
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTantalum ureate complexes for photocatalytic hydroaminoalkylation†
Han
Hao‡
 a,
Manfred
Manßen‡
b and
Laurel L.
Schafer
*c
a,
Manfred
Manßen‡
b and
Laurel L.
Schafer
*c
aDepartment of Chemistry, University of Toronto, Toronto, Ontario M5S 3H6, Canada
bInstitut für Anorganische Chemie, Eberhard Karls Universität Tübingen, Auf der Morgenstelle 18, 72076, Tübingen, Germany
cDepartment of Chemistry, University of British Columbia, Vancouver, British Columbia V6T 1Z4, Canada. E-mail: schafer@chem.ubc.ca
First published on 19th April 2023
Abstract
Using a tantalum ureate pre-catalyst, photocatalytic hydroaminoalkylation of unactivated alkenes with unprotected amines at room temperature is demonstrated. The combination of Ta(CH2SiMe3)3Cl2 and a ureate ligand with a saturated cyclic backbone resulted in this unique reactivity. Preliminary investigations of the reaction mechanism suggest that both the thermal and photocatalytic hydroaminoalkylation reactions begin with N–H bond activation and subsequent metallaaziridine formation. However, a select tantalum ureate complex, through ligand to metal charge transfer (LMCT), results in photocatalyzed homolytic metal–carbon bond cleavage and subsequent addition to unactivated alkene to afford the desired carbon–carbon bond formation. Origins of ligand effects on promoting homolytic metal–carbon bond cleavage are explored computationally to support enhanced ligand design efforts.
Introduction
Advances in homogenous photoredox catalysis for amine and N-heterocycle alkylation have been extensively reported in recent years.1–4 By using the energy of optimized light sources, many photoredox transformations can be achieved at ambient temperatures. This approach is ideal for reactions involving sensitive substrates as elevated temperatures are not required to overcome high-energy barriers. However, among the photocatalytic systems developed to facilitate α-alkylation of amines, most rely on classic noble metal photoredox catalysts such as [Ru(bpy)3]2+- or [Ir(ppy)3].1–3,5–11 Alternatively, there has been recent attention on the use of earth abundant metal complexes in photocatalytic C–C bond formation in arylamine–arylamine dehydrogenative coupling,12,13 but examples using alkylamine substrates are rare.14–16Early transition metal catalyzed hydroaminoalkylation is a reaction for the formation of a new Csp3–Csp3 bond by hydrofunctionalization of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C multiple bonds with a C–H bond α- to an amine.17–21 It is completely atom-economic and avoids pre-functionalized coupling partners in that simple secondary amines and alkenes can be used directly. Also, less toxic and less expensive early transition metals from groups 3, 4 and 5, including earth abundant metals like titanium can be used as catalysts.22–26 In recent years this strategy has proven to be an efficient, thermally promoted reaction. However, like many other C–H functionalization reactions, this transformation often requires very high reaction temperatures, even up to 180 °C.27–31 Even though a variety of early transition metal complexes for hydroaminoalkylation have been reported, room temperature reactivity is rare. To date there is one Ta phosphoramidate complex that can promote reactivity with styrenes and unprotected dialkyl secondary amines at ambient temperature,32 and more recently the Doye group, inspired by earlier work of Hou on scandium-catalyzed hydroaminoalkylation,33,34 reported a cationic titanium-catalyst for hydroaminoalkylation of alkenes with tertiary amines at room temperature.35
C multiple bonds with a C–H bond α- to an amine.17–21 It is completely atom-economic and avoids pre-functionalized coupling partners in that simple secondary amines and alkenes can be used directly. Also, less toxic and less expensive early transition metals from groups 3, 4 and 5, including earth abundant metals like titanium can be used as catalysts.22–26 In recent years this strategy has proven to be an efficient, thermally promoted reaction. However, like many other C–H functionalization reactions, this transformation often requires very high reaction temperatures, even up to 180 °C.27–31 Even though a variety of early transition metal complexes for hydroaminoalkylation have been reported, room temperature reactivity is rare. To date there is one Ta phosphoramidate complex that can promote reactivity with styrenes and unprotected dialkyl secondary amines at ambient temperature,32 and more recently the Doye group, inspired by earlier work of Hou on scandium-catalyzed hydroaminoalkylation,33,34 reported a cationic titanium-catalyst for hydroaminoalkylation of alkenes with tertiary amines at room temperature.35
An alternative low-temperature activation strategy takes advantage of photoredox catalysis for hydroaminoalkylation. Amines undergo single electron oxidation chemistry,36,37 to form α-aminoalkyl radical intermediates that can react further with alkenes. To date, several systems based on late transition metal (LTM) photoredox catalysts have been developed for hydroaminoalkylation. For example, in 2012, the Nishibayashi group first reported hydroaminoalkylation of electron deficient, activated alkenes with tertiary amines via α-aminoalkyl radicals formed by a classic iridium photocatalyst (Scheme 1a).38 Xu and Li demonstrated that the addition of α-aminoalkyl radicals to activated acrylate derivatives could be achieved with [Ru(bpy)3][BF4]2 as a photoredox catalyst.39 The Rovis group showed that dual metal catalysis, using an Ir photocatalyst in combination with a cobalt complex, can be used for the mild hydroaminoalkylation of dienes with tertiary amines (Scheme 1b).40 Furthermore, they demonstrated that this strategy could be extended to the direct alkylation of primary amines by “protection” of the amine with CO2 to form a carbamate functionality in situ.41 Moreover, organic photosensitizers have been reported to facilitate similar chemistry by the Nicewicz group (Scheme 1c), showing the promising potential of organo-photocatalysis.42
