
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA copper-seamed coordination nanocapsule as a semiconductor photocatalyst for molecular oxygen activation†
Xiangquan
Hu
 a,
Meirong
Han
b,
Leicheng
Wang
c,
Li
Shao
a,
Yadav
Peeyush
a,
Jialei
Du
*c,
Steven P.
Kelley
a,
Scott J.
Dalgarno
d,
David A.
Atwood
e,
Sisi
Feng
*b and
Jerry L.
Atwood
*a
a,
Meirong
Han
b,
Leicheng
Wang
c,
Li
Shao
a,
Yadav
Peeyush
a,
Jialei
Du
*c,
Steven P.
Kelley
a,
Scott J.
Dalgarno
d,
David A.
Atwood
e,
Sisi
Feng
*b and
Jerry L.
Atwood
*a
aDepartment of Chemistry, University of Missouri-Columbia, 601 S College Ave, Columbia, MO 65211, USA. E-mail: atwood@missouri.edu
bKey Laboratory of Chemical Biology and Molecular Engineering of Ministry of Education, Institute of Molecular Science, Shanxi University, Taiyuan 030006, P. R. China. E-mail: ssfeng@sxu.edu.cn
cInstitute for Advanced Interdisciplinary Research, University of Jinan, Jinan 250022, P. R. China. E-mail: ifc_dujl@ujn.edu.cn
dInstitute of Chemical Sciences, Heriot-Watt University, Riccarton, Edinburgh EH14 4AS, UK
eDepartment of Chemistry, University of Kentucky, Lexington, KY 40506, USA
First published on 21st March 2023
Abstract
Here we report that a Cu2+-seamed coordination nanocapsule can serve as an efficient semiconductor photocatalyst for molecular oxygen activation. This capsule was constructed through a redox reaction facilitated self-assembly of cuprous bromide and C-pentyl-pyrogallol[4]arene. Photophysical and electrochemical studies revealed its strong visible-light absorption and photocurrent polarity switching effect. This novel molecular solid material is capable of activating molecular oxygen into reactive oxygen species under simulated sunlight irradiation. The oxygen activation process has been exploited for catalyzing aerobic oxidation reactions. The present work provides new insights into designing nonporous discrete metal–organic supramolecular assemblies for solar-driven molecular oxygen activation.
Introduction
The self-assembly of large multicomponent coordination cage-like assemblies with well-defined geometries and cavities such as porous cages and nonporous capsules continues to attract intense interest due to not only their synthetic challenge and aesthetic appeal,1–5 but also their potential application across a range of fields including host-guest chemistry,6–11 catalysis,12–18 gas adsorption,19–21 and biomedicine.22,23 With the aim of expanding the application scope of these fascinating cage compounds, the construction of photoactive metal–organic nanocapsules (MONCs) has recently gathered considerable attention, as they can display emergent photophysical properties, for instance, light harvesting and photoelectronics.24–26 Although that is the case, the use of these functional entities for photochemical reactions remains largely unexplored.Activating molecular oxygen is crucial for many important biological and chemical processes, including cell signaling,27 most catalytic oxidation reactions,28 and oxygen reduction.29 Molecular oxygen can be activated through an electron transfer process, resulting in the formation of reactive oxygen species (ROS) such as the superoxide radical anion (O2˙−), peroxide anion (O22−), and hydroxyl radical (˙OH),30,31 whilst triplet oxygen can transform into active singlet oxygen (1O2) via energy transfer.32–34 Molecular oxygen activation can be accomplished by many physical, chemical, and biological processes.35 Amongst them, solar-driven activation of molecular oxygen was believed to be the most green and sustainable approach.36 Nevertheless, effectively coupling solar energy into molecular oxygen activation remains challenging.
Recent work in our group has focused on the construction of metal–organic nanocapsules (MONCs) using bowl-shaped C-alkyl-pyrogallol[4]arenes (PgCn, where n represents pendant alkyl chain lengths) as building blocks.26,37 Over the past few years we have prepared diverse MONCs containing various metal ions and functionalized PgCn units.37–43 These MONCs feature large enclosed internal cavities and small external voids in the solid state, which can be considered as nonporous materials.44,45 The nonporous nature of such kind of molecular solids greatly limits their heterogeneous application potentials in fields such as gas storage, molecular separation, and catalysis.43
Herein we report the preparation of a MONC assembled from six PgC5 and 24 Cu2+ ions, [Cu24(C48H56O12)6(Br)3(H2O)8(pyridine)4(DMF)4] (1). This MONC is isolated via an in situ redox reaction. Despite the lack of visible-light absorbing components, this new MONC exhibits strong light absorption in the visible region, which originates from the integrated charge transfer from pyrogallol moieties to the copper ions. Photoelectrochemical studies revealed its effective charge separation and potential-dependent photocurrent polarity switching effect. Interestingly, simulated sunlight irradiation of a solid sample of 1 with molecular oxygen effectively generates superoxide radicals and singlet oxygen. Benefiting from the oxygen activation process, 1 exhibited efficient heterogeneous photocatalytic activity towards numerous aerobic oxidation reactions. Notably, this is the first report on MONCs functioning as a semiconducting type of material for photochemical reactions, demonstrating the great application potential of MONCs in the heterogeneous phase.
