
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceChirally and chemically reversible Strecker reaction†
Yutaro
Machida
,
Yudai
Tanaka
,
Yuya
Masuda
,
Aya
Kimura
and
Tsuneomi
Kawasaki
 *
*
Department of Applied Chemistry, Tokyo University of Science, Kagurazaka, Shinjuku-ku, Tokyo 162-8601, Japan. E-mail: tkawa@rs.tus.ac.jp
First published on 28th March 2023
Abstract
In the pursuit of a credible mechanism for the abiotic synthesis of α-amino acids, solid-state asymmetric Strecker/retro-Strecker reactions have been demonstrated. Asymmetric addition of cyanide to enantiomorphic crystals of achiral imines proceeded to produce enantioenriched aminonitriles. Moreover, dehydrocyanation of enantioenriched aminonitriles gave chiral crystals of achiral imines stereoselectively. We found, for the first time to the best of our knowledge, a stereoinversion of the synthetic intermediates imine and aminonitrile in the sequence of reactions including HCN addition and elimination. Thus, the reversible Strecker reaction is expected to be a focus of research on the origin of chirality.
Introduction
Biological systems are composed of molecules with a specific handedness such as seen in L-amino acids and D-sugars. Since the discovery of molecular chirality by Pasteur,1 the origin and amplification of chirality have been long-standing mysteries closely related to the chemical origin of life.2 Several chiral factors and processes have been proposed and investigated as possible candidates for the origin of chirality,3 and chiral crystallization of achiral compounds3b,4 is one of the possibilities. Initiated by the pioneering work of Schmidt et al.,5 stereospecific reactions utilizing a chiral crystal of achiral compounds as a substrate have been demonstrated.6 Their utilization as heterogeneous chiral triggers for asymmetric autocatalysis, i.e., the Soai reaction,7 has also been reported.8 To the best of our knowledge, there are no reports on asymmetric HCN addition using this strategy.Meanwhile, Strecker synthesis has long been considered to be one of the way that α-amino acids are abiotically synthesized.9 Because chiral aminonitrile intermediates10 hydrolyze to amino acids, asymmetric HCN addition11 determines the molecular handedness. Thus, research on chirality of the sequence of reactions involving cyanide addition and elimination would be a challenge but might expand our knowledge of biological homochirality as exemplified by α-amino acids.
We have reported on the spontaneous absolute asymmetric Strecker synthesis based on the spontaneous crystallization of aminonitriles (Fig. 1).12a,b In addition to the total spontaneous resolution of aminonitriles as a result of a three-component Strecker reaction, the enantiopurity of aminonitriles can be improved significantly. Because corresponding amino acids can act as chiral triggers for the amplification of their own chiral intermediate, the process represents the replication of chiral α-amino acids.12c,d The oriented prochirality of imines,12e chiral isotopomers12f and chiral crystal of the related racemate12g can be responsible for the generation and amplification of enantioenriched aminonitriles.
