B6, B8, and B10. The Ac-3Hep peptide, which cannot form covalent linkages, exhibits an intense band corresponding to the monomeric peptide (2522.4 Da; Fig. 3, lane 1).
The effect of concentration on this speciation pattern was analyzed by equilibrating libraries at varied concentrations (Fig. S31 and S32†). Overall, changing the monomer concentration over a range of 50 μM up to 675 μM did not cause significant changes in speciation or relative abundance beyond a slight increase in higher mass band intensity for the highest concentration libraries (Fig. S31, lanes 1, 5, 9, 13; S32,† lanes 1, 4, 7, 10). Given that coiled coil formation is concentration-dependent,2 this slight difference is expected. Consistent with the MALDI data, the B-2.5Hep-Def libraries showed faint bands for B5–B7 in addition to intense bands for B3 and B4 when equilibrated at the highest concentration (Fig. S32,† lane 13). However, in contrast to all B-NHep DCLs designed to form coiled coils, B-2.5Hep-Def does not exhibit preferential formation of any specific higher mass species. In sum, the SDS-PAGE analysis demonstrates that coiled coil formation perturbs the speciation of the DCLs in a length-dependent manner and drives formation of higher-mass oligomers.
Speciation is thermodynamically controlled
We considered the fact that B7 could be formed by the reaction of a B3 species with a B4 species, resulting from a kinetic preference rather than representing an equilibrium mixture. To evaluate whether the speciation of the libraries represents equilibrium or a kinetic trap, a re-equilibration experiment was performed using a 360 μM B-2.5Hep library which had previously equilibrated for over a month. 10 mol% TCEP relative to total monomer concentration was added to partially reduce the library. The library was then allowed to re-equilibrate for one week, and both parent and re-equilibrated samples were analyzed by gel electrophoresis (Fig. 4). Comparing the parent and re-equilibrated library samples (Fig. 4, lanes 1 and 2 respectively), the speciation and abundance are virtually identical, indicating that the higher mass species shown are in fact thermodynamically favored products and that the libraries in this study are fully equilibrated.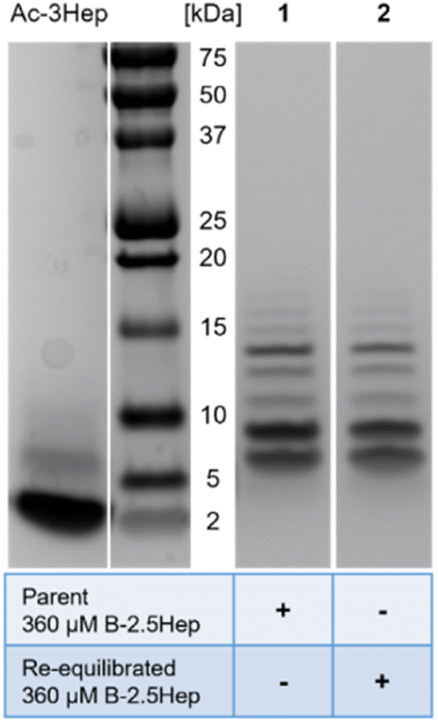 | ||
| Fig. 4 Bis-Tris SDS-PAGE analysis of the parent (lane 1) and re-equilibrated (partially reduced with 10mol% TCEP and allowed to re-equilibrate; lane 2) 360 μM B-2.5Hep library. Ac-3Hep peptide was included as a mass standard. Gels were stained with Coomassie Blue for visualization. | ||
Denaturing conditions confirm that the higher mass species are covalent oligomers
While the SDS in Bis-Tris gels is denaturing, we wanted to ensure that all noncovalent interactions in the libraries were fully disrupted and that the larger mass species are covalent oligomers. To do this, we compared our standard gel system (Bis-Tris SDS-PAGE; Fig. S33b†) to a gel containing 6 M urea (Fig. S33a†), which has previously been shown to be an effective denaturing agent for a variety of parallel, dimeric coiled coils.45–47 Additionally, our own CD analysis showed that adding 6 M urea thoroughly eliminated coiled coil formation for 150 μM B-2.5Hep and Ac-3Hep library samples (Fig. S34, Table S8†). The addition of 6 M urea to the gel did not cause any significant changes to the number or resolution of bands compared to our standard conditions, further validating that the higher mass species are covalent oligomers.Cyclic oligomers are formed
