
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceArsenic binding to human metallothionein-3†
Amelia T.
Yuan
 and
Martin J.
Stillman
*
and
Martin J.
Stillman
*
Department of Chemistry, University of Western Ontario, 1151 Richmond St., London, ON N6A 5B7, Canada. E-mail: martin.stillman@uwo.ca
First published on 4th May 2023
Abstract
Arsenic poisoning is of great concern with respect to its neurological toxicity, which is especially significant for young children. Human exposure to arsenic occurs worldwide from contaminated drinking water. In human physiology, one response to toxic metals is through coordination with the metallochaperone metallothionein (MT). Central nervous system expression of MT isoform 3 (MT3) is thought to be neuroprotective. We report for the first time on the metalation pathways of As3+ binding to apo-MT3 under physiological conditions, yielding the absolute binding constants (log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Kn, n = 1–6) for each sequential As3+ binding event: 10.20, 10.02, 9.79, 9.48, 9.06, and 8.31 M−1. We report on the rate of the reaction of As3+ with apo-MT3 at pH 3.5 with rate constants (kn, n = 1–6) determined for each sequential As3+ binding event: 116.9, 101.2, 85.6, 64.0, 43.9, and 21.0 M−1 s−1. We further characterize the As3+ binding pathway to fully metalated Zn7MT3 and partially metalated Zn-MT3. As3+ binds rapidly with high binding constants under physiological conditions in a noncooperative manner, but is unable to replace the Zn2+ in fully-metalated Zn-MT3. As3+ binding to partially metalated Zn-MT3 takes place with a rearrangement of the Zn-binding profile. Our work shows that As 3+ rapidly and efficiently binds to both apo-MT3 and partially metalated Zn-MT3 at physiological pH.
Kn, n = 1–6) for each sequential As3+ binding event: 10.20, 10.02, 9.79, 9.48, 9.06, and 8.31 M−1. We report on the rate of the reaction of As3+ with apo-MT3 at pH 3.5 with rate constants (kn, n = 1–6) determined for each sequential As3+ binding event: 116.9, 101.2, 85.6, 64.0, 43.9, and 21.0 M−1 s−1. We further characterize the As3+ binding pathway to fully metalated Zn7MT3 and partially metalated Zn-MT3. As3+ binds rapidly with high binding constants under physiological conditions in a noncooperative manner, but is unable to replace the Zn2+ in fully-metalated Zn-MT3. As3+ binding to partially metalated Zn-MT3 takes place with a rearrangement of the Zn-binding profile. Our work shows that As 3+ rapidly and efficiently binds to both apo-MT3 and partially metalated Zn-MT3 at physiological pH.
Introduction
Chronic arsenic poisoning of populations has been reported throughout history and continues today. According to the World Health Organization, there are 200 million people exposed to dangerous levels of arsenic from a variety of sources including air, minerals, water, and soil, and most commonly ingested as a component of food or water.1,2 Other sources of arsenic include occupational exposure from mining and the electronics industry.1 Arsenic can be leached into food and water under reducing conditions from surrounding soil and minerals.3,4 Arsenic can exist in the inorganic form as As(III) or As(V), as well as organic arsenicals of monomethylarsonic acid (MMA), dimethylarsinic acid (DMA), and trimethylarsine oxide.3,5 As3+ is particularly toxic because of its ability to enter cells and bind to thiol groups of cysteine-containing proteins important in human physiology.1,3,5–9Chronic and acute arsenic exposure leads to a multitude of symptoms, including abdominal pain, skin lesions, diabetes, neuropathy, hepatic and renal dysfunction, and reproductive consequences.3,10 One of the concerns raised is its neurotoxicity, which may lead to developmental adverse outcomes, especially with children.11 This could manifest as epilepsy, lower IQ scores, lower vocabulary scores, higher risk of intellectual disability diagnosis, and lower visuospatial skills, as suggested by cross-sectional studies ranging from populations in Bangladesh to the United States.12–14 While symptoms related to As3+ exposure are relatively well-established, the methodology of action is less established. As3+ is known to cross the blood–brain barrier, however, its specific target in the neurological system is unknown, with some suggestion of binding to proteins such as the amyloid beta plaques characteristic of Alzheimer's Disease.12,13,15 The commonly accepted effect of As(III), either as inorganic or organic arsenic, is the production of reactive oxygen species.1,7,12,14,16,17 These reactive oxygen species then produce downstream effects such as the activation of apoptosis, mitochondrial stress, and neurotransmitter imbalances.12,14,15,17
Metallothioneins (MTs) are cysteine-rich proteins that participate in heavy metal detoxification, as well as homeostatic control of physiologically relevant metals such as Cu+ and Zn2+.18–20 MTs are found across species, with mammalian MTs classified as 20-cysteinyl proteins ranging from 6–8 kDa in size.21–23 Mammalian MTs do not contain any aromatic amino acid residues or disulfide bonds, despite a typical 30% of the sequence being cysteines.24 This family of proteins is thought to participate in the regulation of essential metals by acting as a reservoir and donating these metal ions to apo-metalloenzymes when necessary.25–28 MTs also sequester toxic metals such as Cd2+ and As3+ that are excreted in the urine or bile.29–32 Lastly, MTs are antioxidants in that they can neutralize reactive oxygen species to form disulfide bonds between cysteinyl thiols.33–35 There are four isoforms of mammalian MTs.36,37 MT1 and MT2 are inducible by metals and are expressed in all tissues, with primary concentration in the kidneys and liver.20,37–39 MT3 and MT4 are not metal-inducible, and are expressed mostly in the central nervous system and squamous cell tissue, respectively.36,37
MT3 is of particular interest due to its expression in the central nervous system, in particular, in the brain.40–42 It was reported that under normal conditions, it exhibits growth-inhibitory activity and its downregulation may be linked to the progression of neurodegenerative diseases such as Alzheimer's Disease and Parkinson's Disease.43–45 The chemical basis of its protective effects lies in its ability to remove copper from insoluble amyloid-beta plaques characteristic of Alzheimer's Disease and insoluble α-synuclein characteristic of Parkinson's Disease.44,46–49