Results and discussion
Compound 1 was synthesized by dissolving PgC5 and copper(I) bromide in a hot DMF/MeOH/pyridine (10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :15:1, v/v/v) solution. Slow evaporation over 48 hours afforded dark red single crystals of 1 (see Experimental section in ESI† for details). The bulk-phase purity of the crystals was confirmed by comparing experimental and simulated PXRD patterns (Fig. S1†). Crystals of 1 were found to be in a triclinic cell and structural analysis was performed in the space group P
:15:1, v/v/v) solution. Slow evaporation over 48 hours afforded dark red single crystals of 1 (see Experimental section in ESI† for details). The bulk-phase purity of the crystals was confirmed by comparing experimental and simulated PXRD patterns (Fig. S1†). Crystals of 1 were found to be in a triclinic cell and structural analysis was performed in the space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , revealing a discrete MONC containing 6 PgC5 units and 24 copper ions (Fig. 1, S2 and S3†). The overall geometry of this MONC resembles that of an octahedron, of which each face is capped by a six-membered [Cu3O3] array with Cu–O distances ranging from 1.87(1)–1.98(1) Å; Cu–O–Cu angles from 142.8(8)° to 146.8(8)°, and O–Cu–O angles from 93.1(6)° to 97.1(6)°. With respect to the coordination spheres, all equatorial sites of the constituent metal centres are occupied by PgC5 phenolic oxygen atoms, whilst exterior axial sites are occupied by pyridine (four), DMF (four) and aqua (eight) ligands. The difference electron density map of the interior of the MONC shows peaks which could be satisfactorily modeled as partial occupancy bromide ions, totally three Br− ions disordered over ten positions. Bond valence sum (BVS) analysis reveals that all Cu ions are in the 2+ oxidation state (Table S1†), with charge balance provided by 45 deprotonated phenolic groups and 3 Br− ions. The remaining PgC5 hydroxyl groups stabilize the assembly together through intramolecular hydrogen bonding interactions (O⋯O distances in the range of 2.38(2)–2.48(2) Å).
, revealing a discrete MONC containing 6 PgC5 units and 24 copper ions (Fig. 1, S2 and S3†). The overall geometry of this MONC resembles that of an octahedron, of which each face is capped by a six-membered [Cu3O3] array with Cu–O distances ranging from 1.87(1)–1.98(1) Å; Cu–O–Cu angles from 142.8(8)° to 146.8(8)°, and O–Cu–O angles from 93.1(6)° to 97.1(6)°. With respect to the coordination spheres, all equatorial sites of the constituent metal centres are occupied by PgC5 phenolic oxygen atoms, whilst exterior axial sites are occupied by pyridine (four), DMF (four) and aqua (eight) ligands. The difference electron density map of the interior of the MONC shows peaks which could be satisfactorily modeled as partial occupancy bromide ions, totally three Br− ions disordered over ten positions. Bond valence sum (BVS) analysis reveals that all Cu ions are in the 2+ oxidation state (Table S1†), with charge balance provided by 45 deprotonated phenolic groups and 3 Br− ions. The remaining PgC5 hydroxyl groups stabilize the assembly together through intramolecular hydrogen bonding interactions (O⋯O distances in the range of 2.38(2)–2.48(2) Å).
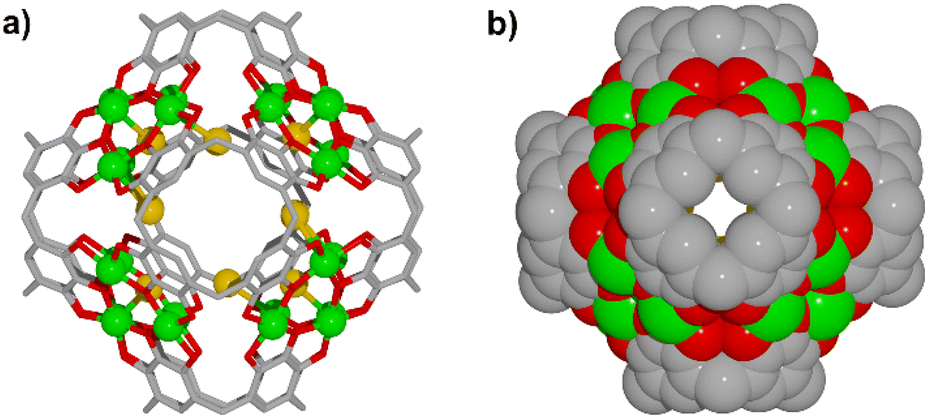 | ||
| Fig. 1 (a) Ball and stick and (b) space-filling representation of 1. Hydrogen atoms, alkyl tails, and some axial ligands have been omitted for clarity. Colour codes: C, grey; O, red; Br, orange; Cu, green. | ||