To confirm that these higher-mass species are cyclic, as has been observed in other DCLs using dithiols, we treated equilibrated libraries with 5-iodoacetamidofluorescein (5-IAF), a fluorescent tag capable of reacting with any remaining free thiol. Any acyclic oligomers would contain two thiols that would react with 5-IAF, resulting in both a shift in the mass of the oligomer and a fluorescent signal for bands corresponding to these species on a gel. In comparison, a cyclic macrocycle would not contain any free thiols and would not react with 5-IAF, leaving the library unchanged. We thus compared the fluorescence tagging of a library of B-2.5Hep with 5-IAF to a control without added 5-IAF (Fig. 5). We also compared the results to several other controls, including B-2.5Hep-Def, reduced B-2.5Hep, and an equilibrated B-2.5Hep library with added oxidant (sodium perborate).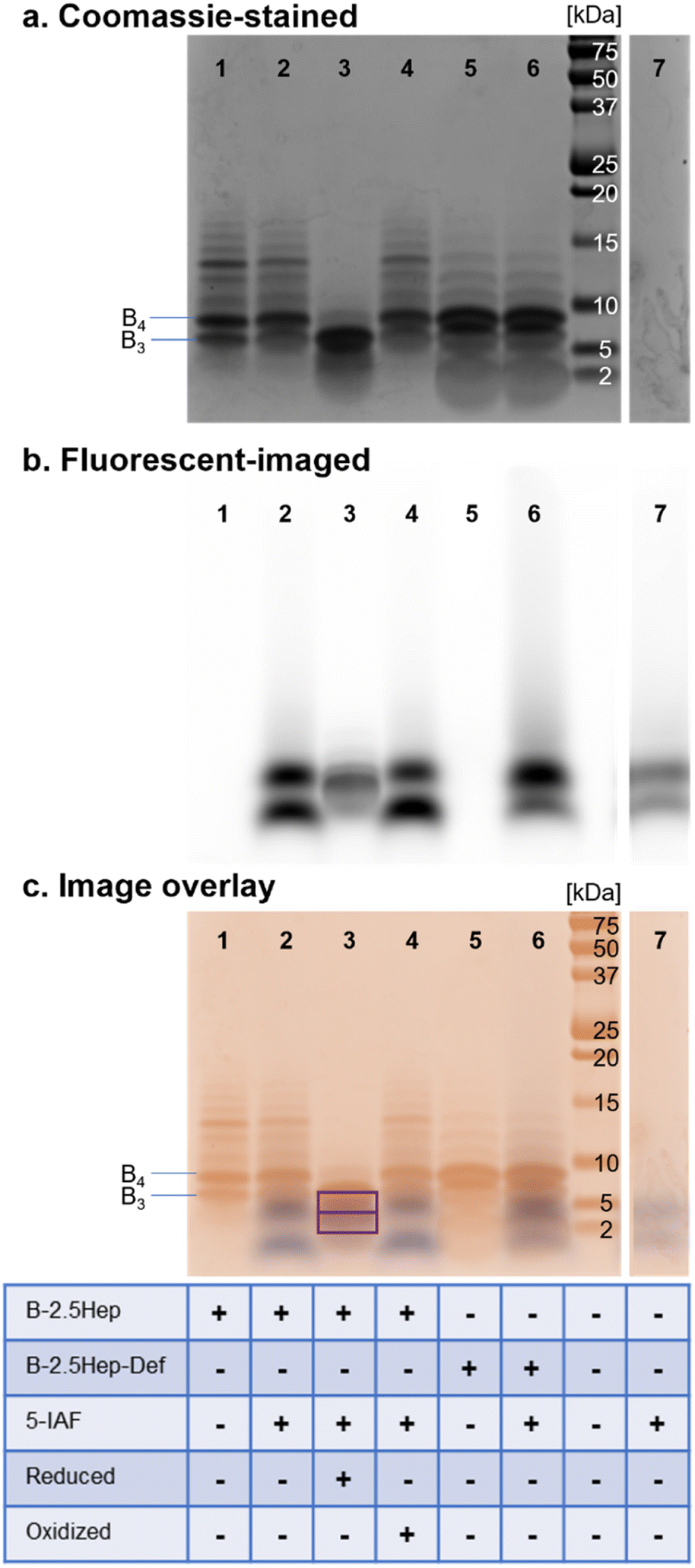 | ||
| Fig. 5 Bis-Tris SDS-PAGE analysis of the B-2.5Hep and B-2.5Hep-Def 450 μM libraries following reaction with 5-IAF. Gels were visualized using (a) Coomassie blue stain and (b) fluorescent-imaging (c) overlay of Coomassie-stained (orange, top) and fluorescent (blue, bottom) images of SDS-PAGE analysis of B-2.5Hep and B-2.5Hep-Def libraries following reaction with 5-IAF. Purple bands are places of overlap (outlined with purple squares), blue bands are fluorescent bands without Coomassie overlap, and orange bands are Coomassie bands without fluorescent overlap. Reduced libraries contained 756 μM TCEP (2.1 equivalents), while oxidized libraries 756 μM sodium perborate (2.1 equivalents). | ||
All 5-IAF labeling experiments utilized equilibrated 450 μM B-2.5Hep and B-2.5Hep-Def libraries which were then diluted to a concentration of 360 μM library with the addition of 90 μM 5-IAF (0.25 equivalents). Incubation of the library with 5-IAF did not cause any measurable shifts in the bands in the Coomassie-stained gel as compared to libraries without 5-IAF (Fig. 5, lanes 1 vs. 2 and 5 vs. 6), suggesting that the bands that correspond to B3 and larger ring sizes are cyclic and thus unreactive toward 5-IAF. The comparison between lanes 1, 2, 5 and 6 also reveals that B-2.5Hep and B-2.5Hep-Def libraries respond similarly to the addition of 5-IAF, indicating that closed macrocycles form regardless of the presence of coiled coils (Fig. 5c). Fluorescence imaging does not show labeling of any higher mass species. The 5-IAF control sample exhibits two fluorescence bands at low molecular weights (∼1.5 kDa and 4 kDa, Fig. 5b and c, lane 7). These same two bands are the only fluorescent bands observed when 5-IAF is mixed with B-2.5Hep or B-2.5Hep-Def (Fig. 5, lanes 2, 4, and 6) indicating that none of the B-2.5Hep species are labeled.
As positive and negative controls, we also investigated the labeling of a reduced library (Fig. 5, lane 3) and an “oxidized” library, in which sodium perborate was added after equilibration in air to ensure that the library was fully oxidized (Fig. 5, lane 4). The reduced sample contained 2.1 equiv. TCEP, while the fully oxidized library contained 2.1 equiv. sodium perborate. An overlay of the Coomassie-stained and fluorescent-imaged gels demonstrates that the reduced library (Fig. 5, lane 3) contains labeled species at low molecular weight that are not seen in standard conditions or in the 5-IAF control (Fig. 5, lane 7). Interestingly, a band for B3 is also apparent, suggesting rapid re-oxidation to B3 after reduction with TCEP. In contrast, the oxidized library (Fig. 5, lane 4) looks identical to the equilibrated libraries with or without 5-IAF (Fig. 5, lanes 1 and 2), further supporting that those libraries were fully oxidized and contain only cyclic species.