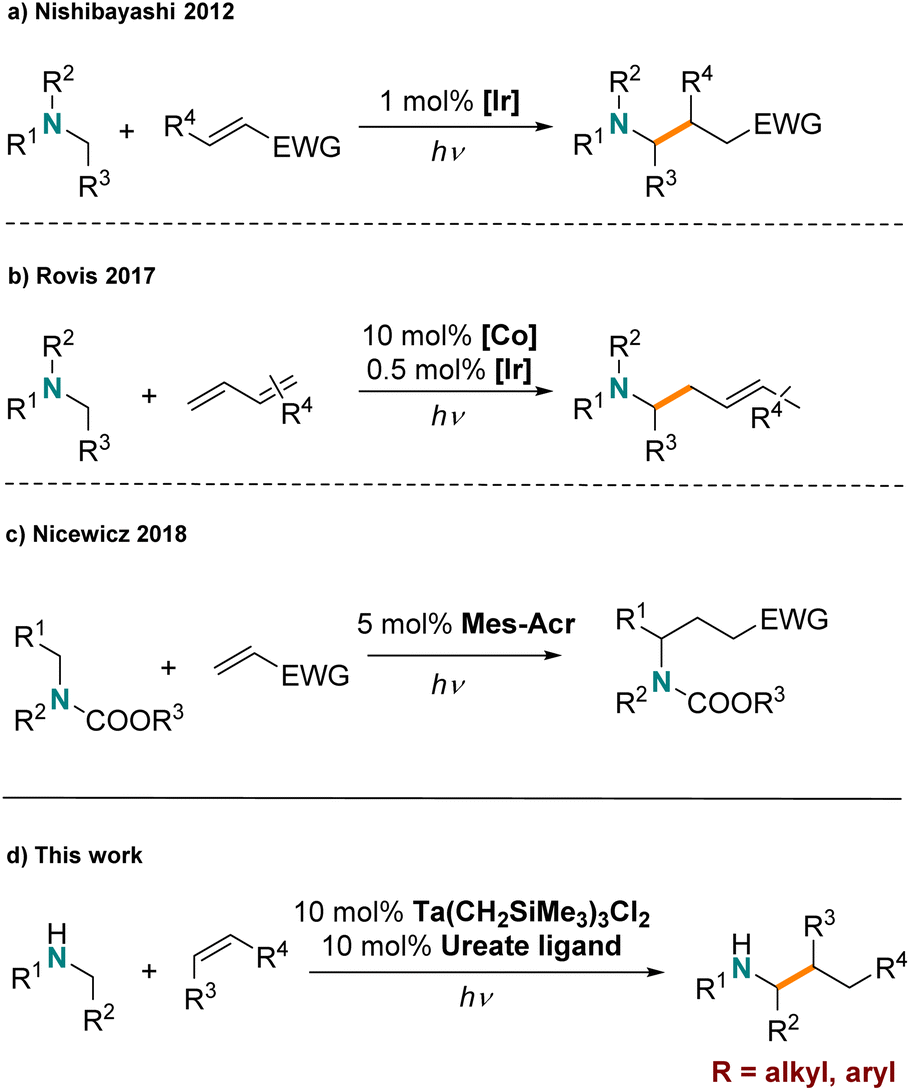 | ||
| Scheme 1 Examples for photocatalyzed hydroaminoalkylations | ||
However established LTM and organic photocatalysts are not effective for the regioselective hydroaminoalkylation of unactivated alkenes. Typically photoredox hydroaminoalkylation reactions are limited to activated alkenes and amines. For example α,β-unsaturated alkenes are used as substrate, which can efficiently harvest amine radicals generated by photoactivation.38,39,42–45 Additionally, primary and secondary amines are easily over-oxidized and are not well tolerated without protection. Recently, the Cresswell group successfully overcame this challenge by using a hydrogen-atom-transfer (HAT) reagent together with organic photocatalysts to achieve styrene hydroaminoalkylation with unprotected primary alkylamines.46
Thermally promoted early-transition-metal catalyzed hydroaminoalkylation is efficient with unactivated alkenes and unprotected amines, but high temperatures are usually required. Meanwhile, early-transition-metal complexes featuring M–Csp3 bonds are known to undergo light induced homolytic cleavage.47–50 For example, the Ta(V)–Ta(IV) single-electron redox couple is readily accessible through such light induced processes.51 We questioned if these reactivity trends could be harnessed to realize inexpensive and abundant early-transition-metal complexes for photocatalytic hydroaminoalkylation of unactivated alkenes under mild conditions. To date the handful of reported early-transition-metal photocatalysts are limited to dehalogenative reduction/homo-coupling and olefin reduction reactions;52–55 amine functionalization chemistry has not yet been disclosed. Herein, we report the first example of early-transition-metal photocatalytic hydroamino-alkylation using an organometallic tantalum ureate complex (Scheme 1d). This reaction features the use of unprotected amines and affords an alternative mechanism for achieving photocatalytic hydroaminoalkylation to include unactivated alkenes.
Result and discussion
Early transition metal complex reactivity screening
Simple homoleptic Ta(NMe2)5 was among the very first reported early-transition-metal complexes as hydroaminoalkylation pre-catalyst under thermal conditions,56 and has been used as Ta source in catalytic hydroaminoalkylation.57–62 Our initial investigation was conducted using 10 mol% Ta(NMe2)5 loading and N-methylaniline/1-octene as the model substrate combination. Notably, there have been no previous reports for the photocatalytic hydroaminoalkylation of simple unactivated alkenes. Here, the reaction mixture was irradiated by a domestic G4 light bulb with UV filter at room temperature for 20 h (Fig. S1†). The reaction progress was monitored by 1H-NMR spectroscopy and the NMR yield of hydroaminoalkylation product formation was determined by comparing the ortho-H integration of the product vs. the 1,3,5-trimethoxybenzene internal standard. Unfortunately, the use of known thermally catalytically active Ta(NMe2)5 offered no observable reactivity after irradiating for 20 h (Table 1, entry 1). Interestingly, by switching to Ta(CH2SiMe3)3Cl2 (Ta1) trace (around 4%) hydroaminoalkylation product could be observed (entry 2). Consequently, we conducted further ligand screening using Ta1 as the Ta source.|
|
||||
|---|---|---|---|---|
| Entry | Precatalyst | Ligand | Loading (mol%) | Yielda (%) |
| a Determined by 1H NMR. b Yield after 72 h. c No light. d Dipp – 2,6-diisopropylphenyl. | ||||
| 1 | Ta(NMe2)5 | — | 10 | 0 |
| 2 | Ta(CH2SiMe3)3Cl2 | — | 10 | 4 |
| 3 | Ta(CH2SiMe3)3Cl2 | L1 | 10 | 30 (91)b |
| 4 | Ta(CH2SiMe3)3Cl2 | L1 | 10 | 0c |
| 5 | Ta(CH2SiMe3)3Cl2 | L1 | 0 | 0 |
| 6 | — | L1H | 10 | 0 |
| 7 | Ta(CH2SiMe3)3Cl2 | L2 | 10 | 10 |
| 8 | Ta(CH2SiMe3)3Cl2 | L3 | 10 | 0 |
| 9 | Ta(CH2SiMe3)3Cl2 | L4 | 10 | 0 |
| 10 | Ta(CH2SiMe3)3Cl2 | L5 | 10 | 0 |
| 11 | Ti(Bn)4 | L1 | 10 | 0 |
| 12 | Ti(CH2SiMe3)4 | L1 | 10 | 0 |
| 13 | Zr(Bn)4 | L1 | 10 | 0 |
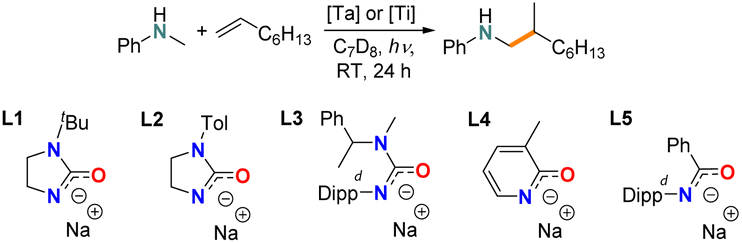
We then tested a N,O-chelated cyclic ureate ligand L1 on Ta1, which offers turnover frequencies of up to 60 h−1 at 110 °C for thermally promoted hydroaminoalkylation.28 A yield of 30% was achieved after 24 h irradiation, and finally reached almost quantitative yield (91%) after 3 days (entry 3). Importantly, the branched product was formed exclusively, which is aligned with thermally promoted early-transition-metal-catalyzed-hydroaminoalkylation.18 In a control reaction, the experiment was conducted under complete exclusion of light by covering the sample with aluminum foil (Entry 4). Here, no conversion was observed, confirming this is a photocatalyzed process. Furthermore, by only using the substrates without the catalyst system Ta1/L1 (Entry 5) or only using the proteo ligand (Entry 6) no conversion was observed, confirming that Ta is crucial to this photocatalytic transformation. With identification of the crucial role of Ta, the 1,3-N,O-chelating ligand L1 and –CH2SiMe3 auxiliary ligands, we then tested several other 1,3-N,O-chelating ligands (L2–L5, entries 7–10) that are known to promote high catalytic activity in tantalum-catalyzed hydroaminoalkylation under thermal conditions.27,28,63,64 Here, the acyclic ureate L3, pyridonate L4 and amidate L5 all showed no activity at all while the cyclic ureate L2, which changes the tBu substituent on the non-ligating nitrogen of L1 to 4-tolyl, shows inferior reactivity towards L1 (10%).
