
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEfficient light harvesting in self-assembled organic luminescent nanotubes†‡
Shubhra Kanti
Bhaumik
 a,
Dibyendu
Maity
b,
Ipsita
Basu
b,
Suman
Chakrabarty
*b and
Supratim
Banerjee
*a
a,
Dibyendu
Maity
b,
Ipsita
Basu
b,
Suman
Chakrabarty
*b and
Supratim
Banerjee
*a
aThe Department of Chemical Sciences, Indian Institute of Science Education and Research Kolkata, Mohanpur, Nadia 741246, India. E-mail: supratim.banerjee@iiserkol.ac.in
bDepartment of Chemical and Biological Sciences, S. N. Bose National Centre for Basic Sciences, Kolkata, India. E-mail: sumanc@bose.res.in
First published on 28th March 2023
Abstract
Luminescent organic nanotubes derived from the co-assembly of cyanostilbene (CS) based cationic supramolecular polymers and bio-polyanion heparin, a known anticoagulant, have been utilized as highly efficient FRET (fluorescence resonance energy transfer) donors in aqueous media resulting in amplified acceptor emission in the orange-red and near-infrared (NIR). Energy transfer efficiencies higher than 80% and an ultra-high antenna effect of 150 were achieved even at high donor/acceptor ratios (500![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1–100:1) translating to emission quenching of several hundred donors by one acceptor. Utilizing the temperature responsiveness of the FRET process, these systems were employed as ratiometric emission thermometers in the temperature range 20–90 °C. Moreover, the energy transfer was very effective in solid and polymer films. This allowed us to generate multi-color emissions ranging from blue to red including white light in solution as well as in solid and polymer films.
:1–100:1) translating to emission quenching of several hundred donors by one acceptor. Utilizing the temperature responsiveness of the FRET process, these systems were employed as ratiometric emission thermometers in the temperature range 20–90 °C. Moreover, the energy transfer was very effective in solid and polymer films. This allowed us to generate multi-color emissions ranging from blue to red including white light in solution as well as in solid and polymer films.
Introduction
In natural photosynthetic systems, sunlight is captured and subsequently transferred through highly efficient light-harvesting ensembles comprising of suitably aligned antenna chromophores.1,2 In recent years the creation of artificial light-harvesting systems has gathered a lot of attention as they are related to a number of practical applications such as solar cells,3 photocatalysis,4,5 optical sensors,6,7 and tunable multi-color light emitting materials.8,9 As artificial light-harvesting ensembles, conjugated polymers and dendritic architectures10,11 as well as micellar and vesicular systems, dye assemblies, host–guest complexes,12–24etc. have been employed. However, the requirement of a close-packed arrangement of the chromophores in artificial systems also increases the chance of self-quenching25 eventually leading to a detrimental effect on the light-harvesting efficiency.In recent years, biomolecular scaffolds such as DNA,26,27 peptides,28 sugars,29,30 viruses31,32etc. with covalently or non-covalently integrated dye molecules have been employed to achieve directional energy transfer (ET). We herein focus on another anionic biopolymer heparin for the design of artificial light-harvesting systems in aqueous media. Heparin is a known anticoagulant that is routinely utilized during surgeries to prevent blood clot formation and also for treating various heart, lung, and circulatory disorders with an escalated chance of blood clotting.33–35 It is the most negatively charge dense biopolymer and because of its therapeutic relevance, a variety of cationic chromophoric molecules have been developed for its detection.36–42 We have recently explored the multivalent binding of heparin to cationic luminescent 1D supramolecular polymers derived from amphiphilic pyridinium-cyanostilbene molecules with peripheral n-alkyl segments (NCSNs, Fig. 1, right panel).43–45 This provided a sensitive and selective method for the detection of the polyanion in aqueous buffer, serum, and plasma.44 Cyanostilbenes (CSs) are known for their enhanced emission properties in the aggregated state (AIE: aggregation-induced emission) and have been employed in functional materials including luminescent assemblies, optoelectronic materials, and bioimaging probes.46–48 In the presence of heparin, the CS-based supramolecular polymers formed larger co-assemblies with enhanced emission44 and rather intriguingly, the originally formed nanofibers were transformed into interconnected nanotubes in the case of two of the amphiphilic derivatives. We envisaged that these luminescent nanotubes would be excellent templates for the design of artificial light-harvesting systems as the CS luminophores are densely packed and yet possessed enhanced emissive properties.49,50 Although a few CS-based light harvesting systems are known in the literature,51,52 heparin-templated CS chromophores for such purpose have not been explored yet.
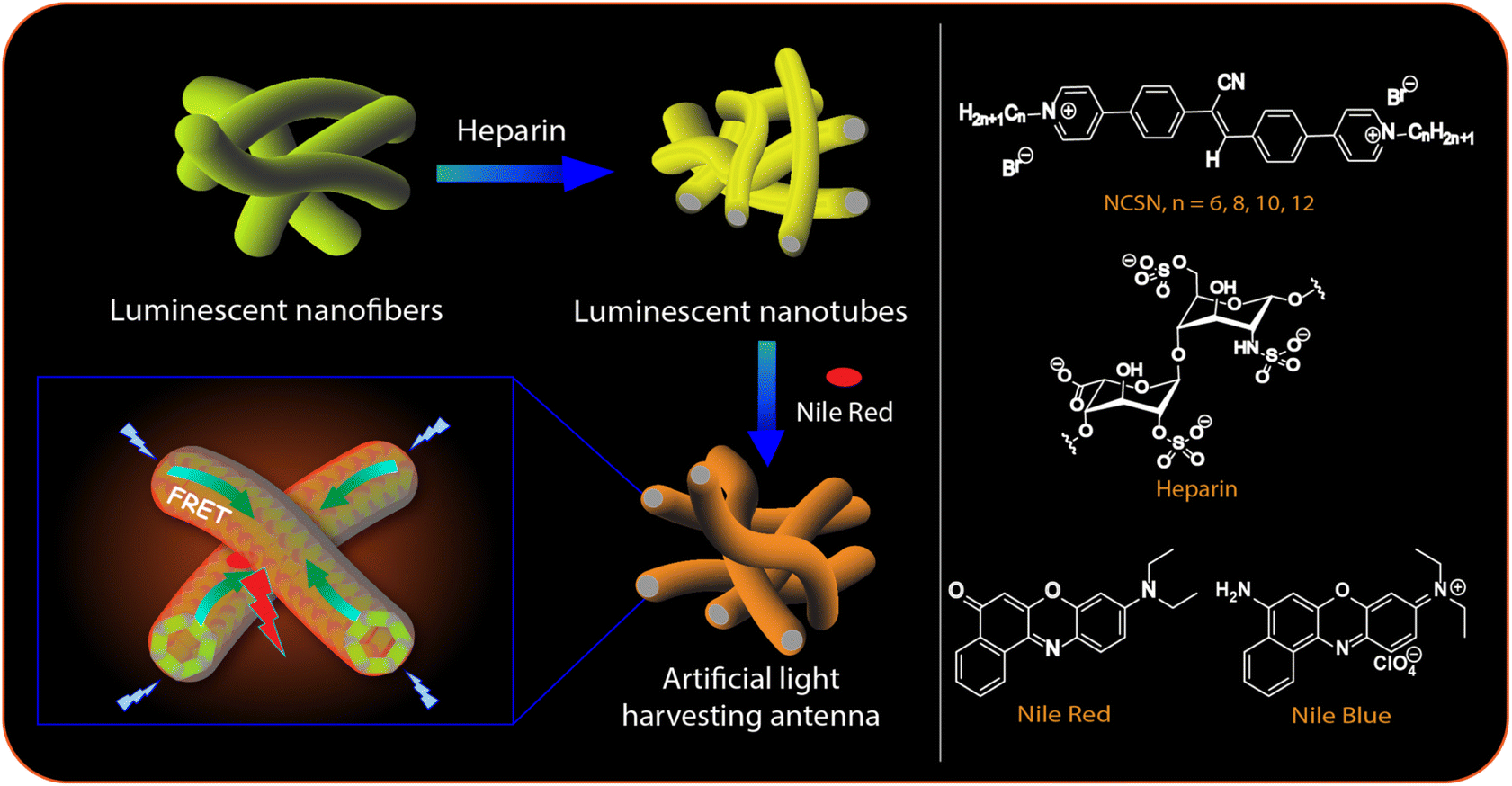 | ||
| Fig. 1 (Left panel) Schematic illustration of the FRET-based artificial light harvesting in NCSN–heparin-dye triad. (Right panel) Chemical structures of NCSNs (n = 6, 8, 10, 12), heparin, Nile Red and Nile Blue. | ||
Taking these features in perspective, we report here a Förster resonance energy transfer (FRET) system in aqueous media in which the CS chromophores in the nanotubes were utilized as donors to transfer the excitation energy to dyes with very high efficiency (up to 80%) and ultrahigh antenna effect (AE) ∼150. Such an excellent combination of high ET efficiency and AE is still uncommon in aqueous-based artificial light-harvesting systems.16–19,22–24 The ET was found to be efficient even in solid and polymer films. Hence, by using a variable composition of the components, we could achieve emission colors from blue to reddish-orange thus covering almost the whole visible range in the solution phase as well as in solid and polymer films. Moreover, the systems exhibited temperature-dependent FRET ON/OFF features enabling the design of ratiometric fluorescence thermometers exhibiting a linear response to temperature in the range 20–90 °C. We also present a detailed Molecular dynamics (MD) simulation study of the assembly formation from the monomers in the absence and presence of heparin and the results suggested the evolution of cylindrical morphology consistent with that observed from electron microscopy studies.
