
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis, bridgehead functionalization, and photoisomerization of 9,10-diboratatriptycene dianions†
Sven E.
Prey
a,
Jannik
Gilmer
a,
Samira V.
Teichmann
a,
Luis
Čaić
a,
Mischa
Wenisch
a,
Michael
Bolte
 a,
Alexander
Virovets
a,
Hans-Wolfram
Lerner
a,
Felipe
Fantuzzi
b and
Matthias
Wagner
*a
a,
Alexander
Virovets
a,
Hans-Wolfram
Lerner
a,
Felipe
Fantuzzi
b and
Matthias
Wagner
*a
aInstitut für Anorganische und Analytische Chemie, Goethe-Universität Frankfurt, Max-von-Laue-Straße 7, D-60438 Frankfurt am Main, Germany. E-mail: matthias.wagner@chemie.uni-frankfurt.de
bSchool of Chemistry and Forensic Science, University of Kent, Park Wood Rd, Canterbury CT2 7NH, UK
First published on 3rd March 2023
Abstract
9,10-Diboratatriptycene salts M2[RB(μ-C6H4)3BR] (R = H, Me; M+ = Li+, K+, [n-Bu4N]+) have been synthesized via [4 + 2] cycloaddition between doubly reduced 9,10-dihydro-9,10-diboraanthracenes M2[DBA] and benzyne, generated in situ from C6H5F and C6H5Li or LiN(i-Pr)2. [HB(μ-C6H4)3BH]2− reacts with CH2Cl2 to form quantitatively the bridgehead-derivatized [ClB(μ-C6H4)3BCl]2−, while twofold H− abstraction with B(C6F5)3 in the presence of SMe2 leads cleanly to the diadduct (Me2S)B(μ-C6H4)3B(SMe2). Photoisomerization of K2[HB(μ-C6H4)3BH] (THF, medium-pressure Hg lamp) provides facile access to diborabenzo[a]fluoranthenes, a little explored form of boron-doped polycyclic aromatic hydrocarbons. According to DFT calculations, the underlying reaction mechanism consists of three main steps: (i) photoinduced di-π-borate rearrangement, (ii) “walk reaction” of a BH unit, and (iii) boryl anion-like C–H activation.
Introduction
The terms “aromatic hydrocarbon” and “planar π system” can be used as synonyms for most representatives of these substance classes. Triptycenes are among the few aromatic hydrocarbons that enter the third dimension, which is one reason why they have fascinated the chemical community for decades. Triptycene is built upon a [2.2.2]-bicyclic system with three 1,2-phenylene blades to generate a D3h-symmetric paddle-wheel scaffold.1,2 Due to its exceptional rigidity, the triptycene framework has been used as a backbone for transition-metal ligands3,4 and as a building block for molecular motors.5 When incorporated into emitting polymers, triptycene units can sterically block the individual polymer chains so that the materials exhibit solution-like luminescence properties in the solid state, with applications in sensor technology.6 The three-dimensional shape of a triptycene building block creates a free volume around the molecule (i.e., between the blades of the paddle-wheel structure), which can be designed such that it confers size selectivity in sensory responses,7 promotes the desired alignment in oriented polymers,8 or creates porous organic materials.9 Of more academic interest – but no less fascinating – is the excited-state reactivity of triptycene and the resulting photorearrangement products (i.e., a norcaradiene and a benzo[a]fluoranthene derivative).10,11A proper means of expanding the chemical space of triptycenes as well as their application potential is heteroatom doping, i.e., the replacement of selected C atoms by other p-block elements. Our group has a long-standing interest in triptycenes featuring B atoms at their bridgehead positions. One example is the (BN)2-triptycene A (Fig. 1). A-type structures are accessible by simple mixing of 9,10-dihydro-9,10-diboraanthracenes (DBAs) with 1,2-diazenes, such as pyridazine. Since the reaction is based on the essentially barrierless formation of a double B–N adduct, A is obtained in very good yields of ≈80%.12–14 An organometallic (BN2)2-triptycene is the pyrazabole-bridged ansa-ferrocene B (Fig. 1).15 Recently, our group succeeded in abstracting one of its bridgehead H− ligands and characterized the resulting free, cationic Lewis superacid [B − H]+ by X-ray crystallography.16 Berionni and coworkers reported a series of boratatriptycenes C (Fig. 1) possessing various different atoms/groups “E” at the second bridgehead position (E = CH, PH+, S+, Se+).17–20 A derivative of C combining E = CH and Ar = 4-t-Bu-C6H4 underwent selective protodeboronation of the exocyclic C–B bond upon treatment with bistriflimidic acid (HNTf2); the resulting boratriptycene Lewis acid still engages in a dynamic association–dissociation equilibrium with the weakly coordinating [NTf2]− anion.18 This finding underscores the potential for enhancing Lewis acidity by designing a rigid organic framework in a way to impose a structural constraint on the BC3 core that forces it out of planarity (see also the Lewis superacidity of [B − H]+ mentioned above16).17