The electronic absorption spectrum of 1 in chloroform showed an absorption band at 525 nm (ε ≈ 5000 M−1 cm−1) which can be assigned to LMCT transitions.46 Indeed, the PgC5 ligand exhibited no absorption in the visible light region (Fig. S4†). Moreover, the UV-vis diffuse reflectance spectrum (DRS) of 1 exhibited a broad absorption throughout the region of 400–700 nm (Fig. S5†). Accordingly, the optical bandgap of 1 was calculated to be 2.39 eV, unveiling its potential to be used as a semiconducting photocatalyst. Mott–Schottky (MS) measurements were conducted to clarify the semiconductor properties of 1. As shown in Fig. S6,† the MS plot of 1 has a positive slope, indicating that the 1 is an n-type semiconductor. Furthermore, the flat band potential (EFB) estimated from the M–S plot is 0.7 V vs. NHE. Since the flat band potential of an n-type semiconductor is very close to its conduction band potential (EVB), the conduction band potential (ECB) and valence band potential (ECB) of 1 is estimated to be 0.7 V and 1.69 V vs. NHE, respectively.47 The CB potential of 1 is sufficiently negative for many photochemical reactions such as the reduction of O2 to O2˙− (−0.33 V vs. NHE).33
Transient photocurrent measurements were performed to investigate the photoinduced charge separation efficiency of 1. As shown in Fig. 2a, remarkable cathodic photocurrent responses were observed upon irradiation of 1 with visible light (λ > 420 nm, 1-sun, 100 mW cm−2) at an applied bias potential of 0 V versus SCE, suggesting the effective separation of photogenerated electron–hole pairs. The photocurrents are highly reversible and reproducible, demonstrating the high photostability of 1 in aqueous solution (Fig. S7 and S8†). Moreover, apparent anodic photocurrent responses were also detected at an applied bias potential of 0.5 V vs. SCE upon chopped light irradiation (Fig. 2b). The potential-dependent photocurrent polarity switching effect has been observed in some inorganic semiconductor materials (e.g., cadmium sulphide) and presents exciting potential applications in photoelectric logic gates and chemosensors.48 To our knowledge, no discrete metal–organic assemblies have been reported that have been shown to possess such an intriguing phenomenon. Unlike those insoluble inorganic semiconductors, assembly 1 is soluble in organic solvents such as chloroform, so can be readily solution processed for use in photoelectric device applications.
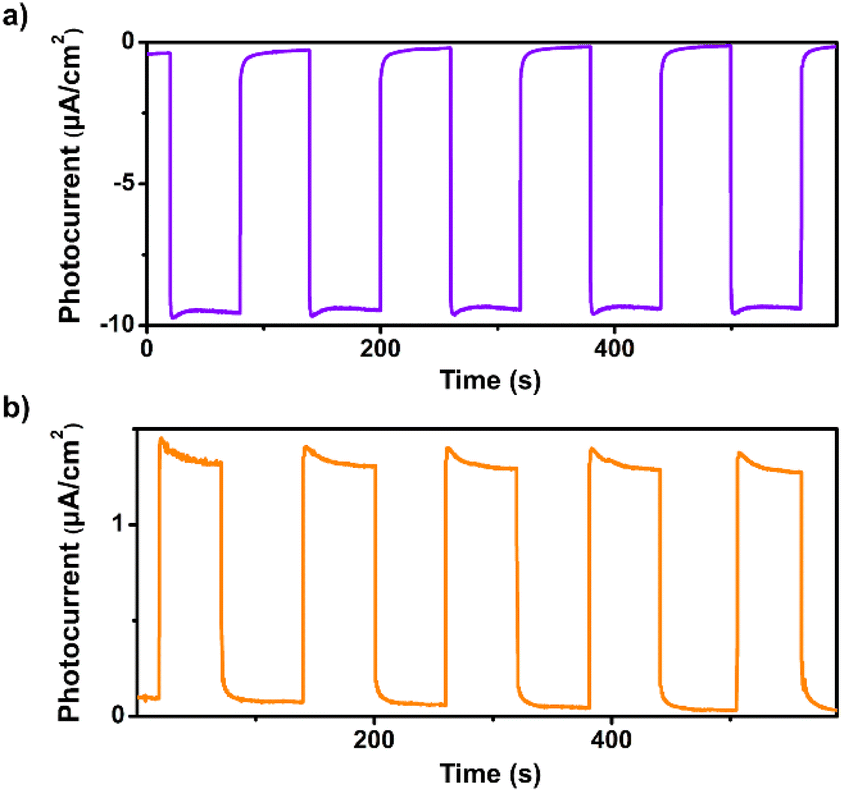 | ||
| Fig. 2 (a) Transient photocurrent response of 1 in 0.1 M air-saturated Na2SO4 aqueous solution under irradiation of λ > 420 nm at (a) 0 V and (b) 0.5 V vs. SCE. | ||
We observed that the magnitudes of the cathodic photocurrents in oxygen-saturated electrolyte solution was ∼3.2 times higher than the photocurrents obtained in air-saturated electrolyte solution (Fig. 3). Given this observation, we deduce that the generation of cathodic photocurrent originated from the single electron transfer from the photogenerated electrons to molecular oxygen. Electron paramagnetic resonance (ESR) experiments were performed to confirm this corollary. 5,5-Dimethyl-1-pyrroline N-oxide (DMPO) was employed as the spin-trapping agent to detect O2˙−. As shown in Fig. 4a, after irradiating the mixed solution of 1 and DMPO by visible light, a 1:2:2:1 signal typical for the DMPO-O2˙− adduct was observed, corroborating that O2˙− was generated under light conditions.30 Furthermore, 2,2,6,6-tetramethylpiperidine (TEMP) was applied to detect the possible generation of 1O2 during the molecular oxygen activation process. As shown in Fig. 4b and S9,† a 1:1:1 triplet signal was observed under visible light irradiation, in accordance with that of 2,2,6,6-tetramethylpiperidine-N-oxyl (TEMPO), demonstrating the production of 1O2.32 In addition, ESR experiments revealed that no ˙OH radical was generated during the molecular oxygen activation process (Fig. S10†). Taken together, these results demonstrate that O2 can be activated into O2˙− and 1O2 by 1 under visible-light irradiation.