Result and discussions
Photophysical properties of the self-assemblies and heparin-based co-assemblies
The synthesis of the CS derivatives (Fig. 1) was earlier reported by us.43–45,53 We have shown previously that in aqueous buffer, 12CS12 and 10CS10 (Fig. 1) aggregated significantly at low micromolar concentrations to produce supramolecular polymeric nanofibers (referred as SP10 and SP12, respectively).43–45 Their aggregation was characterized by the appearance of greenish-yellow excimeric emission (average lifetime: 4.3–6.97 ns) at 540–545 nm (Fig. S1a and b, ESI‡) and this excimeric emission was not observed in the derivatives with n-hexyl or lower chain lengths.53 For example, 6CS6 (Fig. 1), which remained in a molecularly dissolved state in aqueous buffer, exhibited only monomeric emission (average lifetime: 0.45 ns) around 450 nm (Fig. S1d, ESI‡). The derivative, 8CS8, presented a borderline case as both monomeric and excimeric emissions were observed in its aqueous solutions (Fig. S1c, ESI‡). For all the cases, the emission intensity upon the heparin addition got saturated after 1–2 equiv. of heparin (Fig. S1, ESI‡). So, the length of the hydrophobic segment played a key role in the self-assembly of these molecules and dictated their critical aggregation concentrations as typically observed in the aggregation of amphiphilic molecules in water.54 For example, 12CS12 started aggregating ∼0.5–1 μM whereas the onset of aggregation was ∼2.5 μM for 10CS10 in buffer as observed from the dynamic light scattering (DLS) studies.44 When heparin was added to the aqueous solutions of the derivatives with chain lengths N ≥ 8 in fully or partially aggregated state, all of them formed larger co-assemblies (referred to as HSP8, HSP10 and HSP12, respectively) through multivalent mode of binding in which the polyanionic heparin backbone binds to the array of positive charges on the surface of these nanofibrous aggregates (Table S1, ESI‡).44 This was further supported by the fact that the initial positive zeta potential values of the aggregates gradually decreased and eventually became negative upon heparin addition (Table S1, ESI‡).44 This process resembles the electrostatically driven binding of oppositely charged polyelectrolytes in solution leading to a complete or partial neutralization of the charges. In this process, mobile counterions of the polyelectrolytes are released into the bulk solution and this contributes to the increase in translational entropy. This provides the major driving force as known for the association between DNA and cationic surfactants and lipids.55,56 Although there was no appreciable shift in the emission wavelength, the co-assembly formation with heparin led to a significant enhancement in the excimeric emission (Fig. S1a–c, ESI‡) along with noticeably longer lifetimes and higher quantum yields (Table S2, ESI‡). This suggested a more compact packing of the CS chromophores in the co-assemblies leading to a more efficient suppression of the non-radiative pathways compared to the native supramolecular polymers. In order to support the electrostatically driven co-assembly formation, we investigated its formation at lower pH values. The enhancement in the emission intensity of SP12 (5 μM) upon heparin (10 μM) addition at pH 2.0 was found to be significantly lower (Fig. S2, ESI‡) than that observed at pH 7.4 and this indicated the disruption the co-assembly formation at low pH. Interestingly, the thermal stability of the co-assembly HSP12 was found to be significantly higher than the supramolecular polymer SP12 as upon heating up to 90 °C, the co-assembly showed only a partial decrease in the excimeric emission whereas the emission of SP12 practically disappeared (Fig. S3, ESI‡). The images of the HSP12 co-assembly under UV at variable temperatures were found to be very similar (Fig. S3c, ESI‡) and this further suggested a reasonably high thermal stability of the HSP12 co-assembly.Interestingly, the native supramolecular polymeric nanofibers of SP10 and SP12 (25–90 nm diameters) were transformed into nanotubes of 15–40 nm of outer diameters in the co-assemblies HSP10 and HSP12 (Fig. 2a, e and S4, ESI‡) although it was not observed for HSP8. We then looked at the mechanism of the formation of these nanotubes through transmission electron microscopy (TEM). The formation of nanotubular structures from nanofibers typically occurs through a two-step process.57–59 The first step is the formation of nanosheets or nanoribbons owing to the side-by-side association of the nanofibers followed by the rolling up of the nanosheets which eventually transform into nanotubes.57–59 In order to gain a deeper insight into the morphological evolution, we tried to capture the morphology in the initial stage after heparin addition to these nanofibers (within first 5 min) by quenching the samples with liquid nitrogen at different time intervals.60 TEM images indeed revealed the presence of flattened surfaces and some rollup up flattened surfaces at the early stage of the co-assembly (Fig. 2b–d). Although the exact pathway of this morphological evolution is not clear at this moment, heparin seems to play a crucial role in the emergence of nanotubes from the native nanofibers (Fig. 2g) as in its absence, the existence of nanotubes was not detected from an aged sample of nanofibers. The inner diameters and wall thickness of the nanotubes were found to be 10–15 nm and 5–8 nm, respectively (Fig. 2f). Notably, even the addition of a small amount of heparin (1 μM) to the nanofibers of SP12 (5 μM, Fig. S4g, ESI‡) was able to induce this transformation. In order to gain more insight about the co-assembly, we recorded the circular dichroism (CD) spectra of SP12 and HSP12. However, not only the self-assemblies, the heparin-based co-assemblies were also CD silent (Fig. S5a, ESI‡). This suggested that in the co-assemblies, the CS chromophores resided in an environment in which no induction of chirality occurred from the chiral heparin backbone. We furthermore measured the Fourier transform infrared (FT-IR) spectra (freeze-dried samples) and powder X-ray diffraction (PXRD) data (solid films) of SP12 and HSP12 to discern any difference that might exist between them (Fig. S5b and c, ESI‡). Although no significant change in their FT-IR spectra (Fig. S5b, ESI‡) was observed, the PXRD data of HSP12 displayed a new peak at 2θ = 31.66° suggesting the presence of a new crystal plane (Fig. S5c, ESI‡). This indicated a slight variation in the molecular packing of the 12CS12 chromophores in the co-assemblies compared to their self-assemblies.
