
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSterically controlled isodesmic late-stage C–H iodination of arenes†
Mirxan
Farizyan
a,
Rita
de Jesus‡
b,
Jyotirmoy
Dey‡
b and
Manuel
van Gemmeren
 *ab
*ab
aOrganisch-Chemisches Institut, Westfälische Wilhelms-Universität Münster, Corrensstraße 36, 48149 Münster, Germany
bOtto-Diels-Institut für Organische Chemie, Christian-Albrechts-Universität zu Kiel, Otto-Hahn-Platz 4, 24118 Kiel, Germany. E-mail: vangemmeren@oc.uni-kiel.de
First published on 27th March 2023
Abstract
Aryl iodides are key motifs in organic chemistry due to their versatility as linchpins in metal-mediated cross-coupling reactions for synthesis and drug discovery. These scaffolds are typically prepared indirectly from prefunctionalized starting materials or via electrophilic aromatic iodination protocols. These methods are limited to specific regioisomers by their inherent selectivities and/or the availability of the required starting materials. Herein, we describe the sterically controlled iodination of arenes through an isodesmic C–H/C–I bond metathesis approach enabled by our dual ligand-based catalysts for arene-limited nondirected C–H activation. The protocol gives direct access to a complementary product spectrum with respect to traditional methods. Its synthetic utility is demonstrated by a broad scope and the suitability for late-stage modification.
Introduction
Aryl halides are synthetically versatile intermediates with an important role in the preparation of many organic molecules such as pharmaceuticals and agrochemicals.1 Aryl iodides prove to be particularly advantageous as they constitute attractive coupling partners and precursors to organometallic species in cross-coupling reactions with higher reactivity than their chlorinated and brominated counterparts.2 Thus, the direct conversion of C–H into C–I bonds is highly attractive, in order to access linchpins for further diversification. Electrophilic aromatic substitution (SEAr) reactions, widely used to synthesize aryl iodides, rely on the substrates' electronic properties to control the regioselectivity (Fig. 1A(I)).3 However, the iodination of structurally complex arenes remains more challenging than the analogous chlorination and bromination, due to the limited availability of suitable electrophilic iodinating reagents, the typically required harsh reaction conditions leading to poor functional group tolerance.4 Recent research has aimed for milder and more efficient electrophilic C–H iodination enabling the functionalization of challenging and complex molecules.5 Despite these advances, many aryl iodides are prepared indirectly from prefunctionalized arenes, such as amines (Sandmeyer reaction),6 carboxylic acids (via photochemistry),7 aryl bromides (aromatic Finkelstein reaction),8 or iododeborylation (Fig. 1A(II)).9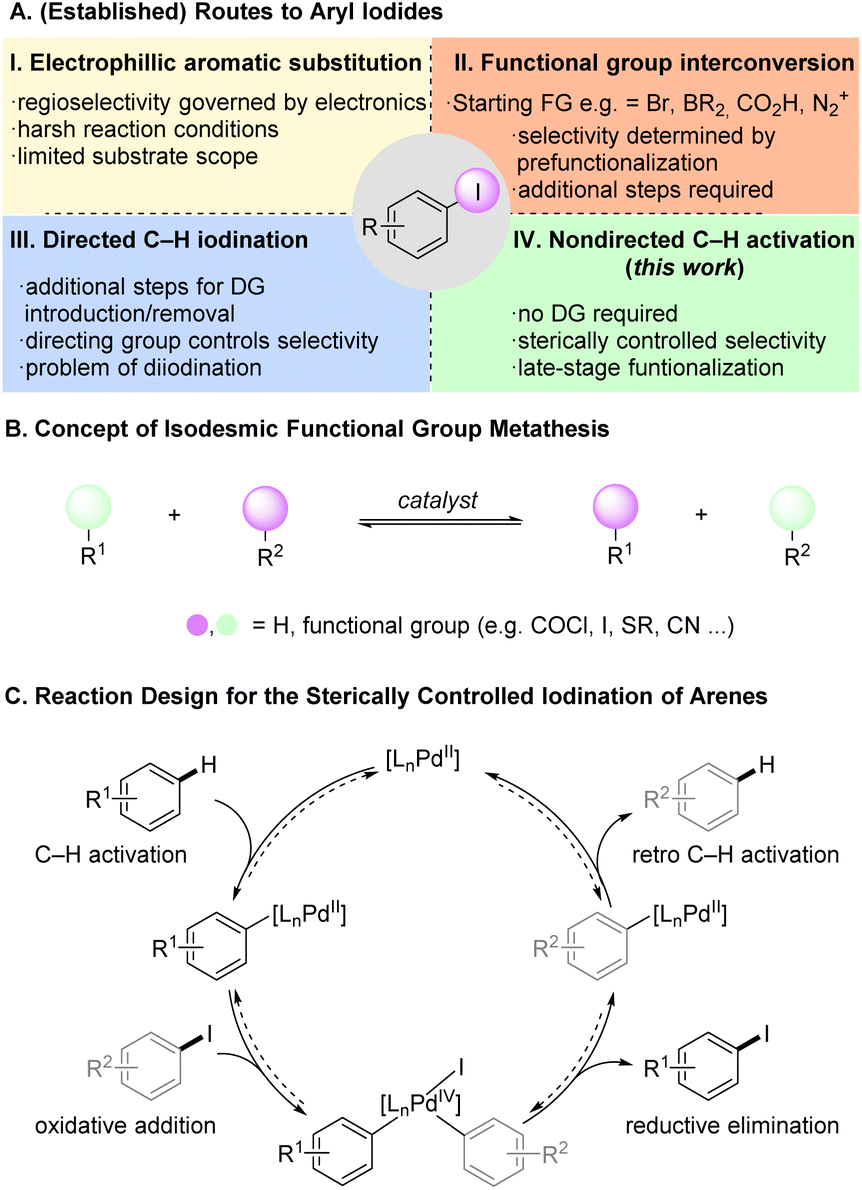 | ||
| Fig. 1 Approaches towards the iodination of arenes. | ||