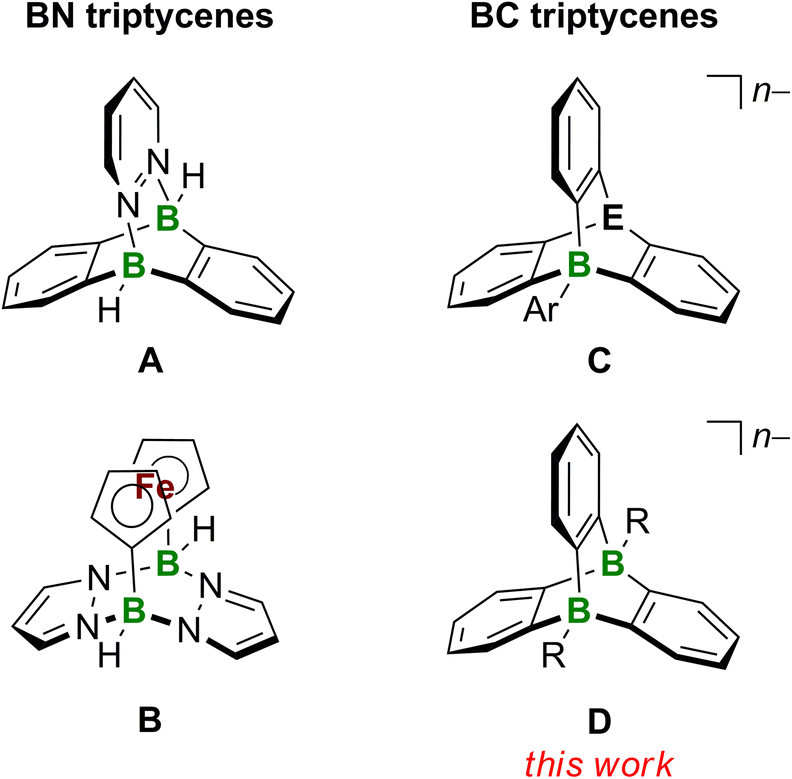 | ||
| Fig. 1 Boron-containing triptycenes. Left: the DBA-pyridazine adduct A and the 1,1′-ferrocenediyl-bridged pyrazabole B as BN derivatives of triptycene. Right: Berionni's BC-triptycenes C featuring one B atom at a bridgehead position (E = CH, n = 1; E = PH+, S+, Se+, n = 0; Ar = aryl) and the dibora(ta)triptycenes D presented in this work (R = H, Me, n = 2; R = SMe2, n = 0). DBA = 9,10-dihydro-9,10-diboraanthracene. | ||
From the application examples of triptycenes, it is obvious that the degree of repulsive or attractive interactions between the paddle-wheels’ blades and their immediate environments play an important role. Since these interactions will be strongly influenced by the overall charge of the triptycene scaffold, we are herein targeting dianionic diboratatriptycenes D (Fig. 1; R = H, Me; n = 2). We will show that H− abstraction in situ releases a Lewis acid with potentially cooperating B atoms21 and allows convenient derivatization of both bridgehead positions. We finally show that the photorearrangement of D (R = H; n = 2) provides access to the little explored compound class of diborabenzo[a]fluoranthenes.22
Results and discussion
Synthesis of 9,10-diboratatriptycenes D
We tested two conceptually different ways to prepare our D-type target compounds: (a) adduct formation on neutral DBA and (b) [4 + 2] cycloaddition on doubly reduced [DBA]2−. The first approach (a) is inspired by the facile synthesis of A and based on the assumption that pyridazine can, in principle, be replaced by the isoelectronic benzene-1,2-diide to assemble the tricyclic scaffold of Dvia a twofold B–C-adduct formation. Unfortunately, the available literature on 1,2-dimetalated benzenes indicates a rather limited synthetic value of these highly reactive species.23 We therefore resorted to a stepwise reaction protocol, starting from in situ-prepared o-iodophenyllithium and 1 (−110 °C, THF/Et2O (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1); Scheme 1).24 Cyclization of the obtained B(sp2)–B(sp3) intermediate Li[4] was subsequently accomplished by stirring with Mg0 (room temperature, THF). Since the NMR-spectroscopic investigation of the crude product still showed the formation of some unwanted side products that later proved difficult to remove, the B–C-adduct approach was eventually abandoned in favor of the Umpolung strategy (b): given that doubly reduced DBAs easily undergo [4 + 2]-cycloaddition reactions with (hetero)olefins or internal alkynes,25 the reaction of DBA salts M2[1]–M2[3] with in situ-generated benzyne should likewise generate diboratatriptycenes M2[5]–M2[7] (Scheme 1). For an initial proof-of-principle experiment, we selected Li2[1], because we have had good experience with cycloadditions on this compound in the past.25 LiN(i-Pr)2 in THF was added with stirring at −78 °C to a mixture of freshly prepared Li2[1] (ref. 26 and 27) and C6H5F in THF (Scheme 1). After warming to room temperature overnight, the reaction mixture had lost the characteristic red color of Li2[1] and turned to pale yellow. 11B NMR spectroscopy showed the selective formation of one new species; its chemical shift value of δ(11B) = −13.2 ppm testified to the presence of tetracoordinated B atoms.28 An 1H NMR spectrum, recorded on the crude product after a solvent change to THF-d8, indicated an essentially quantitative conversion: a signal at 0.56 ppm (6H) and two resonances at 7.24 (br, 6H) and 6.44 ppm (m, 6H) are assignable to two chemically equivalent CH3 groups and three chemically equivalent C6H4 rings, respectively. Also the 13C{1H} NMR spectra supported the proposed D3h-symmetric molecular structure of [5]2− in solution. Purification of Li2[5] through crystallization from THF gave yields of 50–75% (attempts at X-ray analysis were not successful). Next, we moved on from DBA 1 to DBA 2 to also get a hold of the parent diboratatriptycene. K2[6] was prepared in a similar manner as Li2[5] and isolated in 41% yield (δ(11B) = −8.3 ppm, d, 1J(B,H) = 87 Hz); the BH resonance was found at 3.36 ppm in the 1H{11B} NMR spectrum).29 To stress the steric limits of the [4 + 2] cycloaddition, we finally treated dimesitylated K2[3] with benzyne and still obtained K2[7], albeit in negligible yield of only a few crystals.24