To explain the difference in photocatalytic reactivities of Ta1/L1–Ta1/L5, which are all known good hydroaminoalkylation catalyst under thermal conditions, we computationally explored the differences in photo-excited states of these compounds. To interpret the influence of ligand over the photocatalytic reactivity of Ta compounds, we chose TaL1 and TaL4 as examples for computational analysis.
Based on TD-DFT calculations on Ta1/L1, the tris-alkyl-mono-chloro-tantalum ureate pre-catalyst has no absorption in the visible region, which matches the experimentally measured UV-Vis absorption (Fig. S6†). However, a small absorption band at around 483 nm was identified with a tantalaaziridine complex bearing L1 ligand (Fig. S7.2†). This computational observation is close to the experimentally observed wavelength of maximum reactivity (440 nm, see ESI† for determination of wavelength dependency of reactivity). We then further computationally compared the excitation process of tantalaaziridine complexes bearing L1 and L4.
In the case of Ta/L1, which has a ureate ligand on Ta, the excitation process is a ligand to metal charge transfer (LMCT) process (Fig. 1, top),49,50 which has been demonstrated as a unique feature for early transition metal based photocatalysis.65 With the charge transfer from the aziridine ligand to the tantalum metal center, the Ta–C bond is weakened as indicated by an increase in bond length (2.134 vs. 2.236 Å). However in the case of Ta/L4 which has a pyridonate ligand, the excitation is not only LMCT but also has ligand to ligand charge transfer features from amido to pyridonate ligand (Fig. 1, bottom). The resultant elongation of the Ta–C bond length is also not as significant (2.148 vs. 2.219 Å). Furthermore, the singly occupied natural orbitals of Ta/L4 delocalize across the amido and pyridonate ligands, while they are localized on the tantalum aziridine moiety on Ta/L1 (Fig. S4 and S5†). This suggests that the conjugated pyridonate ligand energetically stabilizes the charge transfer species, disfavoring Ta–C bond cleavage. Therefore the excited state has less LMCT character, meaning lower population of the partially antibonding Ta–C molecular orbital.
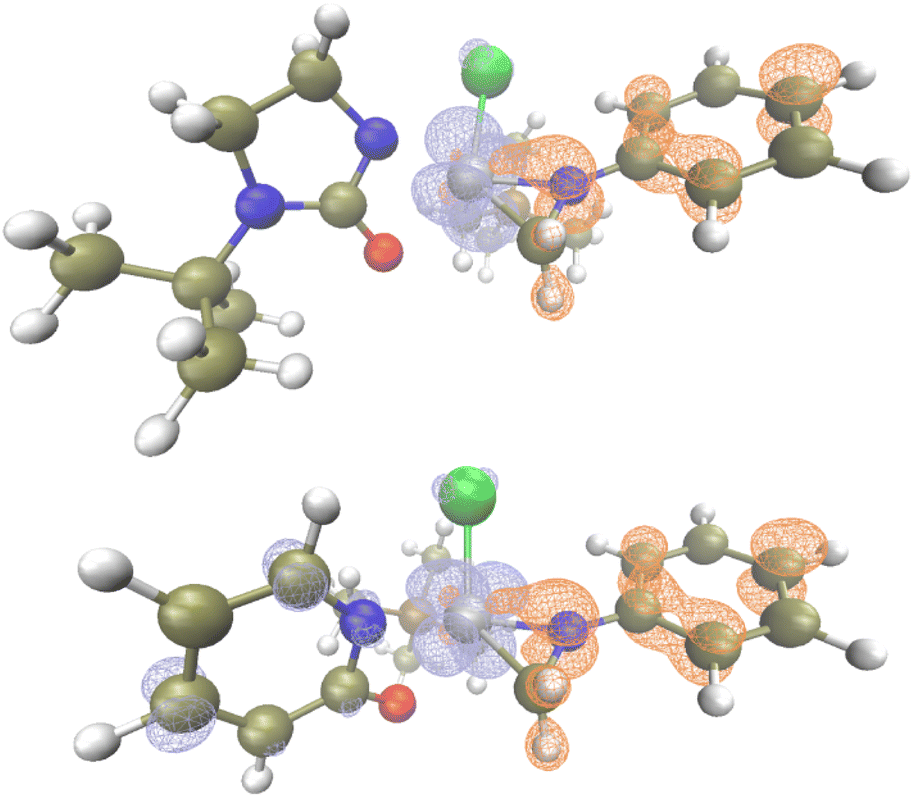 | ||
| Fig. 1 Excitation analysis of TaL1 (top) and TaL2 in toluene, and electrons (blue) and holes (orange) after excitation (opt: M06L-D3/Def2SVP, TD: M06L-D3/Def2TZVPP, isovalue = 0.05). | ||
Finally, we tested several other homoleptic early-transition-metal-alkyl complexes that are known hydroaminoalkylation catalysts, such as Ti(CH2SiMe3)4 and ZrBn4,22,27,66 With L1 ligand, none of them gave observable photoreactivity (entries 11–13), suggesting the unique reactivity of the Ta complex under photonic excitation.
Substrate screening
After confirming the room temperature photocatalytic reactivity of Ta1/L1 system for the hydroaminoalkylation of N-methylaniline and 1-octene, next a comparison to the thermally promoted reaction (>110 °C) was completed with various substrates. Specifically, we are interested in establishing the scope and functional group tolerance of this system, to determine the similarities and differences in substrate scope between the photocatalytic and thermally promoted reactions.For the identification of the substrate scope for photocatalytic hydroaminoalkylation using Ta1/L1 similar NMR-screening techniques were applied (Scheme 2). To maximize yields, the reaction time was increased to 72 h.