While delivering the desired compounds, these approaches require additional steps and remain limited by the availability of the required prefunctionalized starting materials. Intense efforts have been directed towards the transition metal catalyzed synthesis of aryl iodides.10 Various transition metals have been used in conjunction with coordinating directing groups (DGs) on the substrate to enable efficient and broadly applicable C–H iodination protocols (Fig. 1A(III)).11 Most of these protocols lead to ortho iodination, but specialized DGs have been reported that enable meta selectivity.12 Li, Li and coworkers recently described directed ortho and meta C–H iodination reactions using a C–H/C–I metathesis approach with 2-nitro iodobenzene (2) as iodine source.13 The authors proposed a Pd(II)/Pd(IV) cycle where the reductive elimination giving aryl iodide is favored over a competing biaryl formation. The report by Li, Li and coworkers uses the general strategy of isodesmic functional group metathesis, in which hydrogen substituents and/or functional groups are exchanged between two compounds without a change in the overall number and type of bonds (Fig. 1B).14 Prominent examples for this approach are the contemporaneous reports by Morandi and Arndtsen on the interconversion of aroyl chlorides and aryl iodides and related studies.15
Despite these advances in transition metal catalyzed C–H iodination, a direct iodination without DGs has not been reported to date. Recently, Pd-catalysts were developed that enable the nondirected activation/functionalization of unbiased arene C–H bonds resulting in valuable methods for late-stage modification.16 Our group introduced a series of dual ligand-based Pd-catalysts for the nondirected C–H activation of complex (hetero)arenes enabling challenging functionalizations with predominantly sterically controlled regioselectivity.17
Results and discussion
Reaction development
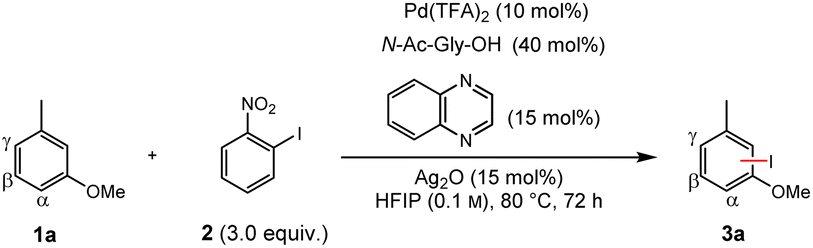
We envisioned that the combination of our dual ligand catalysts and a C–H/C–I metathesis approach (Fig. 1C) could enable a sterically controlled direct iodination of complex arenes through a Pd(II)/Pd(IV) catalytic cycle.18 We decided to investigate the iodination of m-cresol methyl ether (1a) as model substrate since here sterically and electronically preferred positions are orthogonal to one another, enabling us to directly monitor the factor(s) governing the regioselectivity (Table 1). Extensive optimization of the reaction conditions resulted in the use of N-acetylglycine and quinoxaline as suitable ligands, along with catalytic amounts of Ag2O that presumably works as Lewis acid activator and/or halide scavenger. Under these conditions even the strong electronic effect exerted by a methoxy group can be overcome, giving functionalization mainly in the β-position.19 Control experiments confirm that both ligands are required for activity and steric control. The omission of Ag2O likewise leads to a decrease of yield and selectivity (for a discussion regarding the role of silver, see the section “Expanded mechanistic discussion” in the ESI†).|