:1); Scheme 1).24 Cyclization of the obtained B(sp2)–B(sp3) intermediate Li[4] was subsequently accomplished by stirring with Mg0 (room temperature, THF). Since the NMR-spectroscopic investigation of the crude product still showed the formation of some unwanted side products that later proved difficult to remove, the B–C-adduct approach was eventually abandoned in favor of the Umpolung strategy (b): given that doubly reduced DBAs easily undergo [4 + 2]-cycloaddition reactions with (hetero)olefins or internal alkynes,25 the reaction of DBA salts M2[1]–M2[3] with in situ-generated benzyne should likewise generate diboratatriptycenes M2[5]–M2[7] (Scheme 1). For an initial proof-of-principle experiment, we selected Li2[1], because we have had good experience with cycloadditions on this compound in the past.25 LiN(i-Pr)2 in THF was added with stirring at −78 °C to a mixture of freshly prepared Li2[1] (ref. 26 and 27) and C6H5F in THF (Scheme 1). After warming to room temperature overnight, the reaction mixture had lost the characteristic red color of Li2[1] and turned to pale yellow. 11B NMR spectroscopy showed the selective formation of one new species; its chemical shift value of δ(11B) = −13.2 ppm testified to the presence of tetracoordinated B atoms.28 An 1H NMR spectrum, recorded on the crude product after a solvent change to THF-d8, indicated an essentially quantitative conversion: a signal at 0.56 ppm (6H) and two resonances at 7.24 (br, 6H) and 6.44 ppm (m, 6H) are assignable to two chemically equivalent CH3 groups and three chemically equivalent C6H4 rings, respectively. Also the 13C{1H} NMR spectra supported the proposed D3h-symmetric molecular structure of [5]2− in solution. Purification of Li2[5] through crystallization from THF gave yields of 50–75% (attempts at X-ray analysis were not successful). Next, we moved on from DBA 1 to DBA 2 to also get a hold of the parent diboratatriptycene. K2[6] was prepared in a similar manner as Li2[5] and isolated in 41% yield (δ(11B) = −8.3 ppm, d, 1J(B,H) = 87 Hz); the BH resonance was found at 3.36 ppm in the 1H{11B} NMR spectrum).29 To stress the steric limits of the [4 + 2] cycloaddition, we finally treated dimesitylated K2[3] with benzyne and still obtained K2[7], albeit in negligible yield of only a few crystals.24
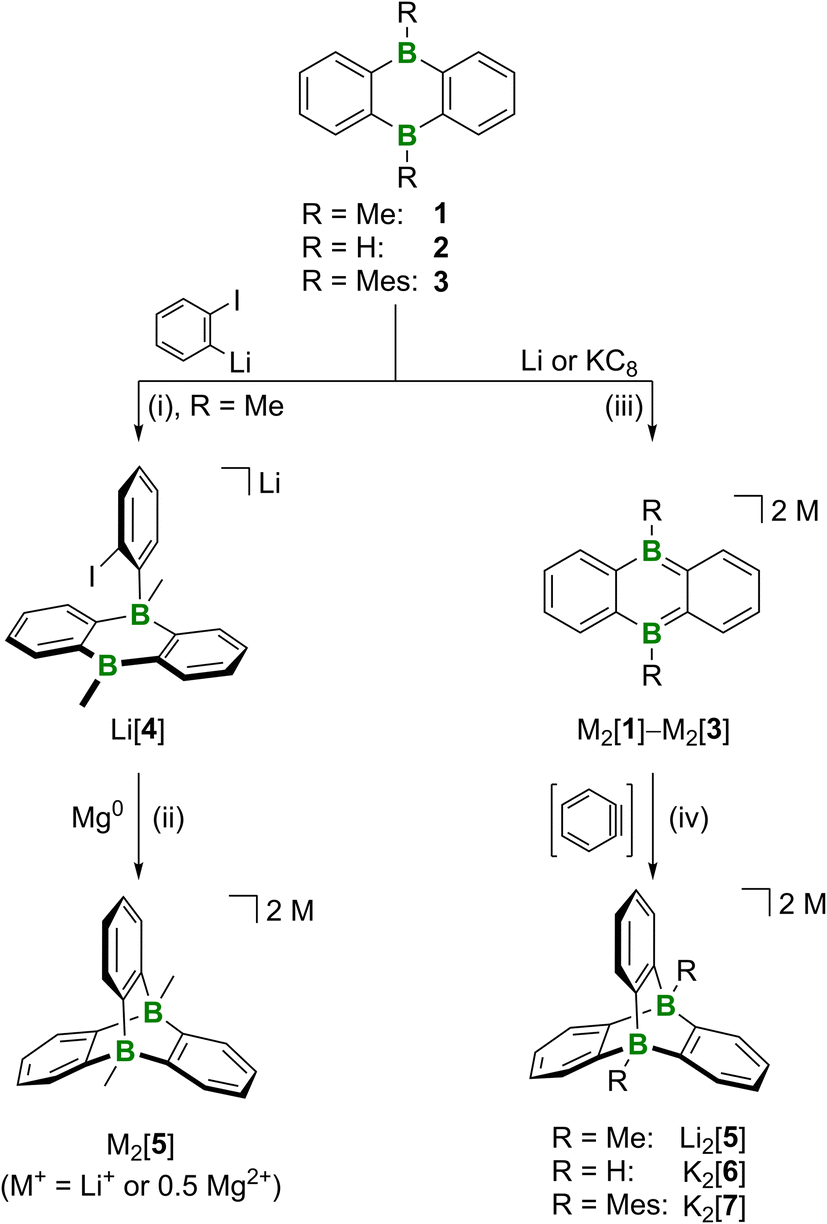 | ||
| Scheme 1 Synthesis of 9,10-diboratatriptycenes by either sequential B–C-adduct formation (left) or [4 + 2] cycloaddition of in situ-generated benzynes with M2[DBA] salts (right). (i) 1,2-C6H4I2, t-BuLi, THF/Et2O (1:1), −110 °C → room temperature, overnight, 21% yield; (ii) 1,2-C2H4Br2 (6 mol%), THF, room temperature, 3 d; (iii) THF, room temperature, 1 h, quant. conv. by NMR; (iv) C6H5F (1–2 equiv.), base (LiN(i-Pr)2 or C6H5Li), THF, −78 °C → room temperature, overnight to 5 d, Li2[5]: 50–75% yield, K2[6]: 41% yield, K2[7]: few crystals. | ||
Crystals of [K(dme)][K(dme)2][6] suitable for X-ray diffraction (XRD) were obtained from a gas-phase diffusion experiment (n-hexane/1,2-dimethoxyethane (DME); Fig. 2). In [K(dme)]-[K(dme)2][6], the diboratatriptycene dianion [6]2− acts as chelating ligand toward both K+ ions (via the π-electron clouds of its 1,2-phenylene rings). The coordination sphere of K(1)+ is completed by two dme ligands; K(2)+ carries only one dme ligand and is further bonded to two phenylene rings of an adjacent [6]2− moiety, thereby creating a coordination polymer in which all three pockets of the paddle-wheel anion are occupied by K+ ions (Fig. S61†). The six B–C-bond lengths of C1-symmetric (in the crystal lattice) [6]2− range from 1.620(3) to 1.643(3) Å (avg. 1.631 Å) and are comparable to those in the structurally unconstrained, acyclic analogue [HBPh3]− (1.628(6) to 1.633(6) Å, avg. 1.631 Å).30 Of the three dihedral angles 107.9°, 124.8°, and 127.3° between phenylene rings, the smallest is associated with the pocket hosting the doubly η6-coordinated K(1)+.