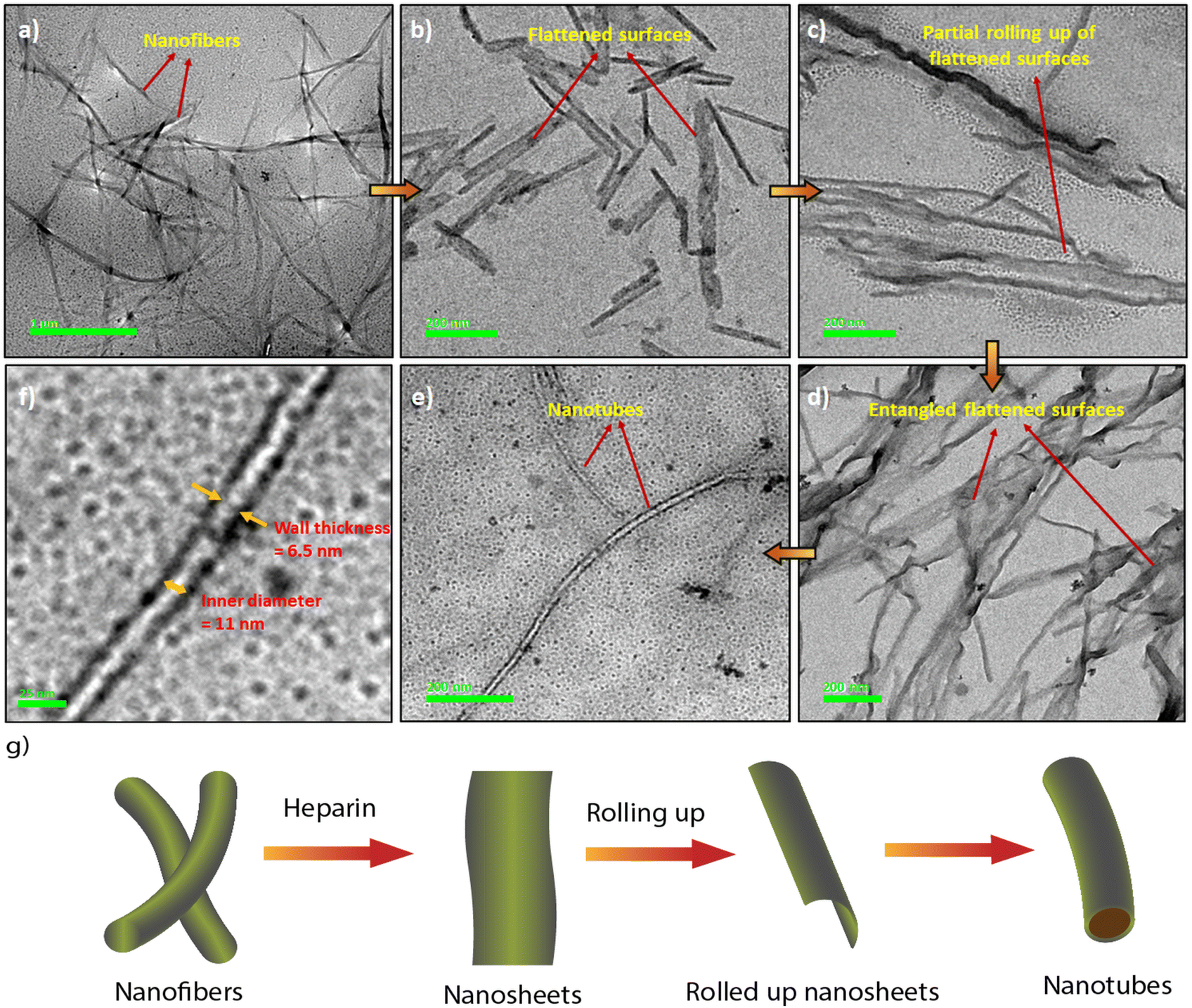 | ||
| Fig. 2 Transmission Electron Microscopic (TEM) images of (a) SP12 nanofibers (12CS12: 5 μM), (b–d) HSP12 (12CS12: 5 μM; heparin: 10 μM, recorded by quenching with liquid N2 at time intervals of 1 min, 3 min, and 5 min, respectively), (e) HSP12 (12CS12: 5 μM; heparin: 10 μM, recorded after air drying and drying under vacuum). (Inset) Illustration of a single nanotube. (f) TEM image of a single nanotube demonstrating the inner diameter and wall thickness. (g) Schematic illustration of the nanotube formation process from the nanofibers in the presence of heparin. | ||
Molecular dynamics (MD) simulation has emerged as a powerful technique to understand the molecular interactions, packing, and dynamic factors that lead to the formation of specific morphologies in self-assembled structures.61–65 In this work, we present atomistic classical MD simulation studies (computational details are provided on page S3, ESI‡) of the formation of self-assembled nanostructures for a few representative systems. Fig. 3 shows the time evolution of the system of 12CS12 molecules randomly dispersed in water both in the absence and presence of heparin. Both systems show the rapid formation of rod-like aggregates within the simulation timescale of 500 ns. Interestingly, for the heparin-containing system, the 12CS12 molecules were found to get attached to the charged heparin polymer rapidly and form a cylindrical structure in which the heparin got fully covered by the 12CS12 molecules. The driving force for the 12CS12 molecules binding to heparin is primarily electrostatic interaction as reported earlier for other charged (cationic) dye molecules.64,65
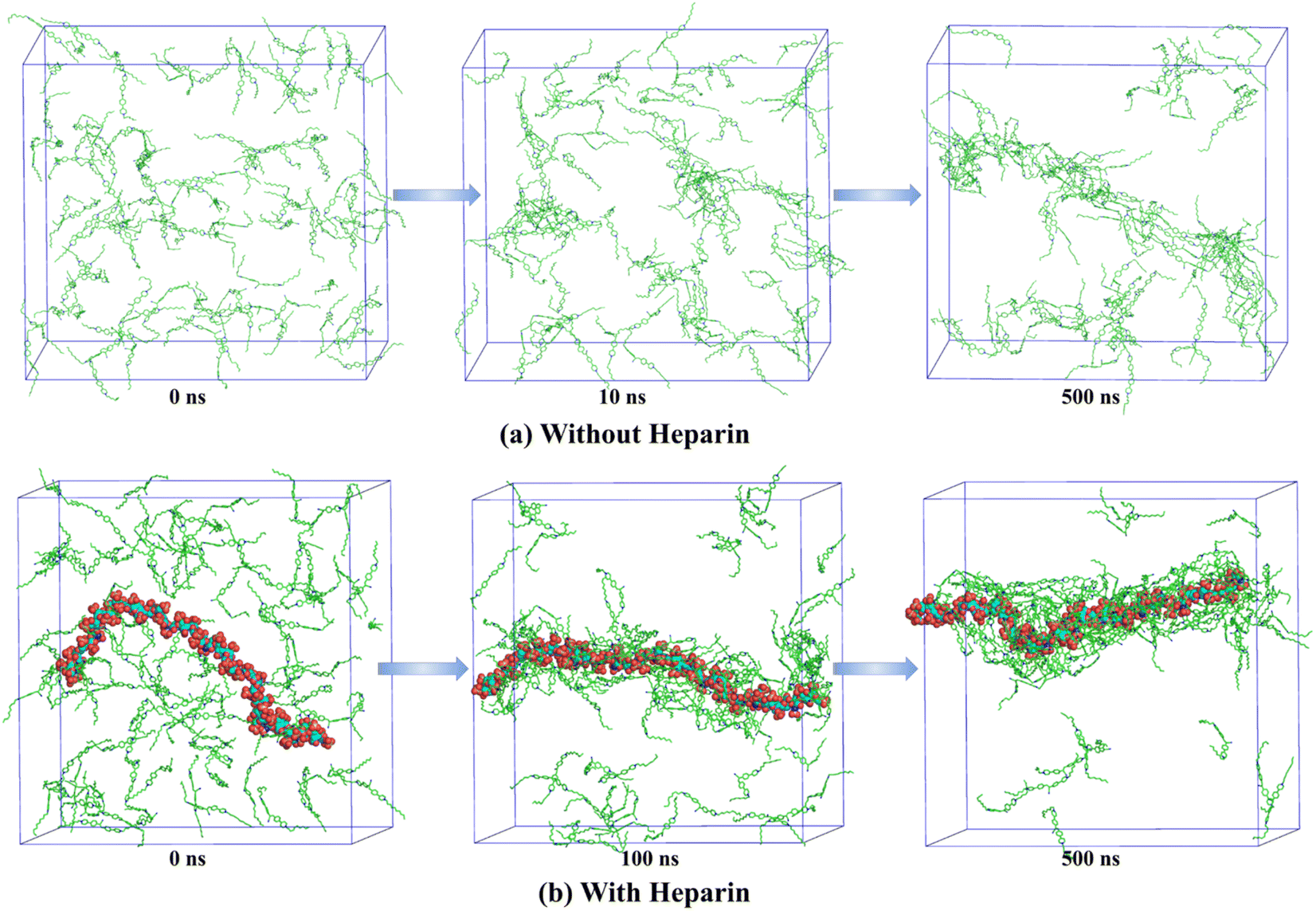 | ||
| Fig. 3 Time evolution of aggregation of 12CS12 dyes in the systems (a) without heparin and (b) with heparin. The water molecules and ions are not shown for visual clarity. | ||
If we further dive down into the structural details of the clusters formed, we can see that they are mostly cylindrical in shape. In order to characterize the shape of these clusters/aggregates, we have used two shape order parameters: asphericity and acylindricity that can be derived from the eigenvalues of gyration tensor as discussed in ESI (page S6‡). For both the systems (with and without heparin), this analysis clearly revealed predominantly cylindrical symmetry in the clusters formed (Fig. S6, ESI‡).
Next, we paid attention to the nature of the packing of the 12CS12 molecules present within the clusters. In order to quantify this, we have utilized two geometric parameters: (i) the relative angle between the aromatic core region of the two neighboring 12CS12 molecules (Fig. 4a), and (ii) the extent of alignment in a parallel stacked configuration (Fig. 4b–d). Fig. 4a represents the probability density of the cosine of the above-mentioned angle, and from Fig. 4a also we can see two sharp peaks at θ = 0° and θ = 180° that correspond to parallel and antiparallel orientations, respectively. These two orientations have equal probability due to the lack of directionality in this molecule, as expected.
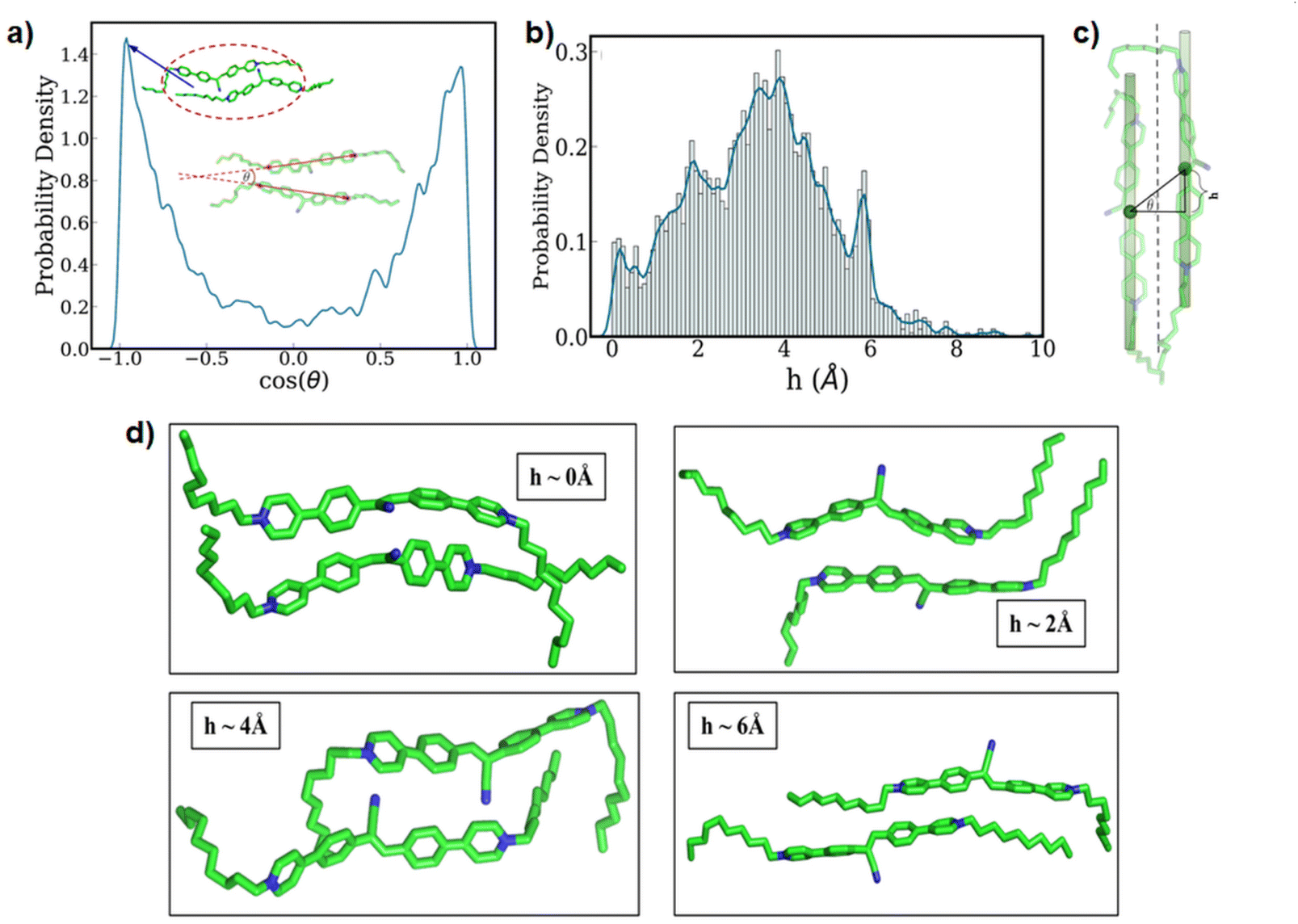 | ||
| Fig. 4 (a) The relative orientation of the neighboring 12CS12 molecules: the probability density of the cosine of the angle between two neighboring molecules is shown here. A representative snapshot of the parallel stacking is marked inside the plot. The definition of the angle used here has been shown pictorially. (b) Probability density of the shift of the center of mass (h) of molecules in the parallel stacking and (c) schematic of the shift. (d) Different snapshots of the molecular packing from the peaks were obtained from the probability density of the shift of the center of mass (h) of molecules. | ||
While the relative orientation confirms the predominantly parallel stacking of the 12CS12 molecules, visual inspection suggests the presence of a lateral shift in the arrangement. We have geometrically quantified the extent of this shift as shown in Fig. 4c, and computed the probability distribution of the same (Fig. 4b). Here h corresponds to the magnitude of the lateral shift. The distribution clearly indicates that the most probable configuration corresponds to a lateral shift of ∼4 Å. Interestingly, the multi-modal nature of the distribution shows the relative population of several possible stacking configurations and representative snapshots of such arrangements corresponding to each peak have been shown in Fig. 4d. We can visually understand that the presence of multiple aromatic rings leads to several distinct possibilities of π-stacking interactions between the 12CS12 molecules in these different arrangements. Overall, the shifted arrangements are always more populated than the perfectly aligned configurations (h = 0). It is pertinent to note here that earlier computational studies have suggested that both stacking and non-stacking binding modes are possible depending on the nature of the specific dye molecule binding to heparin.65
In general, MD simulation has limitations in the length and timescales that can be accessed within a reasonable computer time. The simulation box (length) is normally <10 nm, and the timescale is limited to a few microseconds. Hence, it is not always possible to achieve a direct or exact correspondence between the experimental observations and simulated results. However, we noted that the wall thickness of the nanotubes was around 5–8 nm (from the TEM images) whereas the average diameter of the cylindrical co-assembly as obtained from the MD simulations was 2.46 ± 0.49 nm. This suggested that the wall of the nanotubes was possibly consisted of 2–3 layers of the cylindrical co-assemblies. Additionally, the simulation results have provided significant microscopic insights about the early stages of the self-assembly and co-assembly processes in this study. The local molecular level interactions and packing that leads to the overall anisotropic morphologies can be directly visualised and quantified through our simulations.