|
|||
|---|---|---|---|
| Entry | Deviation from standard conditions | Yield (%) |
α![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :β:γ :β:γ |
| a Reactions performed on a 0.1 mmol scale. b Yields and regioselectivities determined by GC-FID using mesitylene as internal standard. | |||
| 1 | None | 54 | 11:86:3 |
| 2 | No Pd(TFA)2 | 2 | — |
| 3 | No N-Ac-Gly-OH | 12 | 59:0:41 |
| 4 | No quinoxaline | 39 | 46:6:48 |
| 5 | No Ag2O | 18 | 62:25:13 |
Synthetic scope
We proceeded to investigate the substrate scope (Scheme 1). Naphthalene (1b) underwent iodination with excellent selectivity and synthetically useful yield. A wide range of monosubstituted arenes could be applied (1c–h). The iodination is highly sensitive to steric effects, a methyl group being sufficient to completely suppress ortho product formation. In contrast, electronic effects have little to no influence on the regioselectivity.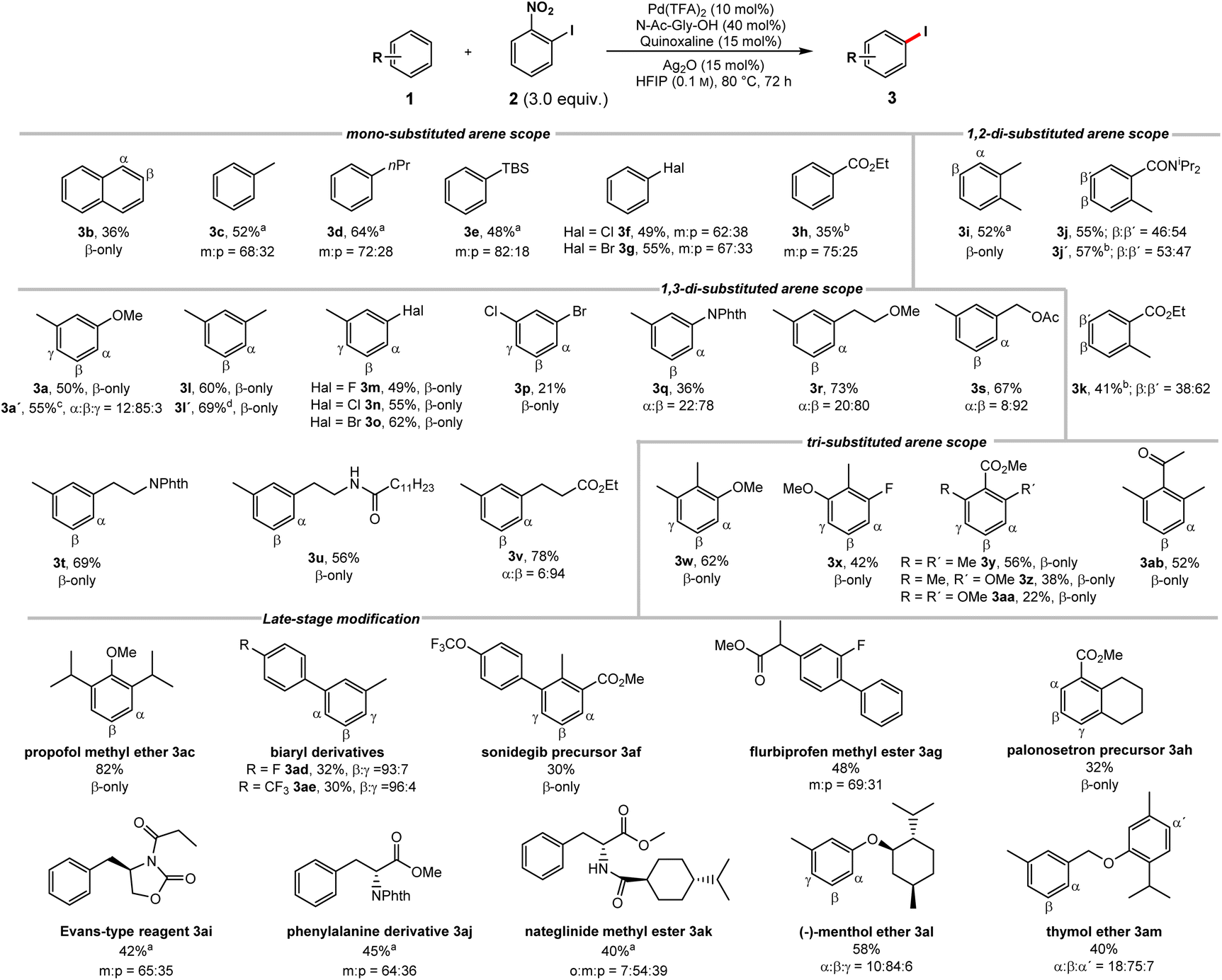 | ||
| Scheme 1 Reaction scope (for unsuccessful substrates, see Scheme S43 in the ESI†). Structures of the respective starting materials shown for simplicity. Reactions conducted on a 0.2 mmol scale; isolated yields; regioselectivities determined by NMR. a 2 = 2.0 equiv., 70 °C, 48 h. b 2 = 4.0 equiv., AgNO3 (30 mol%), 100 °C. c Contains 6% di-iodinated arene. d 3.0 mmol scale. | ||
Using a silylated benzene delivered the iodinated product 3e in good yields. Notably, the C–H iodination of silyl arenes usually requires indirect approaches.20 Aryl halides 1f and 1g show no ortho functionalization, despite being known to exhibit an ortho directing effect in related reports.16d
Importantly, the observed selectivity profile is complementary to electrophilic functionalizations of haloarenes.21 Electron-poor ethyl benzoate (1h) gave meta and para iodination of the arene.22 Despite being a potential DG, no functionalization occurred in ortho position. o-Xylene delivered 3i as single isomer. Nonsymmetric 1,2-disubstituted arenes 1j–k deliver nearly equal mixtures in the sterically accessible yet electronically very different β and β′ positions, showcasing the low sensitivity to directing and electronic effects. 1,3-Disubstituted substrates, where electronically and sterically preferred positions are orthogonal gave preferential iodination in the sterically favored position (3a, 3l–v). m-Xylene underwent iodination exclusively in the less congested β-position complementing electrophilic approaches (3l).20b This transformation could be performed on an increased scale without a detrimental effect. 3-Halide substituted toluene derivatives 1m–o gave the sterically favored products in good yields. These structures are commonly synthesized using indirect approaches,23 while direct approaches, deliver the electronically favored products.24 1,3-Dihalogenated 1p was iodinated exclusively in the β-position. Despite the moderate yield, it should be noted that this molecule was previously only accessible through an indirect approach.9 Phthaloyl protected aniline was likewise