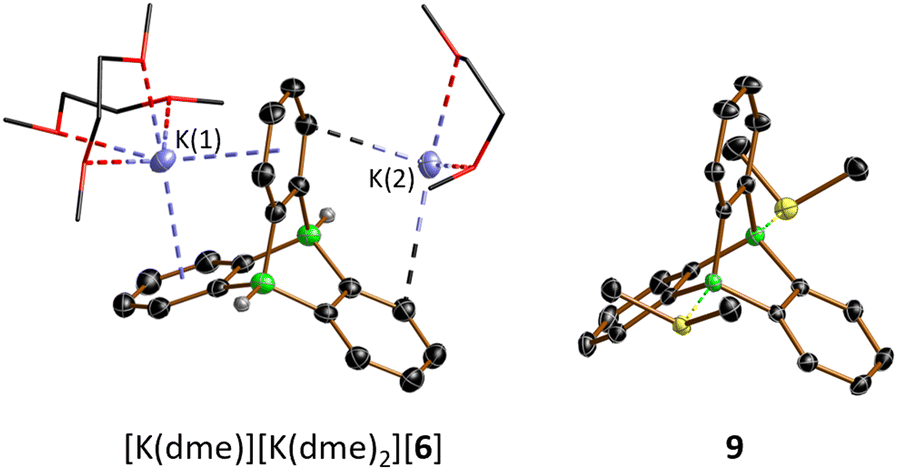 | ||
| Fig. 2 Solid-state structures of [K(dme)][K(dme)2][6] and 9 with thermal ellipsoids shown at the 50% probability level; C-bonded H atoms and a non-coordinating CH2Cl2 molecule in the crystal lattice of 9 are omitted for clarity, dme ligands are depicted as wireframes. H: gray, B: green, C: black, O: red, S: yellow, K: light blue. | ||
Activation of the B–H bonds of the 9,10-diboratatriptycene [6]2−
Having the diboratatriptycene salt [K(dme)][K(dme)2][6] in hand, we next addressed the question of how to activate its B–H bonds to derivatize the bridgehead positions and/or generate the free Lewis acid in situ. The neutral ditopic Lewis acid S1 (Scheme 2) should be among the strongest neutral B-based acids, as judged by its computed F−-ion affinity FIAsolv(S1) of 384 kJ mol−1 (cf. FIAsolv(B(C6F5)3) = 315 kJ mol−1; solv = CH2Cl2; for FIAs of further B2-triptycenes and a more detailed discussion see the ESI†).16 As the dme ligands present in the complex cations could potentially become B-coordinating and thereby interfere with the planned reactivity studies, we first performed a cation exchange with [n-Bu4N]Cl in THF. [n-Bu4N]2[6] precipitates together with KCl from the reaction mixture and was separated from the latter by extraction into CH2Cl2 (93% yield). Care must be taken to remove the solvent from the product in time after workup to avoid B–H/B–Cl exchange.31 On the other hand, the reaction between [n-Bu4N]2[6] and CH2Cl2 can be used for the straightforward synthesis of bridgehead-chlorinated [n-Bu4N]2[8] (14 d at room temperature or 18 h at 50 °C; 89% yield; Scheme 2). In an attempt to release the free monotopic Lewis acid, [n-Bu4N]2[6] was treated with 1 equiv. of B(C6F5)3 in CD2Cl2. NMR spectroscopy showed that B(C6F5)3 had been quantitatively transformed to [n-Bu4N][HB(C6F5)3], even though some unconsumed starting material [n-Bu4N]2[6] remained in the mixture. Several non-identified species and small quantities of a precipitate had formed. When the amount of B(C6F5)3 added was increased to 2 equiv., the H− scavenger was still fully converted, phenylene resonances were no longer detectable in the 1H NMR spectrum, and substantial amounts of precipitate were found in the NMR tube. A similar result was obtained when [Ph3C][B(C6F5)4] was used instead of B(C6F5)3. The precipitate could not be re-dissolved by addition of SMe2. The situation changed when the double H− abstraction was carried out with B(C6F5)3 in the presence of SMe2, which led to the clean formation of the diboratriptycene–SMe2 diadduct 9 (Scheme 2). Based on the computed FIAs of relevant B2-triptycenes, we propose that the formation of 9 proceeds in a stepwise manner without intermediate formation of S1.24 X-ray quality crystals of 9·CH2Cl2 were grown from CH2Cl2 (Fig. 2).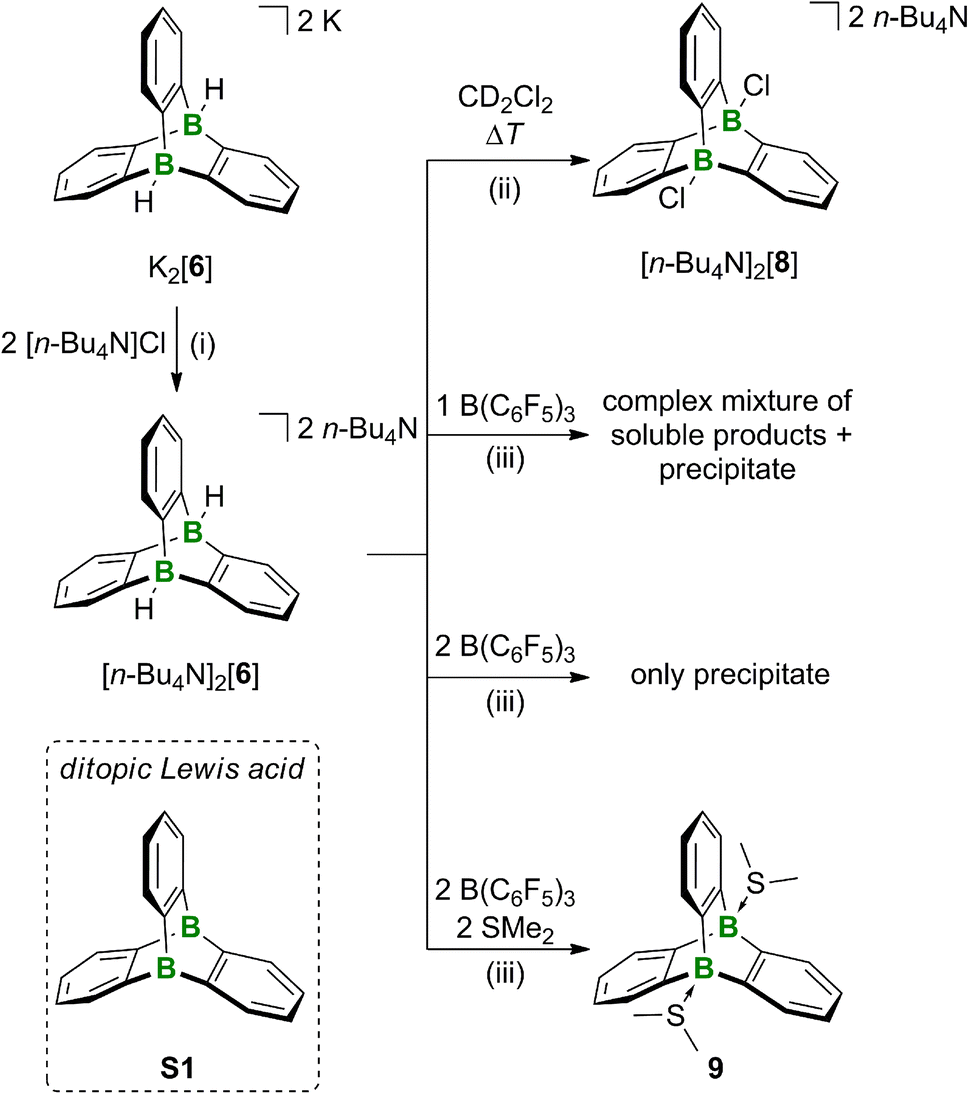 | ||
| Scheme 2 Reactivity of 9,10-diboratatriptycene [6]2−. (i) (1) THF, room temperature, 1 h; (2) extraction into CH2Cl2, 93% yield; (ii) 50 °C, 18 h, 89% yield; (iii) CD2Cl2, room temperature, 5 min, 9: quant. conv. by NMR. | ||
The 11B NMR spectrum of 9 is characterized by a signal at −1.4 ppm, in the region of tetracoordinated B nuclei.28 The 1H/13C resonances of the SMe2 ligands appear at 3.07/21.1 ppm (CD2Cl2), significantly downfield-shifted from those of free SMe2 (2.09/18.4 ppm) and even Me2S–B(C6F5)3 (2.17/20.8 ppm;32 CDCl3), thus indicating a strong S–B-adduct bond. This conclusion is further confirmed by the short S–B-bond lengths of 9 (1.9710(19), 1.9775(18) Å; cf. Me2S–B(C6F5)3: S–B = 2.091(5) Å (ref. 33)).