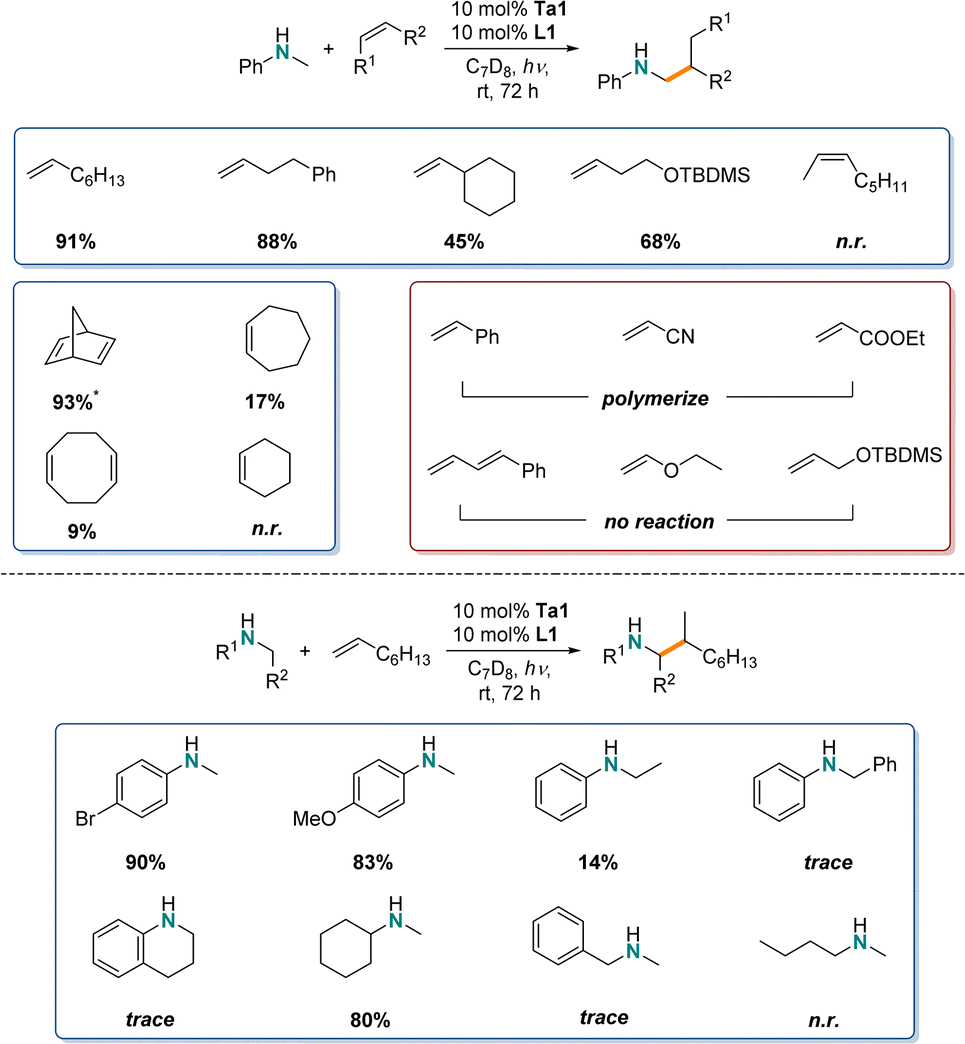 | ||
| Scheme 2 Substrate scope of Ta1 and L1 for photocatalytic hydroaminoalkylation of various amines and alkenes. Yields were determined by integration in 1H-NMR vs. internal standard (*: mixture of both mono- and bis-alkylation, consumption of amine substrate vs. internal standard is reported), trace means detectable by 1H-NMR but below quantification criteria (2%), n.r. means no detected product by both 1H-NMR and GC-MS. | ||
Similar to the reaction of 1-octene, other unactivated terminal alkenes like 4-phenyl-1-butene gave a high yield (88%). By increasing the steric bulk next to the alkene functionality, while maintaining an alkyl substituted alkene e.g. vinylcyclohexane, the reactivity drops significantly (45%). However, with addition of just one methylene unit in the carbon chain, even larger substituents like -OTBDMS are tolerated very well (68%).
Furthermore, we tested internal alkenes, which are generally known to be very challenging substrates in early transition metal catalyzed-hydroaminoalkylation.18,19 Here, acyclic internal 2-octene showed no reactivity, while strained cyclic alkenes like norbornadiene showed near quantitative substrate consumption (93%) (producing mixtures of both mono- and bis-alkylated products as well as ring-opening polymerized product). Less strained cyclic alkene like cycloheptene (17%), 1,5-cyclooctadiene (9%) and cyclohexene (n.r.) showed poor reactivity, as has been observed under thermal conditions.
These first photocatalytic results show reduced catalytic activity in comparison to the thermal variant of tantalum catalyzed hydroaminoalkylation as longer reaction times are required to reach comparable yields,28 but they do show that unactivated alkenes can be used in photocatalyzed hydroaminoalkylation. In our case, unlike LTM photocatalysts which favour Michael acceptors as activated alkene substrates, the Ta1/L1 system did not catalyze hydroaminoalkylation of styrene or α,β-unsaturated alkenes like acrylonitrile or ethyl acrylate but instead caused polymerization, as observed by the formation of either brittle solids inside the NMR tube or broad signals in the 1H-NMR spectra. Additionally, conjugated dienes, vinyl ethers or allyl ethers were unreactive under standard conditions. This photocatalytic hydroaminoalkylation (α-C–H alkylation of amines) using a Ta based photosensitizer offers complementary substrate scope to the work using established LTM based photoredox catalysts.
For the amine scope, both electron withdrawing (–Br, 90%) and electron donating (-OMe, 83%) para-substituents for N-methylanilines showed high reactivity. Here, especially the halide substituents are of interest, because such aryl halides can then be used in further cross-coupling reactions with late-transition metal catalysts.67,68 Varying the N-alkyl substituent led to decreased reactivity: N-ethyl- (14%), N-benzylaniline (trace) and tetrahydroquinoline (trace). Interestingly, by switching to dialkylated amines, the yields for N-methylcyclohexylamine are similar to those of the N-methylaniline derivates (80%). However, sterically less demanding substituents like N-methylbenzylamine (trace) and N-methyl-n-butylamine (n.r.) led to a drastic decrease in reactivity. This suggests that secondary amines with bulky groups are preferred for this photocatalytic reaction. Tertiary amines were also unreactive in these conditions (vide infra).
Finally, we were interested in the intramolecular hydroaminoalkylation of aminoalkenes leading to ring-closure. While there are examples of group 4 metal catalysts for this transformation,62,66,69,70 to date, tantalum has not been reported for this reaction. Here, we show (Scheme 3) that intramolecular hydroaminoalkylation of aminoalkene furnishes the cyclopentylamine derivative using Ta1/L1.