Energy transfer studies using self-assembled nanofibers
For the FRET studies, we employed the native aggregates of 8CS8, 10CS10, and 12CS12 (SP8, SP10, and SP12, respectively) and their heparin-based co-assemblies (HSP8, HSP10, and HSP12, respectively) as the donors (D). As acceptors, we explored four dyes: Rhodamine B (RhB), Rhodamine 6G (Rh6G), Nile Red (NR), and Nile Blue (NB) (Chart S1b and c, ESI‡ and Fig. 1). They displayed absorption maxima at 553 nm, 527 nm, 587 nm, and 635 nm, respectively in aqueous buffer (5 mM tris-HCl, 99:1 water/DMSO, pH 7.40, Fig. S8 and S18, ESI‡), and exhibited excellent spectral overlaps between their absorption spectra and the emission spectra of the supramolecular polymeric nanofibers (SPs) as well as the heparin based co-assembled (HSPs) as provided in Table S3, ESI.‡ We first examined the ET from SP8, SP10, and SP12 in aqueous buffer, with NR and NB as the acceptor dyes, as reference systems. Both NR and NB are weakly emissive in aqueous media,66–68 but they emit more efficiently in hydrophobic environments or in non-aqueous media.69,70 First, we investigated ET from SP10 and SP12 (the concentration of 10CS10 and 12CS12 was 5 μM, Fig. S9a and b, ESI‡) employing NR as the acceptor. A reasonably efficient ET was observed even at high donor–acceptor ratios (500:1 to 100:1). ET led to the emergence of new emission peaks in the range of 616–626 nm which was accompanied by a reduction in the excimeric emission of the SPs (Fig. S9a and b, ESI‡). The ET efficiency at a 100:1 donor/acceptor ratio was found to be 36.7% and 52.4% for SP10 and SP12, respectively (Tables S4 and S5, ESI‡). Similarly, with NB, reasonable ET efficiencies (100:1 D/A ratio) of 40% and 37% were noted for SP10 and SP12, respectively (Fig. S13 and Tables S8, S9, ESI‡). The NB emission appeared at 660–665 nm in the presence of SPs.
The efficiency of a FRET system is also evaluated in terms of AE which reflects the amplification of the acceptor emission upon donor excitation relative to its direct excitation. To calculate the AE values, we excited SP10 and SP12 in the presence of NR or NB at 365 nm (λmax of the donor) as well as at 586 nm (λmax of NR) or 635 nm (λmax of NB) and their corresponding emissions were recorded (see page S4 in ESI‡). The AE values were found to be reasonably high even at high D/A ratios. For example, the AE values at 500:1 D/A ratio were 79 and 95 (for NR) with SP10 and SP12, respectively (Fig. S10, S11 and Tables S4, S5, S7, S8, ESI‡). A simple variation in the emission color from greenish-yellow to orange-red was possible by increasing the concentration of NR in SPs (Fig. S9b, ESI‡). In comparison, for SP8, significantly poorer ET efficiencies were observed (Fig. S14b, S15b and Table S10, ESI‡). As a control, we also studied ET from monomeric 6CS6 (Fig. 1) to NR and NB. Only negligible ET was observed in this case (Fig. S14a, S15a and Table S10, ESI‡). The poor ET efficiencies observed for 8CS8 and 6CS6 clearly indicated the importance of the extended network of aggregates as provided by SP10 and SP12.
Energy transfer studies using heparin-based co-assemblies
We subsequently investigated ET from the co-assemblies HSP8, HSP10, and HSP12 (Fig. 5a and S19b, c, ESI‡). Initial studies with HSP12 (12CS12: 5 μM; heparin: 10 μM) and NR at different D/A ratios showed excellent ET efficiencies (Fig. 5a and Table S15, ESI‡) resulting in the tuning of the emission color from greenish-yellow to reddish-orange (Fig. 5b). Further investigations revealed that the ET efficiency was dependent on the concentration as well as the ratio of 12CS12 and heparin in the HSP12 co-assemblies (Fig. S20 and Table S16, ESI‡). A similar trend was also observed for the HSP8 and HSP10 (Fig. S21 and Table S17, ESI‡). A 1:2 ratio of the CS derivatives and heparin with their respective concentrations at 5 μM and 10 μM were utilized for all the subsequent studies as it provided the best results (see Fig. S20, S21 and Tables S16, S17, ESI‡). When NR was employed as the acceptor, HSP12 was found to be the most efficient in terms of ET efficiency among the three co-assemblies (Fig. 6a and Table S18, ESI‡). At a D/A ratio of 100:1, the ET efficiency for HSP8, HSP10, and HSP12 was 45.75%, 50%, and, 80.3%, respectively (Table 1). The ET phenomenon led to a decrease in the average lifetime of the donor emission in the presence of NR (Table 1 and Fig. S22, ESI‡). For example, the average lifetime of the excimeric band (λem = 542 nm) for HSP12 decreased from an initial value of 9.98 ns to 3.71 ns at a D/A ratio of 100:1 (Table 1). The excitation spectra collected for the emission at 625 nm of the NR-loaded HSP12 co-assembly (D/A ratio of 100:1) displayed only a peak at 370 nm, characteristic of the absorption spectra of the HSP12 (Fig. S23, ESI‡) while no peak was observed at the absorption maxima of NR at 586 nm. This supported the FRET process. Studies with NB as the acceptor dye also displayed efficient ET (Fig. S22, S24 and S25b, ESI‡). Fluorescence lifetime imaging microscopy (FLIM) has been proven to be a useful technique to characterize morphological transformations, to probe FRET, to examine interaction of dyes in lipid vesicles, etc.71–74 In order to obtain temporally and spatially resolved fluorescence profiles, we further performed FLIM for the self-assembly, co-assembly and the co-assembly in the presence of acceptor using 10CS10 (Fig. 5d–f). FLIM images displayed anisotropic nanofibers for SP10 having lifetimes of 3.8 ns and 4.8 ns (Fig. 5d). For the co-assemblies HSP10 and HSP10-NR (D/A = 50:1), cylindrical nanostructures were observed (Fig. 5e and f). Interestingly, we noted that for the HSP10 co-assembly with NR, two lifetimes of 1 ns and 2.3 ns were observed. Presumably, the 1 ns lifetime corresponds to the reduced lifetime of the donor due to the FRET process whereas the higher one corresponds to NR.