iodinated in the sterically favored position (3q). Substrates 1r–u were subjected to the iodination protocol. Despite the presence of coordinating functionalities the reaction proceeded predominantly in the sterically favored position. Hydrocinnamate 1v, gave a high selectivity for the meta position. The functionalization in this position was previously achieved indirectly, by installing a designed meta-directing template on the carboxylate moiety, while the analogous acid gave exclusive ortho-functionalization.13a Finally, we studied 1,2,3-trisubstituted arenes with a strong electronic bias towards the ortho positions (3w-ab). Despite the presence of a methoxy group our protocol addressed the electronically disfavored position (3w).25 The fluorinated derivative 3x gave the β-product in good yield. Substrates 1y-aa highlight the steric control of our catalyst system as only the sterically favored yet electronically disfavored position is iodinated.26 A stronger electronic bias towards the sterically disfavored positions reduces the activity of our system. Nevertheless, an arene bearing a deactivating keto group (1ab) could be iodinated. Next, we probed the late-stage modification of structurally more complex substrates. Propofol methyl ether (3ac) was efficiently iodinated. Biaryls constitute key motifs in organic chemistry,27 rendering their late-stage selective iodination in unprecedented positions an attractive linchpin for further diversification. The iodination of biaryls 1ad and 1ae delivered products in synthetically useful yields and excellent selectivities for the sterically favored C–H bond. Analogously, sonidegib precursor 1af and the flurbiprofen methyl ester 1ag containing a biaryl scaffold were iodinated with remarkable selectivities. Palonosetron precursor 1ah was exclusively iodinated in the β-position. The Evans-type reagent 1ai and the phenylalanine derivative 1aj likewise delivered meta iodinated products as major isomer, which is inaccessible through direct electrophilic approaches.28 The iodination of nateglinide ester 1ak and the (−)-menthol ether of m-cresol 1al likewise gave regioisomers, which would otherwise be challenging to obtain. The thymol ether of m-cresol 1am was mainly iodinated in the sterically most accessible β-position of the m-cresol moiety. Remarkably, the more electron-rich thymol motif barely underwent iodination.
As evidenced by these scope studies, we have developed a broadly applicable protocol for the nondirected late-stage iodination of arenes with predominant steric control of the regioselectivity. To highlight the complementarity of our transformation with traditional approaches, we compared different iodination procedures using substrate 1w (Scheme 2). Subjecting this arene to an electrophilic iodination was reported to deliver the electronically favored γ-product 4.29 The synthesis of the corresponding α-isomer 5 was achieved by an ortho-lithiation step.30 Our approach complements these methods by forming the sterically controlled product 3w.