To further validate these results, we analyzed the libraries corresponding to the standard conditions (Fig. 5, lane 2), reduced library (positive control; Fig. 5, lane 3), and oxidized library (negative control; Fig. 5, lane 4) by LC-MS (Fig. S35†). The LC-MS traces of the oxidized and standard libraries were nearly identical with no identifiable masses corresponding to new 5-IAF labeled oligomers (Fig. S35a and c†), consistent with the Coomassie-stained and fluorescent imaged gels showing no labeling by 5-IAF (Fig. 5, lanes 1 and 2). The LC-MS trace of the reduced sample shows a mixture of unlabeled, 5-IAF-monolabeled, and 5-IAF-dilabeled monomer (Fig. S35b†), consistent with expectations based on the 0.25 equivalents of 5-IAF used and the appearance of overlapping Coomassie-labeled and fluorescent bands on the gel (Fig. 5, lane 3). Taken together, these results show that the oligomers observed in these B-NHep libraries do not possess free thiol groups. This indicates that large discrete macrocycles with up to 13 subunits are formed.
Characterization of DCLs by circular dichroism (CD) spectroscopy
The importance of coiled coil formation on speciation has been suggested by all analyses. Thus, we used CD to characterize the extent of coiled coil formation in the DCLs, as monomeric peptides adopt a random coil conformation whereas binding induces a helical structure, forming a coiled coil. α-Helical coiled coil peptides exhibit a maximum at 190 nm and minima at 208 and 222 nm, whereas random coil structures exhibit a minimum at about 195 nm.48As controls for the B-NHep DCLs, we utilized acetyl-capped peptides (Ac-NHep) to characterize the degree of coiled coil formation in the absence of a covalent linkage and Ac-NHep-Def peptides as negative controls for peptides that cannot form coiled coils. An equilibrated R-2.5Hep DCL was used to characterize coiled coil formation with only a single disulfide, which exclusively forms a dimer. Thus, comparing the degree of helicity of Ac-2.5Hep to the R-2.5Hep dimer and the B-2.5Hep DCL correlates with the effect of adding a disulfide linkage and forming a macrocycle, respectively.
As expected, Ac-3Hep-Def CD analysis shows a random coil minimum at 198 nm (Fig. S37†). Analysis of the acetylated peptides shows that only the Ac-3Hep is able to form coiled coils at 150 μM, whereas the shorter acetylated peptides remain unfolded as random coils, as expected (Fig. 6a).43 The 4-residue difference between the Ac-2.5Hep and Ac-3Hep appears to have a significant influence on folding under these conditions, in agreement with similar sequences that have previously been reported.43
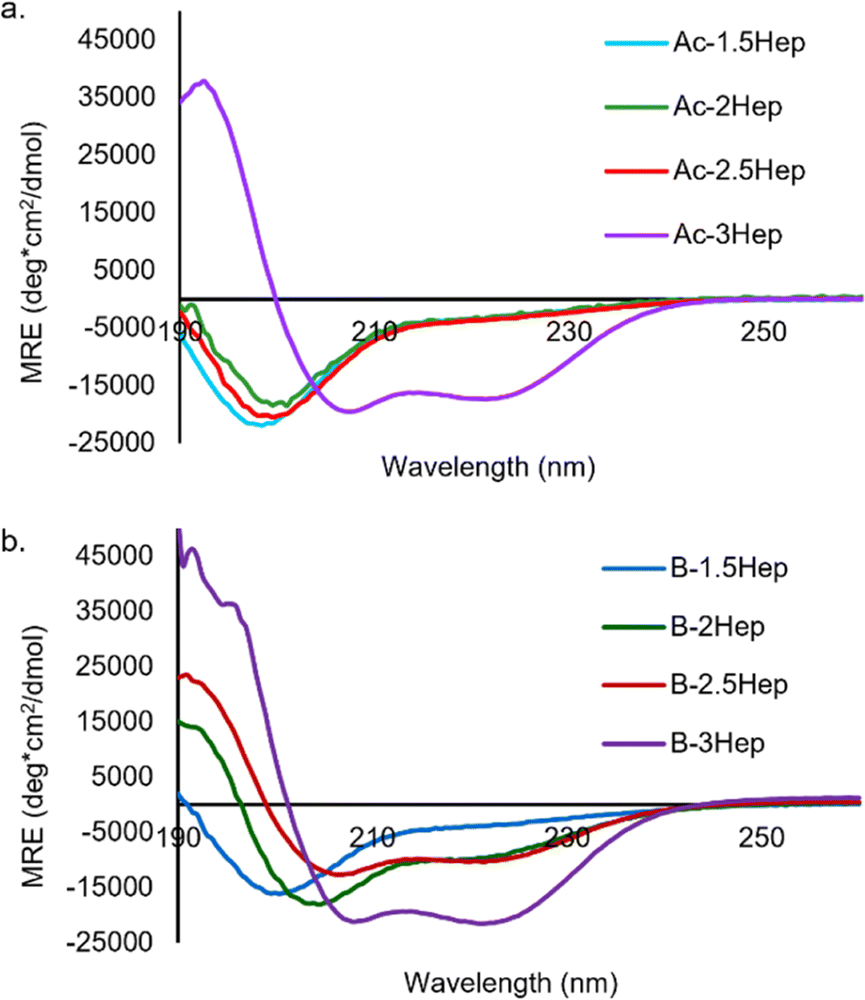 | ||
| Fig. 6 (a) CD spectra of acetylated coiled coil peptides. (b) CD spectra of B-NHep libraries. Concentration of peptide is 150 mM, 50 mM borate buffer, pH 8.5. Spectra measured at 20 °C. | ||
To determine the influence of covalent templation arising from the macrocycles formed via DCC on coiled coil formation, we compared the CD spectra of equilibrated B-NHep DCLs with the appropriate Ac-NHep control peptide (Fig. 6). The B-NHep DCLs represent a mixture of species, so the CD spectra represent the average signal arising from the speciation of the library. Comparison of the B-1.5Hep to the Ac-1.5Hep peptide indicates that covalent templation has little effect on the extent of coiled coil formation at this length (blue lines, Fig. 6a and b), which is consistent with the speciation studies that show that, like unmodified B, B-1.5Hep forms only B3 and B4. However, as the peptide length increases, coiled coil formation is stabilized in the B-NHep peptides (Fig. 6b). This is consistent with increased effective molarity of the covalently linked peptides and has been observed in other covalently templated coiled coils.25,49–56B-2.5Hep exhibits the greatest improvement in folding/binding relative to Ac-2.5Hep, indicating that it is the most cooperative at this concentration. In contrast, B-3Hep only exhibits a modest increase in folding relative to Ac-3Hep because Ac-3Hep forms a dimer in the absence of a covalent linkage at this concentration. As expected, the B-2Hep-Def and B-2.5Hep-Def samples (Fig. S38 and S39†) remain random coil in the context of the library, indicating that increased effective molarity alone is incapable of inducing coiled coil formation.