To summarize, we assume that after H− abstraction on [6]2−, the corresponding free Lewis acid S1 is not stable but rather forms the insoluble solid by rearrangement or ring-opening polymerization. Although the structure of the solid remains to be confirmed, a plausible candidate is a polymer composed of 1,2-phenylene-bridged DBAs (Fig. S15†). The formation of a precipitate is suppressed if SMe2 is present from the beginning to stabilize the diboratriptycene.
Photoisomerization of the 9,10-diboratatriptycene [6]2−
In contrast to the thermal rearrangement of free neutral diboratriptycene, photoisomerization of the diboratatriptycene salt K2[6] results in the well-defined, soluble product K2[10] (Scheme 3). K2[10] can be viewed as a molecule in which a DBA moiety shares one B atom and one phenylene ring with a 9H-9-borafluorene unit.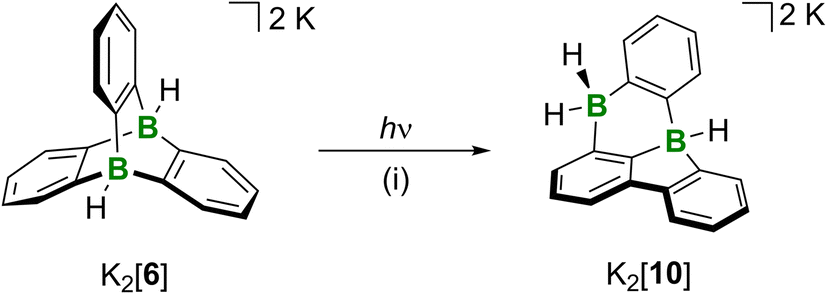 | ||
| Scheme 3 Photoisomerization of K2[6] to the 8,12b-dihydro-8,12b-diborabenzo[a]fluoranthene derivative K2[10]. (i) Medium-pressure Hg lamp, THF, room temperature, 1 h, quant. conv. by NMR. | ||
Irradiation of a THF solution of K2[6] with a medium-pressure Hg lamp in a quartz vessel for 1 h led to a color change from colorless to yellow.34 NMR spectroscopy on a THF-d8 solution revealed the highly selective formation of K2[10]. Its 11B NMR spectrum shows a doublet (δ(11B) = −13.7 ppm, 1J(B,H) = 73 Hz) and a pseudo-triplet (δ(11B) = −16.7 ppm, 1J(B,H) = 78 Hz), assignable to a BH- and a BH2 group, respectively. The corresponding BHn resonances were found in the 1H{11B} NMR spectrum at 2.65 (BH) and 2.52/2.40 ppm (BH2). The 13C{1H} NMR spectrum supports the proposed C1-symmetric molecular structure, since we observe 18 signals belonging to three different six-membered rings (according to 1H-13C-HSQC and -HMBC experiments). Of those signals, five are severely broadened due to 1J(B,C) coupling and the quadrupolar effect of the 10/11B nuclei and therefore correspond to the five required B-bonded C atoms; deshielded, quaternary 13C atoms giving rise to NMR resonances at 151.1/144.3 ppm point toward the presence of a biphenyl fragment in the molecule. Moreover, the DFT-computed 13C shift values of [10]2− agree well with the experimentally observed ones for K2[10] (avg. deviation: 2.0 ppm).24 In order to gain more information on the mechanism underlying the photoisomerization of [6]2− to [10]2−, we performed corresponding quantum-chemical calculations.
Assessment of the photoisomerization of K2[6] by theory
All energies were calculated at the SMD(THF)35/ωB97XD36/6-311++G** level of theory, with optimized structures at SMD(THF)/ωB97XD/6-31+G**. Vertical excitation energies were calculated with the respective unrestricted functional (T1 states) or by TD-DFT calculations (S1 states). The calculations were performed on the bare dianionic species (Fig. 3a).37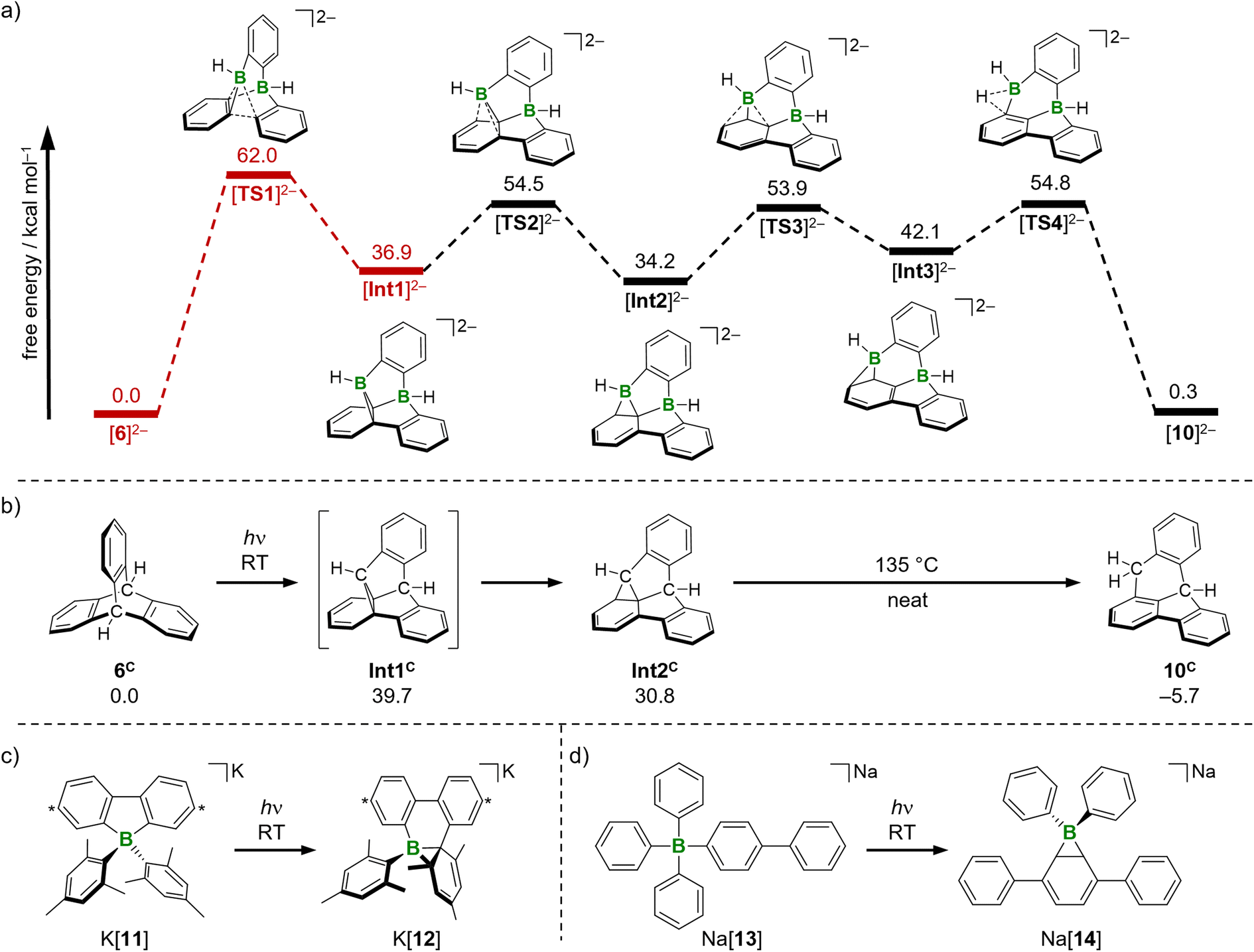 | ||
| Fig. 3 (a) Computed mechanism of the photoisomerization [6]2− → [10]2− and free energies of intermediates and transition states in their S0 states. The energetically unfavorable and thus photochemically conducted step [6]2− → [Int1]2− is marked in red. (b) Literature-reported intermediates of the photoisomerization of triptycene 6C and their calculated free energies (this work). (c) and (d) Known boratanorcaradienes K[12] and Na[14] obtained from photoreactions of tetraarylborates K[11] and Na[13], respectively. Carbon atoms marked with asterisks bear t-Bu substituents. | ||
We took the known photoisomerization pathway of carbonaceous triptycene 6C and the di-π-borate rearrangement of the tetraarylborate K[11] as blueprints for our mechanistic studies (Fig. 3b and c).10,11,38 The first reaction step [6]2− → [Int1]2− involves C–C coupling between two aryl rings with simultaneous extrusion of one BH unit. In line with experiment, the associated energy barrier of 62.0 kcal mol−1 is prohibitively high for a reaction under ambient conditions (in the S0 states). To account for the actually applied photochemical protocol, this crucial step was investigated in detail also for the excited T1 and S1 states of the molecules involved.