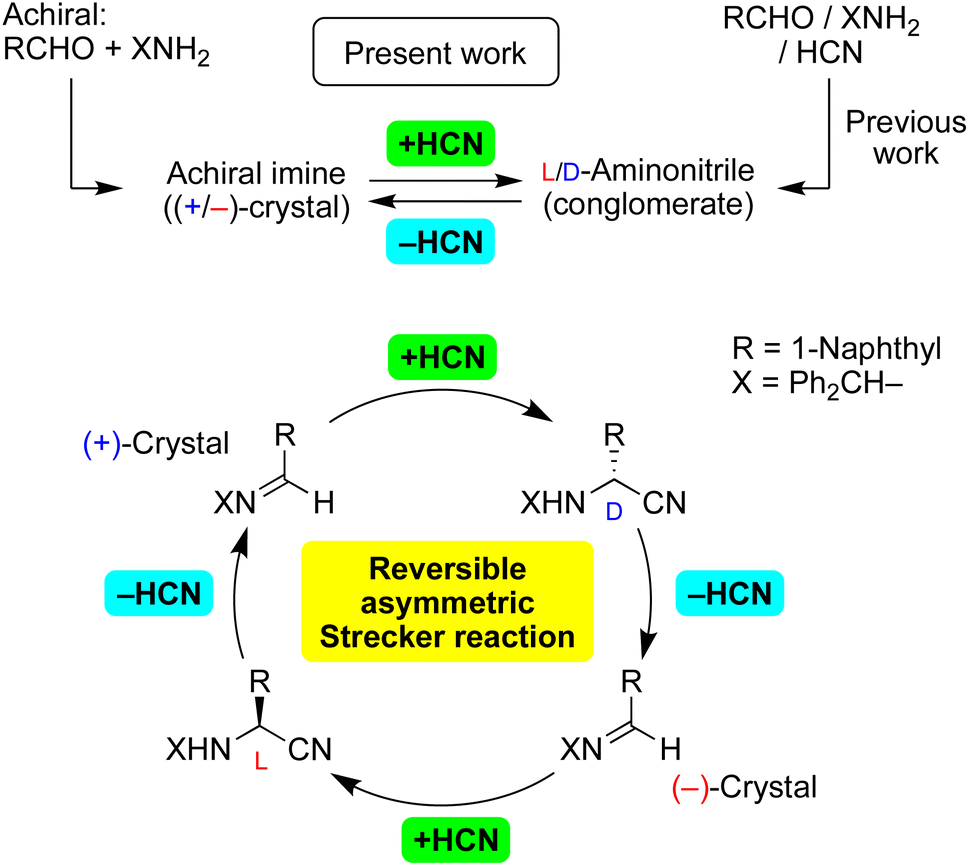 | ||
| Fig. 1 Concept of the current work. | ||
Here, we report on asymmetric addition of HCN to a chiral crystal of achiral imines to afford highly enantioenriched aminonitriles in conjunction with an amplification of chirality (Fig. 1). In the reverse reaction, dehydrocyanation13 proceeded stereoselectively with the loss of a stereocenter to give the corresponding enantiomorphic crystals of achiral imines. Furthermore, the inversion of the enantiomorphic crystals of an achiral compound was observed for the first time; therefore, the (+)-crystal of the imine was transformed to its enantiomorphic (−)-crystal by carrying out HCN addition and then elimination and vice versa. In addition, the sequence of stereoselective reactions without stereoinversion was also investigated using other substrates. Note a requirement of the current methods being chiral crystallization (conglomerate formation) of achiral or racemic compounds, observed in only ca. 10% of crystalline compounds.
Results and discussion
We carried out a chiral crystallization of achiral imine 1 synthesized from 1-naphthaldehyde and benzhydrylamine (Fig. 2a). The handedness of crystal 1 was determined from solid-state circular dichroism (CD) spectroscopy with a KBr matrix; one crystal showed a positive Cotton effect at a wavelength of 340 nm (CD(+)340KBr) and the other showed a negative effect (CD(−)340KBr) (Fig. 2b). When the dehydrative condensation of 1-naphthaldehyde and benzhydrylamine proceeded, enantioenriched powder-like crystal 1 formed spontaneously under stirred conditions3c,14 (Table S1†). The crystal was determined to belong to the chiral space group P21 (Fig. 2c) and the relationship between the absolute configuration of the crystal 1 and its CD was determined (Table S2†).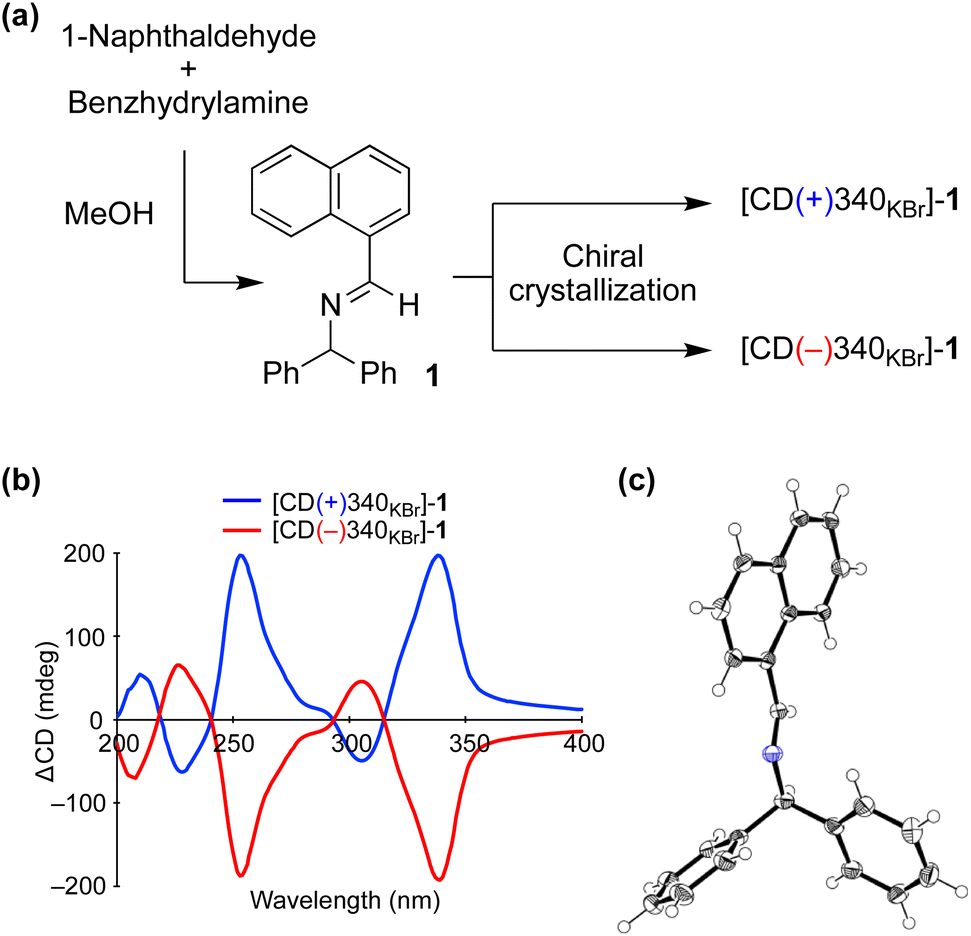 | ||
| Fig. 2 (a) Chiral crystallization of achiral imine 1. (b) Solid-state CD spectrum of 1. (c) Single-crystal X-ray structure of 1 (CCDC 2224018†). | ||
Because the solid-state CD of 1 was quite reproducible when carefully preparing a fixed and translucent KBr disc, the enantioenrichment of 1 could be characterized from the value of ΔCD at 340 nm. Because the enantiomeric excess (ee) of suspended solid 1 can be enhanced by vigorously stirring the mixture3d,15 (Fig. S1†), the maximum ΔCD value of 201 mdeg at 340 nm was considered as that with 100% ee. The linearity between the ΔCD and ee was checked by mixing enantiomorphic crystals at various proportions (Fig. S2†).16
The chiral crystal of achiral imine 1 was subjected to an asymmetric HCN addition by exposing the crystal to HCN vapor. Due to dissolution of 1 causing the disappearance of the chirality, HCN and 1 were stored separately in sealed reaction vials (Fig. S3†). We then added DBU, HCN, methanol and toluene to the product of the vapor-phase reaction to prepare a suspension of 2 with an enantiomeric imbalance corresponding to the asymmetric hydrocyanation. Due to the tendency of aminonitrile 2 to form a conglomerate, asymmetric amplification was applied to this suspension as in the previous report (see the figure in Table 1).12d The results are summarized in Tables 1 and S3.† When a powder of [CD(−)340KBr]-1 was treated with HCN, L-aminonitrile 2 with >99% ee was synthesized in 67% yield (Table 1, series I, entry 1). In contrast, addition of HCN to [CD(+)340KBr]-1 gave D-2 with >99% ee in 60% yield (entry 2). The stereochemical relationships were reproducible as confirmed in entries 3 and 4.