To determine whether the formation of macrocycles in B-NHep influences the extent of coiled coil formation, we compared the helicity of the B-2.5Hep DCL to that of the R-2.5Hep disulfide-linked dimer. Overlays of the CD spectra are virtually identical, indicating that the templation effect in the acyclic R-2.5Hep dimer is equivalent to that in the mixture of cyclic species formed in the DCL of B-2.5Hep (Fig. 7). Importantly, this observation confirms that coiled coil formation is optimized in the DCL.
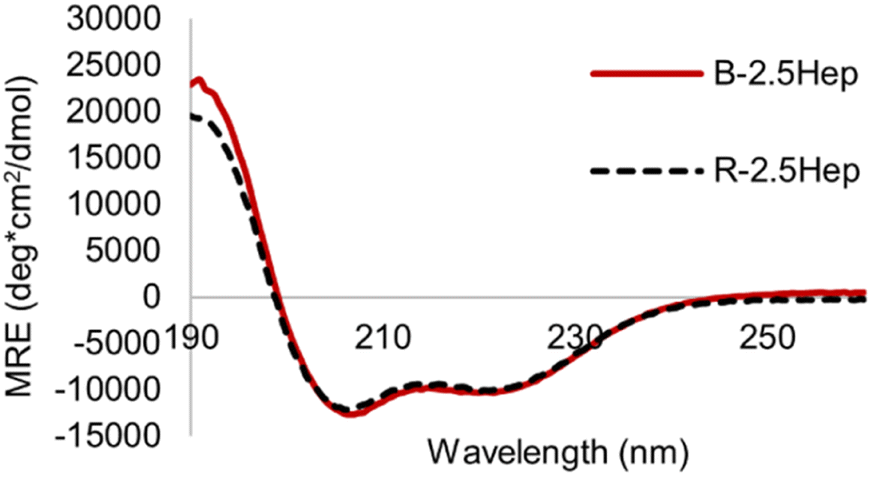 | ||
| Fig. 7 CD spectrum comparing R-2.5Hep and B-2.5Hep. Concentration of peptides is 150 μM, scans taken at 20 °C in 50 mM borate buffer, pH 8.5. DCLs were equilibrated for at least 7 days. | ||
Taken together, the CD data suggests a scenario in which the magnitude of the coiled coil interaction, as defined by its length, and the effective molarity due to covalent bond formation, are maximized in B-2.5Hep to give the greatest improvement in folding due to maximized cooperativity. If the inherent strength of the coiled coil interaction is too low, as in B-1.5Hep, the covalent linkage will have little effect and if the coiled coil interaction is too strong, as in B-3Hep, the covalent linkage is not necessary to induce folding.
To complement the urea denaturation gel experiments, we measured the concentration dependance of the CD spectrum of B-2Hep, B-2.5Hep and R-2.5Hep from 30–200 mM (Fig. 8, S40, and S41†) to verify that helicity arises from intramolecular interactions. If coiled coil formation is intermolecular, it would be expected to be concentration dependent. However, no change in the CD spectrum was observed, consistent with folding being driven by intramolecular interactions in the B-NHep peptides.
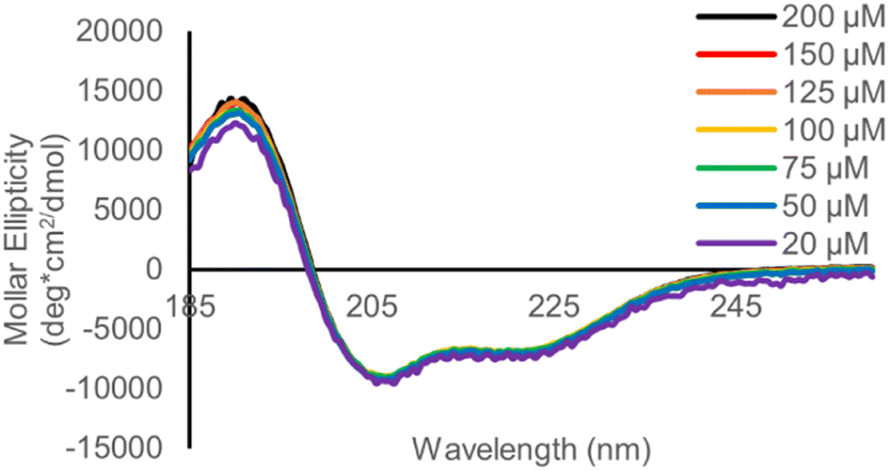 | ||
| Fig. 8 CD spectrum comparing B-2.5Hep from 20–200 μM, demonstrating lack of dependence on concentration. Scans taken at 20 °C in 50 mM borate buffer, pH 8.5. DCLs were equilibrated for at least 7 days. | ||
Thermal denaturation studies
To evaluate the effect of covalent linkage on the stability of the coiled coil assemblies, thermal denaturation experiments were run on the DCLs by monitoring the MRE signal at 222 nm over a range of temperatures. Melts were performed on Ac-3Hep, B-3Hep, and B-2.5Hep, as each exhibit significant folding at room temperature, as well as the R-2.5Hep to compare dimer to oligomer folding. The B-3Hep library is the most thermally stable (Fig. 9) with a TM > 60 °C, which is a ∼30 °C shift in TM relative to Ac-3Hep. Interestingly, the B-3Hep library appears to exhibit another thermal transition between 5 °C and 35 °C, which may arise from different thermal stabilities of different species in the library. Comparison of the melting curves of B-2.5Hep and R-2.5Hep indicates that the coiled coil stability of these two libraries is virtually identical (Fig. 9). This suggests that, on average across the species, macrocyclization does not template coiled coil formation any better than the single disulfide bond in the R-2.5Hep dimer. Furthermore, the similarity between Ac-3Hep and B-2.5Hep suggests that covalently linking the 2.5-heptad peptide through disulfide linkages stabilizes the coiled coil approximately as much as adding a half of a heptad in length (Fig. 9).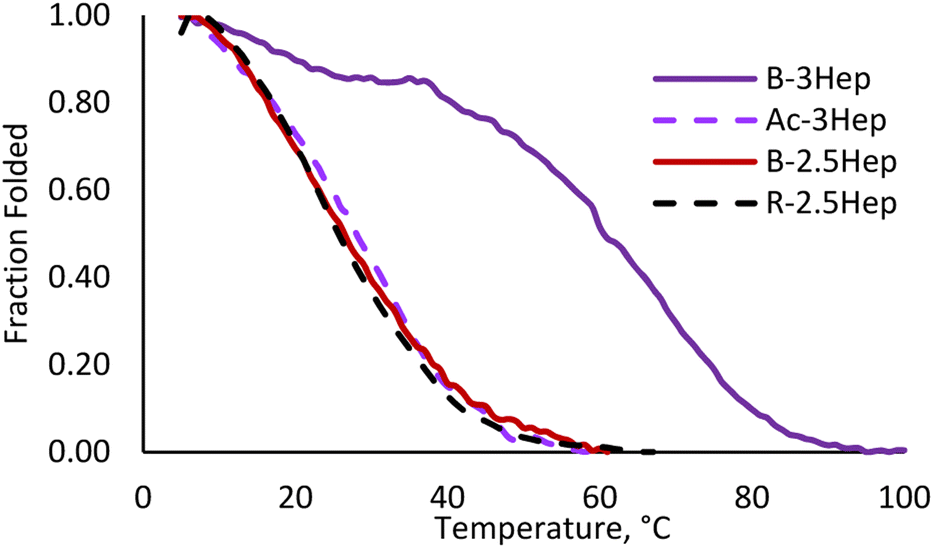 | ||
| Fig. 9 Melting curve for Ac-3Hep (purple dashed), B-3Hep (dark purple), B-2.5Hep (red), and R-2.5Hep (black dashed). Measurements taken at 222 nm, 150 μM equilibrated samples. | ||
Discussion and conclusions