First, we verified the basic premise, namely that the Hg lamp employed (emission range: 200–600 nm) does indeed provide the wavelength required to overcome the computed S0 → S1 energy gap of [6]2− (ΔES0→S1 = 5.068 eV, corresponding to λexc = 244.7 nm; fosc = 0.1010).
We scanned the T1 and S1 energy surface topographies along the reaction coordinate between [6]2− and [Int1]2− based on ground-state structures obtained from the [TS1]2−-IRC scan (Fig. 4; IRC = intrinsic reaction coordinate): the energy curves for both excited states have minima located downstream of the [TS1]2− structure along the reaction coordinate. It is reasonable that – regardless of a potential intersystem crossing – both these minima may provide access back to the ground state.39 From that on, a barrierless relaxation leads to [Int1]2− at 36.9 kcal mol−1 relative to [6]2−. We note that boratanorcaradiene substructures, such as the one present in the key intermediate [Int1]2−, have been characterized by XRD in the form of K[12] and Na[14] (Fig. 3c and d).38,40
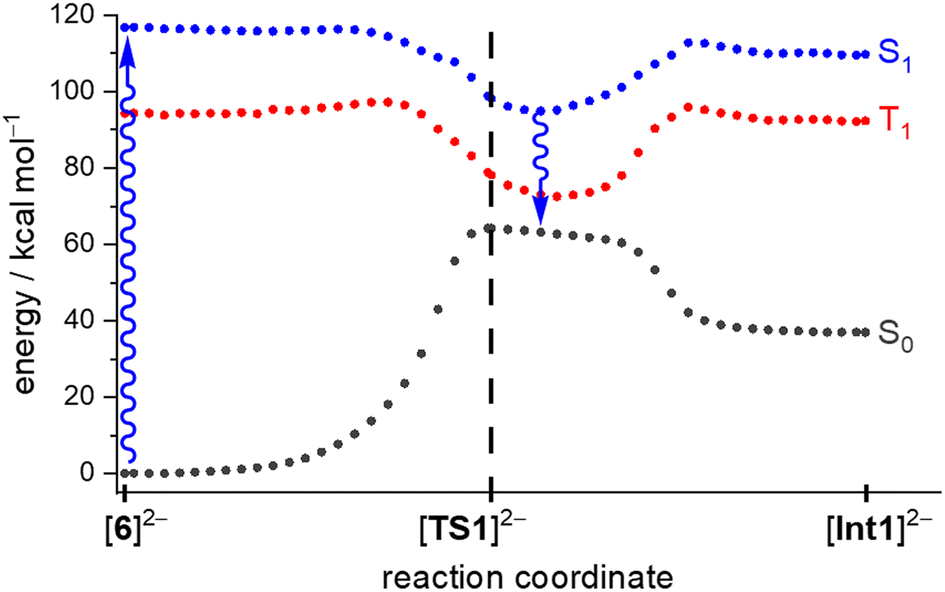 | ||
| Fig. 4 Computed energy profiles of the photoinduced reaction [6]2− → [Int1]2− in the S0 (gray), T1 (red), and S1 (blue) states. The dashed black line marks the position of [TS1]2− in the ground state; the blue arrows mark the electronic excitation and relaxation (the overall conclusion would remain the same if the relaxation occurred after a putative intersystem crossing from T1). | ||
All barriers of the following “walk reaction” on the S0 surface are reachable at ambient conditions:41 [Int1]2− undergoes a [1,5]sigmatropic shift of the boryl group to form [Int2]2−, which is 2.7 kcal mol−1 more stable than [Int1]2−. The corresponding barrier [Int1]2− → [TS2]2− amounts to ΔG‡ = 17.6 kcal mol−1. A second, slightly endergonic [1,5]sigmatropic shift via [TS3]2− gives [Int3]2− (ΔG‡ = 19.7 kcal mol−1, ΔG = 7.9 kcal mol−1). In the last step, C–H activation from [Int3]2− leads to [10]2−.42 The corresponding [TS4]2− lies 12.7 kcal mol−1 above [Int3]2−, so that the free-energy span to be covered from [Int2]2− to [TS4]2− is 20.6 kcal mol−1.43 The final product [10]2− is essentially isoenergetic to the starting material [6]2−. Nevertheless, we consider a reverse reaction unlikely for the following reasons: (i) the energy barrier for the step [Int1]2− → [TS1]2− in the reverse reaction is significantly higher (25.1 kcal mol−1; Fig. 3) than any barrier downstream [Int1]2− for the forward reaction. (ii) Assuming that the barrier [Int1]2− → [TS1]2− could be overcome photochemically, the system would again be in the state described in Fig. 4 and should therefore preferentially fall back to [Int1]2−.