|
|
|||
|---|---|---|---|
| Entrya | Imine 1 | Aminonitrile 2 | |
| % ee (config.)b | Yieldc | ||
| a Asymmetric amplification of 2 was carried out after the vapor-phase reaction in series I was carried out. b Determined using HPLC on a chiral stationary phase. The absolute configuration of 2 (CCDC 2224019) was reconfirmed by derivatizing it to α-(1-naphthyl)glycine (5) (Fig. S5). c Each value in series I indicates the isolated yield (%) of 2, whereas each value in series II indicates the reaction conversion (%), which was determined using 1H NMR from the molar ratio of 1 to 2. d Chiral crystal 1 was ground into a fine powder together with Na2SO4. e Chiral crystal 1 was ground into a fine powder together with KCN and then the mixture was exposed to acetic acid vapor. | |||
| Series I | |||
| 1 | CD(−)340KBr | >99 (L) | 67 |
| 2 | CD(+)340KBr | >99 (D) | 60 |
| 3 | CD(−)340KBr | >99 (L) | 53 |
| 4 | CD(+)340KBr | >99 (D) | 56 |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
|||
| Series II | |||
| 5 | CD(−)340KBr | 7 (L) | 18 |
| 6 | CD(+)340KBr | 7 (D) | 23 |
| 7e | CD(−)340KBr | 2 (L) | 34 |

The enantioenrichment of 2 was monitored as shown in Fig. S4;† the initially slight enantioimbalance of suspended 2 continuously increased to finally achieve an ee >99% when carrying out thermal cycling. In this heating–cooling cycles,12c nearly equimolar amounts of D- and L-2 dissolved during heating to afford a reduced amount of suspended 2 with amplified ee. Then, cooling-induced deracemization of 2 occurred, i.e., the crystal recovered during gradual crystal growth without a decrease in the amplified ee.
In series II, the enantioselectivity of the HCN addition was examined without asymmetric amplification. In this series, chiral crystal 1 was finely ground together with Na2SO4 to expand the reactive chiral surface, which was exposed to HCN. When [CD(−)340KBr]-1 was subjected to HCN addition, L-2 with 7% ee formed with an 18% reaction conversion. In contrast, the [CD(+)340KBr]-1 enantiomorph gave the oppositely configured D-2 (entries 5 and 6). As shown in entry 7, when [CD(−)340KBr]-1 was ground with KCN and the resulting mixture was exposed to acetic acid vapor, L-2 was obtained with 2% ee12f with the same stereoselectivity. Because fine powder 1 was exposed to HCN vapor, the addition reaction occurred under heterogeneous conditions of a random orientation of 1. Thus, enantiomorphic crystals 1 were responsible for the present asymmetric Strecker reaction.
The ability of enantioenriched 2 to be hydrolyzed to the corresponding amino acid, α-(1-naphthyl)glycine (5),12d allowed chiral crystallization of achiral imine to be connected with production of enantioenriched α-amino acid via a Strecker reaction and asymmetric amplification.