This investigation demonstrates that an interplay between the mismatched energetic preferences of reversible covalent (a DCC monomer) and noncovalent components (coiled coil peptides) leads to complex behavior. Rather than resulting in the simplest outcome – templation of tetramer B4 through formation of two coiled coils–the equilibrium is shifted to a series of macrocyclic species from B4 up to B13, despite the entropic cost of forming larger macrocycles. In addition to B4, there is also a notable preference for the B7 macrocycle, which is unexpected as an odd-numbered macrocycle prevents all peptides from having a binding partner. The importance of the coiled coil formation in these species is evident by the significant differences in the abundance and range of higher mass covalent assemblies between the B-2.5Hep and B-2.5Hep-Def DCLs (Fig. 3), correlating with increased helicity compared to the acetylated control peptides. Concentration dependence studies support that the macrocyclic species are intramolecularly templated. These data, in addition to the fluorescent-labeling experiments (Fig. 5), identify the higher mass species as discrete, internally templated macrocycles.CD analysis of libraries containing these larger macrocycles does not reveal any significant change to helicity compared to the dimeric R-NHep controls (Fig. 7), indicating that the peptides access similar coiled coil dimer structures within the context of the B-NHep libraries. The thermal denaturation data indicate increased stability of the coiled coil in B-NHep libraries when compared to the corresponding acetylated peptides (Fig. 9) and nearly identical denaturation to the R-NHep dimer (Fig. S40†). These data demonstrate that the macrocyclic covalent linkages stabilize the coiled coils due to the higher effective concentration of the peptides from the covalent linkages. However, the similar degree of helicity in the B-NHep macrocycles as in the R-NHep dimers indicates that formation of higher-mass macrocycles is not driven by an increase in coiled coil formation beyond that of a simple dimer.
As the degree of helicity is no different in the R-NHep dimer versusB-NHep DCL, suggesting that larger macrocycles are not driven by better-templated coiled coils, we considered other factors that may stabilize larger macrocycles. One possibility is that in a small macrocycle, the environment is too crowded or the orientation of the B-monomers is unfavorable for optimal coiled coil formation, driving the equilibrium towards larger macrocycles to optimize coiled coils. An additional source of stability may come from dynamic multivalency, which is known to contribute to the binding interactions of some intrinsically disordered proteins.57–59 In such protein–protein interactions, dynamic multivalency arises when there is more than one degenerate binding epitope that can bind to a single partner protein. Similarly, in B-macrocycles reported here, the ability to form multiple degenerate folded states may provide an additional driving force for forming large macrocycles. Coiled coils are known to have rapid folding and unfolding rates.60 Thus, each peptide likely exists in a rapid equilibrium of dimeric partners with the peptide on either side. This results in increased dynamic multivalency as ring size and peptide components in the ring increase. Making the simplifying assumption that the peptides can only form coiled coils with adjacent peptides and that each coiled coil is energetically identical, this dynamic multivalency results in each macrocycle having at least two enthalpically degenerate states in even-numbered macrocycles, resulting in a more entropically favored assembly (Fig. 10). Odd -numbered rings can exist in more than 2 degenerate states due to the presence of an unpaired peptide (Fig. 10b), increasing their entropic favorability. Furthermore, specific macrocycles may be favored due to optimization of intramolecular interactions within the aromatic core, as it has been shown that such disulfide-linked macrocycles can take on specific folded structures mediated by π–π stacking between the subunits.33,61,62 Furthermore, cross-macrocycle coiled coils between non-adjacent monomers may also be possible. Together, these combined factors contribute to the increased stability of large macrocycles.