Structurally, MT3 forms two domains when fully metalated with a divalent metal such as Zn2+ or Cd2+.22 These domains are conserved across all isoforms of MTs, with an N-terminal β domain and a C-terminal α domain.19,23 MT3 has a TCPCP conserved sequence (AA 5–9) in the N-terminal domain, which has been associated with its growth inhibitory activity.50 In addition, it has an acidic loop insert (AA 58–68) in the C-terminal domain, which has been suggested to allow increased flexibility of the protein.51 The sequence of the MT3 used in this paper is shown in Fig. 1. The fully-metalated Cd7MT3 has been partially characterized by NMR methods, with the β-domain structure undetermined due to fluxionality.52 For MTs, in general, divalent metals (Cd2+, Zn2+) bind in a tetrahedral coordination, whereas monovalent metals (Cu+) bind trigonally or digonally, and trivalent metals (Bi3+, As3+) bind trigonally.22,53–57 For MT3, the only structure reported has been the Cd7MT3 species, however, titrations with Pb2+, Zn2+ and Cu+ have been shown to result in well-defined species.43,44,48,58–60
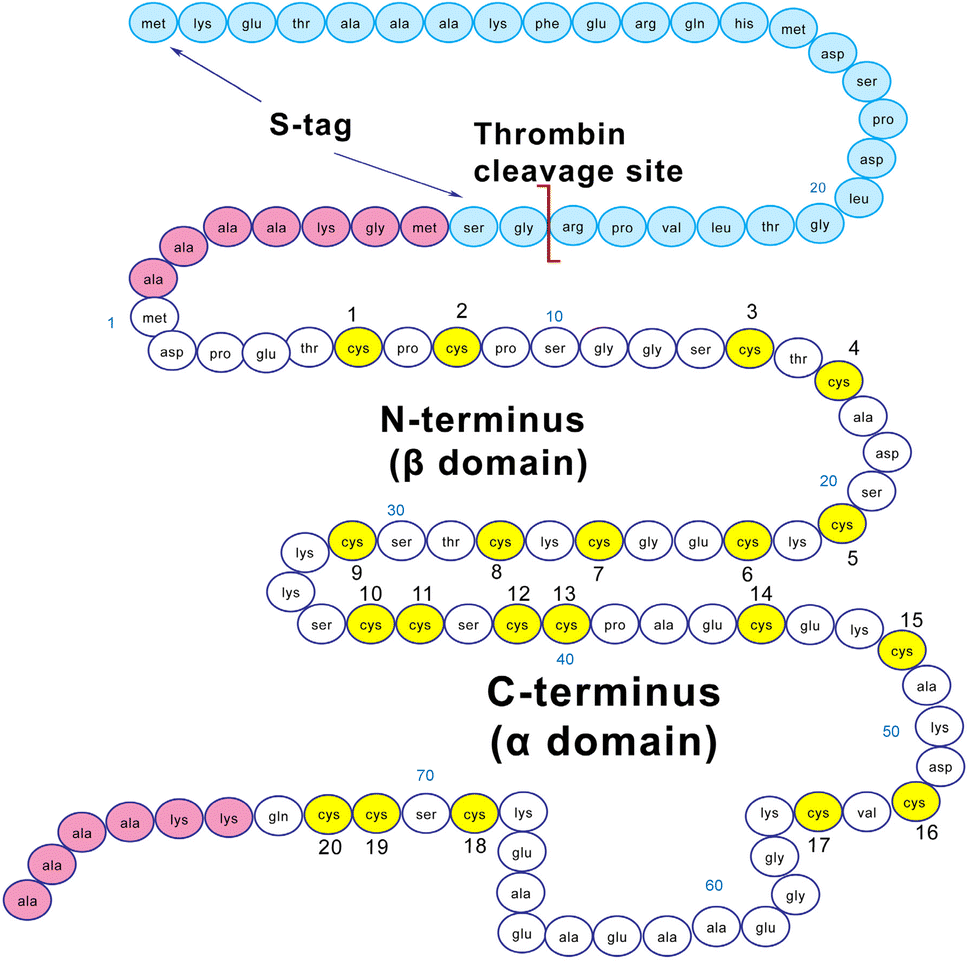 | ||
| Fig. 1 Amino acid sequence of MT3 used in this study. Amino acids (AAs) 1 to 31 form the β domain, 32 to 34 form the linker region, and 35 to 73 form the α domain. The domains are labelled for the clusters formed when 7 equivalents of Zn2+ or Cd2+ bind to the cysteines (yellow). The AAs in pink are included in the recombinant protein for increased stability during the expression step. The AAs in blue are the S-tag for increased stability, removed in the protein purification process. | ||
MTs have multiple binding pathways: a cooperative pathway where clusters are formed and bridging thiols are used to coordinate the metal ions and a noncooperative pathway where metal ions are coordinated by terminal cysteines, and thus no bridging thiols are present.61 It is important to note that these two distinct pathways impact the stability of the metal-thiolate network. We have also found that these binding pathways change depending on conditions such as pH and the presence of interacting proteins.28,61
Detailed descriptions of As3+ binding to MTs has only been reported for MT1 and MT2. Thus far, As3+ binding to MTs have been defined by noncooperative binding and trigonal-pyramidal coordination by cysteinyl thiols.57,62–64 This was investigated at physiological pH for MT2 and acidic pH for MT1.57,63,65 For MT1, the individual stepwise speciation was used to calculate the corresponding relative binding constants and rate constants consistent with noncooperative, terminally-coordinated arsenic binding.57 The distribution of Asn-MT (n = 1–6) species were Normally distributed and centered upon the average number of As3+ bound to MT, characteristic of noncooperative binding.22,57,63,65 Slightly more complicated metalation properties have been reported for the related Bi3+ metalation reactions. Bi3+ is also coordinated in a trigonal-pyramidal coordination by cysteinyl thiols in MTs.55,66 For Bi3+, noncooperative binding occurs under acidic conditions but a cooperative Bi2MT species forms under physiological conditions for MT1.55
In this paper, we report detailed spectroscopic analysis of As3+ binding to apo-MT3 and Zn-MT3 under physiological and acidic conditions. We calculated the relative binding constants for each As3+ binding event from the ESI-mass spectral data. We report the rate constants for each of the six As3+ binding events under acidic conditions and determined that the relative rate of As3+ binding under physiological conditions was too fast to be measured by mass spectrometric methods. We were able to further support these conclusions with time-dependent absorption spectra. Using ESI-MS and time-dependent absorption spectra, we investigated how As3+ bound to the physiologically relevant fully and partially Zn-metalated MT3 species.
Results and discussion
Arsenic binding to metallothionein 3 under physiological conditions
Apo-MT3 concentrations at pH 7.4 were determined using UV-visible spectroscopy. Apo-MT3 was confirmed to be reduced from the ESI-mass spectral data, with the corresponding mass recorded as 8211 Da. To determine the rate of As3+ binding to apo-MT3 at pH 7.4, 8 molar equivalents of As3+ was added to a solution of apo-MT3 and the ESI-mass spectral data recorded. In Fig. 2A and B, we show that within 1 minute, 6 As3+ ions have bound to the MT3, with no intermediates isolated.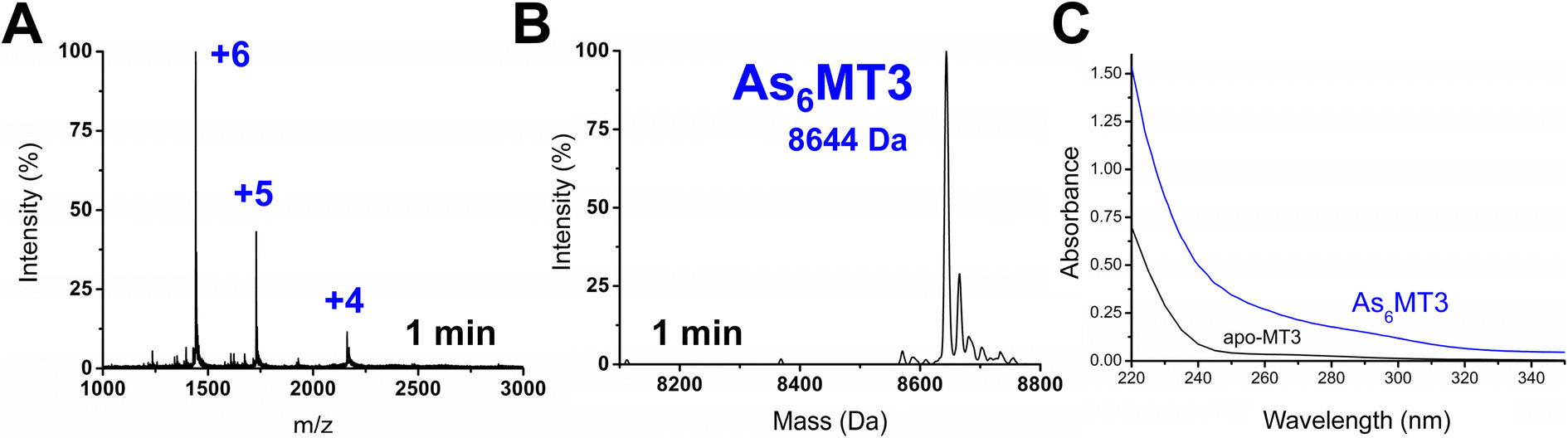 | ||
| Fig. 2 As3+ metalation of apo-MT3 at pH 7.4, monitored using ESI-MS. The concentration of apo-MT3 used was 50 μM and 8 molar equivalents of As3+ from As2O3 were added. (A) Charge state spectrum at 1 minute post metalation. (B) Deconvoluted spectrum 1 minute post metalation. (C) UV-visible absorption spectrum of apo-MT3 and As6MT3. The As6MT3 was confirmed to be the sole species using ESI-MS. | ||
The mass spectral data show that there is only one species in solution, As6MT3 at a mass of 8644 Da, even though 8 molar equivalents of As3+ were added to the solution. The As3+ are likely coordinated with 3 cysteinyl thiols each, with no evidence of no bridging cysteines.57 Any remaining cysteines are reduced as no disulfide bonds were noted by a 280 nm absorption band. This is comparable to the data illustrating As3+ binding to MT1.57 Further support of the non-bridging noncooperative binding pathway was reported for MT1 in the past by linking three α domains together to form ααα-MT1, where there were 33 cysteine sites available.67 In this case, only 11 As3+ bound to the protein, likely with each As3+ coordinated by 3 cysteinyl thiols.67 This coordination is also consistent with that of trialkyl trithioarsenite compounds, As(SR)3, and As(GS)3 where trigonal-pyramidal geometry has been reported.68–73
The absorption spectrum of the As6MT3 solution shows a band at 290 nm (Fig. 2C). This band is relatively weak and has low molar absorptivity compared to the S–Cd charge transfer band. We identify 290 nm as the S–As charge-transfer band for MT3, similar to the previously reported 270 nm S–As charge-transfer band for As(GS)3.74
Fig. 2 shows that at physiological pH, As3+ intermediates cannot be isolated due to the rapid binding of As3+ under these conditions. Stopped flow methods were considered for further analysis of the rate of As3+ binding to the apo-MT3, however, stopped flow data only provides an average rate constant dependant on the absorption at the 290 nm charge transfer band. The implication of rapid binding of As3+ to apo-MT3 indicates potential dangers in arsenic poisoning, as it is not hindered kinetically from binding to apo-MTs at physiological pH; this is in contrast to the slow rates observed under acidic conditions.57 This rapid binding of As3+ to apo-MT3 is similar to that observed in the early stages of Zn2+ and Cd2+ binding to apo-MTs, when the non-clustered terminally coordinated beads form.65 However, to obtain more information on the partially metalated As-MT3 intermediates that form, we used stepwise As3+ metalation.