Next, the retro-Strecker reaction of enantioenriched aminonitrile 2 was examined (Table 2). Guillemin et al. reported carrying out the dehydrocyanation of aminonitriles to give imines by utilizing KOH in vacuo.13 After the investigation, we found K2CO3 to be an appropriate solid base for the present transformation of N-benzhydryl substrates. Thus, enantioenriched 2 was ground into a fine powder with K2CO3 and the mixture was heated at 50 °C in vacuo. It was found to give imine 1 with the elimination of HCN and without any side reaction detected. After the removal of K2CO3, the optical properties and crystal structure of the resulting 1 were analyzed. Solid-state CD spectra (see the figure in Table 2) and a powder XRD pattern (Fig. S6†) indicated the formation of essentially the same crystal of 1 as that obtained from recrystallization from the solvent.
|
|
||||
|---|---|---|---|---|
| Entrya | % ee (config) of 2 | Aminonitrile 2 | ||
| ΔCDb | % eec | Yieldd | ||
| a Aminonitrile 2 was ground into a fine powder together with K2CO3 and heated at 50 °C in vacuo (<0.2 kPa). b Sign and value (mdeg) of the solid-state CD at 340 nm. c Each value was obtained from the corresponding calibration curve (Fig. S2). d Isolated yield (%) of solid 1. | ||||
| 1 | >99 (L) | +90 | 45 | 71 |
| 2 | 89 (D) | −89 | 45 | 70 |
| 3 | 67 (L) | +48 | 24 | — |
| 4 | 67 (D) | −55 | 28 | — |
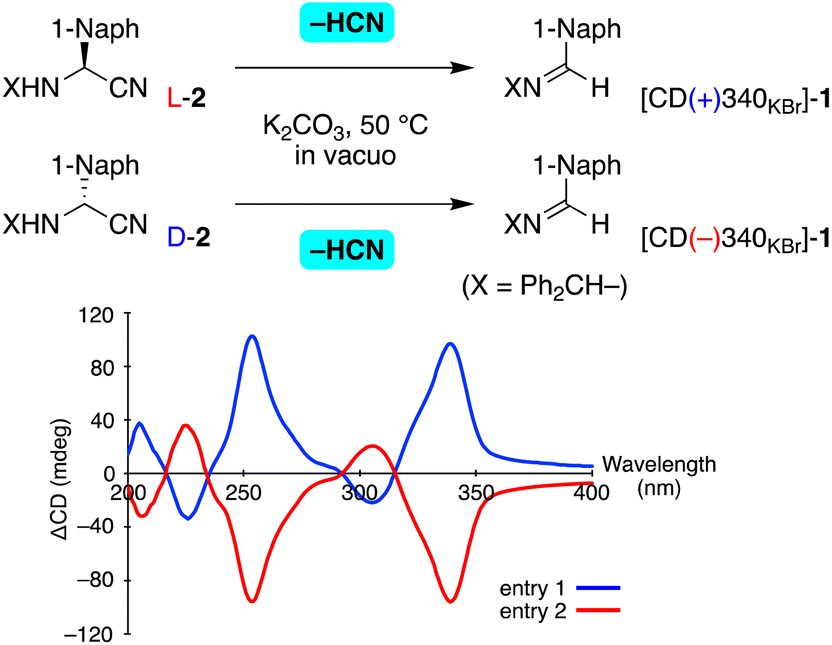
The stereochemical outcomes of this retro-Strecker reaction are summarized in Tables 2 and S4.† When L-2 with >99% ee was subjected to dehydrocyanation, imine 1 with positive CD at 340 nm was isolated in 71% yield (Table 2, entry 1). From the value of ΔCD (+90 mdeg), the crystal ee was calculated to be 45% (see also Fig. S2†). In contrast, [CD(−)340KBr]-1 with a ΔCD value of −89 mdeg (45% ee) was formed from D-2 with 89% ee (entry 2). As seen in entries 3 and 4, dehydrocyanation occurred stereoselectively to produce chiral crystal 1, even when using, respectively, L- and D-2, each with a slightly lower ee value of 67%. Thus, a chiral crystal of achiral imine 1 could be synthesized enantioselectively by carrying out a dehydrocyanation of enantioenriched 2.
An unprecedented reversal of chirality was observed in the present transformations (Fig. 1). That is, addition of HCN to [CD(−)340KBr]-imine 1 gave L-aminonitrile 2, and then dehydrocyanation of L-2 gave the opposite [CD(+)340KBr]-1. Thus, chiral crystallization-induced stereoinversion was realized between enantiomorphs of 1, and of course, between L- and D-2.
The current asymmetric cyanide addition and dehydrocyanation were checked using substrates 3 and 4 with the 2-furyl substituent17 (Fig. 3). Achiral imine 3 formed from furfural and benzhydrylamine (Fig. 3a) and crystallized in the chiral space group P212121 (Fig. 3b). The enantiomorphs of 3 were identified from a solid-state CD study, in which large positive and negative Cotton effects were observed at a wavelength of 300 nm (Fig. 3c).