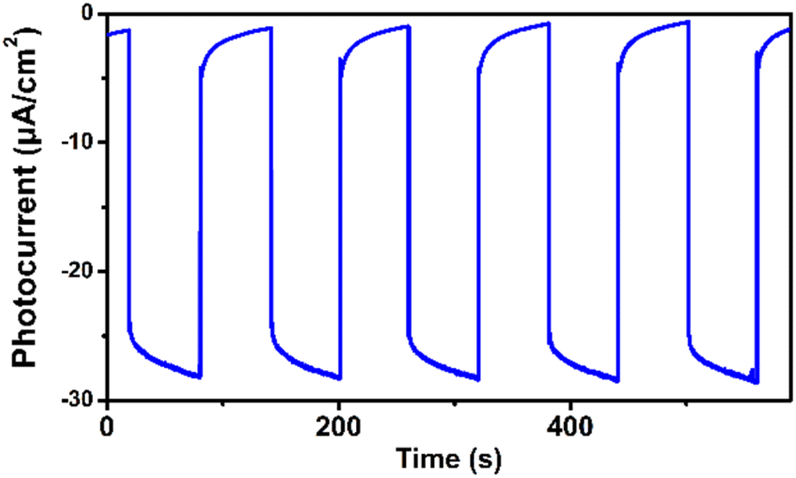 | ||
| Fig. 3 Transient photocurrent response of 1 in 0.1 M oxygen-saturated Na2SO4 aqueous solution under irradiation of λ > 420 nm at 0 V vs. SCE. | ||
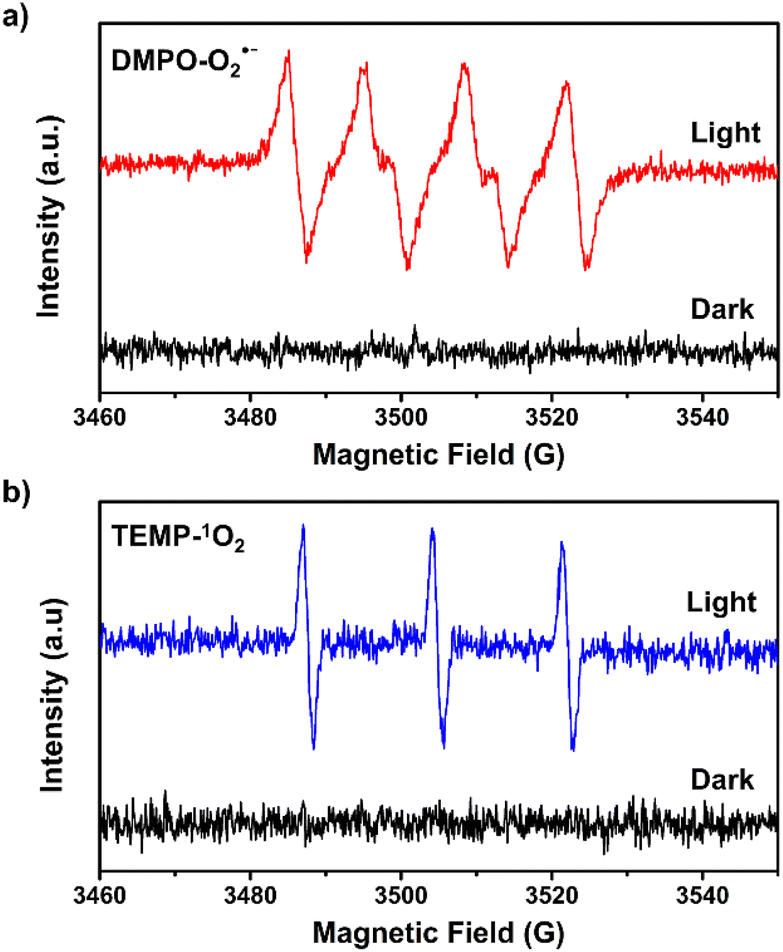 | ||
| Fig. 4 ESR spectra of 1 in the presence of (a) DMPO and (b) TEMP under dark and light conditions in methanol. | ||
The ability for 1 to efficiently generate ROS under visible light irradiation prompted us to investigate its heterogenous photocatalytic activity towards a ROS-involved oxidation reaction. For this purpose, the O2˙−-mediated oxidative hydroxylation of boronic acids has been examined.49,50 In a typical performance, 1 was employed as the photocatalyst, triethylamine (TEA) as the electron donor, oxygen as the oxidizing agent, water as the solvent, and 4-cyanophenylboronic acid as the model substrate. Pleasingly, the desired reaction occurred to give the phenol in 93% yield after 5 hours (Table 1, entry 1). A series of control experiments revealed that the catalyst 1, light, oxygen are all essential components for this transformation, clearly indicating that this is a photocatalytic oxygen-activation-mediated process (Table 1, entries 2–7). The yield of product decreased to 34% when 2 equiv. of 1,4-benzoquinone (BQ) was added to the reaction mixture as a scavenger of O2˙−, which is consistent with the O2˙−-mediated reaction mechanism (Table 1, entries 8 and 9). As a stable heterogeneous catalyst, 1 can be easily isolated from the reaction mixture by centrifugation and reused at least five times without a significant decrease in photocatalytic activity (Fig. S11†).