To determine the absolute binding constants for As3+ binding to apo-MT3 at pH 7.4, we used a competitive ligand with a known binding constant in solution with apo-MT3 (Fig. 3). In our case, we used the peptide glutathione (GSH), which binds to As3+ similarly to MT3 with a trigonal-pyramidal coordination with 3 molar equivalents of glutathione per As3+ ion.70,74,75 The reported binding constant for the As(GS)3 complex is logβ = 7, therefore, this value was used in modelling the experimental data of binding of As3+ to apo-MT3 and GSH.74 A sample of the ESI-mass spectral data collected (Fig. 3A–D) as well as speciation (Fig. 3E) and modelling data (Fig. 3F) are shown in Fig. 3.76 The absolute binding constants (logKn, n = 1–6) for each As3+ binding step (Scheme 1) were determined: 10.20, 10.02, 9.79, 9.48, 9.06, and 8.31 M−1. Simulated mass spectra are generated using this model and compared to experimental spectra in ESI Fig. S1.† In addition, the first binding site was confirmed to be reasonable with the chelate effect equation: logK(polydentate) = logβn(unidentate) + (n − 1)log55.5 where logK(polydentate) is the binding constant of a n-dentate polydentate ligand (MT3, in this case) and logβn(unidentate) is the binding constant of the complex with n unidentate analogues (GSH) and 55.5 is the molarity of water, first described by Hancock et al.77
 | ||
| Fig. 3 As3+ stepwise titration into apo-MT3 with 3 molar equivalents of reduced GSH at pH 7.4. A sample of charge state spectra (left panels) and deconvoluted spectra (right panels) for As3+ added at 1.15 (A), 2.30 (B), 4.60 (C), and 5.75 (D) molar equivalents. All spectra were recorded for 2 minutes at ambient temperature. (E) Speciation of AsnMT3 species as a function of mol. eq. As3+ added to apo-MT3 in solution with GSH at pH 7.4. Symbols represent experimental data and solid lines represent model fitted to data. The model was calculated using HySS software. (F) Corresponding logK inputted to HySS. The logK values determined for each As3+ binding event are as follows: 10.20, 10.02, 9.79, 9.48, 9.06, and 8.31 M−1. | ||
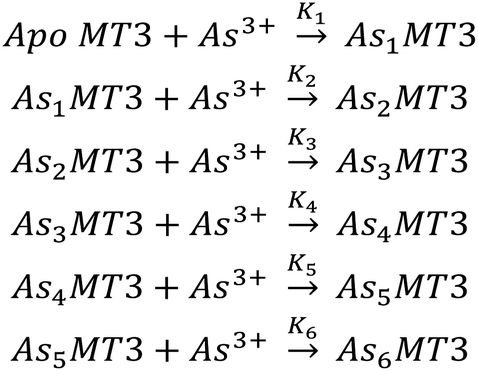 | ||
| Scheme 1 The sequential binding pathway for As3+ binding to apo-MT3. The equilibrium constants for each step are indicated by K1–6 for each As3+ binding event. | ||
To rule out protein–protein interactions, apo-MT3 in the absence of GSH was metalated with increasing molar equivalents of As3+ at pH 7.4 (Fig. S2†). The metalation profile shown in Fig. S1† is similar to that reported for MT1 metalated by As3+ under acidic pH conditions. For both proteins, we see noncooperative binding, or a normal distribution among AsnMT3 (n = 1–6) species centered on the average As3+ bound.65 This means that there are no AsnMT3 species that are thermodynamically favoured or more stable than the rest. From the speciation diagram calculated from the raw data, we can model this stepwise titration with HySS software.76 The same binding constants (logKn, n = 1–6) as determined above with GSH acting as a competitive ligand can be fit to the experimental data (Fig. S3†).
We report that the metalation data recorded for As3+ binding to apo-MT3 in the presence and absence of GSH are essentially identical, as noted by the speciation diagrams shown in Fig. 3E and S3A.† This indicates that the presence of the competing GSH in solution with the apo-MT3 did not introduce additional interactions between the protein and the peptide, and thus, did not change the metalation pathway. We note that GSH does not effectively compete for the As3+ until MT3 has reached saturation. This is confirmed using the logK values for As3+ binding to MT3 and noting the free As3+ at an arbitrary point and comparing it to what was calculated for As3+ binding to GSH (Table S1†).
The binding constants obtained through this competition experiment are in the range of logK = 8–10, which is surprisingly low compared with those of Zn2+ and Cd2+. For Zn2+, the logK values for each individual metalation step ranges from logK = 11–12, whereas for Cd2+, the values range from logK = 14–15 for 7 binding steps. Since we see As3+ binding to apo-MT1 at pH 3.5, a condition that does not allow Cd2+ or Zn2+ binding as the protons outcompete the metal ions for the cysteinyl thiolates, we expected that the binding constants would be higher for As3+ binding. However, larger logK values do not accurately fit the As3+ binding data and the possibility of cluster formation and tetrahedral coordination may also further stabilize Zn2+ and Cd2+ bound MTs, and thus, resulting in a higher binding constant.22
The binding constants for each subsequent As3+ binding event decrease, which is as a result of the statistical loss of available free thiols as the protein is sequentially metalated. This means that the last As3+ bound shows the least affinity, and therefore, may be outcompeted by other metals in the solution.
Because the pathway is noncooperative, As3+ can bind to apo-MT3 resulting in partially metalated As-MT3 species even with just one As3+ bound. In this manner the As3+ binding is similar to the initial stages of Zn2+ and Cd2+ binding, in which terminal thiolate coordination dominates. This property of As3+ binding also implies that partially metalated As-MT3 species are stable in solution, therefore, may not release As3+ into the cellular environment. Even though As3+ binding to MT3 shows decreased affinity after each subsequent binding event, the overall affinity of each of these reactions is relatively high and greater than the As3+ binding constant to glutathione as a result of the effect of multiple cysteines and the chelate effect.67,74 As MTs contain 20× the thiols of glutathione, the overall logβ is much greater even though each As3+ involves 3 thiolates from the MT3 similar to the As(GS)3 structure.
The trend in Fig. 3F shows that the logK values do not linearly decrease, suggesting the presence of additional factors that impact As3+ binding. One possible reason for this non-linear trend is the rearrangement of bound As3+ in existing MT3 sites to accommodate the incoming As3+. Because of the trigonal pyramidal coordination with no evidence of bridging cysteines, As3+ binding thiolates are likely arranged in sequential order, therefore, if the first As3+ ions bound do not follow this pattern, rearrangement is necessary to bind the 6 As3+ ions that use the 18 cysteinyl thiolates.