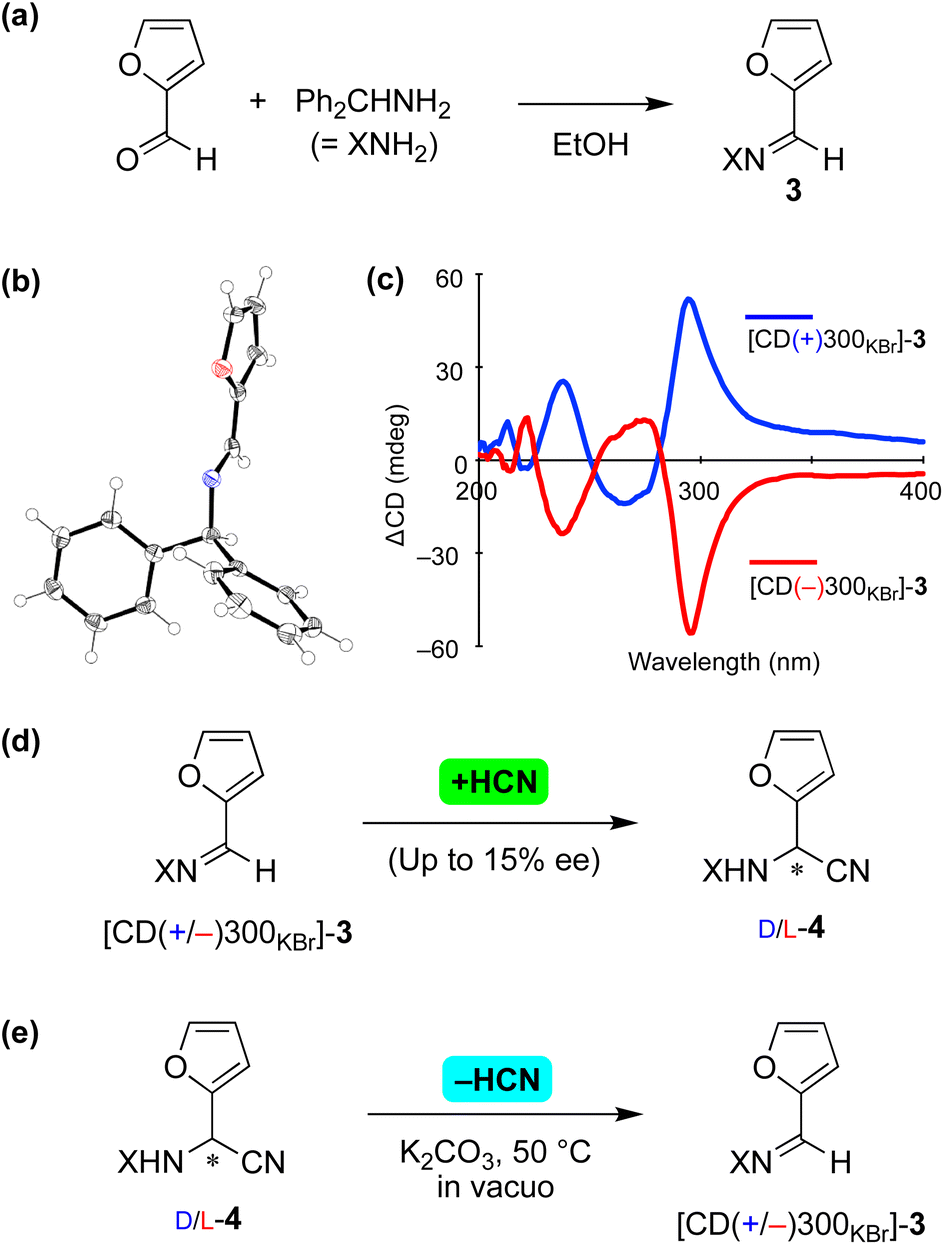 | ||
| Fig. 3 Reversible asymmetric Strecker reaction between imine 3 and aminonitrile 4 with a 2-furyl substituent. (a) Synthesis of imine 3. (b) Single-crystal X-ray structure of [CD(+)300KBr]-3 (CCDC 2224020†). (c) Solid-state CD spectrum of 3. (d) Asymmetric vapor-phase addition of HCN to imine 3. (e) Stereoselective retro-Strecker reaction of 4 (CCDC 2224021†). | ||
A chiral enantioenriched crystal of achiral 3 ([CD(−)300KBr]-3) was subjected to a reaction with vapor-phase HCN (Fig. 3d). When the reaction conversion was 6%, L-aminonitrile 4 with an ee of up to 15% was found to have formed. While allowing the reaction to continue further appeared to have resulted in a decrease in enantioselectivity, the stereochemical relationships between the enantiomorphic crystals 3 and the absolute configuration of 4 remained constant and reproducible, as shown in Table S5.†
Next, retro-Strecker reactions were investigated (Fig. 3e and Table S6†) using asymmetrically synthesized 4 (ref. 18) (Fig. S7†). Dehydrocyanations of L- and D-4 were found to give [CD(−)300KBr]- and [CD(+)300KBr]-3, respectively. The solid-state CD spectra (see the figure in Table S6†) and XRD pattern (Fig. S8†) of product 3 indicated the formation of essentially the same crystal 3 (P212121) as that obtained as a result of recrystallization from the solvent. Note that stereoinversion was not observed in the reactions between 3 and 4, even though cyanide addition and elimination occurred in an enantioselective manner.