|
|
||||
|---|---|---|---|---|
| Entry | Catalyst | Light | Atmosphere | Yieldb (%) |
| a Reaction conditions: catalyst (10 mg), 4-cyanophenylboroic acid (0.4 mmol), TEA (0.8 mmol), O2 (1 atm), 3 mL water, 300 W xenon lamp with visible light (λ > 420 nm, 100 mW cm−2), room temperature, 5 hours. b Isolated yield. c Additive: 2 equiv. of BQ. d Additive: 2 equiv. of NaN3. | ||||
| 1 | 1 | + | O2 | 93 |
| 2 | PgC5 | + | O2 | Trace |
| 3 | CuBr | + | O2 | Trace |
| 4 | CuBr2 | + | O2 | Trace |
| 5 | None | + | O2 | Trace |
| 6 | 1 | — | O2 | n.d. |
| 7 | 1 | + | N2 | Trace |
| 8c | 1 | + | O2 | 34 |
| 9d | 1 | + | O2 | 91 |

The IR and MALDI-TOF MS spectra of 1 after six cycles showed no significant structural change (Fig. S12–S14, Table S2†). The scope of this oxidative hydroxylation reaction was also evaluated, and the results are summarized in Table S3.† Boronic acids bearing electron-neutral, electron-withdrawing and electron-donating substituents were successfully converted into corresponding phenols in 84–93% yields. Given that superoxide species could catalyse the oxidative dehydrogenation of hydrazobenzenes to azobenzenes,51,52 we then examined the photocatalytic activity of 1 in the oxidative dehydrogenation of 1,2-diphenylhydrazine (Table 2).
|
|
||||
|---|---|---|---|---|
| Entry | Catalyst | Light | Atmosphere | Yieldb (%) |
| a Reaction conditions: catalyst (10 mg), 1,2-diphenylhydrazine (0.1 mmol), O2 (1 atm), 2 mL methanol, 0.5 mL water, 300 W xenon lamp with visible light (λ > 420 nm, 100 mW cm−2), room temperature, 2 hours. b Isolated yield. c Additive: 2 equiv. of BQ. d Additive: 2 equiv. of sylvan. | ||||
| 1 | 1 | + | O2 | >99 |
| 2 | PgC5 | + | O2 | Trace |
| 3 | CuBr | + | O2 | Trace |
| 4 | CuBr2 | + | O2 | Trace |
| 5 | None | + | O2 | n.d. |
| 6 | 1 | − | O2 | n.d. |
| 7 | 1 | + | N2 | Trace |
| 8c | 1 | + | O2 | 32 |
| 9d | 1 | + | O2 | 93 |
Remarkably, irradiation of the mixture of 1 and hydrazobenzene in MeOH/H2O (4:1, v/v) under oxygen atmosphere with visible light gave the desired azobenzene in quantitative yield after 2 hours (Table 2, entry 1). Control experiments demonstrated that catalyst 1, light, and oxygen were indispensable for the dehydrogenation reaction (Table 2, entries 2–7). It is worth noting that this is first example of heterogeneous photocatalytic transformation of hydrazobenzene to azobenzene. Moreover, the introduction of 2 equiv. of BQ significantly hindered the reaction, indicating that O2˙− is responsible for this transformation (Table 2, entry 8). No significant decrease of yield was observed upon the addition of 2 equiv. of 2-methylfuran (sylvan) as a scavenger of 1O2, suggesting that 1O2 is not the key active oxygen species in this reaction (Table 2, entry 9). The catalyst was also recovered from the reaction mixture by centrifugation and reused five times without loss of activity (Fig. S15†). The IR and MALDI-TOF MS spectra of the recovered catalyst indicated that it maintained its structural integrity after six cycle reactions (Fig. S16 and S17†).
Encouraged by above results, we next tested the capability of 1 in the photocatalytic oxidative coupling of benzylamine.53,54 As shown in Fig. 5 and Table S4,† >99% benzylamine in methanol could be converted into N-benzylidenebenzylamine with the help of 1 within 2 hours under oxygen atmosphere upon irradiation with visible light. No benzaldehyde (the possible byproduct) was detected, demonstrating a high degree of selectivity of this reaction. Control experiments confirmed the essential role of the catalyst 1, light, and oxygen in this reaction (Table S4†). The addition of 2 equiv. of BQ and sylvan into the reaction mixture gave 38% and 87% conversion of benzylamine, respectively, demonstrating the more important role of O2˙− instead of 1O2 in the reaction process (Fig. 5b). The stability of the 1 has also been examined by performing a recycling test, which manifests its activity and selectivity can be well retained after six cycle reactions (Fig. 5c). IR and MALDI-TOF MS spectra confirmed that structural integrity of the recovered catalyst is maintained (Fig. S18 and S19†).