With reference to the charge states in Fig. 3A–D, we note that the distribution of charge states for each spectrum as a function of As3+ metal loading shifts to that of a lower weighted mean. For example, we see that the initial As3+ loading of MT3 (Fig. 3A) has charge states of mostly 5+ and 6+ with some 7+ and 4+, whereas As5MT3 and As6MT3 (Fig. 3D) has charge states of mostly 5+ and 4+ species. When looking at the weighted mean of the charge state present in the spectra, the value is +5.4 for apo-MT3, 4.8+ for As6MT3, and 4.9+ for Cd7MT3 (unpublished data). We note that the average weighted charge states for all these species are relatively similar, with apo-MT3 only slightly larger, indicating a larger surface area.78 This is mostly consistent with As-MT1 species, however, at physiological pH, we see that 5+ instead of 6+ is the dominant species.65 This means that for MT3 under physiological conditions, we may see a greater percentage of more compact structures than we see for MT1 at pH 3. In comparison to Cd7MT species, the 5+ charge state is similarly favoured under fully metalated conditions at physiological pH.22,79 From a structural point of view, it is clear that the two-cluster domains of Cd7MT3 is likely as compact as the MT3 can be. However, it is evident from the data presented here that the As6MT3 comprises 6 terminally coordinated As3+ that we associated with the term “beads” is also compact, which suggests that MTs generally adopt structures that limit exposure of the coordinating cysteines to the solvent. Our molecular dynamics calculations described below illustrate how compact the fully As-bound MT3 is (vide infraFig. 6).
In addition, the elucidation of the binding mechanism of As3+ under physiological conditions further supports the mechanism of binding under acidic conditions as previously determined.57 The kinetic parameters, as well as the thermal stability measurements, reported previously at pH 3.5 for MT1 may therefore be applicable in relative terms to the physiological pathway. This is surprising in some regard because of the changes in binding pathways seen for most other metals binding to MTs, for example, Bi3+ binds cooperatively at physiological pH but not under acidic conditions, and Zn2+ initially binds cooperatively under acidic conditions to form clusters but not under physiological conditions, where beads predominate initially.55,61
The logKn values for As3+ binding to apo-MT3 under physiological conditions reported here are especially important in the context of MT3 because MT3 is constitutively expressed and not metal-inducible. Therefore, it follows that the de novo apo-MT3 exists in cellular systems without Zn2+ protection at certain time points and is susceptible to As3+ binding in the event that As3+ is transported into the neural environment.
As3+ reaction rate when binding to apo-MT3 is faster than previously reported for apo-MT1
To further elucidate the As3+ binding pathway, we measured the As3+ binding kinetics using acidic pH to slow down the reaction. Each of the AsnMT3 (n = 0–6) species were observed via ESI-mass spectral data as a function of time (Fig. 4). Fig. 4A–F show a reduced selection of charge state spectra on the left panels and corresponding deconvoluted spectra on the right for different time points during the reaction. Fig. 4G shows the experimental points and corresponding fitted speciation as determined by COPASI using the Scheme 2 set of equations. The corresponding fitted bimolecular rate constants are plotted in Fig. 4H with error bars representing the standard deviation from the fitting of 6 separate experimental replicates. Each step of the As3+ metalation of apo-MT3 follows the second order reaction with the rate law: Rate = kn[Asn−1MT3][As3+]. The bimolecular rate constants determined for each As3+ binding to apo-MT3 at pH 3.5 are as follows: 116.9, 101.2, 85.6, 64.0, 43.9, and 21.0 M−1 s−1. | ||
| Fig. 4 As3+ binding to apo-MT3 as a function of time. 8 molar equivalents of As3+ were added to 20 μM apo-MT3 at pH 3.5. A total of 6 replicates were analyzed to ensure accuracy. (A–F) Charge state (left) and deconvoluted (right) mass spectra of As3+ metalation kinetics taken at 2 minutes (A), 4 minutes (B), 10 minutes (C), 16 minutes (D), 24 minutes (E), and 45 minutes (F) as a sample of data collected. Charge states are labelled on the first spectra and apply to the spectra below. Apo-MT3 and AsnMT3 species are labelled and applies to all spectra. (G) Speciation of AsnMT3 species as a function of time, with experimentally determined data represented by points and modelled speciation curves fitted by COPASI as solid lines. (H) Fitted rate constants for each As3+ binding event to MT3, with standard error represented by error bars. | ||
 | ||
| Scheme 2 The sequential binding pathway for As3+ binding to apo-MT3. The biomolecular rate constants for each step are indicated by k1–6 for each As3+ binding event. | ||
One observation we can make about the kinetic data is that the trend in each stepwise rate constant is approximately linear (Fig. 4H). It follows that as the thiolate sites are used up for sequential As3+ binding that the rate decreases due to the lack of accessibility for subsequent binding. This is similar to the result noted for MT1.57,65 However, MT3 differs from MT1 in terms of the relation between k1 and k2. For MT1, both the full protein and the α domain fragment exhibited a reduction in the magnitude of k1 with respect to k2, which suggested that the first As3+ was binding in the alpha domain region. For MT3, the rate constants of k1 with respect to k2 show the opposite pattern. This may be due to the α domain region being significantly different in structure because of the acidic loop insert that is located before the final three cysteines (Fig. 1). Another possible explanation for the absence of this inhibited first As3+ binding event is the fluxionality of the β domain of MT3, as suggested by Wang et al.52 Although the structure is compact, if the first binding site is located in the β domain, then As3+ may be able to access the thiols of that domain without significant interactions that reduce the rate.
Another observation we can make about the data is the similar shift of charge states as seen for the stepwise addition of As3+ under physiological conditions. Interestingly, the 5+ charge state remains the dominant species of As6MT3 even at pH 3.5. This is indicative that the compact structure persists despite the expectation that at the acidic pH, the structure of MT3 would be more greatly expanded, and thus, would reveal higher charge states. We can also compare these data to those for MT1, where the 6+ charge state is the dominant species, indicating that MT3 is more compact when fully metalated.80 This could have implications in the stability of the As6MT3 as a possible protective mechanism against brain located As3+ species.
Lastly, the data in Fig. 4 show that the rate constants for As3+ binding at pH 3.5 are significantly faster than those measured for MT1. For MT1, the rate constant values ranged from 4–25 M−1 s−1 at room temperature.65 This is already significantly faster than observed for the single domain fragments, which ranged from 1–7 M−1 s−1 for both the β and α domains.57 In contrast, the rate constants for MT3 range from 20 to 117 M−1 s−1. Considering the similarity between the sequence and available cysteines of apo-MT3 and apo-MT1, this is surprising.
However, there is a major difference in the structural positioning of the final 3 cysteines in MT3 compared with MT1. Based on the analysis of Ngu et al. that the first As3+ bound in α domain, we can consider whether the last 3 cysteines are significantly more exposed to the solvent and therefore allow more rapid binding. The rate of the first As3+ bound controls the subsequent 5 reactions. Ngu et al. suggested that the evolution of a two-domain structure of MT1 was important in that the probability of collision with correct orientation between the thiols and As3+ was increased as a function of the number of thiols in the protein, thus, providing evidence for its efficacy as a metal scavenger.65 In MT3, we now see also that the availability of those thiols can change the rate of reaction, where in this case the acidic loop changes the overall structure of the protein.
To introduce a secondary source of kinetic data, we monitored the As3+ binding mechanism using UV-visible spectroscopy and compared the overall rate determined with the ESI-mass spectral data under the same conditions (Fig. 5). We used the 290 nm S–As charge transfer band as an indicator of As3+ binding and determined an overall rate constant of 16.6 M−1 s−1. Using ESI-MS data outlined in Fig. 4, we can determine the average As3+ bound to MT3 as a function of time and plot it against the kinetic trace determined using UV-visible spectroscopy (Fig. 5C). these data confirm that the mass spectral data are correlated with the solution reactions obtained from the absorption spectroscopy, and that the overall kinetic parameters determined from ESI-mass spectral data can be reliably used in the case of As3+ binding to MT3.