In the crystal structures of 1 and 3, a weak interaction between the imine and adjacent aromatic rings of neighboring molecules19 was observed (Fig. 4). Thus, HCN might approach preferentially from the side of the molecule opposite these interactions, consistent with the stereochemical outcomes observed for the additions of cyanide to 1 and 3. Comparing the patterns of the CD curves of 1 and 3 indicated a similarity of their crystal systems, a feature supporting the assumption that asymmetric induction occurred in similar manners for the additions of HCN to 1 and 3.
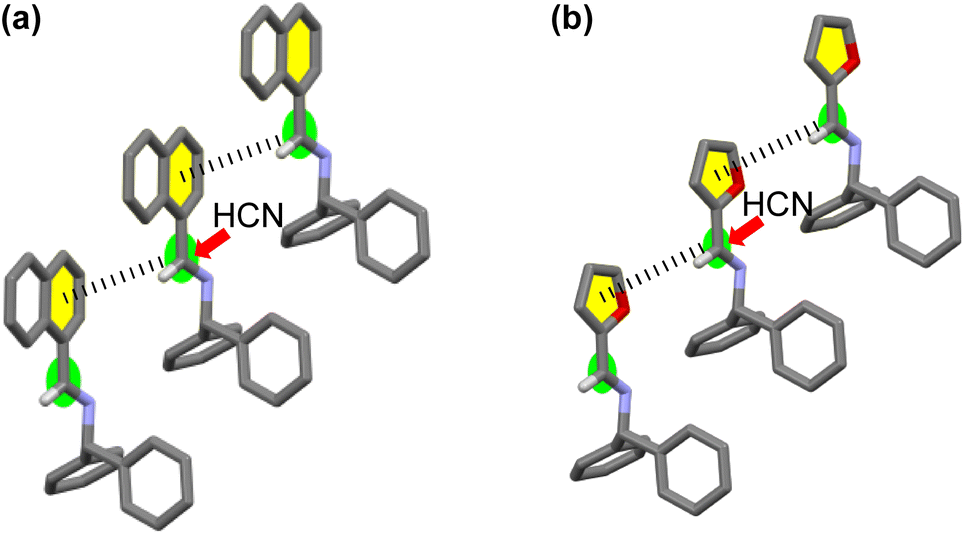 | ||
| Fig. 4 Enantioselectivity of the addition of HCN to (a) [CD(+)340KBr]-1 and (b) [CD(+)300KBr]-3 to form D-2 and -4, respectively. | ||
When dehydrocyanation of 2 was performed in the presence of 10 mol% of 1 with mismatched enantiomorphic crystals as seeds, enantiomorphic crystals of imine 1 with the same chirality as that of the seeds was formed in a highly enantioselective manner (Fig. S9†). Thus, the initial selective formation of the crystal of the imine may have occurred on the chiral surface of the aminonitrile, with this surface acting as a chiral seed for further dehydrocyanation. The mechanism of dehydrocyanations as a selective process is now under investigation.
Conclusions
In conclusion, asymmetric addition of cyanide to a chiral crystal of achiral imines has been demonstrated for the first time to the best of our knowledge. Moreover, asymmetric dehydrocyanation of enantioenriched aminonitrile was realized and stereoinversion of a chiral crystal formed from an achiral compound has also been achieved for the first time to the best of our knowledge. The present reversible asymmetric transformations, while requiring chiral crystallization (conglomerate formation) of the substrate, would nevertheless be expected to help investigators better understand the mechanism of abiotic synthesis of enantioenriched amino acids.Author contributions
T. K. conceptualized the project and designed the experiments. Y. M., Y. T., Y. M. and A. K. carried out the experiments and analyzed the data. T. K. wrote the manuscript with contributions from all authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
T. K. acknowledges support from JSPS KAKENHI (Grant Numbers JP16K05692 & JP19K22190). The authors thank Dr Yukana Terasawa for single-crystal X-ray structure analysis.References
- L. Pasteur, Ann. Chim. Phys., 1848, 24, 442–459 Search PubMed.