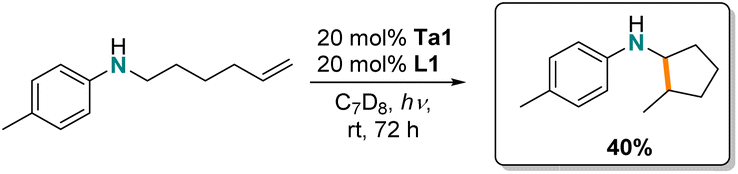 | ||
| Scheme 3 Photocatalytic intramolecular hydroamino-alkylation of aminoalkenes. | ||
Mechanistic probes
α-Aminoalkyl radicals are involved in LTM catalyzed photoredox hydroaminoalkylation,2 and tertiary amines, such as N,N-dimethylaniline, are preferred substrates due the increased stability of tertiary amine radical.71 In contrast to that, early-transition-metal-catalyzed thermal hydroaminoalkylation requires the use of secondary or primary amines as substrates to access the requisite metallaaziridine reactive intermediates.17,18,72,73For our photocatalytic process at room temperature, N,N-dimethylaniline gave no conversion, suggesting that tantalum amido intermediates are involved in the mechanistic cycle (Scheme 4a). To test the involvement of radicals, the irradiation of N-(cyclopropylmethyl)toluidine with 20 mol% tantalum ureate complex for 20 h, in the absence of alkene substrate showed alkene signals forming in the 1H NMR spectrum, consistent with ring-opening of the cyclopropyl unit (Scheme 4b). However, a similar experiment with only the alkene substrate 2-cyclopropylpropene (Scheme 4c) resulted in no observable ring-opening. While the addition of the same substrate to N-methylaniline under photocatalytic conditions resulted in one single product (identified by GC-MS, ESI 8.2†). NMR spectroscopy showed that the cyclopropyl group was untouched. Based on these mechanism probes, formation of metal-amido based radicals as a key intermediate prior to the C–C bond forming step is proposed to be involved.
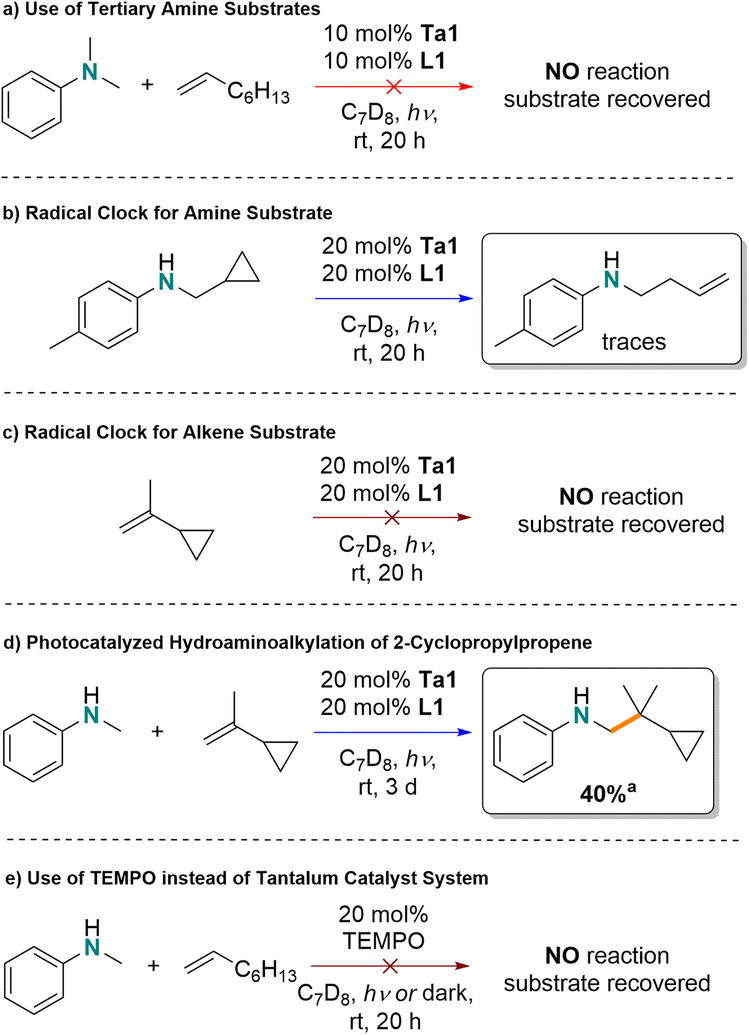 | ||
| Scheme 4 Mechanism probes evaluating the photocatalyzed hydroaminoalkylation by Ta1/L1; aNMR yield. | ||
Finally, in a control reaction, TEMPO was explored as an alternative radical source to promote the reaction of N-methylaniline and 1-octene (Scheme 4e). Here, no conversion was observed either in the dark or under light irradiation, suggesting that the reaction is not just initiated by radicals. Additionally, irradiating a reaction containing both Ta1/L1 and TEMPO led to no product formation, which may be due to the formation of an inactive Ta–TEMPO complex.
Based on these control experiments and our computational observations, we postulate the formation of a metallaaziridine species that undergoes a metal–carbon homolytic cleavage (see Scheme 6, B) as the key species that adds to coordinated alkene for photocatalytic hydroaminoalkylation. We demonstrated in the ligand screening (Table 1) that some of the N,O-chelating ligands with conjugated moieties such as pyridonate L4 or the phenyl substituted ureate L2 exhibit no or inferior reactivity compared to the alkyl substituted cyclic ureate L1. Consequently, we hypothesized that ligand aromatic moieties may stabilize such photogenerated radicals and inhibit radical addition to the alkene substrate.
After these initial experiments testing our mechanistic hypothesis, we performed isotope labeling experiments for a direct comparison to the thermal hydroaminoalkylation process. For this, we used again the model reaction of 1-octene with partially or fully deuterated versions of N-methylaniline in crucial N–H and/or –CH3 positions using our photocatalytic conditions. Here, for PhNDCH3 and PhNHCD3 proton/deuteron scrambling in the substrates as well as in the products was observed (Scheme 5a and b). This indicates reversible formation of a tantalaaziridine key intermediate, which is the same catalytically active species for thermally promoted group 5 metal catalyzed hydroaminoalkylations.18,59,72–74 Finally, no ortho-deuteration on the phenyl ring of the hydroaminoalkylation product was found. This behavior is different from the thermal version of hydroaminoalkylation catalyzed by cyclic ureate complexes of tantalum and other group 5 complexes,28 and consequently indicates that a side reaction of reversible aromatic C–H bond activation does not occur here.