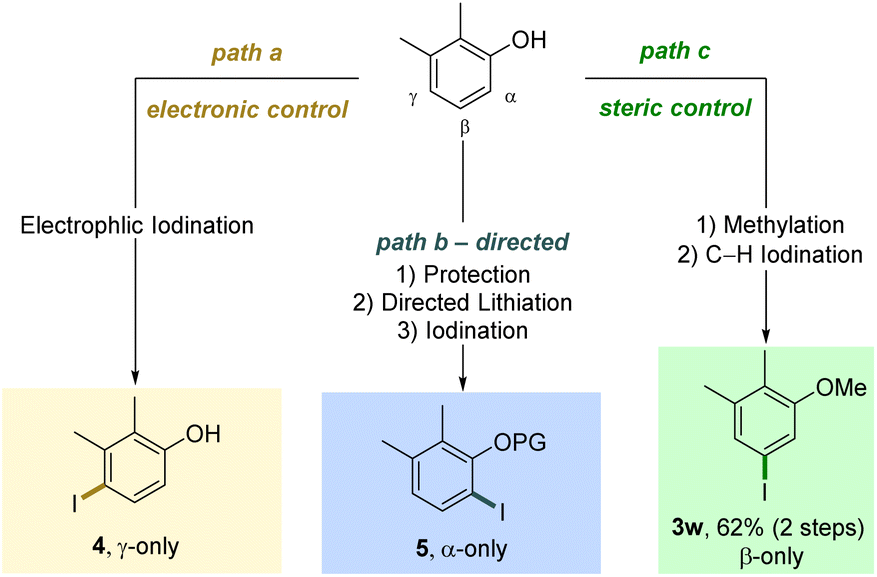 | ||
| Scheme 2 Complementarity with traditional approaches. | ||
Mechanistic studies
We proceeded to interrogate the mechanism of our protocol (Scheme 3). Based on the general pathway shown in Fig. 1C, all steps of the reaction could in principle be reversible. We thus investigated if the process is thermodynamically or kinetically controlled. Scrambling experiments were performed, in which isomerically pure (disfavored isomer) aryl iodides were added to a reaction forming a different product (Scheme 3A). While the formation of the expected products shows that catalysis took place under these conditions, the absence of scrambling in the additives clearly demonstrates that the overall reaction occurs irreversibly (i.e. kinetically controlled).31 Parallel (kH/kD = 1.9) and competition (kH/kD = 1.7–1.8) KIE-experiments indicated that the C–H activation step is turnover-limiting and thus responsible for the kinetically controlled regioselectivity (Scheme 3B). The kinetic orders with respect to arene 1 (0.5) and iodine reagent 2 (0.2) were determined (Scheme 3C). These broken kinetic orders indicate that besides the C–H activation the subsequent oxidative addition also contributes to the overall rate of the reaction, which implies that the C–H activation may be partially reversible. Reversibility was probed using 1h-d5 (Scheme 3D). In the absence of reagent 2a substantial H/D exchange occurred showing that the C–H activation is reversible when no reaction partner is available (some H/D exchange was observed in the ortho position, which is not iodinated in the overall process). In the presence of reagent 2 H/D exchange in the remaining substrate was strongly reduced. This demonstrates that when product formation is possible, it can outcompete the retro-C–H activation. These results show that the observed regioselectivity is predominantly defined by the kinetically controlled C–H activation step. The selectivity is further increased, since in case of sterically hindered positions retro-C–H activation can efficiently outcompete product formation.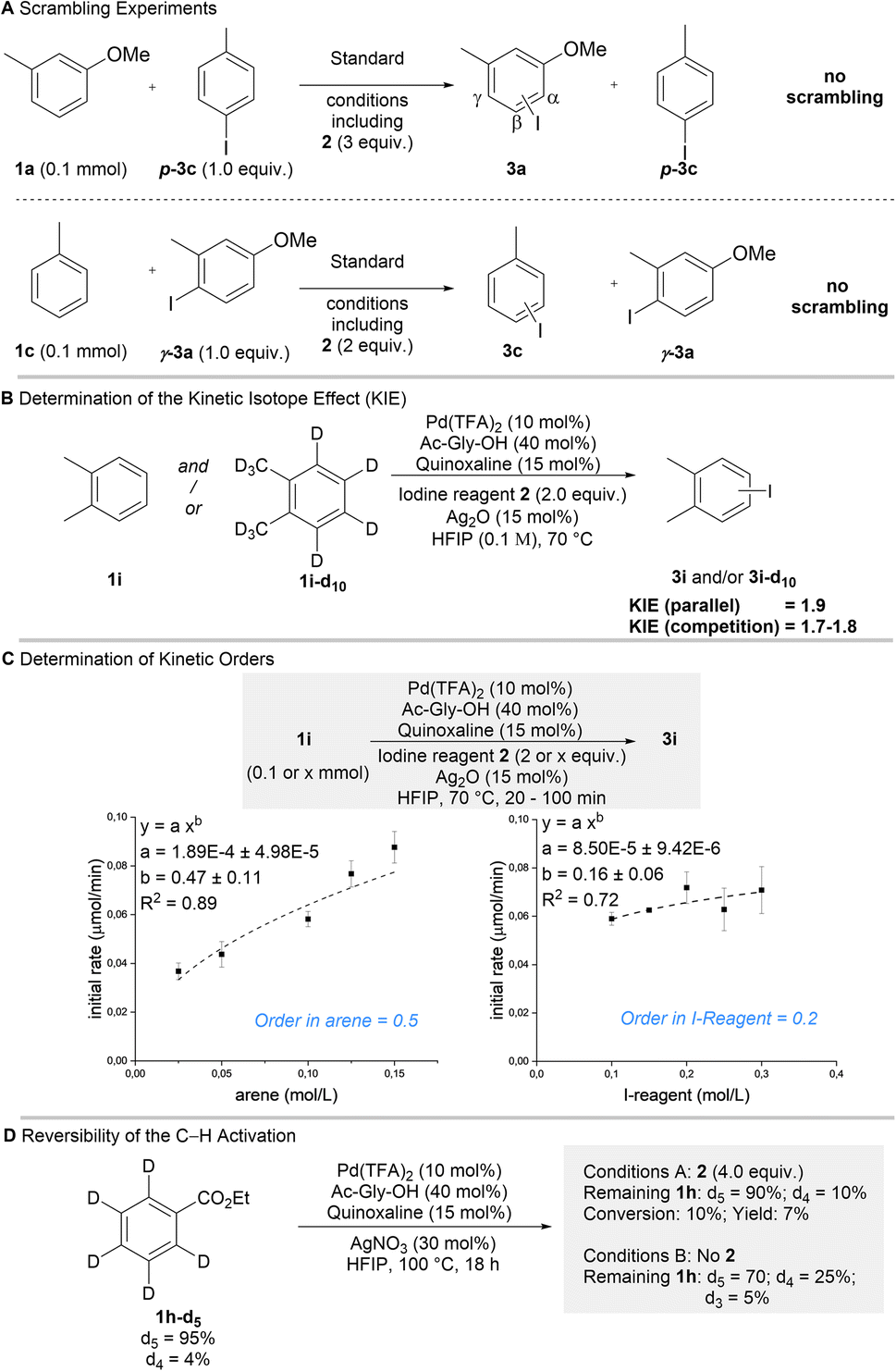 | ||
| Scheme 3 Preliminary Mechanistic Studies. a All Reactions performed on a 0.1 mmol scale. | ||
Conclusions
In summary, we have developed a sterically controlled C–H iodination of arenes. Our protocol is widely applicable tolerating a broad range of functional groups and substitution patterns. The sensitivity towards steric factors, renders our approach complementary to established iodination strategies. We have demonstrated the utility of this reaction for late-stage functionalization, providing valuable linchpins for further diversification with otherwise challenging substitution patterns.Data availability