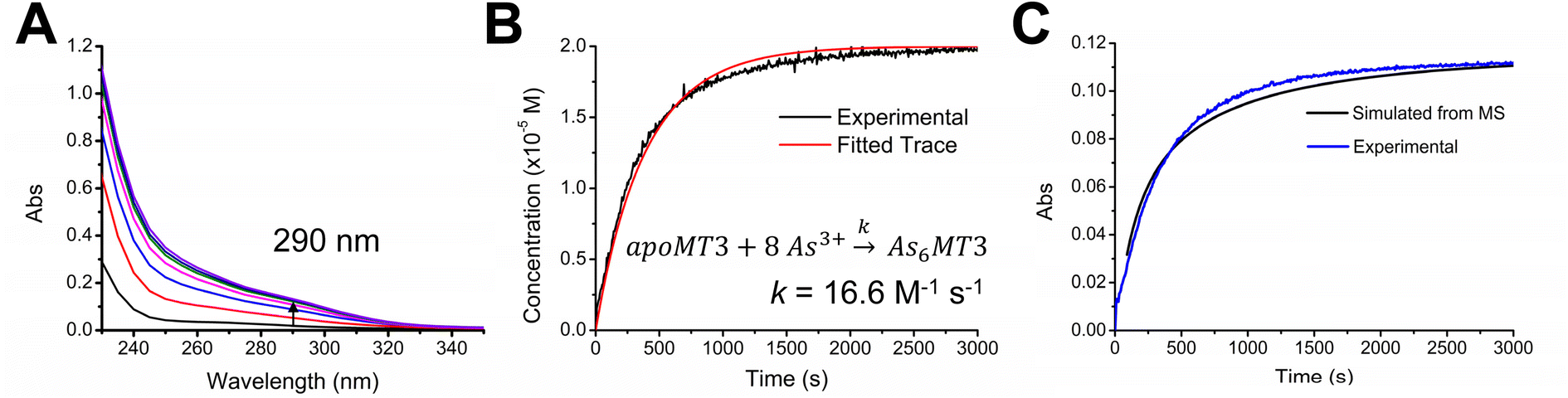 | ||
| Fig. 5 As3+ binding to apo-MT3 at pH 3.5 monitored at 290 nm as a function of time. (A) Sample of absorption spectra taken at various time point intervals during the reaction. The arrow shows the 290 nm absorption band increasing in intensity as As3+ binds to apo-MT3. (B) Kinetic trace of As3+ binding determined by the 290 nm band (black line) and fitted trace determined by COPASI (red line). The fitted k = 16.6 M−1 s−1. (C) Kinetic trace of As3+ binding experimentally determined by UV-visible spectroscopy (blue line) compared with the time-dependence of As3+ binding calculated from mass spectral data (black line). | ||
 | ||
| Fig. 6 Ribbon and ball-and-stick molecular models of representative As-MT3 species. The energy-minimized structures were calculated for 300 K conditions for 1000 ps with 0.02 ps equilibration time. The protein backbone is represented by the grey ribbon, the S atoms in yellow, the As3+ ions in purple, and the rest of the protein in brown. (A) The As3+ was bound to three cysteinyl thiolates sequentially starting at the N-terminal β domain (Cys1–3, Cys4–6, Cys7–9, Cys10–12, Cys13–15, Cys18–20), with the last As3+ bound by the last three thiolates due to the sequential thiolates occurring on either side of the acidic loop of the α domain. (B) The As3+ was bound to three cysteinyl thiolates sequentially starting at the C-terminal α domain, in reverse order to (A). | ||
Modelling the structure of AsnMT3 (n = 0–6)
The structures of the As3+ bound to apo-MT3 can be modelled using Scigress Modelling Software (Fig. 6). There are two steps in this calculation: the first being the molecular mechanics minimization of the structure as constructed and the second being the use of molecular dynamics to search for the lowest energy structure. The conditions used in the molecular dynamics step were 300 K and the calculation was performed for 1000 ps with 0.02 ps equilibration time to select for the energy-minimized structure of each AsnMT3 structure. We showed As3+ sequential metalation both starting in the N-terminal β domain (Fig. 6A) and C-terminal α domain (Fig. 6B).The series of structures in Fig. 6 show that binding As3+ does not significantly change the overall surface area of MT3. This is confirmed using solvent accessible surface area (SASA) calculations, where all structures show SASAs of 5500–6300 Å2.81 As an apoprotein, MT3 is still relatively compact, as determined previously.80 The addition of As3+ to the MT3 does not result in a specific structure when compared, for example, to the structure of Zn7MT with its two cluster domains. The As3+ bound to the protein, however, is relatively shielded and located in the interior space of the protein. This may explain the minimal change to lower charge states upon metalation (Fig. 3 and 4).78 Our models also demonstrate that since the structure of MT3 is not greatly impacted by metalation, the subsequent As3+ binding sites are not significantly different or shielded, therefore explaining the linearly decreasing rate constants of As3+ binding attributed to the decreasing availability of sites illustrated in Fig. 4.
As3+ does not outcompete fully metalated Zn7MT3
With the knowledge of the binding constants of As3+ binding to apo-MT3 as well as the binding pathway involved in the metalation process, we further extended our work to include Zn7MT3, which is fully metalated with no cysteines available for binding additional metals. Essentially, we were interested in determining whether As3+ would be able to displace Zn2+ in MT3 under physiological pH, as it could further provide information on the conditions under which As3+ could be disruptive to the functions of MT3. The results obtained by the addition of 10 molar equivalents of As3+ to Zn7MT3 at pH 7.4 are summarized in Fig. 7. ESI-mass spectral data show the continued presence of Zn7MT3 before the reaction and the changes in the spectrum obtained after As3+ addition can be attributed to adduct formation (Fig. 7A and B). This reaction can be further analyzed using UV-visible spectroscopy, where we note the minimal change in absorption after the addition of As3+ even after 12 hours of reaction time (Fig. 7C). Our interpretation is that the As3+ added does not displace Zn2+, as the absorption that we noted for the S–As charge transfer band at 290 nm does not appear in Fig. 7C. | ||
| Fig. 7 As3+ binding to Zn7MT3 at pH 7.4. (A and B) Charge state (left) and deconvoluted (right) spectra of Zn7MT3 (A) and Zn7MT3 with 10 molar equivalents of As3+ added (B). The charge states are labelled in the first panel and apply to the charge state spectrum below. The mass of Zn7MT3 is labelled in the deconvoluted spectra. (C) Absorption spectra of Zn7MT3 (black line) and Zn7MT3 with 10 molar equivalents of As3+ added after 12 hours of reaction time. | ||
This result can be explained by the lower binding constants obtained for As3+ metalation of apo-MT3, as it was in the range of logK = 8–10, which, for the first As3+ bound, would be an order of magnitude lower than that of Zn2+ binding, which is in the range of logK = 11–12.
For Zn7MT3, the Zn2+ is coordinated by bridging cysteines to form stable Zn4S11 and Zn3S9 clusters in the α and β domains, respectively.22 These clusters are formed cooperatively at low pH and in the presence of over 5 molar equivalents of Zn2+.22,61,82 Interestingly, it has been suggested that the clusters of the individual domains of MT3 may be less stable compared with those of the other MT isoforms. This is illustrated by pH-induced unfolding, where fully metalated MT2 shows a two step unfolding process for each domain in comparison to the absent two step distinction for metalated MT3.83 In addition, 113Cd NMR studies show minor resonances for the 113Cd bound to the β domain compared to that of the α domain and 15N NMR studies illustrate the fluxionality of the β domain, suggesting the instability of this domain in comparison to other MT isoforms.52,83 However, even with the fluxional structure of fully metalated Zn7MT3, full metalation offers protection from As3+ metalation. Due to the coordination of the Zn2+ in fully metalated MT proteins, the As3+ would have to disrupt the clustered Zn4S11 and Zn3S9 structures in order to bind in a noncooperative manner.