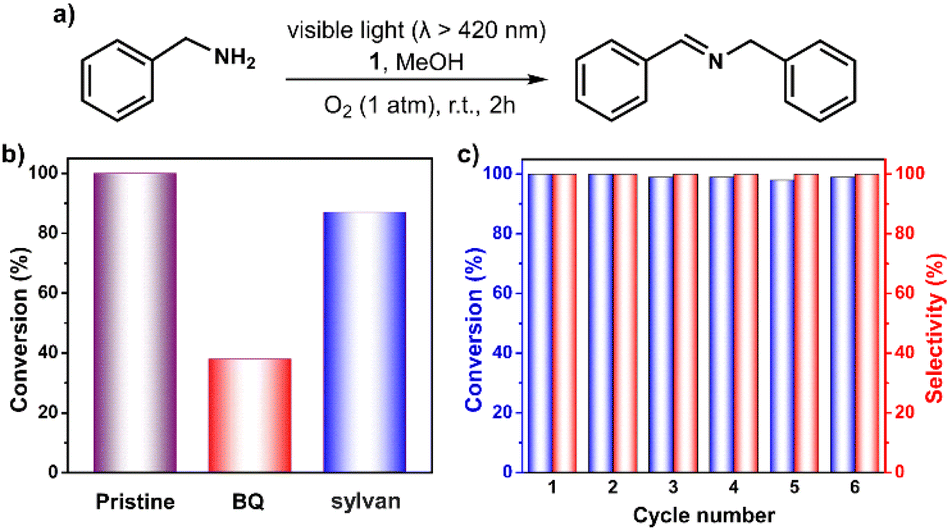 | ||
| Fig. 5 (a) Photocatalytic oxidative coupling of benzylamine. (b) Yield of target product using 1 in the absence (pristine) or presence of different scavengers (2 equiv.). (c) Yield and selectivity of oxidation of benzylamine with 1 in six repeating cycles. | ||
These catalytic experiments clearly indicate that 1 can harvest solar energy in the visible region and utilize it to generate ROS from molecular oxygen. Consequently, it can serve as an efficient heterogeneous catalyst for ROS-involved aerobic oxidation reactions. To the best of our knowledge, no MONCs have been reported that have been shown to possess heterogeneous photocatalytic activities toward organic reactions. Since 1 is a nonporous solid catalyst, the direct contact between the MONC catalysts and reactants will be severely hindered by the diffusion problem, which is supposed to show a poor catalytic performance.55,56 Our findings demonstrate an important proof of concept that nonporous discrete metal–organic supramolecular assemblies can serve as an efficient heterogeneous photocatalyst for organic transformations. These results may ignite new research interest toward exploration of the photocatalytic properties of nonporous discrete metal–organic supramolecular assemblies in the heterogeneous phase.
Conclusions
In summary, we have constructed a copper-seamed MONC via an in situ redox reaction facilitated self-assembly approach. The resultant MONC exhibited strong visible light absorption and potential-dependent photocurrent polarity switching effect. It can serve as an efficient semiconductor photocatalyst for molecular oxygen activation, exemplified in aerobic oxidation reactions. Considering that various metal ions and functional pyrogallol[4]arenes could be employed to construct this incredibly versatile MONC system, a vast range of new photoactive MONCs with novel functionality (solar energy conversion, photoconductivity, heterogeneous catalysis, etc.) may be accessible through coordination chemistry. The investigation of photocatalytic properties of other related MONCs is underway, the results of which will be reported in due course.Data availability
All experimental data associated with this work are available in the ESI.†Author contributions
X. H., J. D., S. F., and J. A. conceived the project, X. H., H. M., L. W., L. S., Y. P., and S. K. performed the experiments; X. H., H. M., J. D., S. F., S. D., D. A., and J. A. analysed the data, prepared the figures, and provided conceptual contributions; X. H., S. D., D. A., and J. A. wrote the manuscript through contributions from all authors. All authors have given their approval to the final version of the manuscript.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
We thank University of Missouri for financial and research facility support of this work. We thank the U. S. National Science Foundation for support of this work (1825352). This work was also supported by the National Natural Science Foundation of China (52002146) and Natrual Science Foundation of Shanxi Province (201701D121039).Notes and references
- T. R. Cook, Y. R. Zheng and P. J. Stang, Chem. Rev., 2013, 113, 734–777 CrossRef CAS PubMed.
- D. Fujita, Y. Ueda, S. Sato, N. Mizuno, T. Kumasaka and M. Fujita, Nature, 2016, 540, 563–566 CrossRef CAS PubMed.
- Y. Zhang, H. Gan, C. Qin, X. Wang, Z. Su and M. J. Zaworotko, J. Am. Chem. Soc., 2018, 140, 17365–17368 CrossRef CAS PubMed.
- S. Chakraborty and G. R. Newkome, Chem. Soc. Rev., 2018, 47, 3991–4016 RSC.
- S. J. Dalgarno, N. P. Power and J. L. Atwood, Coord. Chem. Rev., 2008, 252, 825–841 CrossRef CAS.
- T. D. Hamilton, G. S. Papaefstathiou, T. Friščić, D.-K. Bučar and L. R. MacGillivray, J. Am. Chem. Soc., 2008, 130, 14366–14367 CrossRef CAS PubMed.