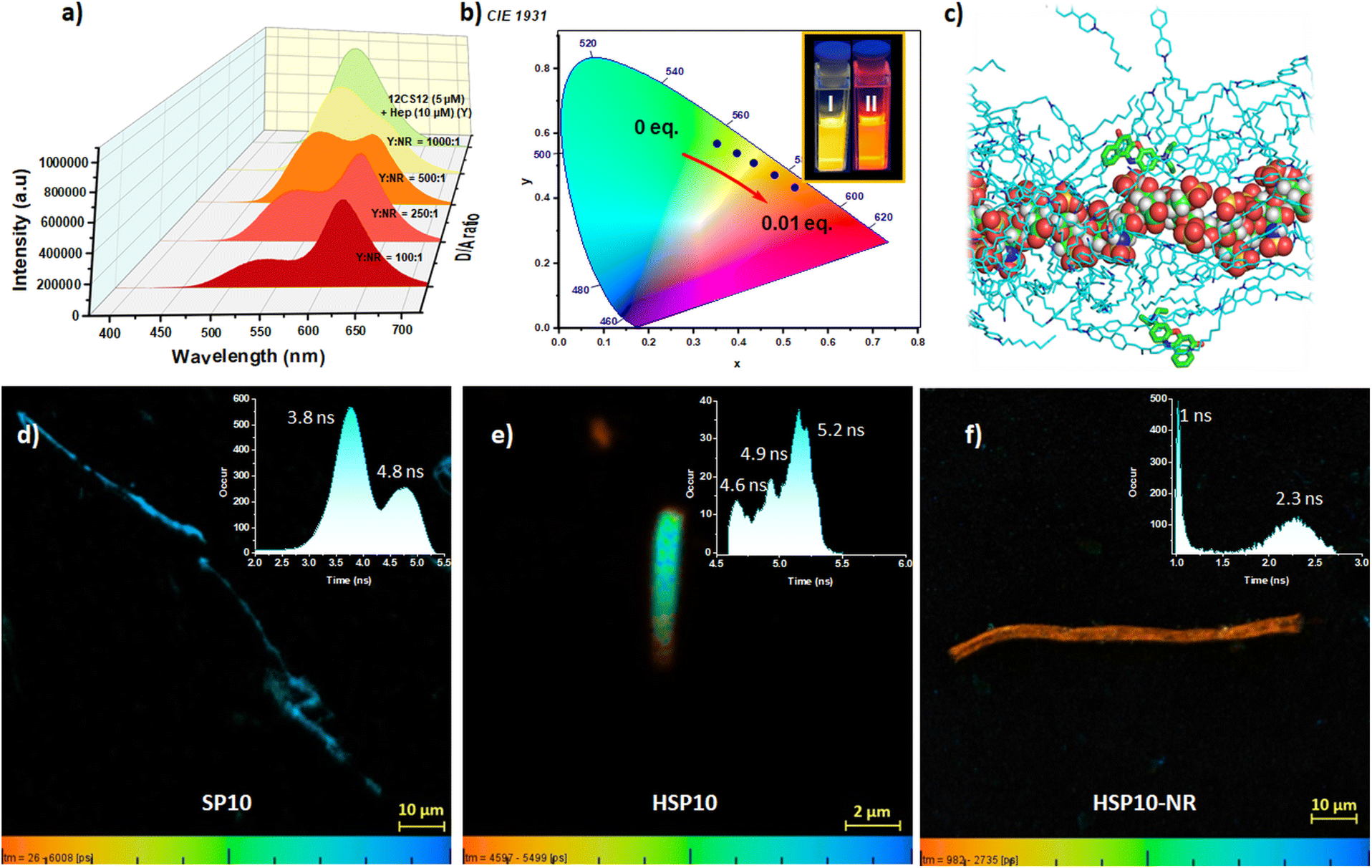 | ||
| Fig. 5 (a) Emission spectral changes of HSP12 [12CS12 (5 μM)–heparin (10 μM), Y] upon addition of Nile Red (NR) in aqueous buffer (λex = 365 nm). (b) CIE diagram displaying the transition of emission colour of HSP12 [12CS12 (5 μM)–heparin (10 μM), Y] from greenish yellow to orange red in the presence of NR (0 to 0.01 eq.). (Inset) Image of HSP12 [12CS12 (5 μM)–heparin (10 μM), Y] (I) without and (II) with NR under UV 365 nm. (c) A snapshot showing the location of the Nile Red molecules bound around heparin in the 12CS12–heparin co-assembly. Here the heparin and 12CS12 molecules are shown in sphere and stick representations, respectively. FLIM of (d) SP10 (40 μM), (e) HSP10 [10CS10: 40 μM; heparin: 80 μM] and (f) HSP10-NR [10CS10: 40 μM; heparin: 80 μM; NR: 0.8 μM; D/A = 50:1] in aqueous buffer (λex = 405 nm). Insets show corresponding lifetime histograms. | ||
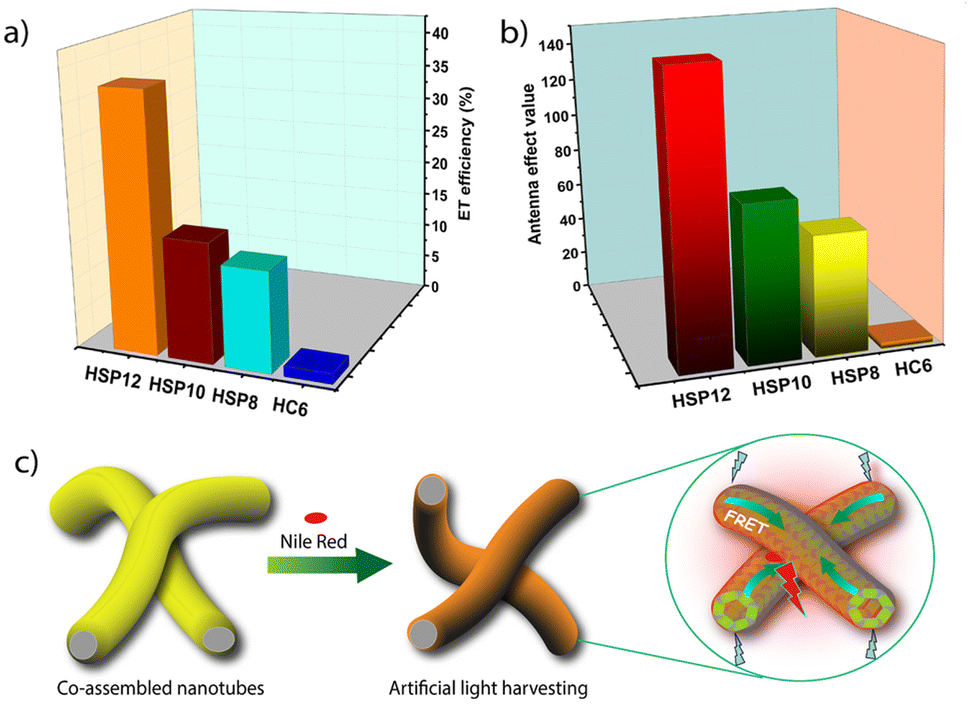 | ||
| Fig. 6 Bar diagram comparing (a) ET efficiencies and (b) AE values in the energy transfer process from HC6, HSP8, HSP10 and HSP12 to NR (D/A ratio = 500:1) in aqueous buffer. (c) A schematic representation of the light harvesting process. | ||
| NCSN–heparin conjugate | τ avg w/o-NRa (ns) | τ avg w/-NRa,b (ns) | ET efficiencyb (%) | AEc |
|---|---|---|---|---|
|
a
λ
em = 540 nm.
b Donor/acceptor ratio = 100:1.
c Donor/acceptor ratio = 500:1.
|
||||
| 6CS6 (5 μM)–heparin (10 μM) | 19.73 | 19.7 | 11.40 | 3 |
| 8CS8 (5 μM)–heparin (10 μM) | 19.74 | 16.51 | 45.75 | 62 |
| 10CS10 (5 μM)–heparin (10 μM) | 17.36 | 11.78 | 50.00 | 81 |
| 12CS12 (5 μM)–heparin (10 μM) | 9.98 | 3.71 | 80.30 | 149 |
The AE values were also found to be higher for HSP12 compared to the other co-assemblies. Notably, at a D/A ratio of 500:1, the AE value for HSP12-NR was found to be 149 whereas the corresponding values for the HSP8-NR and HSP10-NR were 62 and 81, respectively (Table 1 and Fig. 6b). Similarly, high AE values were obtained for HSP-NB triads (Table S22, ESI‡). So, both in terms of the ET efficiency and AE, the co-assembled nanotubes showed much better ET efficiency and AE than the native supramolecular polymeric nanofibers. Other than a few reports on light-harvesting in dye-encapsulated polymeric nanoparticles,14,15 these are some of the highest values reported so far in aqueous self-assembled systems.16–19,22–24 The derivative 6CS6, which essentially stayed in a monomeric state, formed small clusters on the heparin surface exhibiting similar excimeric emissions as observed for the co-assemblies as found from our earlier investigations.53 As a reference system, we employed the HC6 clusters (6CS6: 5 μM and heparin: 10 μM) as the energy donor (Fig. S19a and S25a, ESI‡). The ET efficiencies were found to be noticeably lower (Tables 1 and S18, ESI‡).
Energy transfer studies with the rhodamine dyes
We subsequently employed the two Rh dyes RhB and Rh6G as the acceptors. Although they displayed comparable theoretical values of the spectral overlap integral as that of NR and NB (Fig. S8, S18 and Tables S3, S14, ESI‡), much poorer ET efficiencies were observed for them. Only at a D/A ratio of 5:1, moderate ET efficiencies of 28.6% and 34.8% were observed for RhB and Rh6G with their characteristic emission peaks appearing at 574 nm and 558 nm, respectively (Fig. S32, S33 and S26, ESI‡). The poor AE values suggested that other than FRET to the acceptor, the donor emission was quenched through other non-radiative pathways in the presence of the acceptors (Table S27, ESI‡). As a result, the emission color of HSP12 could only be tuned from greenish-yellow to yellow in the presence of RhB and Rh6G (Fig. S34, ESI‡).