As3+ can change Zn2+ distribution in partially metalated Zn-MT3 species
We used partially metalated Zn-MT3 species as a starting point to better emulate physiological conditions when As3+ may bind to existing Zn-MT3. We believe this is a better representation of how MT3 exists in the cellular environment because of the presence of additional MTs that can be induced by Zn2+ presence.22,82 Therefore, it is unlikely for MTs to be fully metalated at any point in time as it would further trigger the production of additional MT.We chose to start with 4 molar equivalents of Zn2+, which forms multiple species including Zn2MT3, Zn3MT3, Zn4MT3, and Zn5MT3 using terminal thiolates in the “bead” structural model (Fig. 8A). Adding even 0.5 molar equivalents of As3+ changes the distribution of Zn2+ species, meaning that the Zn2+ ions are able to rearrange (Fig. 8B). This is especially evident if we look at the ratio of Zn3MT3 to Zn4MT3, where Zn3MT3 became less favored as a species in solution with the addition of As3+, even with the Zn4MT3 species forming mixed As3+ species (Zn4As1MT3). Adding more As3+ molar equivalents to the solution results in additional rearrangement of Zn2+ with predominantly Zn3 and Zn4 species existing as mixed Zn,As-MT3 species in solution (Fig. 8B–H). However, due to the overlap in masses of mixed Zn,As-MT3 species, not all peaks were identified. However, of the identified peaks, it is important to note the prevalence of Zn4As3 species, which we believe is representative of 4 Zn2+ coordinated by the α domain of the protein in the classic cluster conformation and 3 As3+ terminally coordinated by the 9 cysteines of the β domain. Therefore, in the presence of Zn2+, we speculate that As3+ preferentially binds to the N-terminal β domain in MT3. We note that this is contrary to our interpretation of the binding pathway for apo-MT3 above and suggests that the presence of Zn2+ in MT3 may significantly influence the binding of xenobiotic metals by forming substitution-resistant clusters in the α domain.
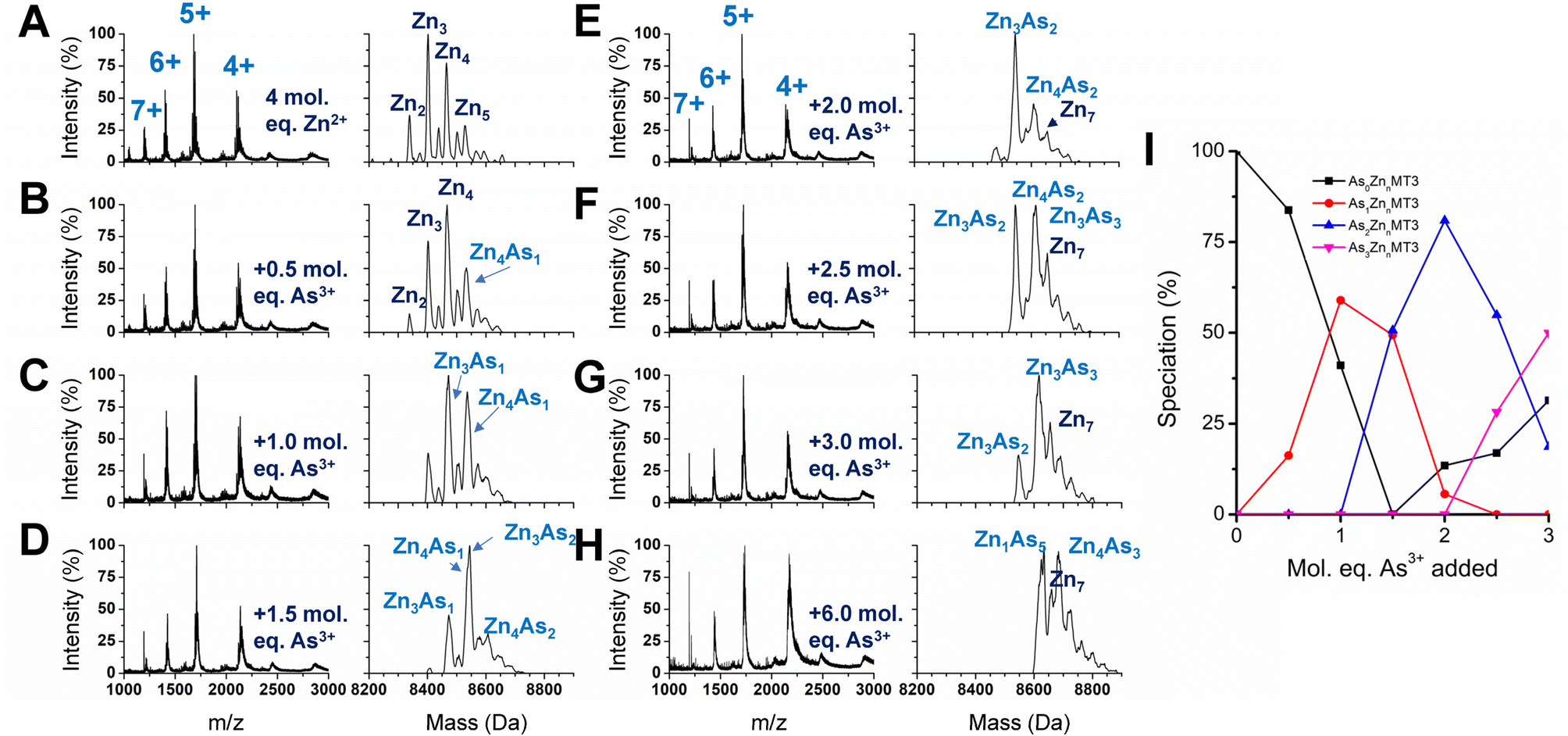 | ||
| Fig. 8 As3+ titration into partially metalated Zn-MT3 at pH 7.4. (A–H) Charge state (left) and deconvoluted (right) spectra of As3+ added to the Zn-MT3 solution formed by adding 4 molar equivalents of Zn2+ to apo-MT3 with As3+ molar ratios of 0 molar equivalents (A), 0.5 molar equivalents (B), 1.0 molar equivalents (C), 1.5 molar equivalents (D) 2.0 molar equivalents (E), 2.5 molar equivalents (F), 3.0 molar equivalents (G), and 6.0 molar equivalents (H). (I) Speciation diagram of AsnMT3 species as a function of molar equivalents of As3+ added to the Zn-MT3 solution formed by adding 4 molar equivalents of Zn2+ to apo-MT3. | ||
If we look specifically at the As3+ bound MT3 and track the number of As3+ bound as a function of As3+ added, it is similar to the As3+ titration at pH 7.4, where As3+ binds sequentially (Fig. 8I). This means that all sites for As3+ binding in partially metalated Zn-MT3 are similar to sites in apo-MT3 – the partial metalation with Zn2+ does not impede additional As3+ binding significantly until the As5 point associated with Zn1As5MT3.
We note that the binding constants of As3+ binding to MT3 are lower than the comparable binding constants for Zn2+ binding to MT3. It appears that when As3+ binds to partially metalated Zn-MT3, the species that form maximize the use of 20 cysteines so that Zn1As5MT3 forms, using 19 cysteines, and Zn4As3MT3 forms, using 20 cysteines, and Zn7MT3 using 20 cysteines forms. This indicates that the metals bound in MT3 rearrange to maximize the number of cysteines involved in the structures. What this suggests in that because mammalian MTs are not considered to be fully metalated under normal conditions, xenobiotic metals such as As3+, Bi3+ and Pt2+ can take advantage of the cysteine availability to form mixed-metal structures with no displacement being required of the existing Zn2+.
This experiment with partially metalated Zn-MT3 further probes the properties of As3+ binding under physiological conditions, where MT3 may not be fully protected as a fully metalated Zn7MT3 protein, but instead exists as multiple partially metalated species. This may imply that Zn2+ supplementation could be beneficial to As3+ poisoning and Zn2+ deficiency exacerbates As3+ poisoning effects, which has been suggested in the past in rat and mouse model systems.3,84–87
Conclusions
Arsenic poisoning has long been a reoccurring problem worldwide due to water contamination. Amongst the effects of arsenic poisoning are neurological issues, thus, it is important to study the brain-located metallochaperone, MT3. This paper reports novel information regarding As3+ binding to MT3, including absolute binding constants of each individual As3+ to apo-MT3 under physiological conditions, as well as ESI-mass spectral data of fast As3+ binding to apo-MT3 at pH 7.4. In addition, we have provided evidence that the binding pathway determined kinetically at pH 3.5 follows the binding pathway determined with stepwise metalation at pH 7.4. There appears to be no specific pH dependence in binding pathway for MT3. In addition, we have shown that fully metalated Zn7MT3 impedes As3+ binding at pH 7.4, but partial metalation with Zn2+ does not significantly impact As3+ binding. This provides support that Zn2+ supplementation will be protective against As3+ poisoning of MTs.Data availability
All experimental details and data supporting the findings of this study are available in the paper and in the ESI.† Any additional data are available from the corresponding author upon request.Author contributions
A. T. Y. and M. J. S. designed the experiments and conceived the project. A. T. Y. carried out the experiments and analysis with the guidance of M. J. S. All authors contributed to the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the Natural Sciences and Engineering Research Council of Canada for a Canada Graduate Scholarship Doctoral (CGS-D) to A. T. Y. and a Discovery Grant (06545-2020) to M. J. S. We would like to thank Mr John Vanstone, Ms Melanie Glover, and Mr Barakat Misk of the Electronics Shop at the University of Western Ontario for the outstanding maintenance of our instruments.References
- IARC, Arsenic, Metals, Fibres, and Dusts, International Agency for Research on Cancer, Lyon, France, 2012 Search PubMed.