- (a) A. Guijarro and M. Yus, The origin of chirality in the molecules of life, The Royal Society of Chemistry, Cambridge, 2009; (b) K. Soai, Proc. Jpn. Acad., Ser. B, 2019, 95, 89–110 CrossRef CAS PubMed; (c) Q. Sallembien, L. Bouteiller, J. Crassous and M. Raynal, Chem. Soc. Rev., 2022, 51, 3436–3476 RSC; (d) D. G. Blackmond, Chem. Rev., 2020, 120, 4831–4847 CrossRef CAS PubMed; (e) A. J. Bissette and S. P. Fletcher, Angew. Chem., Int. Ed., 2013, 52, 12800–12826 CrossRef CAS PubMed; (f) C. Lee, J. M. Weber, L. E. Rodriguez, R. Y. Sheppard, L. M. Barge, E. L. Berger and A. S. Burton, Symmetry, 2022, 14, 460 CrossRef CAS.
- (a) J. L. Bada, Nature, 1995, 374, 594–595 CrossRef CAS PubMed; (b) I. Weissbuch and M. Lahav, Chem. Rev., 2011, 111, 3236–3267 CrossRef CAS PubMed; (c) D. K. Kondepudi, R. J. Kaufman and N. Singh, Science, 1990, 250, 975–976 CrossRef CAS PubMed; (d) C. Viedma, Phys. Rev. Lett., 2005, 94, 065504 CrossRef PubMed; (e) W. L. Noorduin, E. Vlieg, R. M. Kellogg and B. Kaptein, Angew. Chem., Int. Ed., 2009, 48, 9600–9606 CrossRef CAS PubMed; (f) T. Buhse, J.-M. Cruz, M. E. Noble-Teran, D. Hochberg, J. M. Ribó, J. Crusats and J.-C. Micheau, Chem. Rev., 2021, 121, 2147–2229 CrossRef CAS PubMed; (g) C. Viedma, J. M. McBride, B. Kahr and P. Cintas, Angew. Chem., Int. Ed., 2013, 52, 10545–10548 CrossRef CAS PubMed; (h) K.-H. Ernst, Phys. Status Solidi B, 2012, 249, 2057–2088 CrossRef CAS; (i) G. L. Rikken and E. Raupach, Nature, 2000, 405, 932–935 CrossRef CAS PubMed; (j) R. Naaman, Y. Paltiel and D. H. Waldeck, Nat. Rev. Chem., 2019, 3, 250–260 CrossRef CAS; (k) Y. Inoue, Chem. Rev., 1992, 92, 741–770 CrossRef CAS; (l) B. L. Feringa and R. A. van Delden, Angew. Chem., Int. Ed., 1999, 38, 3418–3438 CrossRef; (m) T. Kawasaki, M. Sato, S. Ishiguro, T. Saito, Y. Morishita, I. Sato, H. Nishino, Y. Inoue and K. Soai, J. Am. Chem. Soc., 2005, 127, 3274–3275 CrossRef CAS PubMed; (n) J. R. Cronin and S. Pizzarello, Science, 1997, 275, 951–955 CrossRef CAS PubMed; (o) I. Myrgorodska, D. Meinert, S. V. Hoffmann, N. C. Jones, L. Nahon and U. J. Meierhenrich, ChemPlusChem, 2017, 82, 74–87 CrossRef CAS PubMed; (p) V. A. Soloshonok, H. Ueki, M. Yasumoto, S. Mekala, J. S. Hirschi and D. A. Singleton, J. Am. Chem. Soc., 2007, 129, 12112–12113 CrossRef CAS PubMed; (q) G. Storch and O. Trapp, Nat. Chem., 2017, 9, 179–187 CrossRef CAS PubMed; (r) T. Kawasaki, Y. Matsumura, T. Tsutsumi, K. Suzuki, M. Ito and K. Soai, Science, 2009, 324, 492–495 CrossRef CAS PubMed.
- (a) T. Matsuura and H. Koshima, J. Photochem. Photobiol., C, 2005, 6, 7–24 CrossRef CAS; (b) M. Sakamoto, Chem.–Eur. J., 1997, 3, 684–689 CrossRef CAS.
- K. Penzien and G. M. J. Schmidt, Angew. Chem., Int. Ed., 1969, 8, 608–609 CrossRef CAS.
- (a) L. Addadi, J. Van Mil and M. Lahav, J. Am. Chem. Soc., 1982, 104, 3422–3429 CrossRef CAS; (b) M. Hasegawa, C. M. Chung, N. Muro and Y. Maekawa, J. Am. Chem. Soc., 1990, 112, 5676–5677 CrossRef CAS; (c) M. Sakamoto, M. Takahashi, K. Kamiya, K. Yamaguchi, T. Fujita and S. Watanabe, J. Am. Chem. Soc., 1996, 118, 10664–10665 CrossRef CAS; (d) H. Koshima, K. Ding, Y. Chisaka and T. Matsuura, J. Am. Chem. Soc., 1996, 118, 12059–12065 CrossRef CAS; (e) M. Vestergren, J. Eriksson and M. Håkansson, Chem.–Eur. J., 2003, 9, 4678–4686 CrossRef CAS PubMed.
- K. Soai, T. Shibata, H. Morioka and K. Choji, Nature, 1995, 378, 767–768 CrossRef CAS.