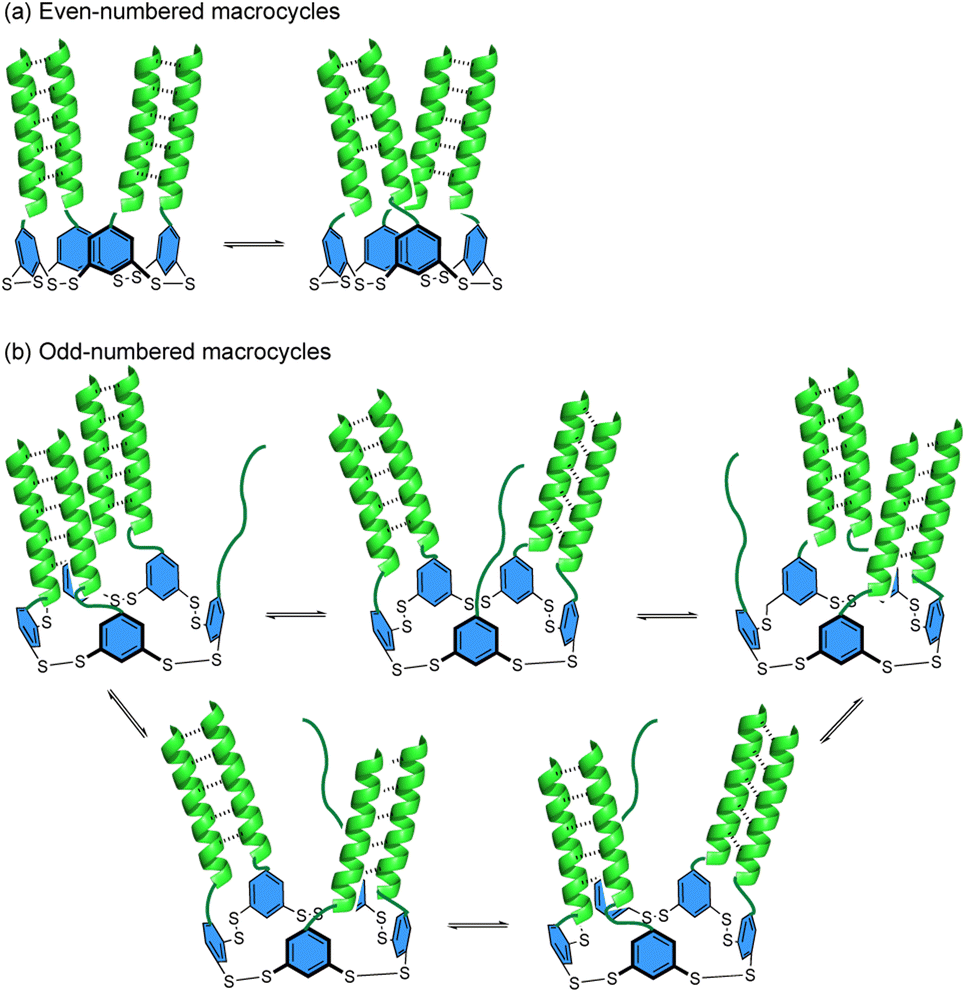 | ||
| Fig. 10 Cartoon depiction of the proposed dynamic multivalency in macrocycles with (a) even and (b) odd numbers of monomers. Dashed lines show intramolecular coiled coil formation between peptides. | ||
In summary, we describe the coupling of mismatched covalent and noncovalent templation that results in emergent behavior beyond the individual preferences of each component. Although the peptide forms coiled coil dimers, differences in conformational preferences of different ring size that influence interactions between neighboring peptides as well as dynamic exchange between coiled coils within the covalent macrocycle may provide an additional driving force for assembly into larger rings. The size and speciation of the macrocycles correlates with the length, and hence extent of folding and binding, of the coiled coil peptides, resulting in a greater shift towards higher-mass species with more stable coiled coils. Moreover, specific ring sizes are favored over others, presumably due to other conformational preferences. The outcome of this mismatched templation is access to discrete yet complex protein-like assemblies not readily accessible using other methods.12 The changes in assembly based on heptad length and coiled coil stability suggest a wealth of novel behavior still to be accessed, given the broad array of coiled coil designs that can be incorporated. Work towards exploring this line of inquiry is currently underway.
Data availability
All experimental procedures and characterization data are available in the ESI.†Author contributions
M. L. W. conceived of the project and obtained funding. K. J. S., B. A. C., and K. M. K. contributed to collecting the data. K. J. S., B. A. C., K. M. K., and M. L. W. contributed to analyzing the data. K. J. S. wrote the original draft of the manuscript. K. M. K and M. L. W. edited the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge funding from NSF grant CHE-2107685 to M. L. W. and NIH grant K12-GM000678 to K. M. K. This work was also supported by the National Cancer Institute of the National Institutes of Health under award number P30CA016086 to support the UNC Macromolecular Interactions Facility. We thank Brandie Ehrmann and E. Diane Weatherspoon in the University of North Carolina's Department of Chemistry Mass Spectrometry Core Laboratory for their assistance with mass spectrometry analysis. We thank Dr Ashutosh Tripathy for assistance with CD.Notes and references
- A. R. Thomson, C. W. Wood, A. J. Burton, G. J. Bartlett, R. B. Sessions, R. L. Brady and D. N. Woolfson, Science, 2014, 346, 485–488 CrossRef CAS PubMed.
- D. N. Woolfson, in Fibrous Proteins: Structures and Mechanisms, ed. D. A. D. Parry and J. M. Squire, Springer International Publishing, Cham, 2017, pp. 35–61 Search PubMed.