- S. Rahman, K.-H. Kim, S. K. Saha, A. M. Swaraz and D. K. Paul, J. Environ. Manage., 2014, 134, 175–185 CrossRef CAS PubMed.
- Q. Y. Chen and M. Costa, Annu. Rev. Pharmacol. Toxicol., 2021, 61, 47–63 CrossRef CAS PubMed.
- J. Y. Chung, S. D. Yu and Y. S. Hong, J. Prev. Med. Public Health, 2014, 47, 253–257 CrossRef PubMed.
- S. Shen, X.-F. Li, W. R. Cullen, M. Weinfeld and X. C. Le, Chem. Rev., 2013, 113, 7769–7792 CrossRef CAS PubMed.
- Y. Y. Chang, T. C. Kuo, C. H. Hsu, D. R. Hou, Y. H. Kao and R. N. Huang, Arch. Toxicol., 2012, 86, 911–922 CrossRef CAS PubMed.
- A. Vahidnia, G. B. van der Voet and F. A. de Wolff, Hum. Exp. Toxicol., 2007, 26, 823–832 CrossRef CAS PubMed.
- R. K. Virk, R. Garla, N. Kaushal, M. P. Bansal, M. L. Garg and B. P. Mohanty, Chemosphere, 2023, 316, 137735 CrossRef CAS PubMed.
- K. Rehman and H. Naranmandura, Metallomics, 2012, 4, 881–892 CrossRef CAS PubMed.
- R. N. Ratnaike, Postgrad. Med. J., 2003, 79, 391 CrossRef CAS PubMed.
- G. A. Wasserman, X. Liu, F. Parvez, P. Factor-Litvak, H. Ahsan, D. Levy, J. Kline, A. van Geen, J. Mey, V. Slavkovich, A. B. Siddique, T. Islam and J. H. Graziano, Neurotoxicology, 2011, 32, 450–457 CrossRef CAS PubMed.
- M. Tolins, M. Ruchirawat and P. Landrigan, Ann. Glob. Health, 2014, 80, 303–314 CrossRef PubMed.
- J. S. Tsuji, M. R. Garry, V. Perez and E. T. Chang, Toxicology, 2015, 337, 91–107 CrossRef CAS PubMed.
- H. Mochizuki, Int. J. Mol. Sci., 2019, 20(14), 3418 CrossRef CAS PubMed.
- M. Thakur, M. Rachamalla, S. Niyogi, A. K. Datusalia and S. J. Flora, Int. J. Mol. Sci., 2021, 22(18), 10077 CrossRef CAS PubMed.
- L. P. Chandravanshi, R. Gupta and R. K. Shukla, Biol. Trace Elem. Res., 2018, 186, 185–198 CrossRef CAS PubMed.
- C. Prakash, M. Soni and V. Kumar, J. Appl. Toxicol., 2016, 36, 179–188 CrossRef CAS PubMed.
- P. Coyle, J. C. Philcox, L. C. Carey and A. M. Rofe, Cell. Mol. Life Sci., 2002, 59, 627–647 CrossRef CAS PubMed.
- P. Babula, M. Masarik, V. Adam, T. Eckschlager, M. Stiborova, L. Trnkova, H. Skutkova, I. Provaznik, J. Hubalek and R. Kizek, Metallomics, 2012, 4, 739–750 CrossRef CAS PubMed.
- C. D. Klaassen, J. Liu and S. Choudhuri, Annu. Rev. Pharmacol. Toxicol., 1999, 39, 267–294 CrossRef CAS PubMed.
- M. Namdarghanbari, W. Wobig, S. Krezoski, N. M. Tabatabai and D. H. Petering, J. Biol. Inorg. Chem., 2011, 16, 1087 CrossRef CAS PubMed.
- D. E. K. Sutherland and M. J. Stillman, Metallomics, 2011, 3, 444–463 CrossRef CAS PubMed.
- M. J. Stillman, Coord. Chem. Rev., 1995, 144, 461–511 CrossRef CAS.
- J. H. R. Kägi, S. R. Himmelhoch, P. D. Whanger, J. L. Bethune and B. L. Vallee, J. Biol. Chem., 1974, 249, 3537–3542 CrossRef.
- A. Z. Mason, N. Perico, R. Moeller, K. Thrippleton, T. Potter and D. Lloyd, Mar. Environ. Res., 2004, 58, 371–375 CrossRef CAS PubMed.
- A. Z. Mason, R. Moeller, K. A. Thrippleton and D. Lloyd, Anal. Biochem., 2007, 369, 87–104 CrossRef CAS PubMed.
- T. B. Pinter and M. J. Stillman, Biochemistry, 2014, 53, 6276–6285 CrossRef CAS PubMed.
- A. T. Yuan, N. C. Korkola, D. L. Wong and M. J. Stillman, Metallomics, 2020, 12, 767–783 CrossRef CAS PubMed.
- M. Nakajima, E. Kobayashi, Y. Suwazono, M. Uetani, M. Oishi, T. Inaba, T. Kido, Z. A. Shaikh and K. Nogawa, Biol. Trace Elem. Res., 2005, 108, 17–31 CrossRef CAS PubMed.
- N. Sugawara, D. Li and C. Sugawara, Arch. Toxicol., 1994, 68, 520–523 CrossRef CAS PubMed.
- E. M. Kenyon, M. F. Hughes, B. M. Adair, J. H. Highfill, E. A. Crecelius, H. J. Clewell and J. W. Yager, Toxicol. Appl. Pharmacol., 2008, 232, 448–455 CrossRef CAS PubMed.
- M. S. Samuel, S. Datta, R. S. Khandge and E. Selvarajan, Sci. Total Environ., 2021, 775, 145829 CrossRef CAS.
- T. Miura, S. Muraoka and T. Ogiso, Life Sci., 1997, 60, 301–309 CrossRef PubMed.
- K.-S. Min, F. Morishita, N. Tetsuchikawahara and S. Onosaka, Toxicol. Appl. Pharmacol., 2005, 204, 9–17 CrossRef CAS PubMed.
- W. Maret, Neurochem. Int., 1995, 27, 111–117 CrossRef CAS PubMed.
- T. Kimura and T. Kambe, Int. J. Mol. Sci., 2016, 17(3), 336 CrossRef PubMed.
- M. Vašák and G. Meloni, J. Biol. Inorg. Chem., 2011, 16, 1067 CrossRef PubMed.
- M. Vašák and D. W. Hasler, Curr. Opin. Chem. Biol., 2000, 4, 177–183 CrossRef PubMed.
- A. T. Miles, G. M. Hawksworth, J. H. Beattie and V. Rodilla, Crit. Rev. Biochem. Mol. Biol., 2000, 35, 35–70 CrossRef CAS PubMed.
- I. Hozumi, J. S. Suzuki, H. Kanazawa, A. Hara, M. Saio, T. Inuzuka, S. Miyairi, A. Naganuma and C. Tohyama, Neurosci. Lett., 2008, 438, 54–58 CrossRef CAS PubMed.
- Y. Uchida, K. Takio, K. Titani, Y. Ihara and M. Tomonaga, Neuron, 1991, 7, 337–347 CrossRef CAS PubMed.
- A. Krężel and W. Maret, Chem. Rev., 2021, 121, 14594–14648 CrossRef PubMed.
- M. Vašák and G. Meloni, Int. J. Mol. Sci., 2017, 18, 1117 CrossRef PubMed.