- (a) K. Soai, A. Matsumoto and T. Kawasaki, Isr. J. Chem., 2021, 61, 507–516 CrossRef CAS; (b) T. Kawasaki, K. Jo, H. Igarashi, I. Sato, M. Nagano, H. Koshima and K. Soai, Angew. Chem., Int. Ed., 2005, 44, 2774–2777 CrossRef CAS PubMed; (c) A. Matsumoto, H. Ozaki, S. Tsuchiya, T. Asahi, M. Lahav, T. Kawasaki and K. Soai, Org. Biomol. Chem., 2019, 17, 4200–4203 RSC.
- (a) A. Strecker, Ann. Chem. Pharm., 1850, 75, 27–45 CrossRef; (b) S. L. Miller, Science, 1953, 117, 528 CrossRef CAS PubMed; (c) K. Harada, Nature, 1963, 200, 1201 CrossRef CAS PubMed.
- (a) M. Shoji, N. Watanabe, Y. Hori, K. Furuya, M. Umemura, M. Boero and Y. Shigeta, Astrobiology, 2022, 22, 1129–1142 CrossRef CAS PubMed; (b) V. V. Kouznetsov and C. E. P. Galvis, Tetrahedron, 2018, 74, 773–810 CrossRef CAS; (c) P. Canavelli, S. Islam and M. W. Powner, Nature, 2020, 578, E22 CrossRef CAS PubMed; (d) A. J. Wagner, D. Yu. Zubarev, A. Aspuru-Guzik and D. G. Blackmond, ACS Cent. Sci., 2017, 3, 322–328 CrossRef CAS PubMed; (e) L. Legnani, A. Darù, A. X. Jones and D. G. Blackmond, J. Am. Chem. Soc., 2021, 143, 7852–7858 CrossRef CAS PubMed.
- J. Wang, X. Liu and X. Feng, Chem. Rev., 2011, 111, 6947–6983 CrossRef CAS PubMed.
- (a) T. Kawasaki, N. Takamatsu, S. Aiba and Y. Tokunaga, Chem. Commun., 2015, 51, 14377–14380 RSC; (b) S. Miyagawa, S. Aiba, H. Kawamoto, Y. Tokunaga and T. Kawasaki, Org. Biomol. Chem., 2019, 17, 1238–1244 RSC; (c) S. Aiba, N. Takamatsu, T. Sasai, Y. Tokunaga and T. Kawasaki, Chem. Commun., 2016, 52, 10834–10837 RSC; (d) S. Aiba, Y. Tanaka, Y. Tokunaga and T. Kawasaki, Bull. Chem. Soc. Jpn., 2019, 92, 1656–1661 CrossRef CAS; (e) S. Miyagawa, K. Yoshimura, Y. Yamazaki, N. Takamatsu, T. Kuraishi, S. Aiba, Y. Tokunaga and T. Kawasaki, Angew. Chem., Int. Ed., 2017, 56, 1055–1058 CrossRef CAS PubMed; (f) T. Kawasaki, H. Kubo, S. Nishiyama, T. Saijo, R. Yokoi and Y. Tokunaga, J. Am. Chem. Soc., 2021, 143, 19525–19531 CrossRef CAS PubMed; (g) Y. Yoshimura, Y. Tanaka, R. Kobayashi, K. Niikura and T. Kawasaki, Org. Biomol. Chem., 2023, 21, 520–524 RSC.
- (a) J. C. Guillemin and J. M. Denis, J. Chem. Soc., Chem. Commun., 1985, 951–952 RSC; (b) J. C. Guillemin, W. Nasraoui and H. Gazzeh, Chem. Commun., 2019, 55, 5647–5650 RSC.
- D. K. Kondepudi, J. Laudadio and K. Asakura, J. Am. Chem. Soc., 1999, 121, 1448–1451 CrossRef CAS.
- A. V. Tarasevych, A. E. Sorochinsky, V. P. Kukhar, L. Toupet, J. Crassous and J.-C. Guillemin, CrystEngComm, 2015, 17, 1513–1517 RSC.
- A. Lennartson, M. Vestergren and M. Håkansson, Chem.–Eur. J., 2005, 11, 1757–1762 CrossRef CAS PubMed.
- Synthesis of related compounds, see: N. Takamatsu, S. Aiba, T. Yamada, Y. Tokunaga and T. Kawasaki, Chem.–Eur. J., 2018, 24, 1304–1310 CrossRef CAS PubMed.
- V. Banphavichient, W. Mansawat, W. Bhanthumnavian and T. Vilavan, Tetrahedron, 2004, 60, 10559–10568 CrossRef.
- A. L. Pickering, G. Seeber, D.-L. Long and L. Cronin, CrystEngComm, 2005, 7, 504–510 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Additional experimental data, methods, synthetic and experimental procedures, characterization of synthesized compounds, supplementary tables and figures. CCDC 2224018–2224021. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3sc00359k |
| This journal is © The Royal Society of Chemistry 2023 |