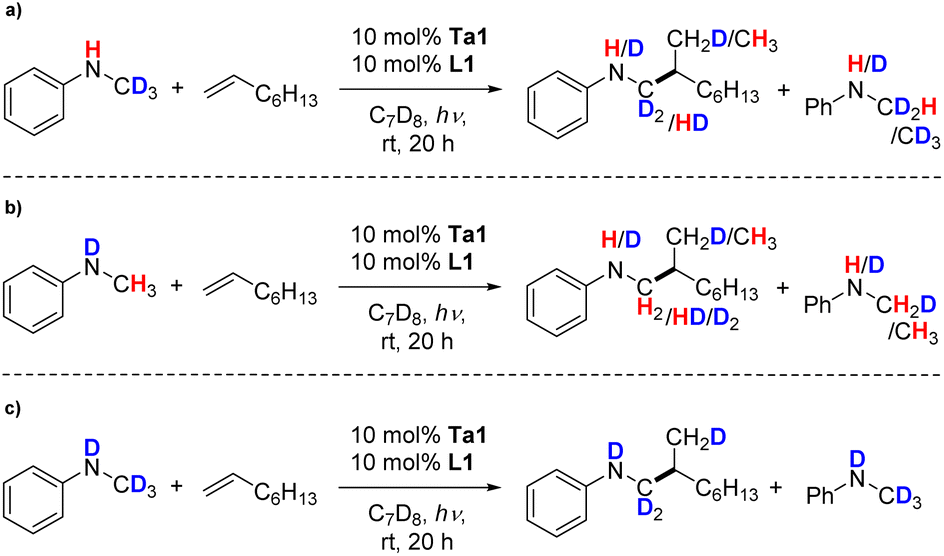 | ||
| Scheme 5 Kinetic isotope labelling experiments of the hydroaminoalkylation of (a) C-, (b) N- and (c) C,N-deuterated N-methylaniline with 1-octene using Ta1/L1. | ||
An evaluation of kinetic isotope effects (KIE) (Fig. S2†) found that small KIEs were observed for both kNH/kND and kCH3/kCD3 (values were found to be 1.5 and 1.9 respectively), which is different from the thermally promoted reaction, where only the N–H bond displayed a KIE > 1.28 Additionally, long induction periods were observed. Therefore, neither C–H nor N–H activation is invoked as being involved in the turnover limiting step.
Based on these investigations, we propose the following reaction mechanism (Scheme 6) that is similar to the mechanism of the thermal hydroaminoalkylation reaction by early transition metals. First, the organometallic tantalum pre-catalyst undergoes protonolysis with a secondary amine to generate the catalytically active tantalum(V) aziridine (A) via N–H/C–H activation and elimination of tetramethylsilane, as observed by 1H NMR spectroscopy. Due to the fact that alkene based radicals are ruled out (see Scheme 4c and d) but tantalum amido radicals are likely to be involved we propose a tantalum(IV) amido diradical (B) is formed by photolytically induced homolytic cleavage of the Ta–C bond of A. This can undergo addition to coordinated alkene (C) to give the tantalum(V) azacyclopentane (D). D is a common intermediate to the thermally promoted hydroaminoalkylation mechanism, and rationalizes the same branched regioselectivity of thermally promoted Ta-catalyzed hydroaminoalkylation. Finally, and in accordance to the labeling experiments, D reacts via protonolysis of the Ta–C bond of the metallacycle, resulting in the formation of a new bisamido intermediate (E). After another C–H activation step, the product is released from the metal center and the starting catalytically active tantalaaziridine (A) is formed again.
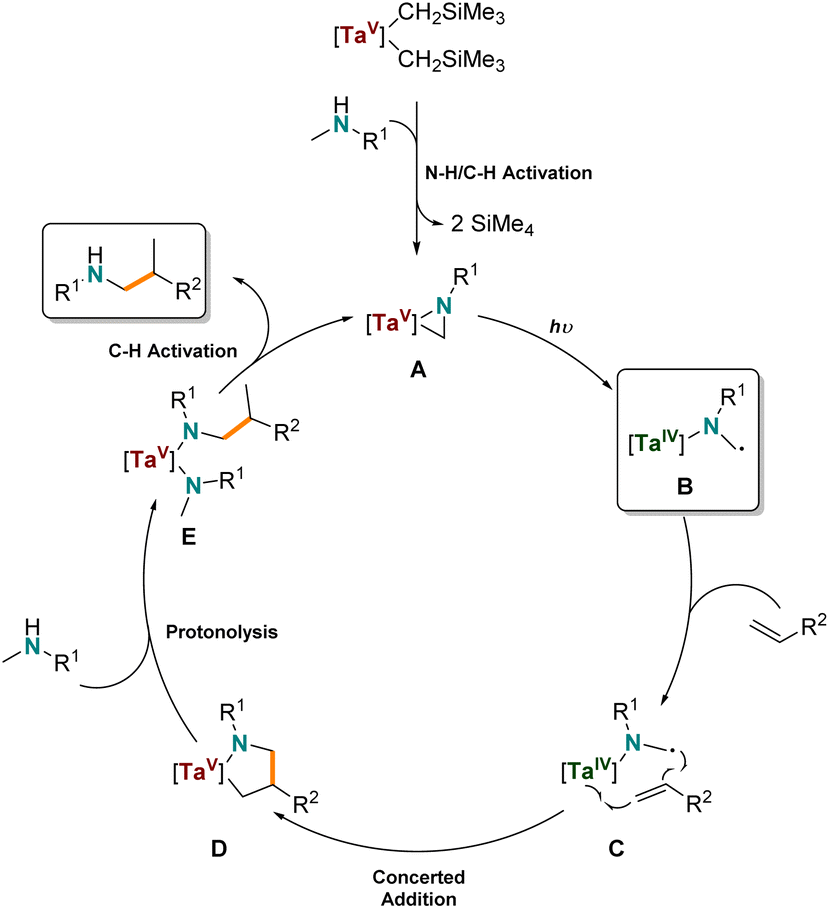 | ||
| Scheme 6 Proposed mechanism for the photocatalytic tantalum-catalyzed intermolecular hydroaminoalkylation of alkenes. | ||
Conclusions
In summary, here we report the first example of photocatalyzed hydroaminoalkylation using an early-transition-metal catalyst. The system of Ta1 and L1 undergoes simple hydroaminoalkylation reactions using a G4 tungsten halogen light bulb at room temperature instead of temperatures at 110 °C or higher, which are commonly used for early-transition-metal-catalyzed hydroaminoalkylation. We demonstrated the reactivity of this catalyst with a series of unprotected amines and unactivated alkene substrates, showing that the reaction can proceed with even the most challenging unactivated substrates that are unknown for established LTM photoredox catalyzed hydroaminoalkylation. Furthermore, we showed the first example of cyclization of an aminoalkene using a tantalum-based catalyst. Using reaction kinetics, and mechanistic probe experiments we propose a mechanism for tantalum catalyzed radical reactions for hydroaminoalkylation and using computational approaches we rationalize the observed ligand dependence of this reactivity. We propose that LMCT resulting in homolytic Ta–C bond cleavage is key to this light-promoted variant, while the other steps are comparable to the known mechanistic cycle for early transition metal catalyzed hydroaminoalkylation. We propose that these insights can be leveraged to realize improved catalyst development featuring inexpensive and low-toxicity early-transition-metal catalysts for increased photocatalytic reactivity in hydroaminoalkylation and other challenging C–E bond forming reactions at low temperatures.Data availability
ESI† is available; including experimental details, computational, photophysical and spectroscopic characterization data.Author contributions
H. H. conceived the project; H. H. and M. M. conducted experiments in consultation with L. L. S.; H. H. conducted computations; H. H. and M. M. prepared the draft of the manuscript; L. L. S. contributed with an advisory role in the project and manuscript.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
Funding from NSERC and the Feodor Lynen Postdoctoral Fellowship of the Humboldt-Foundation: Germany, Funder ID: 100005156 to M. M. is gratefully acknowledged. This work has been supported in part by the Canada Research Chairs program.Notes and references
- R. C. DiPucchio, K. E. Lenzen, P. Daneshmand, M. B. Ezhova and L. L. Schafer, J. Am. Chem. Soc., 2021, 143, 11243–11250 CrossRef CAS PubMed.