Full experimental results are available in the ESI.†Author contributions
M. v. G. and M. F. conceived the project; M. F., R. d. J. and J. D. performed the experiments and analyzed the data. M. F. and M. v. G. wrote the manuscript with contributions from all authors; M. v. G. supervised the project.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge generous financial support by the DFG (Emmy Noether Programme), the WWU Münster and CAU Kiel. This project has received funding from the European Research Council (ERC) under the European Union's Horizon 2020 Research and Innovation Programme (grant agreement no. 946044). We thank all members of our MS and NMR department for their excellent service. We thank F. Deufel and A. Mondal for helpful scientific discussions. Furthermore, we thank Prof. Dr Frank Glorius for his generous support.Notes and references
- (a) K. C. Nicolaou, P. G. Bulger and D. Sarlah, Angew. Chem., Int. Ed., 2005, 44, 4442 CrossRef CAS PubMed; (b) J. Magano and J. R. Dunetz, Chem. Rev., 2011, 111, 2177 CrossRef CAS PubMed; (c) D. J. Weix, Acc. Chem. Res., 2015, 48, 1767 CrossRef CAS PubMed.
- (a) N. Miyaura and A. Suzuki, Chem. Rev., 1995, 95, 2457 CrossRef CAS; (b) P. Knochel, W. Dohle, N. Gommermann, F. F. Kneisel, F. Kopp, T. Korn, I. Sapountzis and V. A. Vu, Angew. Chem., Int. Ed., 2003, 42, 4302 CrossRef CAS PubMed; (c) T. D. Sheppard, Org. Biomol. Chem., 2009, 7, 1043 RSC.
- (a) R. Taylor, Electrophilic aromatic substitution; Wiley, 1990 Search PubMed; (b) J. R. Hanson, J. Chem. Res., 2006, 277 CrossRef CAS; (c) S. Stavber, M. Jereb and M. Zupan, Synthesis, 2008, 1487 CrossRef CAS; (d) M. Bergström, G. Suresh, V. R. Naidu and C. R. Unelius, Eur. J. Org. Chem., 2017, 3234 CrossRef; (e) N. L. Sloan and A. Sutherland, Synthesis, 2016, 48, 2969–2980 CrossRef.
- (a) J. Barluenga, J. M. Gonzalez, M. A. Garcia-Martin, P. J. Campos and G. Asensio, J. Org. Chem., 1993, 58, 2058 CrossRef CAS; (b) L. Kraszkiewicz, M. Sosnowski and L. Skulski, Tetrahedron, 2004, 60, 9113 CrossRef CAS; (c) L. Kraszkiewicz, M. Sosnowski and L. Skulski, Synthesis, 2006, 1195 CAS.
- (a) L. Tanwar, J. Börgel, J. Lehmann and T. Ritter, Org. Lett., 2021, 23, 5024 CrossRef CAS PubMed; (b) W. Wang, X. Yang, R. Dai, Z. Yan, J. Wei, X. Dou, X. Qiu, H. Zhang, C. Wang, Y. Liu, S. Song and N. Jiao, J. Am. Chem. Soc., 2022, 144, 13415 CrossRef CAS PubMed; (c) R.-J. Tang, T. Milcent and B. Crousse, J. Org. Chem., 2018, 83, 930–938 CrossRef CAS PubMed; (d) R. C. Samanta and H. Yamamoto, Eur. J. Chem., 2015, 21, 11976–11979 CrossRef CAS PubMed; (e) K. Iida, S. Ishida, T. Watanabe and T. Arai, J. Org. Chem., 2019, 84, 7411–7417 CrossRef CAS PubMed; (f) D. T. Racys, C. E. Warrilow, S. L. Pimlott and A. Sutherland, Org. Lett., 2015, 17, 4782–4785 CrossRef CAS PubMed.