- A. J. Scott, J. Vallejo, A. Sarkar, L. Smythe, E. R. Martí, G. S. Nichol, W. T. Klooster, S. J. Coles, M. Murrie, G. Rajaraman, S. Piligkos, P. J. Lusby and E. K. Brechin, Chem. Sci., 2021, 12, 5134–5142 RSC.
- E.-S. M. EI-Sayed, Y. Yuan, D. Zhao, Y. Di Yuan, D. Zhao and D. Yuan, Acc. Chem. Res., 2022, 55, 1546–1560 CrossRef PubMed.
- S. Pullen, J. Tessarolo and G. H. Clever, Chem. Sci., 2021, 12, 7269–7293 RSC.
- D. Zhang, T. K. Ronson, Y.-Q. Zou and J. R. Nitschke, Nat. Rev. Chem., 2021, 5, 168–182 CrossRef CAS.
- H. Kumari, C. A. Deakyne and J. L. Atwood, Acc. Chem. Res., 2014, 47, 3080–3088 CrossRef CAS PubMed.
- M. Yoshizawa, J. K. Klosterman and M. Fujita, Angew. Chem., Int. Ed., 2009, 48, 3418–3438 CrossRef CAS PubMed.
- C. J. Brown, F. D. Toste, R. G. Bergman and K. N. Raymond, Chem. Rev., 2015, 115, 3012–3035 CrossRef CAS PubMed.
- D. Zhang, T. K. Robson and J. R. Nitschke, Acc. Chem. Res., 2018, 51, 2423–2436 CrossRef CAS PubMed.
- C. Tan, J. Jiao, Z. Li, Y. Liu and Y. Cui, Angew. Chem., Int. Ed., 2017, 57, 2085–2090 CrossRef PubMed.
- D.-Y. Yan, L.-X. Cai, P.-M. Cheng, S.-J. Hu, L.-P. Zhou and Q.-F. Sun, J. Am. Chem. Soc., 2021, 143, 16087–16094 CrossRef CAS PubMed.
- Y. Fang, J. A. Powell, E. Li, Q. Wang, Z. Perry, A. Kirchon, X. Yang, Z. Xiao, C. Zhu, L. Zhang, F. Huang and H.-C. Zhou, Chem. Soc. Rev., 2019, 48, 4707–4730 RSC.
- M. Yoshizawa and L. Catti, Acc. Chem. Res., 2019, 52, 2392–2404 CrossRef CAS PubMed.
- Z. Niu, L. Wang, S. Feng, P. C. Lan, B. Aguila, J. Perman, J.-G. Ma, P. Cheng, X. Li and S. Ma, Chem. Sci., 2019, 10, 661–6665 Search PubMed.
- Z. Niu, S. Feng, X. Liu, J.-G. Ma, S. Ma and P. Cheng, J. Am. Chem. Soc., 2015, 137, 14873–14876 CrossRef CAS PubMed.
- T.-F. Liu, Y.-P. Chen, A. A. Yakovenko and H.-C. Zhou, J. Am. Chem. Soc., 2012, 134, 17358–17361 CrossRef CAS PubMed.
- Y. Xu, C. Li, S. Lu, Z. Wang, S. Liu, X. Yu, X. Li and Y. Su, Nat. Commun., 2022, 13, 2009 CrossRef CAS PubMed.
- H. Sepehrpour, X. Fu, Y. Sun and P. J. Stang, J. Am. Chem. Soc., 2019, 141, 14005–14020 CrossRef CAS PubMed.
- O. Chepelin, J. Ujma, X. Wu, A. M. Z. Slawin, M. B. Pitak, S. J. Coles, J. Michel, A. C. Jones, P. E. Barran and P. J. Lusby, J. Am. Chem. Soc., 2012, 134, 19334–19337 CrossRef CAS PubMed.
- P. D. Frischmann, V. Kunz and F. Würthner, Angew. Chem., Int. Ed., 2015, 54, 7285–7289 CrossRef CAS PubMed.
- X. Hu, M. Han, L. Shao, C. Zhang, L. Zhang, S. P. Kelley, C. Zhang, J. Lin, S. J. Dalgarno, D. A. Atwood, S. Feng and J. L. Atwood, Angew. Chem., Int. Ed., 2021, 60, 10516–10520 CrossRef CAS PubMed.
- T. Finkel, J. Cell Biol., 2011, 194, 7–15 CrossRef CAS PubMed.
- X. Huang and J. T. Groves, Chem. Rev., 2018, 118, 2491–2553 CrossRef CAS PubMed.
- Z. Peng, Y. Chen, P. G. Bruce and Y. Xu, Angew. Chem., Int. Ed., 2015, 54, 8165–8168 CrossRef CAS PubMed.
- C. Pan, C. Wang, X. Zhao, P. Xu, F. Mao, J. Yang, Y. Zhu, R. Yu, S. Xiao, Y. Fang, H. Deng, Z. Luo, J. Wu, J. Li, S. Liu, S. Xiao, L. Zhang and Y. Guo, J. Am. Chem. Soc., 2022, 144, 4942–4951 CrossRef CAS PubMed.
- X. Sun, X. Luo, X. Zhang, J. Xie, S. Jun, H. Wang, X. Zheng, X. Wu and Y. Xie, J. Am. Chem. Soc., 2019, 141, 3797–3801 CrossRef CAS PubMed.