Rationalization of the observed energy transfer
The aforementioned observations led to a few important questions: (a) why NR and NB were better acceptors than the Rh dyes? and (b) why did the co-assemblies show better ET efficiency than the native supramolecular polymers? In order to rationalize the first query, we looked at different factors which are expected to influence the ET event. The proximity of the dyes to the CS chromophores in the assemblies is an important factor. In order to examine the extent of the integration of the dyes in the assemblies, we recorded NR and NB emission spectra at their direct excitation (λex = 586 nm and 635 nm, respectively) with or without the co-assemblies HSPs. The original emission peaks of NR (655 nm) and NB (670 nm) in the buffer underwent clear hypsochromic shifts along with significant enhancements in their intensity in the presence of HSPs and the most pronounced enhancement was noted in the case of HSP12 (Fig. S30, ESI‡). The negative solvatofluorochromism of the NR and NB emission75,76 reflected their embedding in a comparatively more hydrophobic environment provided by the co-assemblies. We also recorded the steady-state emission anisotropy values of NR and NB (direct excitation at their corresponding absorption maxima) in the presence of HSPs. The values were noticeably higher in the presence of HSP8, HSP10, and HSP12 (Table S23, ESI‡) compared to those observed in the buffer. For example, in the presence of HSP12, the anisotropy values of NR and NB were 0.176 and 0.186, respectively whereas the corresponding values were 0.066 and 0.055 in the buffer. This suggested that NR and NB were integrated well in the co-assembled nanotubes and resided in a much more rigid environment. MD simulation also suggested that the NR molecules were intermittently bound around heparin, and they are mostly trapped within the 12CS12 clusters (Fig. 5c and S7, ESI‡).77 We also observed that HSP10 and HSP12 showed reasonably high emission anisotropy values for their excimeric emissions (λex = 365 nm) which remained almost unchanged or increased only slightly in the presence of NR and NB (Table S24, ESI‡). This suggested a collinearity between the dipole moment of the electronic transition and the long axis of the CS aggregate.78–80 in the HSP co-assemblies. However, the emission anisotropy values of NR and NB were low when excited at the donor absorption maximum (λex = 365 nm, Table S25, ESI‡). This suggested that the NR and NB were not arranged in a co-linear fashion with the CS chromophores leading to a depolarization of the emission during the ET process. The Rh dyes, on the other hand, displayed poor emission anisotropy values even in the presence of HSP12 (exited at their absorption maxima, Table S28, ESI‡) possibly due to their higher water solubility. Therefore, these experiments suggested that a better integration of NR and NB in the co-assemblies compared to Rh dyes presumably led to a higher ET efficiency in their cases. We subsequently tried to understand why the co-assemblies acted as better donors than the native supramolecular polymers. The spectral overlap integral values (J(ε)) between the absorption spectrum of NR and the emission spectrum of the co-assembly HSP12SP12 was only slightly higher than that observed for SP12. However, the radiative rate constant (kr) for HSP12 was found to be 1.5-fold higher than SP12 (see pages S8 and S14 in ESI‡ for the calculation of kr).81,82 This, among other factors, might have contributed to a better ET efficiency for the HSPs compared to native SPs.The calculated Förster radii (R0) for the ET from HSP12 and SP12 to NR were found to be 3.9 nm and 3.4 nm, respectively (see pages S8 and S14 in ESI‡ for the calculation of R0). However, the excellent ET efficiency even at high D/A ratios suggested that FRET occurred far beyond the Förster radius (R0) and one acceptor was able to quench the emission of multiple donor molecules. The self-aggregate and the co-assembly formed highly entangled networks which in principle, would assist in a long-range exciton migration.83 This would involve a significant homo-ET among the CS donors before the hetero-ET to the acceptor (NR) took place. Therefore, to determine the number of CS donors (n) taking part in ET per antenna, we employed a mathematical model combining both dynamic quenching (for homo-ET) and static quenching (hetero-ET) mechanisms.84,85 We assumed that n donor molecules were quenched by one acceptor and the (donor)n was treated as a single unit. By modelling the quenching by a 1:1 binding isotherm and the non-linear fitting of the curve of the emission intensities of HSP12vs. concentration of NR (Fig. S31, ESI‡), a value of 516 ± 182 was obtained as the number of CS donors per antenna.84,85
Application as fluorescent thermometers
Fluorescent materials with temperature-dependent dual emission are of great interest as ratiometric fluorescent thermometers.86–88 As a consequence of FRET, HSP12-NR and HSP12-NB displayed dual emissions and we studied their response against temperature in the range 20–90 °C. Interestingly, with increasing temperature, the donor emission at 545 nm gradually increased whereas a significant reduction in the acceptor emission was observed (Fig. 7a, b and S35a, b, ESI‡). As mentioned earlier that the co-assemblies were reasonably robust and only partial disassembly was observed in going to higher temperatures. Hence, this behaviour can be attributed to the temperature-induced change in the transition dipole orientations of the acceptors89 which diminished the FRET appreciably leading to an apparent gain in the donor emission. Moreover, a partial disintegration of the acceptor dyes from the co-assemblies could be another reason for the reduced FRET efficiencies at higher temperatures. Linearly fitted curves of I545/(I545 + I635) vs. temperature (for HSP12-NR) and I545/(I545 + I677) vs. temperature (for HSP12-NB) were obtained (Fig. 7c and S36, ESI‡). For HSP12-NR, the curve consisted of two linear regions, and thus for this system, the temperature sensitivities were different. The temperature sensitivity between 20 °C and 40 °C was calculated to be ∼0.2% whereas the value increased to ∼0.8% between 40 °C and 90 °C.88 For HSP12-NB, a value of ∼0.7% temperature sensitivity was obtained in the range 20 °C to 90 °C.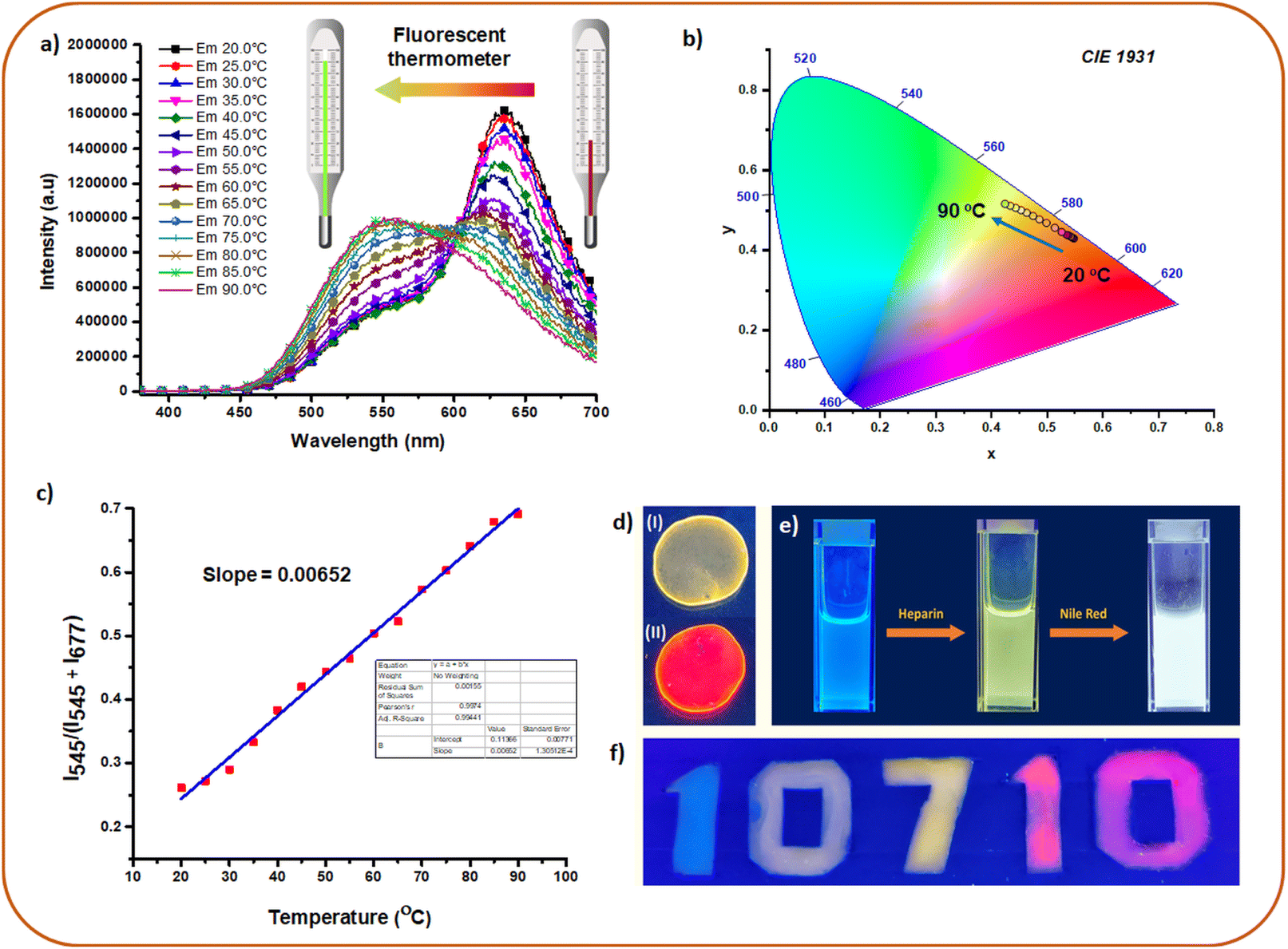 | ||
| Fig. 7 (a) Emission changes of HSP12-NR [12CS12: 5 μM; heparin: 10 μM; NR: 100 nM] at different temperatures demonstrating fluorescence thermometer application. (b) CIE co-ordinates for the plots in (a). (c) Linear fitted plot of I545/(I545 + I677) vs. temperature (for HSP12-NB) in aqueous buffer. (d) Images of solid films of (I) HSP12 [12CS12 (20 μM)–heparin (40 μM)] and (II) HSP12-NR [12CS12 (20 μM)–heparin (40 μM)-NR (200 nM)] under UV 365 nm. (e) Generation of white light emission from blue emitting 8CS8 (10 μM) in PAA (10 mg mL−1 in buffer) using heparin followed by NR. (f) Demonstration of fluorescent ink. For exact composition of each of these samples, see pages S25 and S26 in the ESI.‡ | ||
Application in multicolor luminescence in solution and solid state
The advantage of our system was further demonstrated through the generation of multi-color emissions in the solid state and polymer films. This was possible since the ET was also found to be efficient in the film state. Thus, the greenish-yellow emission of HSP12 could be tuned to red in the presence of NR (D/A ratio 100:1, Fig. 7d) in its solid film. The obtained AE value was 33 in this case (Fig. S37, ESI‡). Moreover, ET from the native SP12 (12CS12: 20 μM) to NR in its solid film was also found to be efficient while exhibiting an AE value of 21 (Fig. S39, ESI‡). Lower AE values in the solid films compared to the aqueous solutions of SPs and HSPs can be attributed partly to the self-aggregation of NR molecules leading to quenching of its emission.70,90,91 For generating a wide array of emission colors in the solution as well as in films, we chose the weakly aggregating solution of 8CS8 (SP8) and its co-assembly with heparin (HSP8) in the presence of polyacrylic acid (PAA, 10 mg mL−1) taken in buffer. SP8 exhibited a cyan-blue fluorescence (Fig. 7e) in the PAA solution which was retained even in the dried PAA film (Fig. S40c, ESI‡). A blue fluorescent solution of SP8 (8CS8: 10 μM) was transformed into a greenish-yellow emitting solution of HSP8 upon the addition of heparin (8 μM) (Fig. 7f). The progressive addition of NR further transformed it into a white light emitting solution (NR: 200 nM, D/A = 500:1) exhibiting CIE coordinate values of 0.26, 0.34 (Fig. 7e and S40a, b, ESI‡). Interestingly, the greenish-yellow emitting solution of HSP8 upon drying was converted to a cyan-blue fluorescent polymer film. Presumably much lower dielectric constant of the PAA matrix prevented the aggregation of the molecules and hence the greenish-yellow excimeric emission was absent. Moreover, a wide range of fluorescent colors from blue to red including green, yellow and orange was obtained in solution as well in the solid state (Fig. S40c, ESI‡). The fluorescent colors of the dried films were more or less regained upon the addition of water (Fig. S40c, bottom panel, ESI‡). The use of different fluorescent colors as fluorescent ink was also demonstrated as shown in Fig. 7f (the detailed procedure for the preparation of all these colors has been described in ESI, pages S25 and S26‡).