- H. H. Hodgson, Chem. Rev., 1947, 40, 251 CrossRef CAS PubMed.
- T. Q. Chen, P. S. Pedersen, N. W. Dow, R. Fayad, C. E. Hauke, M. C. Rosko, E. O. Danilov, D. C. Blakemore, A.-M. Dechert-Schmitt, T. Knauber, F. N. Castellano and D. W. C. MacMillan, J. Am. Chem. Soc., 2022, 144, 8296 CrossRef CAS PubMed.
- A. Klapars and S. L. Buchwald, J. Am. Chem. Soc., 2002, 124, 14844 CrossRef CAS PubMed.
- B. M. Partridge and J. F. Hartwig, Org. Lett., 2013, 15, 140 CrossRef CAS PubMed.
- D. A. Petrone, J. Ye and M. Lautens, Chem. Rev., 2016, 116, 8003 CrossRef CAS PubMed.
- (a) D. Kalyani, A. R. Dick, W. Q. Anani and M. S. Sanford, Org. Lett., 2006, 8, 2523 CrossRef CAS PubMed; (b) J.-J. Li, T.-S. Mei and J.-Q. Yu, Angew. Chem., Int. Ed., 2008, 47, 6452 CrossRef CAS PubMed; (c) T.-S. Mei, R. Giri, N. Maugel and J.-Q. Yu, Angew. Chem., Int. Ed., 2008, 47, 5215 CrossRef CAS PubMed; (d) T.-S. Mei, D.-H. Wang and J.-Q. Yu, Org. Lett., 2010, 12, 3140 CrossRef CAS PubMed; (e) N. Schröder, J. Wencel-Delord and F. Glorius, J. Am. Chem. Soc., 2012, 134, 8298 CrossRef PubMed; (f) X.-C. Wang, Y. Hu, S. Bonacorsi, Y. Hong, R. Burrell and J.-Q. Yu, J. Am. Chem. Soc., 2013, 135, 10326 CrossRef CAS PubMed; (g) L. Wang and L. Ackermann, Chem. Commun., 2014, 50, 1083 RSC.
- (a) L. Chu, M. Shang, K. Tanaka, Q. Chen, N. Pissarnitski, E. Streckfuss and J.-Q. Yu, ACS Cent. Sci., 2015, 1, 394 CrossRef CAS PubMed; (b) G. M. Reddy, N. S. Rao and H. Maheswaran, Org. Chem. Front., 2018, 5, 1118 RSC; (c) Z. Jin, L. Chu, Y.-Q. Chen and J.-Q. Yu, Org. Lett., 2018, 20, 425 CrossRef CAS PubMed; (d) M. Liu, L.-J. Li, J. Zhang, H. Xu and H.-X. Dai, Chin. Chem. Lett., 2020, 31, 1301 CrossRef CAS.
- (a) S. Li, C. Zhang, L. Fu, H. Wang, L. Cai, X. Chen, X. Wang and G. Li, CCS Chem., 2022, 4, 1889 CrossRef CAS; (b) N. Wang, Z. Chi, X. Wang, Z. Gao, S. Li and G. Li, Org. Lett., 2022, 24, 3657 CrossRef CAS PubMed; (c) Z. Gao, H. Wang, C. Zhou, N. Wang, S. Li and G. Li, Org. Lett., 2022, 24, 3926 CrossRef CAS PubMed.
- (a) B. N. Bhawal and B. Morandi, Isr. J. Chem., 2018, 58, 94 CrossRef CAS; (b) B. N. Bhawal and B. Morandi, Angew. Chem., Int. Ed., 2019, 58, 10074 CrossRef CAS PubMed.
- (a) Y. H. Lee and B. Morandi, Nat. Chem., 2018, 10, 1016 CrossRef CAS PubMed; (b) M. de La Higuera Macias and B. A. Arndtsen, J. Am. Chem. Soc., 2018, 140, 10140 CrossRef CAS PubMed; (c) R. G. Kinney, J. Zgheib, P.-L. Lagueux-Tremblay, H. Yang, D. Gauthier Jr and B. Arndtsen, Res. Sq. , 2022 DOI:10.21203/rs.3.rs-1709579/v1; (d) P. Boehm, P. Müller, P. Finkelstein, M. A. Rivero-Crespo, M. Ebert, N. Trapp and B. Morandi, J. Am. Chem. Soc., 2022, 144, 13096 CrossRef CAS PubMed.