Conclusions
We achieved the synthesis of the parent dianionic 9,10-diboratatriptycene [6]2− and its bridgehead derivatization. In the salt [K(dme)][K(dme)2][6], [6]2− acts as a chelating ligand toward both K+ ions, suggesting a possible future use as a tris-benzene ligand for mono- to trinuclear transition metal complexes.3 Abstraction of a bridgehead H− substituent releases the free Lewis acid, which is sufficiently long-lived to be trapped by SMe2; this transformation can be carried out at both bridgehead positions and eventually leads to the neutral boratriptycene-diadduct 9. By using ditopic Lewis bases, this derivatization chemistry should provide an entry point to polymeric dibora(ta)triptycenes for materials science. The facile photoisomerization of K2[6] furnishes the diborabenzo[a]fluoranthene derivative K2[10], which combines the two prominent structural motifs of 9,10-dihydro-9,10-diboraanthracene and 9H-9-borafluorene and belongs to a virtually unexplored class of B-doped polycyclic aromatic hydrocarbons.Data availability
The datasets supporting this article have been uploaded as part of the ESI.†Author contributions
S. E. P. performed the experimental studies and characterized all new compounds. S. E. P. and J. G. performed the quantum-chemical calculations. S. V. T., L. C., and M. We. assisted with the synthesis of compounds 9, [n-Bu4N]2[8], and Li2[5], respectively. M. B. performed the X-ray crystal structure analyses of the compounds [K2(dme)3][3], [Li(thf)2][4], [K(dme)]-[K(dme)2][6], and [K(dme)][K(dme)2][7]. A. V. performed the X-ray crystal structure analysis of 9·CH2Cl2. H.-W. L., F. F., and M. Wa. supervised the project. The manuscript was written by S. E. P., J. G., and M. Wa. and edited by all co-authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
J. G. thanks the German Academic Exchange Service for a research grant. A. V. thanks Dr Matthias Meyer (Rigaku Oxford Diffraction) for his help with the implementation of the CrysAlisPro software for the STOE IPDS II diffraction data.Notes and references
- C.-F. Chen and Y.-X. Ma, Iptycenes Chemistry, Springer Verlag, Berlin, Heidelberg, 2013 Search PubMed.
- M. Woźny, A. Mames and T. Ratajczyk, Molecules, 2022, 27, 250 CrossRef PubMed.
- (a) R. A. Gancarz, J. F. Blount and K. Mislow, Organometallics, 1985, 4, 2028–2032 CrossRef CAS; (b) P. J. Fagan, M. D. Ward and J. C. Calabrese, J. Am. Chem. Soc., 1989, 111, 1698–1719 CrossRef CAS; (c) H. Schmidbaur, T. Probst and O. Steigelmann, Organometallics, 1991, 10, 3176–3179 CrossRef CAS; (d) S. Toyota, H. Okuhara and M. Ōki, Organometallics, 1997, 16, 4012–4015 CrossRef CAS; (e) M. Wen, M. Munakata, Y.-Z. Li, Y. Suenaga, T. Kuroda-Sowa, M. Maekawa and M. Anahata, Polyhedron, 2007, 26, 2455–2460 CrossRef CAS.
- L. Bini, C. Müller, J. Wilting, L. von Chrzanowski, A. L. Spek and D. Vogt, J. Am. Chem. Soc., 2007, 129, 12622–12623 CrossRef CAS PubMed.
- T. R. Kelly, H. De Silva and R. A. Silva, Nature, 1999, 401, 150–152 CrossRef CAS PubMed.
- T. M. Swager, Acc. Chem. Res., 2008, 41, 1181–1189 CrossRef CAS PubMed.
- J.-S. Yang and T. M. Swager, J. Am. Chem. Soc., 1998, 120, 11864–11873 CrossRef CAS.
- F. Araoka, K.-C. Shin, Y. Takanishi, K. Ishikawa, H. Takezoe, Z. Zhu and T. M. Swager, J. Appl. Phys., 2003, 94, 279–283 CrossRef CAS.
- R. Bera, M. Ansari, A. Alam and N. Das, ACS Appl. Polym. Mater., 2019, 1, 959–968 CrossRef CAS.
- T. D. Walsh, J. Am. Chem. Soc., 1969, 91, 515–516 CrossRef CAS.
- N. J. Turro, M. Tobin, L. Friedman and J. B. Hamilton, J. Am. Chem. Soc., 1969, 91, 516 CrossRef CAS.
- A. Lorbach, M. Bolte, H.-W. Lerner and M. Wagner, Chem. Commun., 2010, 46, 3592–3594 RSC.
- Ö. Seven, S. Popp, M. Bolte, H.-W. Lerner and M. Wagner, Dalton Trans., 2014, 43, 8241–8253 RSC.
- Wegner et al. have postulated such (BN)2-triptycenes as key intermediates of DBA-catalyzed inverse electron-demand Diels–Alder (IEDDA) reactions on higher 1,2-diazenes: (a) S. N. Kessler and H. A. Wegner, Org. Lett., 2010, 12, 4062–4065 CrossRef CAS PubMed; (b) S. N. Kessler, M. Neuburger and H. A. Wegner, J. Am. Chem. Soc., 2012, 134, 17885–17888 CrossRef CAS PubMed; (c) L. Schweighauser, I. Bodoky, S. N. Kessler, D. Häussinger, C. Donsbach and H. A. Wegner, Org. Lett., 2016, 18, 1330–1333 CrossRef CAS PubMed; (d) L. Schweighauser and H. A. Wegner, Chem.–Eur. J., 2016, 22, 14094–14103 CrossRef CAS PubMed; (e) L. Hong, S. Ahles, M. A. Strauss, C. Logemann and H. A. Wegner, Org. Chem. Front., 2017, 4, 871–875 RSC; (f) S. Ahles, S. Götz, L. Schweighauser, M. Brodsky, S. N. Kessler, A. H. Heindl and H. A. Wegner, Org. Lett., 2018, 20, 7034–7038 CrossRef CAS PubMed; (g) S. Ahles, J. Ruhl, M. A. Strauss and H. A. Wegner, Org. Lett., 2019, 21, 3927–3930 CrossRef CAS PubMed.
- (a) F. Jäkle, T. Priermeier and M. Wagner, J. Chem. Soc., Chem. Commun., 1995, 1765–1766 RSC; (b) E. Herdtweck, F. Jäkle, G. Opromolla, M. Spiegler, M. Wagner and P. Zanello, Organometallics, 1996, 15, 5524–5535 CrossRef CAS; (c) F. Jäkle, T. Priermeier and M. Wagner, Organometallics, 1996, 15, 2033–2040 CrossRef; (d) E. Herdtweck, F. Jäkle and M. Wagner, Organometallics, 1997, 16, 4737–4745 CrossRef CAS.
- M. Henkelmann, A. Omlor, M. Bolte, V. Schünemann, H.-W. Lerner, J. Noga, P. Hrobárik and M. Wagner, Chem. Sci., 2022, 13, 1608–1617 RSC.
- A. Ben Saida, A. Chardon, A. Osi, N. Tumanov, J. Wouters, A. I. Adjieufack, B. Champagne and G. Berionni, Angew. Chem., Int. Ed., 2019, 58, 16889–16893 CrossRef CAS PubMed.