- D. Zhang, P. Wang, J. Wang, Y. Li, Y. Xia and S. Zhan, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2114729118 CrossRef CAS PubMed.
- Y. Qian, D. Li, Y. Han and H.-L. Jiang, J. Am. Chem. Soc., 2020, 142, 20763–20771 CrossRef CAS PubMed.
- H. I. Hamoud, F. Douma, M. Lafjah, F. Djafri, O. Lebedev, V. Valtchev and M. EI-Roz, ACS Appl. Nano Mater., 2022, 5, 3866–3877 CrossRef CAS.
- J. M. Anglada, M. Martins-Costa, J. S. Francisco and M. F. Ruiz-López, Acc. Chem. Res., 2015, 48, 575–583 CrossRef CAS PubMed.
- N. Zhang, X. Li, H. Ye, S. Chen, H. Ju, D. Liu, Y. Lin, W. Ye, C. Wang, Q. Xu, J. Zhu, L. Song, J. Jiang and Y. Xiong, J. Am. Chem. Soc., 2016, 138, 8928–8935 CrossRef CAS PubMed.
- L. Shao, B. Hua, X. Hu, D. Stalla, S. P. Kelley and J. L. Atwood, J. Am. Chem. Soc., 2020, 142, 7270–7275 CrossRef CAS PubMed.
- X. Hu, S. Feng, J. Du, L. Shao, J. Lang, C. Zhang, S. P. Kelley, J. Lin, S. J. Dalgarno, D. A. Atwood and J. L. Atwood, Chem. Sci., 2020, 11, 12547–12552 RSC.
- D. W. Wagle, S. P. Kelley, G. A. Baker, K. Sikligar and J. L. Atwood, Angew. Chem., Int. Ed., 2020, 59, 8062–8065 CrossRef CAS PubMed.
- X. Hu, J. Chai, C. Zhang, J. Lang, S. P. Kelley, S. Feng, B. Liu, D. A. Atwood and J. L. Atwood, J. Am. Chem. Soc., 2019, 141, 9151–9154 CrossRef CAS PubMed.
- H. Kumari, C. L. Dennis, S. R. Kline, A. W. Mossine, C. A. Deakyne and J. L. Atwood, Angew. Chem., Int. Ed., 2022, 134, e202203010 CrossRef.
- Y. Xie, C. Zhang, X. Hu, C. Zhang, S. P. Kelley, J. L. Atwood and J. Lin, J. Am. Chem. Soc., 2020, 142, 1475–1481 CrossRef CAS PubMed.
- C. Zhang, F. Wang, R. S. Patil, C. L. Barnes, T. Li and J. L. Atwood, Chem. – Eur. J., 2018, 24, 14335–14340 CrossRef CAS PubMed.
- L. Barbour, Chem. Commun., 2006, 11, 1163–1168 RSC.
- P. K. Thallapally, B. Peter McGrail, S. J. Dalagrno, H. T. Schaef, J. Tian and J. L. Atwood, Nat. Mater., 2008, 7, 146–150 CrossRef CAS PubMed.
- M. J. Sever and J. J. Wilker, Dalton Trans., 2004, 7, 1061–1072 RSC.
- Y. Bai, S. Zhang, S. Feng, M. Zhu and S. Ma, Dalton Trans., 2020, 49, 10745–10754 RSC.
- S. Gawęda, A. Podborska, W. Macyk and K. Szaciłowski, Nanoscale, 2009, 1, 299–316 RSC.
- Y.-Q. Zhou, J.-R. Chen, X.-P. Liu, L.-Q. Lu, R. L. Davis, K. A. Jøgensen and W.-J. Xiao, Angew. Chem., Int. Ed., 2012, 124, 808–812 CrossRef.
- E. Jin, S. Fu, H. Hanayama, M. A. Addicoat, W. Wei, Q. Chen, R. Graf, K. Landfester, M. Bonn, K. A. I. Zhang, H. I. Wang, K. Müllen and A. Narita, Angew. Chem., Int. Ed., 2022, 61, e202114059 CAS.
- X. Wang, X. Wang, C. Xia and L. Wu, Green Chem., 2019, 21, 4189–4193 RSC.
- M. E. Czaikowski, A. J. McNeece, J.-N. Boyn, K. A. Jesse, S. W. Anferov, A. S. Filatov, D. A. Mazziotti and J. S. Anderson, J. Am. Chem. Soc., 2022, 144, 15569–15580 CrossRef CAS PubMed.
- F. Jin, E. Lin, T. Wang, D. Yan, Y. Yang, Y. Chen, P. Cheng and Z. Zhang, Chem, 2022, 8, 2899–2901 Search PubMed.
- C. Liu, K. Liu, C. Wang, H. Liu, H. Wang, H. Su, X. Li, B. Chen and J. Jiang, Nat. Commun., 2020, 11, 1047 CrossRef CAS PubMed.
- S. Goel, Z. Wu, S. I. Zones and E. Iglesia, J. Am. Chem. Soc., 2012, 134, 17688–17695 CrossRef CAS PubMed.
- J.-K. Sun, W.-W. Zhan, T. Akita and Q. Xu, J. Am. Chem. Soc., 2015, 137, 7063–7066 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2174024. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3sc00318c |
| This journal is © The Royal Society of Chemistry 2023 |