- (a) P. Wang, P. Verma, G. Xia, J. Shi, J. X. Qiao, S. Tao, P. T. W. Cheng, M. A. Poss, M. E. Farmer, K.-S. Yeung and J.-Q. Yu, Nature, 2017, 551, 489 CrossRef CAS PubMed; (b) P. Wedi and M. van Gemmeren, Angew. Chem., Int. Ed., 2018, 57, 13016 CrossRef CAS PubMed; (c) D. Zhao, P. Xu and T. Ritter, Chem, 2019, 5, 97 CrossRef CAS; (d) A. Mondal, H. Chen, L. Flämig, P. Wedi and M. van Gemmeren, J. Am. Chem. Soc., 2019, 141, 18662 CrossRef CAS PubMed; (e) M. Farizyan, A. Mondal, S. Mal, F. Deufel and M. van Gemmeren, J. Am. Chem. Soc., 2021, 143, 16370 CrossRef CAS PubMed; (f) C. Santiago, H. Chen, A. Mondal and M. van Gemmeren, Synlett, 2022, 33, 357 CrossRef CAS.
- (a) H. Chen, P. Wedi, T. Meyer, G. Tavakoli and M. van Gemmeren, Angew. Chem., Int. Ed., 2018, 57, 2497 CrossRef CAS PubMed; (b) H. Chen, M. Farizyan, F. Ghiringhelli and M. van Gemmeren, Angew. Chem., Int. Ed., 2020, 59, 12213 CrossRef CAS PubMed; (c) A. Mondal and M. van Gemmeren, Angew. Chem., Int. Ed., 2021, 60, 742 CrossRef CAS PubMed; (d) P. Wedi, M. Farizyan, K. Bergander, C. Mück-Lichtenfeld and M. van Gemmeren, Angew. Chem., Int. Ed., 2021, 60, 15641 CrossRef CAS PubMed.
- (a) M. Catellani, E. Motti, N. Della cá and R. Ferraccioli, Eur. J. Org. Chem., 2007, 4153 CrossRef CAS; (b) O. Daugulis, H.-Q. Do and D. Shabashov, Acc. Chem. Res., 2009, 42, 1074 CrossRef CAS PubMed; (c) K. Muniz, Angew. Chem., Int. Ed., 2009, 48, 9412 CrossRef CAS PubMed; (d) L.-M. Xu, B.-J. Li, Z. Yang and Z.-J. Shi, Chem. Soc. Rev., 2010, 39, 712 RSC; (e) A. J. Hickman and M. S. Sanford, Nature, 2012, 484, 177 CrossRef CAS PubMed.
- R. C. Samanta and H. Yamamoto, Eur. J. Chem., 2015, 21, 11976 CrossRef CAS PubMed.
- (a) J. B. G. Gluyas, C. Burschka, S. Dörrich, J. Vallet, H. Gronemeyer and R. Tacke, Org. Biomol. Chem., 2012, 10, 6914 RSC; (b) E. J. Emmett, B. R. Hayter and M. C. Willis, Angew. Chem., Int. Ed., 2014, 53, 10204 CrossRef CAS PubMed.
- T. Sugita, M. Idei, Y. Ishibashi and Y. Takegami, Chem. Lett., 1982, 11, 1481 CrossRef.
- P. Lulinski and L. Skulski, Bull. Chem. Soc. Jpn., 1999, 72, 115 CrossRef CAS.
- J. Lv, Q. Liu, J. Tang, F. Perdih and K. Kranjc, Tetrahedron Lett., 2012, 53, 5248 CrossRef CAS.
- S. N. Joshi, S. M. Vyas, H. Wu, M. W. Duffel, S. Parkin and H.-J. Lehmler, Tetrahedron, 2011, 67, 7461 CrossRef CAS PubMed.
- C. A. Panetta, Z. Fang and D. L. Mattern, J. Org. Chem., 1995, 60, 7953 CrossRef CAS.
- (a) M. L. dos Santos, G. C. de Magalhães and R. Braz Filho, J. Organomet. Chem., 1996, 526, 15 CrossRef CAS; (b) H.-G. Cheng, Z. Yang, R. Chen, L. Cao, W.-Y. Tong, Q. Wei, Q. Wang, C. Wu, S. Qu and Q. Zhou, Angew. Chem., Int. Ed., 2021, 60, 5141 CrossRef CAS PubMed.
- L. Yet, Privileged Structures in Drug Discovery; John Wiley & Sons, Inc, 2018 Search PubMed.
- J. Barluenga, J. M. Alvarez-Gutiérrez, A. Ballesteros and J. M. González, Angew. Chem., Int. Ed., 2007, 46, 1281 CrossRef CAS PubMed.
- (a) N. Tsuji, Tetrahedron, 1968, 24, 1765 CrossRef CAS PubMed; (b) A. R. Hajipour and H. Adibi, J. Chem. Res., 2004, 2004, 294 CrossRef.
- C. Morice, M. Domostoj, K. Briner, A. Mann, J. Suffert and C.-G. Wermuth, Tetrahedron Lett., 2001, 42, 6499 CrossRef CAS.
- Further scrambling experiments and an expanded discussion of all mechanistic experiments are provided in the ESI.†.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sc00801k |
| ‡ Equal contribution. |
| This journal is © The Royal Society of Chemistry 2023 |