- G. Meloni, V. Sonois, T. Delaine, L. Guilloreau, A. Gillet, J. Teissié, P. Faller and M. Vašák, Nat. Chem. Biol., 2008, 4, 366–372 CrossRef CAS PubMed.
- Y. Manso, J. Carrasco, G. Comes, G. Meloni, P. A. Adlard, A. I. Bush, M. Vasak and J. Hidalgo, Cell. Mol. Life Sci., 2012, 69, 3683–3700 CrossRef CAS PubMed.
- A. Binolfi, G. R. Lamberto, R. Duran, L. Quintanar, C. W. Bertoncini, J. M. Souza, C. Cerveñansky, M. Zweckstetter, C. Griesinger and C. O. Fernández, J. Am. Chem. Soc., 2008, 130, 11801–11812 CrossRef CAS PubMed.
- Y. Irie and W. M. Keung, Brain Res., 2003, 960, 228–234 CrossRef CAS PubMed.
- G. Meloni and M. Vašák, Free Radic. Biol. Med., 2011, 50, 1471–1479 CrossRef CAS PubMed.
- J. S. Calvo, N. V. Mulpuri, A. Dao, N. K. Qazi and G. Meloni, Free Radic. Biol. Med., 2020, 158, 149–161 CrossRef CAS PubMed.
- J. Y. Koh and S. J. Lee, Mol. Brain, 2020, 13, 116 CrossRef CAS PubMed.
- Q. Zheng, W. M. Yang, W. H. Yu, B. Cai, X. C. Teng, Y. Xie, H. Z. Sun, M. J. Zhang and Z. X. Huang, Protein Eng. Des. Sel., 2003, 16, 865–870 CrossRef CAS PubMed.
- H. Wang, Q. Zhang, B. Cai, H. Li, K.-H. Sze, Z.-X. Huang, H.-M. Wu and H. Sun, FEBS Lett., 2006, 580, 795–800 CrossRef CAS PubMed.
- J. S. Scheller, G. W. Irvine, D. L. Wong, A. Hartwig and M. J. Stillman, Metallomics, 2017, 9, 447–462 CrossRef CAS PubMed.
- A. Melenbacher, N. C. Korkola and M. J. Stillman, Metallomics, 2020, 12, 1951–1964 CrossRef CAS PubMed.
- N. C. Korkola, P. M. Scarrow and M. J. Stillman, Metallomics, 2020, 12, 435–448 CrossRef CAS PubMed.
- N. C. Korkola, E. Hudson and M. J. Stillman, Metallomics, 2021, 13 Search PubMed.
- T. T. Ngu and M. J. Stillman, J. Am. Chem. Soc., 2006, 128, 12473–12483 CrossRef CAS PubMed.
- M. R. Mehlenbacher, R. Elsiesy, R. Lakha, R. L. E. Villones, M. Orman, C. L. Vizcarra, G. Meloni, D. E. Wilcox and R. N. Austin, Chem. Sci., 2022, 13, 5289–5304 RSC.
- C. Pérez-Zúñiga, À. Leiva-Presa, R. N. Austin, M. Capdevila and Ò. Palacios, Metallomics, 2019, 11, 349–361 CrossRef PubMed.
- J. S. Calvo, R. L. E. Villones, N. J. York, E. Stefaniak, G. E. Hamilton, A. L. Stelling, W. Bal, B. S. Pierce and G. Meloni, J. Am. Chem. Soc., 2022, 144, 709–722 CrossRef CAS PubMed.
- G. W. Irvine, T. B. Pinter and M. J. Stillman, Metallomics, 2016, 8, 71–81 CrossRef CAS PubMed.
- R. Garla, N. Kaur, M. P. Bansal, M. L. Garg and B. P. Mohanty, J. Mol. Model., 2017, 23, 78 CrossRef PubMed.
- M. Toyama, M. Yamashita, N. Hirayama and Y. Murooka, J. Biochem., 2002, 132, 217–221 CrossRef CAS PubMed.
- Y. He and J. Guo, Comput. Theor. Chem., 2015, 1058, 54–60 CrossRef CAS.
- T. T. Ngu, A. Easton and M. J. Stillman, J. Am. Chem. Soc., 2008, 130, 17016–17028 CrossRef CAS PubMed.
- T. T. Ngu, S. Krecisz and M. J. Stillman, Biochem. Biophys. Res. Commun., 2010, 396, 206–212 CrossRef CAS PubMed.
- T. T. Ngu, J. A. Lee, T. B. Pinter and M. J. Stillman, J. Inorg. Biochem., 2010, 104, 232–244 CrossRef CAS PubMed.
- J.-H. Chou and M. G. Kanatzidis, Inorg. Chem., 1994, 33, 1001–1002 CrossRef CAS.
- B. T. Farrer, C. P. McClure, J. E. Penner-Hahn and V. L. Pecoraro, Inorg. Chem., 2000, 39, 5422–5423 CrossRef CAS PubMed.
- N. A. Rey, O. W. Howarth and E. C. Pereira-Maia, J. Inorg. Biochem., 2004, 98, 1151–1159 CrossRef CAS PubMed.
- K. Tani, S.-i. Hanabusa, S. Kato, S.-y. Mutoh, S.-i. Suzuki and M. Ishida, J. Chem. Soc., Dalton Trans., 2001, 518–527, 10.1039/B008702P.
- G. C. Pappalardo, R. Chakravorty, K. J. Irgolic and E. A. Meyers, Acta Crystallogr. C, 1983, 39, 1618–1620 CrossRef.
- B. F. Hoskins, E. R. T. Tiekink and G. Winter, Inorg. Chim. Acta, 1985, 99, 177–182 CrossRef CAS.
- A. M. Spuches, H. G. Kruszyna, A. M. Rich and D. E. Wilcox, Inorg. Chem., 2005, 44, 2964–2972 CrossRef CAS PubMed.
- N. Scott, K. M. Hatlelid, N. E. MacKenzie and D. E. Carter, Chem. Res. Toxicol., 1993, 6, 102–106 Search PubMed.
- L. Alderighi, P. Gans, A. Ienco, D. Peters, A. Sabatini and A. Vacca, Coord. Chem. Rev., 1999, 184, 311–318 CrossRef CAS.
- R. D. Hancock and A. E. Martell, Chem. Rev., 1989, 89, 1875–1914 CrossRef CAS.
- I. A. Kaltashov and A. Mohimen, Anal. Chem., 2005, 77, 5370–5379 CrossRef CAS PubMed.
- D. E. Sutherland and M. J. Stillman, Biochem. Biophys. Res. Commun., 2008, 372, 840–844 CrossRef CAS PubMed.
- A. T. Yuan, N. C. Korkola and M. J. Stillman, J. Biol. Chem., 2023, 299(3), 102899 CrossRef CAS PubMed.
- R. Fraczkiewicz and W. Braun, J. Comput. Chem., 1998, 19, 319–333 CrossRef CAS.
- D. E. K. Sutherland, K. L. Summers and M. J. Stillman, Biochemistry, 2012, 51, 6690–6700 CrossRef CAS PubMed.
- D. W. Hasler, L. T. Jensen, O. Zerbe, D. R. Winge and M. Vašák, Biochemistry, 2000, 39, 14567–14575 CrossRef CAS PubMed.
- R. Ganger, R. Garla, B. P. Mohanty, M. P. Bansal and M. L. Garg, Biol. Trace Elem. Res., 2016, 169, 218–229 CrossRef CAS PubMed.
- M. Ahmad, M. A. M. Wadaan, M. Farooq, M. H. Daghestani and A. S. Sami, Biol. Res., 2013, 46, 131–138 CrossRef PubMed.
- C. P. Wong, E. J. Dashner-Titus, S. C. Alvarez, T. T. Chase, L. G. Hudson and E. Ho, Biol. Trace Elem. Res., 2019, 191, 370–381 CrossRef CAS PubMed.
- L. M. Beaver, L. Truong, C. L. Barton, T. T. Chase, G. D. Gonnerman, C. P. Wong, R. L. Tanguay and E. Ho, PLoS One, 2017, 12, e0183831 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sc00400g |
| This journal is © The Royal Society of Chemistry 2023 |