- J. M. Fletcher, A. L. Boyle, M. Bruning, G. J. Bartlett, T. L. Vincent, N. R. Zaccai, C. T. Armstrong, E. H. C. Bromley, P. J. Booth, R. L. Brady, A. R. Thomson and D. N. Woolfson, ACS Synth. Biol., 2012, 1, 240–250 CrossRef CAS PubMed.
- A. L. Boyle and D. N. Woolfson, Chem. Soc. Rev., 2011, 40, 4295–4306 RSC.
- J. M. Fletcher, K. A. Horner, G. J. Bartlett, G. G. Rhys, A. J. Wilson and D. N. Woolfson, Chem. Sci., 2018, 9, 7656–7665 RSC.
- D. N. Woolfson, J. Mol. Biol., 2021, 433, 167160 CrossRef CAS PubMed.
- W. M. Rink and F. Thomas, Chem. - Eur. J., 2019, 25, 1665–1677 CrossRef CAS PubMed.
- I. V. Korendovych and W. F. DeGrado, Q. Rev. Biophys., 2020, 53, e3 CrossRef CAS PubMed.
- T. D. Samdin, A. G. Kreutzer and J. S. Nowick, Curr. Opin. Chem. Biol., 2021, 64, 106–115 CrossRef CAS PubMed.
- S. Haerianardakani, A. G. Kreutzer, P. J. Salveson, T. D. Samdin, G. E. Guaglianone and J. S. Nowick, J. Am. Chem. Soc., 2020, 142, 20708–20716 CrossRef CAS PubMed.
- S. Zhang, S. Yoo, D. T. Snyder, B. B. Katz, A. Henrickson, B. Demeler, V. H. Wysocki, A. G. Kreutzer and J. S. Nowick, Biochemistry, 2022, 61, 252–264 CrossRef CAS PubMed.
- F. Sheehan, D. Sementa, A. Jain, M. Kumar, M. Tayarani-Najjaran, D. Kroiss and R. V. Ulijn, Chem. Rev., 2021, 121, 13869–13914 CrossRef CAS PubMed.
- M. Nepal, M. J. Sheedlo, C. Das and J. Chmielewski, J. Am. Chem. Soc., 2016, 138, 11051–11057 CrossRef CAS PubMed.
- M. Nambiar, L.-S. Wang, V. Rotello and J. Chmielewski, J. Am. Chem. Soc., 2018, 140, 13028–13033 CrossRef CAS PubMed.
- J. M. Galloway, H. E. V. Bray, D. K. Shoemark, L. R. Hodgson, J. Coombs, J. M. Mantell, R. S. Rose, J. F. Ross, C. Morris, R. L. Harniman, C. W. Wood, C. Arthur, P. Verkade and D. N. Woolfson, Small, 2021, 17, 2100472 CrossRef CAS PubMed.
- H. Tsutsumi, K. Tanaka, J. Y. Chia and H. Mihara, Pept. Sci., 2021, 113, e24214 CAS.
- C. Shao, Y. Tian, Z. Dong, J. Gao, Y. Gao, X. Jia, G. Guo, X. Wen, C. Jiang and X. Zhang, Am. J. Biomed. Sci., 2012, 4, 85–101 CrossRef CAS PubMed.
- J. Wang, C. Wang, Y. Ge, Y. Sun, D. Wang and H. Xu, Pept. Sci., 2021, 113, e24208 CAS.
- K. S. Hellmund, B. von Lospichl, C. Böttcher, K. Ludwig, U. Keiderling, L. Noirez, A. Weiß, D. J. Mikolajczak, M. Gradzielski and B. Koksch, Pept. Sci., 2021, 113, e24201 CAS.
- R. S. Giri, S. Pal, S. Roy, G. Dolai, S. R. Manne, S. Paul and B. Mandal, Pept. Sci., 2021, 113, e24176 CAS.
- H. E. Distaffen, C. W. Jones, B. L. Abraham and B. L. Nilsson, Pept. Sci., 2021, 113, e24224 CAS.
- J. N. Sloand, M. A. Miller and S. H. Medina, Pept. Sci., 2021, 113, e24184 CAS.
- R. W. Curtis and J. Chmielewski, Pept. Sci., 2021, 113, e24190 CAS.
- H. Jędrzejewska, M. Wierzbicki, P. Cmoch, K. Rissanen and A. Szumna, Angew. Chem., Int. Ed., 2014, 53, 13760–13764 CrossRef.
- M. Mutter and S. Vuilleumier, Angew. Chem., Int. Ed. Engl., 1989, 28, 535–554 CrossRef.
- J. O. Freeman, W. C. Lee, M. E. P. Murphy and J. C. Sherman, J. Am. Chem. Soc., 2009, 131, 7421–7429 CrossRef CAS PubMed.
- P. T. Corbett, J. Leclaire, L. Vial, K. R. West, J.-L. Wietor, J. K. M. Sanders and S. Otto, Chem. Rev., 2006, 106, 3652–3711 CrossRef CAS PubMed.
- M.-K. Chung, P. S. White, S. J. Lee, M. R. Gagné and M. L. Waters, J. Am. Chem. Soc., 2016, 138, 13344–13352 CrossRef CAS.
- M.-K. Chung, P. S. White, S. J. Lee, M. L. Waters and M. R. Gagné, J. Am. Chem. Soc., 2012, 134, 11415–11429 CrossRef CAS PubMed.
- M.-K. Chung, S. J. Lee, M. L. Waters and M. R. Gagné, Chem. Commun., 2016, 52, 8103–8106 RSC.
- M.-K. Chung, S. J. Lee, M. L. Waters and M. R. Gagné, J. Am. Chem. Soc., 2012, 134, 11430–11443 CrossRef CAS PubMed.
- C. G. Pappas, B. Liu, I. Marić, J. Ottelé, A. Kiani, M. L. van der Klok, P. R. Onck and S. Otto, J. Am. Chem. Soc., 2021, 143, 7388–7393 CrossRef CAS PubMed.