Conclusions
In conclusion, we have constructed FRET-based artificial light-harvesting ensembles in aqueous buffer using the co-assembly of bio-polyanion heparin and cyanostilbene (CS) based cationic supramolecular polymers. The amphiphilic CS molecules formed cylindrical nanofibers in aqueous buffer as revealed by TEM images and molecular dynamics (MD) simulations. These nanofibers were transformed into nanotubes upon the formation of co-assemblies via intermediate nanosheet formation. MD simulation also suggested clusters of CS molecules on the heparin surface having a cylindrical shape. The CS donors in the heparin-based co-assemblies were found to act as very potent donors and highly efficient FRET was observed for acceptor dyes Nile Red (NR) and Nile Blue (NB) resulting in amplified emission in the orange-red and NIR range. The entangled network of the co-assemblies allowed an efficient exciton energy migration among the CS chromophore before the excitation energy was finally delivered to the acceptor. Approximately, the excitation energy of several hundred CS donors was transferred to a single acceptor leading to ultrahigh antenna effects of 150 even at high D/A ratios. To the best of our knowledge, this is the first report in which heparin has been employed as a bio-template to design such a FRET system. Other than a few reports on the polymer-based NPs, the energy transfer efficiency and antenna effect values reported in this work are some of the highest values reported for aqueous-based self-assembled systems. The FRET-systems were sensitive to temperature variation and thus acted as ratiometric fluorescence thermometers. Furthermore, the energy transfer was found to be very effective in solid and polymer films. This suggested the possible applications of these systems in data encryption-decryption.Data availability
Experimental procedures, characterization data, titration procedures, sample preparation protocols, computational details, and supportive figures and tables are available in the ESI.‡Author contributions
Experiments were designed by S. B. and S. K. B., and S. K. B. performed the experimental measurements and applications. Molecular Dynamics simulation studies were performed by D. M. and I. B in consultation with S. C. The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge Science and Engineering Research Board (SERB), Government of India (project: CRG/2021/004705) and IISER Kolkata for support of this work. SKB thanks Innovation in Science Pursuit for Inspired Research (INSPIRE) for a research fellowship. DM, IB and SC thank SNBNCBS, Kolkata for the high performance computational facility under Technical Research Centre (TRC). The DST-FIST TEM facility at IISER Kolkata is acknowledged for the TEM measurements. We also thank Rakesh Biswas, Nitish Kumar, Arunavo Chatterjee and Prof. Pradipta Purkayastha for the help with FLIM studies.Notes and references
- J. Barber, Chem. Soc. Rev., 2009, 38, 185–196 RSC.
- G. D. Scholes, G. R. Fleming, A. Olaya-Castro and R. Van Grondelle, Nat. Chem., 2011, 3, 763–774 CrossRef CAS PubMed.
- M. Freitag, J. Teuscher, Y. Saygili, X. Zhang, F. Giordano, P. Liska, J. Hua, S. M. Zakeeruddin, J.-E. Moser and M. Grätzel, Nat. Photonics, 2017, 11, 372–378 CrossRef CAS.
- J. Li, Y. Liu, Z. Zhu, G. Zhang, T. Zou, Z. Zou, S. Zhang, D. Zeng and C. Xie, Sci. Rep., 2013, 3, 1–6 Search PubMed.
- Z. Wang, W. Qiao, M. Yuan, N. Li and J. Chen, J. Phys. Chem. Lett., 2020, 11, 4644–4648 CrossRef CAS PubMed.
- T. Mayr, S. M. Borisov, T. Abel, B. Enko, K. Waich, G. Mistlberger and I. Klimant, Anal. Chem., 2009, 81, 6541–6545 CrossRef CAS.
- S. E. Ochmann, C. Vietz, K. Trofymchuk, G. P. Acuna, B. Lalkens and P. Tinnefeld, Anal. Chem., 2017, 89, 13000–13007 CrossRef CAS PubMed.
- Q. Wang, L. Yu, J. Sun, Y. Guan, Z. Zhou, Y. Shin, H. Yang, J. West and D. Yang, Adv. Opt. Mater., 2020, 8, 2001207 CrossRef CAS.
- W. Li, Q. Huang, Z. Mao, Q. Li, L. Jiang, Z. Xie, R. Xu, Z. Yang, J. Zhao and T. Yu, Angew. Chem., Int. Ed., 2018, 57, 12727–12732 CrossRef CAS PubMed.
- H. Yeo, K. Tanaka and Y. Chujo, Macromolecules, 2013, 46, 2599–2605 CrossRef CAS.
- A. Adronov and J. M. J. Fréchet, Chem. Commun., 2000, 1701–1710 RSC.
- H. Sun, X. Zhang, L. Miao, L. Zhao, Q. Luo, J. Xu and J. Liu, ACS Nano, 2016, 10, 421–428 CrossRef CAS PubMed.
- C. B. Winiger, S. Li, G. R. Kumar, S. M. Langenegger and R. Häner, Angew. Chem., Int. Ed., 2014, 53, 13609–13613 CrossRef CAS PubMed.
- L. Xu, Z. Wang, R. Wang, L. Wang, X. He, H. Jiang, H. Tang, D. Cao and B. Z. Tang, Angew. Chem., Int. Ed., 2020, 59, 9908–9913 CrossRef CAS PubMed.
- M. Hao, G. Sun, M. Zuo, Z. Xu, Y. Chen, X. Hu and L. Wang, Angew. Chem., Int. Ed., 2020, 59, 10095–10100 CrossRef CAS PubMed.
- Y. Li, Y. Dong, L. Cheng, C. Qin, H. Nian, H. Zhang, Y. Yu and L. Cao, J. Am. Chem. Soc., 2019, 141, 8412–8415 CrossRef CAS PubMed.
- N. Melnychuk, S. Egloff, A. Runser, A. Reisch and A. S. Klymchenko, Angew. Chem., Int. Ed., 2020, 59, 6811–6818 CrossRef CAS PubMed.
- K. Trofymchuk, A. Reisch, P. Didier, F. Fras, P. Gilliot, Y. Mely and A. S. Klymchenko, Nat. Photonics, 2017, 11, 657–663 CrossRef CAS PubMed.
- Q. Song, S. Goia, J. Yang, S. C. L. Hall, M. Staniforth, V. G. Stavros and S. Perrier, J. Am. Chem. Soc., 2020, 143, 382–389 CrossRef PubMed.
- J. Li, Y. Chen, J. Yu, N. Cheng and Y. Liu, Adv. Mater., 2017, 29, 1701905 CrossRef PubMed.
- S. Guo, Y. Song, Y. He, X. Hu and L. Wang, Angew. Chem., Int. Ed., 2018, 57(57), 3163–3167 CrossRef CAS PubMed.
- X. Chen, Q. Cao, H. K. Bisoyi, M. Wang, H. Yang and Q. Li, Angew. Chem., Int. Ed., 2020, 59, 10493–10497 CrossRef CAS PubMed.
- T. Gorai and U. Maitra, Angew. Chem., Int. Ed., 2017, 56, 10730–10734 CrossRef CAS PubMed.
- S. Kuila and S. J. George, Angew. Chem., Int. Ed., 2020, 59, 9393–9397 CrossRef CAS PubMed.
- G. v. Bünau, Ber. Bunsenges. Phys. Chem., 1970, 74, 1294–1295 Search PubMed.
- Y. N. Teo and E. T. Kool, Chem. Rev., 2012, 112, 4221–4245 CrossRef CAS PubMed.
- P. K. Dutta, R. Varghese, J. Nangreave, S. Lin, H. Yan and Y. Liu, J. Am. Chem. Soc., 2011, 133, 11985–11993 CrossRef CAS PubMed.
- R. J. Brea, M. J. Pérez-Alvite, M. Panciera, M. Mosquera, L. Castedo and J. R. Granja, Chem.–Asian J., 2011, 6, 110–121 CrossRef CAS PubMed.
- B. Valeur, E. Bardez, J. Lehn, L. Jullien and J. Canceill, Angew. Chem., Int. Ed. Engl., 1995, 33, 2438–2439 CrossRef.
- L. Jullien, J. Canceill, B. Valeur, E. Bardez, J.-P. Lefevre, J.-M. Lehn, V. Marchi-Artzner and R. Pansu, J. Am. Chem. Soc., 1996, 118, 5432–5442 CrossRef CAS.
- H. N. Barnhill, S. Claudel-Gillet, R. Ziessel, L. J. Charbonnière and Q. Wang, J. Am. Chem. Soc., 2007, 129, 7799–7806 CrossRef CAS PubMed.
- M. T. Dedeo, K. E. Duderstadt, J. M. Berger and M. B. Francis, Nano Lett., 2010, 10, 181–186 CrossRef CAS PubMed.
- D. Bergqvist, G. Agnelli, A. T. Cohen, A. Eldor, P. E. Nilsson, A. Le Moigne-Amrani and F. Dietrich-Neto, N. Engl. J. Med., 2002, 346, 975–980 CrossRef CAS PubMed.