- A. Trowbridge, S. M. Walton and M. J. Gaunt, Chem. Rev., 2020, 120, 2613–2692 CrossRef CAS PubMed.
- Z. Dong, Z. Ren, S. J. Thompson, Y. Xu and G. Dong, Chem. Rev., 2017, 117, 9333–9403 CrossRef CAS PubMed.
- S. Zhu and D. Wang, Adv. Energy Mater., 2017, 7, 1700841 CrossRef.
- J. J. Murphy, D. Bastida, S. Paria, M. Fagnoni and P. Melchiorre, Nature, 2016, 532, 218–222 CrossRef CAS PubMed.
- R. A. Aycock, C. J. Pratt and N. T. Jui, ACS Catal., 2018, 8, 9115–9119 CrossRef CAS.
- Y. Miyake, Y. Ashida, K. Nakajima and Y. Nishibayashi, Chem. Commun., 2012, 48, 6966–6968 RSC.
- L. Ruiz Espelt, I. S. McPherson, E. M. Wiensch and T. P. Yoon, J. Am. Chem. Soc., 2015, 137, 2452–2455 CrossRef CAS PubMed.
- S.-X. Lin, G.-J. Sun and Q. Kang, Chem. Commun., 2017, 53, 7665–7668 RSC.
- X. Shen, Y. Li, Z. Wen, S. Cao, X. Hou and L. Gong, Chem. Sci., 2018, 9, 4562–4568 RSC.
- K. Miyazawa, T. Koike and M. Akita, Adv. Synth. Catal., 2014, 356, 2749–2755 CrossRef CAS.
- B. M. Hockin, C. Li, N. Robertson and E. Zysman-Colman, Catal. Sci. Technol., 2019, 9, 889–915 RSC.
- C. B. Larsen and O. S. Wenger, Chem.–Eur. J., 2018, 24, 2039–2058 CrossRef CAS PubMed.
- S. Parisien-Collette, A. C. Hernandez-Perez and S. K. Collins, Org. Lett., 2016, 18, 4994–4997 CrossRef CAS PubMed.
- J.-R. Jiménez, B. Doistau, C. Besnard and C. Piguet, Chem. Commun., 2018, 54, 13228–13231 RSC.
- T. P. Nicholls, G. E. Constable, J. C. Robertson, M. G. Gardiner and A. C. Bissember, ACS Catal., 2016, 6, 451–457 CrossRef CAS.
- M. Manßen and L. L. Schafer, Trends Chem., 2021, 3, 428–429 CrossRef.
- M. Manßen and L. L. Schafer, Chem. Soc. Rev., 2020, 49, 6947–6994 RSC.
- P. M. Edwards and L. L. Schafer, Chem. Commun., 2018, 54, 12543–12560 RSC.
- L. L. Schafer, M. Manßen, P. M. Edwards, E. K. J. Lui, S. E. Griffin and C. R. Dunbar, in Advances in Organometallic Chemistry, Elsevier, 2020, vol. 74, pp. 405–468 Search PubMed.
- M. G. Clerici and F. Maspero, Synthesis, 2002, 1980, 305–306 CrossRef.
- T. Kaper, D. Geik, F. Fornfeist, M. Schmidtmann and S. Doye, Chem.–Eur. J., 2022, 28, e202103931 CrossRef CAS PubMed.
- M. Rosien, I. Töben, M. Schmidtmann, R. Beckhaus and S. Doye, Chem.–Eur. J., 2020, 26, 2138–2142 CrossRef CAS PubMed.
- J. Dörfler, T. Preuß, A. Schischko, M. Schmidtmann and S. Doye, Angew. Chem., Int. Ed., 2014, 53, 7918–7922 CrossRef PubMed.
- J. Bielefeld and S. Doye, Angew. Chem., Int. Ed., 2020, 59, 6138–6143 CrossRef CAS PubMed.
- M. Manßen, D. Deng, C. H. M. Zheng, R. C. DiPucchio, D. Chen and L. L. Schafer, ACS Catal., 2021, 11, 4550–4560 CrossRef.
- R. C. DiPucchio, S.-C. Roşca and L. L. Schafer, Angew. Chem., Int. Ed., 2018, 57, 3469–3472 CrossRef CAS PubMed.
- P. Daneshmand, S.-C. Roşca, R. Dalhoff, K. Yin, R. C. DiPucchio, R. A. Ivanovich, D. E. Polat, A. M. Beauchemin and L. L. Schafer, J. Am. Chem. Soc., 2020, 142, 15740–15750 CrossRef CAS PubMed.
- J. Bielefeld and S. Doye, Angew. Chem., Int. Ed., 2017, 56, 15155–15158 CrossRef CAS PubMed.
- E. N. Bahena, S. E. Griffin and L. L. Schafer, J. Am. Chem. Soc., 2020, 142, 20566–20571 CrossRef CAS PubMed.
- M. Manßen, N. Lauterbach, J. Dörfler, M. Schmidtmann, W. Saak, S. Doye and R. Beckhaus, Angew. Chem., Int. Ed., 2015, 54, 4383–4387 CrossRef PubMed.
- P. Garcia, Y. Y. Lau, M. R. Perry and L. L. Schafer, Angew. Chem., Int. Ed., 2013, 52, 9144–9148 CrossRef CAS PubMed.
- A. E. Nako, J. Oyamada, M. Nishiura and Z. Hou, Chem. Sci., 2016, 7, 6429–6434 RSC.
- F. Liu, G. Luo, Z. Hou and Y. Luo, Organometallics, 2017, 36, 1557–1565 CrossRef CAS.
- D. Geik, M. Rosien, J. Bielefeld, M. Schmidtmann and S. Doye, Angew. Chem., Int. Ed., 2021, 60, 9936–9940 CrossRef CAS PubMed.