- C. G. Pappas, P. K. Mandal, B. Liu, B. Kauffmann, X. Miao, D. Komáromy, W. Hoffmann, C. Manz, R. Chang, K. Liu, K. Pagel, I. Huc and S. Otto, Nat. Chem., 2020, 12, 1180–1186 CrossRef CAS PubMed.
- Y. Altay, M. Tezcan and S. Otto, J. Am. Chem. Soc., 2017, 139, 13612–13615 CrossRef CAS PubMed.
- B. Liu, C. G. Pappas, E. Zangrando, N. Demitri, P. J. Chmielewski and S. Otto, J. Am. Chem. Soc., 2019, 141, 1685–1689 CrossRef CAS PubMed.
- M. Malakoutikhah, J. J.-P. Peyralans, M. Colomb-Delsuc, H. Fanlo-Virgós, M. C. A. Stuart and S. Otto, J. Am. Chem. Soc., 2013, 135, 18406–18417 CrossRef CAS PubMed.
- B. Bartolec, M. Altay and S. Otto, Chem. Commun., 2018, 54, 13096–13098 RSC.
- G. Wołczański, M. Cal, M. Waliczek, M. Lisowski and P. Stefanowicz, Chem. - Eur. J., 2018, 24, 12869–12878 CrossRef PubMed.
- L. Roy and M. A. Case, J. Am. Chem. Soc., 2010, 132, 8894–8896 CrossRef CAS PubMed.
- J. M. A. Carnall, C. A. Waudby, A. M. Belenguer, M. C. A. Stuart, J. J.-P. Peyralans and S. Otto, Science, 2010, 327, 1502–1506 CrossRef CAS PubMed.
- J. Li, J. M. A. Carnall, M. C. A. Stuart and S. Otto, Angew. Chem., Int. Ed., 2011, 50, 8384–8386 CrossRef CAS PubMed.
- L. Roy and M. A. Case, J. Phys. Chem. B, 2011, 115, 2454–2464 CrossRef CAS PubMed.
- F. Thomas, A. L. Boyle, A. J. Burton and D. N. Woolfson, J. Am. Chem. Soc., 2013, 135, 5161–5166 CrossRef CAS PubMed.
- J. F. Reuther, J. L. Dees, I. V. Kolesnichenko, E. T. Hernandez, D. V. Ukraintsev, R. Guduru, M. Whiteley and E. V. Anslyn, Nat. Chem., 2018, 10, 45–50 CrossRef CAS PubMed.
- O. D. Monera, N. E. Zhou, C. M. Kay and R. S. Hodges, J. Biol. Chem., 1993, 268, 19218–19227 CrossRef CAS.
- O. D. Monera, C. M. Kay and R. S. Hodges, Protein Sci., 1994, 3, 1984–1991 CrossRef CAS PubMed.
- H. Gradišar and R. Jerala, J. Pept. Sci., 2011, 17, 100–106 CrossRef PubMed.
- M. D. Bruch, M. M. Dhingra and L. M. Gierasch, Proteins: Struct., Funct., Bioinf., 1991, 10, 130–139 CrossRef CAS PubMed.
- M. G. Wuo, A. B. Mahon and P. S. Arora, J. Am. Chem. Soc., 2015, 137, 11618–11621 CrossRef CAS PubMed.
- M. G. Wuo, S. H. Hong, A. Singh and P. S. Arora, J. Am. Chem. Soc., 2018, 140, 16284–16290 CrossRef CAS PubMed.
- L. K. Henchey, A. L. Jochim and P. S. Arora, Curr. Opin. Chem. Biol., 2008, 12, 692–697 CrossRef CAS PubMed.
- P. E. Dawson and S. B. H. Kent, J. Am. Chem. Soc., 1993, 115, 7263–7266 CrossRef CAS.
- M. Mutter, P. Dumy, P. Garrouste, C. Lehmann, M. Mathieu, C. Peggion, S. Peluso, A. Razaname and G. Tuchscherer, Angew. Chem., Int. Ed. Engl., 1996, 35, 1482–1485 CrossRef CAS.
- B. Dang, H. Wu, V. K. Mulligan, M. Mravic, Y. Wu, T. Lemmin, A. Ford, D.-A. Silva, D. Baker and W. F. DeGrado, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 10852–10857 CrossRef CAS PubMed.
- A. J. Doerr and G. L. McLendon, Inorg. Chem., 2004, 43, 7916–7925 CrossRef CAS PubMed.
- M. L. Zastrow and V. L. Pecoraro, Coord. Chem. Rev., 2013, 257, 2565–2588 CrossRef CAS PubMed.
- J. Weng and W. Wang, Curr. Opin. Struct. Biol., 2020, 62, 9–13 CrossRef CAS PubMed.
- L. M. Stevers, P. J. de Vink, C. Ottmann, J. Huskens and L. Brunsveld, J. Am. Chem. Soc., 2018, 140, 14498–14510 CrossRef CAS PubMed.
- H. W. Ooi, J. M. M. Kocken, F. L. C. Morgan, A. Malheiro, B. Zoetebier, M. Karperien, P. A. Wieringa, P. J. Dijkstra, L. Moroni and M. B. Baker, Biomacromolecules, 2020, 21, 2208–2217 CrossRef CAS PubMed.
- H. Chao, E. Houston Michael, S. Grothe, C. M. Kay, M. O'Connor-McCourt, R. T. Irvin and R. S. Hodges, Biochemistry, 1996, 35, 12175–12185 CrossRef CAS PubMed.
- L. Vial, F. Perret and J. Leclaire, Eur. J. Org. Chem., 2022, 2022, e202101274 CAS.
- P.-T. Skowron, M. Dumartin, E. Jeamet, F. Perret, C. Gourlaouen, A. Baudouin, B. Fenet, J.-V. Naubron, F. Fotiadu, L. Vial and J. Leclaire, J. Org. Chem., 2016, 81, 654–661 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Additional experimental details, LC-MS and MALDI methods and data, SDS-PAGE procedures and data, and CD conditions and data. See DOI: https://doi.org/10.1039/d3sc00231d |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2023 |