- N. Mackman, Nature, 2008, 451, 914–918 CrossRef CAS PubMed.
- H. Bussey, J. L. Francis and H. C. Group, Pharmacotherapy, 2004, 24, 103S–107S CrossRef CAS PubMed.
- A. T. Wright, Z. Zhong and E. V Anslyn, Angew. Chem., Int. Ed., 2005, 44, 5679–5682 CrossRef CAS PubMed.
- H. A. Behanna, K. Rajangam and S. I. Stupp, J. Am. Chem. Soc., 2007, 129, 321–327 CrossRef CAS PubMed.
- Q. Dai, W. Liu, X. Zhuang, J. Wu, H. Zhang and P. Wang, Anal. Chem., 2011, 83, 6559–6564 CrossRef CAS PubMed.
- K.-Y. Pu and B. Liu, Macromolecules, 2008, 41, 6636–6640 CrossRef CAS.
- S. Wang and Y.-T. Chang, Chem. Commun., 2008, 1173–1175 RSC.
- T. Noguchi, B. Roy, D. Yoshihara, J. Sakamoto, T. Yamamoto and S. Shinkai, Angew. Chem., Int. Ed., 2016, 55, 5708–5712 CrossRef CAS PubMed.
- D.-H. Kim, Y. J. Park, K. H. Jung and K.-H. Lee, Anal. Chem., 2014, 86, 6580–6586 CrossRef CAS PubMed.
- S. K. Bhaumik and S. Banerjee, Chem. Commun., 2020, 56, 655–658 RSC.
- S. K. Bhaumik, Y. S. Patra and S. Banerjee, Chem. Commun., 2020, 56, 9541–9544 RSC.
- S. K. Bhaumik and S. Banerjee, ACS Appl. Mater. Interfaces, 2022, 14, 36936–36946 CrossRef CAS PubMed.
- X. Li, M. Jiang, J. W. Y. Lam, B. Z. Tang and J. Y. Qu, J. Am. Chem. Soc., 2017, 139, 17022–17030 CrossRef CAS PubMed.
- A. Lavrenova, D. W. R. Balkenende, Y. Sagara, S. Schrettl, Y. C. Simon and C. Weder, J. Am. Chem. Soc., 2017, 139, 4302–4305 CrossRef CAS PubMed.
- H. Wu, Z. Chen, W. Chi, A. K. Bindra, L. Gu, C. Qian, B. Wu, B. Yue, G. Liu and G. Yang, Angew. Chem., Int. Ed., 2019, 58, 11419–11423 CrossRef CAS PubMed.
- Y. Hong, J. W. Y. Lam and B. Z. Tang, Chem. Soc. Rev., 2011, 40, 5361–5388 RSC.
- R. T. K. Kwok, C. W. T. Leung, J. W. Y. Lam and B. Z. Tang, Chem. Soc. Rev., 2015, 44, 4228–4238 RSC.
- L. Ji, Y. Sang, G. Ouyang, D. Yang, P. Duan, Y. Jiang and M. Liu, Angew. Chem., Int. Ed., 2019, 58, 844–848 CrossRef CAS PubMed.
- Y. Liu, H. Wang, P. Zhang, Z. Wang, X. Dong, M. Du, Y. Wang and L. Ji, Colloids Surf., A, 2022, 665, 13029 Search PubMed.
- S. K. Bhaumik and S. Banerjee, Analyst, 2021, 146, 2194–2202 RSC.
- K. R. Bickel, A. E. Timm, D. Nattland and R. Schuster, Langmuir, 2014, 30, 9085–9090 CrossRef CAS PubMed.
- J. Carlstedt, D. Lundberg, R. S. Dias and B. Lindman, Langmuir, 2012, 28, 7976–7989 CrossRef CAS PubMed.
- K. Wagner, D. Harries, S. May, V. Kahl, J. O. Rädler and A. Ben-Shaul, Langmuir, 2000, 16, 303–306 CrossRef CAS.
- E. Pouget, N. Fay, E. Dujardin, N. Jamin, P. Berthault, L. Perrin, A. Pandit, T. Rose, C. Valéry and D. Thomas, J. Am. Chem. Soc., 2010, 132, 4230–4241 CrossRef CAS PubMed.
- L. Ziserman, H.-Y. Lee, S. R. Raghavan, A. Mor and D. Danino, J. Am. Chem. Soc., 2011, 133, 2511–2517 CrossRef CAS PubMed.
- Z. Shen, T. Wang and M. Liu, Chem. Commun., 2014, 50, 2096–2099 RSC.
- S. Li, R. Liu, D. Bekana, Y. Lai and J. Liu, Nanoscale, 2018, 10, 20804–20812 RSC.
- P. W. J. M. Frederix, I. Patmanidis and S. J. Marrink, Chem. Soc. Rev., 2018, 47, 3470–3489 RSC.
- F. Haverkort, A. Stradomska, A. H. de Vries and J. Knoester, J. Phys. Chem. B, 2013, 117, 5857–5867 CrossRef CAS PubMed.
- C. Friedl, T. Renger, H. v Berlepsch, K. Ludwig, M. Schmidt am Busch and J. Megow, J. Phys. Chem. C, 2016, 120, 19416–19433 CrossRef CAS PubMed.
- S. M. Bromfield, A. Barnard, P. Posocco, M. Fermeglia, S. Pricl and D. K. Smith, J. Am. Chem. Soc., 2013, 135, 2911–2914 CrossRef CAS PubMed.
- M. Maszota-Zieleniak, F. Zsila and S. A. Samsonov, Carbohydr. Res., 2021, 507, 108390 CrossRef CAS PubMed.
- P. Greenspan, E. P. Mayer and S. D. Fowler, J. Cell Biol., 1985, 100, 965–973 CrossRef CAS PubMed.
- I. N. Kurniasih, H. Liang, P. C. Mohr, G. Khot, J. P. Rabe and A. Mohr, Langmuir, 2015, 31, 2639–2648 CrossRef CAS PubMed.
- A. Ray, S. Das and N. Chattopadhyay, ACS omega, 2019, 4, 15–24 CrossRef CAS PubMed.
- V. Martinez and M. Henary, Chem.–Eur. J., 2016, 22, 13764–13782 CrossRef CAS PubMed.
- X.-L. Wang, R. Sun, W.-J. Zhu, X.-L. Sha and J.-F. Ge, J. Fluoresc., 2017, 27, 819–827 CrossRef CAS PubMed.
- M. Y. Berezin and S. Achilefu, Chem. Rev., 2010, 110, 2641–2684 CrossRef CAS PubMed.
- X. Chen, P. Jagadesan, S. Valandro, J. T. Hupp, K. S. Schanze and S. Goswami, ACS Mater. Lett., 2022, 4, 370–377 CrossRef CAS.
- A. Chatterjee, A. K. Sharma and P. Purkayastha, Nanoscale, 2022, 14, 6570–6584 RSC.
- S. Das and P. Purkayastha, Langmuir, 2017, 33, 7281–7287 CrossRef CAS PubMed.
- A. Ya. Freidzon, A. A. Safonov, A. A. Bagaturyants and M. V Alfimov, Int. J. Quantum Chem., 2012, 112, 3059–3067 CrossRef CAS.
- H. Tajalli, A. G. Gilani, M. S. Zakerhamidi and P. Tajalli, Dyes Pigm., 2008, 78, 15–24 CrossRef CAS.
- T. Shimizu, M. Masuda and H. Minamikawa, Chem. Rev., 2005, 105, 1401–1444 CrossRef CAS PubMed.
- T. E. Kaiser, H. Wang, V. Stepanenko and F. Würthner, Angew. Chem., Int. Ed., 2007, 46, 5541–5544 CrossRef CAS PubMed.
- J. B. Matson, C. J. Newcomb, R. Bitton and S. I. Stupp, Soft Matter, 2012, 8, 3586–3595 RSC.
- A. Ajayaghosh, V. K. Praveen, C. Vijayakumar and S. J. George, Angew. Chem., Int. Ed., 2007, 46, 6260–6265 CrossRef CAS PubMed.
- P. Andrew and W. L. Barnes, Science, 2000, 290, 785–788 CrossRef CAS PubMed.
- T. Nakamura, M. Fujii, K. Imakita and S. Hayashi, Phys. Rev. B, 2005, 72, 235412 CrossRef.
- R. Camacho, D. Täuber and I. G. Scheblykin, Adv. Mater., 2019, 31, 1805671 CrossRef PubMed.
- H. Peng, Y. Chen, Y. Zhao, Q. Yang, L. Wu, C. Tung, L. Zhang and Q. Tong, Angew. Chem., Int. Ed., 2012, 51, 2088–2092 CrossRef CAS PubMed.
- Y. Han, X. Zhang, Z. Ge, Z. Gao, R. Liao and F. Wang, Nat. Commun., 2022, 13, 1–10 Search PubMed.
- J.-H. Tang, Y. Sun, Z.-L. Gong, Z.-Y. Li, Z. Zhou, H. Wang, X. Li, M. L. Saha, Y.-W. Zhong and P. J. Stang, J. Am. Chem. Soc., 2018, 140, 7723–7729 CrossRef CAS PubMed.
- Z. Zhang, Z. Zhao, L. Wu, S. Lu, S. Ling, G. Li, L. Xu, L. Ma, Y. Hou and X. Wang, J. Am. Chem. Soc., 2020, 142, 2592–2600 CrossRef CAS PubMed.
- K. Rani and S. Sengupta, Chem. Sci., 2021, 12, 15533–15542 RSC.
- E. J. McLaurin, L. R. Bradshaw and D. R. Gamelin, Chem. Mater., 2013, 25, 1283–1292 CrossRef CAS.
- P. Greenspan and S. D. Fowler, J. Lipid Res., 1985, 26, 781–789 CrossRef CAS.
- A. K. Dutta, Photochem. Photobiol., 1996, 93, 57–64 CrossRef CAS.
Footnotes |
| † Dedicated to Prof. Uday Maitra on the occasion of his 65th birthday. |
| ‡ Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sc00375b |
| This journal is © The Royal Society of Chemistry 2023 |