- G. J. Choi, Q. Zhu, D. C. Miller, C. J. Gu and R. R. Knowles, Nature, 2016, 539, 268–271 CrossRef CAS PubMed.
- A. J. Musacchio, B. C. Lainhart, X. Zhang, S. G. Naguib, T. C. Sherwood and R. R. Knowles, Science, 2017, 355, 727–730 CrossRef CAS PubMed.
- Y. Miyake, K. Nakajima and Y. Nishibayashi, J. Am. Chem. Soc., 2012, 134, 3338–3341 CrossRef CAS PubMed.
- X. Dai, D. Cheng, B. Guan, W. Mao, X. Xu and X. Li, J. Org. Chem., 2014, 79, 7212–7219 CrossRef CAS PubMed.
- S. M. Thullen and T. Rovis, J. Am. Chem. Soc., 2017, 139, 15504–15508 CrossRef CAS PubMed.
- J. Ye, I. Kalvet, F. Schoenebeck and T. Rovis, Nat. Chem., 2018, 10, 1037–1041 CrossRef CAS PubMed.
- J. B. McManus, N. P. R. Onuska and D. A. Nicewicz, J. Am. Chem. Soc., 2018, 140, 9056–9060 CrossRef CAS PubMed.
- X. Ju, D. Li, W. Li, W. Yu and F. Bian, Adv. Synth. Catal., 2012, 354, 3561–3567 CrossRef CAS.
- S. Zhu, A. Das, L. Bui, H. Zhou, D. P. Curran and M. Rueping, J. Am. Chem. Soc., 2013, 135, 1823–1829 CrossRef CAS PubMed.
- X. Dai, R. Mao, B. Guan, X. Xu and X. Li, RSC Adv., 2015, 5, 55290–55294 RSC.
- H. E. Askey, J. D. Grayson, J. D. Tibbetts, J. C. Turner-Dore, J. M. Holmes, G. Kociok-Kohn, G. L. Wrigley and A. J. Cresswell, J. Am. Chem. Soc., 2021, 143, 15936–15945 CrossRef CAS PubMed.
- R. Gazzi, F. Perazzolo, S. Sostero, A. Ferrari and O. Traverso, J. Organomet. Chem., 2005, 690, 2071–2077 CrossRef CAS.
- E. Samuel, P. Maillard and C. Giannotti, J. Organomet. Chem., 1977, 142, 289–298 CrossRef CAS.
- H. B. Abrahamson, K. L. Brandenburg, B. Lucero, M. E. Martin and E. Dennis, Organometallics, 1984, 3, 1379–1386 CrossRef CAS.
- H. B. Abrahamson and M. E. Martin, J. Organomet. Chem., 1982, 238, C58–C62 CrossRef CAS.
- L. R. Chamberlain and I. P. Rothwell, J. Chem. Soc., Dalton Trans., 1987, 163–167 RSC.
- Y. Zhang, J. L. Petersen and C. Milsmann, J. Am. Chem. Soc., 2016, 138, 13115–13118 CrossRef CAS PubMed.
- Y. Zhang, D. C. Leary, A. M. Belldina, J. L. Petersen and C. Milsmann, Inorg. Chem., 2020, 59, 14716–14730 CrossRef CAS PubMed.
- Y. Zhang, J. L. Petersen and C. Milsmann, Organometallics, 2018, 37, 4488–4499 CrossRef CAS.
- Y. Zhang, T. S. Lee, J. L. Petersen and C. Milsmann, J. Am. Chem. Soc., 2018, 140, 5934–5947 CrossRef CAS PubMed.
- S. B. Herzon and J. F. Hartwig, J. Am. Chem. Soc., 2007, 129, 6690–6691 CrossRef CAS PubMed.
- P. Eisenberger, R. O. Ayinla, J. M. P. Lauzon and L. L. Schafer, Angew. Chem., Int. Ed., 2009, 48, 8361–8365 CrossRef CAS PubMed.
- A. L. Reznichenko, T. J. Emge, S. Audörsch, E. G. Klauber, K. C. Hultzsch and B. Schmidt, Organometallics, 2011, 30, 921–924 CrossRef CAS.
- J. M. Lauzon, P. Eisenberger, S.-C. Roşca and L. L. Schafer, ACS Catal., 2017, 7, 5921–5931 CrossRef CAS.
- J. Dörfler and S. Doye, Eur. J. Org. Chem., 2014, 2014, 2790–2797 CrossRef.
- G. Zi, F. Zhang and H. Song, Chem. Commun., 2010, 46, 6296–6298 RSC.
- P. Garcia, P. R. Payne, E. Chong, R. L. Webster, B. J. Barron, A. C. Behrle, J. A. R. Schmidt and L. L. Schafer, Tetrahedron, 2013, 69, 5737–5743 CrossRef CAS.
- R. C. DiPucchio, S.-C. Rosca, G. Athavan and L. L. Schafer, ChemCatChem, 2019, 11, 3871–3876 CrossRef CAS.
- E. Chong, J. W. Brandt and L. L. Schafer, J. Am. Chem. Soc., 2014, 136, 10898–10901 CrossRef CAS PubMed.
- Y. Zhang, T. S. Lee, J. M. Favale, D. C. Leary, J. L. Petersen, G. D. Scholes, F. N. Castellano and C. Milsmann, Nat. Chem., 2020, 12, 345–352 CrossRef CAS PubMed.
- I. Prochnow, R. Kubiak, O. N. Frey, R. Beckhaus and S. Doye, ChemCatChem, 2009, 1, 162–172 CrossRef CAS.
- Z. Zhang, J.-D. Hamel and L. L. Schafer, Chem.–Eur. J., 2013, 19, 8751–8754 CrossRef CAS PubMed.
- M. Weers, L. H. Lühning, V. Lührs, C. Brahms and S. Doye, Chem.–Eur. J., 2017, 23, 1237–1240 CrossRef CAS PubMed.
- J. A. Bexrud, P. Eisenberger, D. C. Leitch, P. R. Payne and L. L. Schafer, J. Am. Chem. Soc., 2009, 131, 2116–2118 CrossRef CAS PubMed.
- J. Dörfler, B. Bytyqi, S. Hüller, N. M. Mann, C. Brahms, M. Schmidtmann and S. Doye, Adv. Synth. Catal., 2015, 357, 2265–2276 CrossRef.
- J. Hioe, D. Šakić, V. Vrček and H. Zipse, Org. Biomol. Chem., 2015, 13, 157–169 RSC.
- I. Prochnow, P. Zark, T. Müller and S. Doye, Angew. Chem., Int. Ed., 2011, 50, 6401–6405 CrossRef CAS PubMed.
- D. J. Gilmour, J. M. P. Lauzon, E. Clot and L. L. Schafer, Organometallics, 2018, 37, 4387–4394 CrossRef CAS.
- A. L. Reznichenko and K. C. Hultzsch, J. Am. Chem. Soc., 2012, 134, 3300–3311 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sc00042g |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2023 |