- A. Chardon, A. Osi, D. Mahaut, T.-H. Doan, N. Tumanov, J. Wouters, L. Fusaro, B. Champagne and G. Berionni, Angew. Chem., Int. Ed., 2020, 59, 12402–12406 CrossRef CAS PubMed.
- A. Osi, D. Mahaut, N. Tumanov, L. Fusaro, J. Wouters, B. Champagne, A. Chardon and G. Berionni, Angew. Chem., Int. Ed., 2022, 61, e202112342 CrossRef CAS PubMed.
- A. Osi, N. Tumanov, J. Wouters, A. Chardon and G. Berionni, Synthesis, 2023, 55, 347–353 CrossRef CAS.
- (a) A. Hübner, A. M. Diehl, M. Diefenbach, B. Endeward, M. Bolte, H.-W. Lerner, M. C. Holthausen and M. Wagner, Angew. Chem., Int. Ed., 2014, 53, 4832–4835 CrossRef PubMed; (b) A. Hübner, T. Kaese, M. Diefenbach, B. Endeward, M. Bolte, H.-W. Lerner, M. C. Holthausen and M. Wagner, J. Am. Chem. Soc., 2015, 137, 3705–3714 CrossRef PubMed.
- Very recently, Marder et al. disclosed that thermal rearrangement of 1,2-bis-(9-borafluorenyl)benzene leads to a diborabenzo[a]fluoranthene derivative: J. Krebs, A. Häfner, S. Fuchs, X. Guo, F. Rauch, A. Eichhorn, I. Krummenacher, A. Friedrich, L. Ji, M. Finze, Z. Lin, H. Braunschweig and T. B. Marder, Chem. Sci., 2022, 13, 14165–14178 RSC.
- (a) G. Wittig and F. Bickelhaupt, Chem. Ber., 1958, 91, 883–894 CrossRef CAS; (b) H. J. S. Winkler and G. Wittig, J. Org. Chem., 1963, 28, 1733–1740 CrossRef CAS; (c) M. A. G. M. Tinga, O. S. Akkerman, F. Bickelhaupt, E. Horn and A. L. Spek, J. Am. Chem. Soc., 1991, 113, 3604–3605 CrossRef CAS.
- See the ESI† for more details..
- (a) E. von Grotthuss, S. E. Prey, M. Bolte, H.-W. Lerner and M. Wagner, Angew. Chem., Int. Ed., 2018, 57, 16491–16495 CrossRef CAS PubMed; (b) E. von Grotthuss, S. E. Prey, M. Bolte, H.-W. Lerner and M. Wagner, J. Am. Chem. Soc., 2019, 141, 6082–6091 CrossRef CAS PubMed; (c) S. E. Prey and M. Wagner, Adv. Synth. Catal., 2021, 363, 2290–2309 CrossRef CAS; (d) S. E. Prey, C. Herok, F. Fantuzzi, M. Bolte, H.-W. Lerner, B. Engels and M. Wagner, Chem. Sci., 2023, 14, 849–860 RSC.
- A. Lorbach, M. Bolte, H.-W. Lerner and M. Wagner, Organometallics, 2010, 29, 5762–5765 CrossRef CAS.
- E. von Grotthuss, F. Nawa, M. Bolte, H.-W. Lerner and M. Wagner, Tetrahedron, 2019, 75, 26–30 CrossRef CAS.
- H. Nöth and B. Wrackmeyer, Nuclear magnetic resonance spectroscopy of boron compounds, in NMR Basic Principles and Progress, ed. P. Diehl, E. Fluck and R. Kosfeld, Springer Verlag, Berlin, Heidelberg, New York, 1978 Search PubMed.
- We performed the cycloaddition reaction of M2[2] with M+ = Li+, Na+, and K+. Since crystallization is the key purification step and K2[6] gave the most reproducible yields, we focused on this compound throughout this work..
- Values refer to [K(Me6-tren)][HBPh3] (Me6-TREN = N,N,N-tris(2-(dimethylamino)ethyl)amine): D. Mukherjee, H. Osseili, T. P. Spaniol and J. Okuda, J. Am. Chem. Soc., 2016, 138, 10790–10793 CrossRef CAS PubMed.
- The reaction was monitored by 1H NMR spectroscopy in CD2Cl2, and the resonances of the byproducts CD2HCl and CD2H2 were detected as characteristic 1:2:3:2:1 quintets. Comparable chlorination of a siladodecahedrane in CD2Cl2 was recently reported: M. Bamberg, M. Bursch, A. Hansen, M. Brandl, G. Sentis, L. Kunze, M. Bolte, H.-W. Lerner, S. Grimme and M. Wagner, J. Am. Chem. Soc., 2021, 143, 10865–10871 CrossRef CAS PubMed.
- S. J. Lancaster and D. L. Hughes, Dalton Trans., 2003, 1779–1789 RSC.
- J.-M. Denis, H. Forintos, H. Szelke, L. Toupet, T.-N. Pham, P.-J. Madec and A.-C. Gaumont, Chem. Commun., 2003, 54–55 RSC.
- To compare the photochemical responses of K2[6] and its all-carbon congener 6C, both molecules were irradiated under identical conditions (see the ESI†). We observed a significantly faster reaction in the case of K2[6] and have shown that the reactions lead to different scaffolds: while irradiation of K2[6] cleanly furnishes K2[10], the reaction of 6C stops at the stage of Int2C. The isomerization to 10C requires heating, cf. ref. 10 and 11..
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2009, 113, 6378–6396 CrossRef CAS PubMed.
- J.-D. Chai and M. Head-Gordon, Phys. Chem. Chem. Phys., 2008, 10, 6615–6620 RSC.
- The exclusion of the counter cations is justified by two facts: (a) Unlike Li+ ions, K+ ions do not form strong contact ion pairs in solution that influence the reactivity of the compounds, see ref. 25b; (b) Omitting K+ ions and their coordinating thf ligands in the calculations reduces the computational cost significantly, see ref. 25d..
- J. Radtke, S. K. Mellerup, M. Bolte, H.-W. Lerner, S. Wang and M. Wagner, Org. Lett., 2018, 20, 3966–3970 CrossRef CAS PubMed.
- S. Wang, K. Yuan, M.-F. Hu, X. Wang, T. Peng, N. Wang and Q.-S. Li, Angew. Chem., Int. Ed., 2018, 57, 1073–1077 CrossRef CAS PubMed.
- J. D. Wilkey and G. B. Schuster, J. Am. Chem. Soc., 1991, 113, 2149–2155 CrossRef CAS.
- A comparable [1,5]sigmatropic shift, also known as “walk reaction”, has been studied for norcaradiene: (a) F.-G. Klärner, Angew. Chem., Int. Ed., 1974, 13, 268–270 CrossRef; (b) A. Kless, M. Nendel, S. Wilsey and K. N. Houk, J. Am. Chem. Soc., 1999, 121, 4524–4525 CrossRef CAS.
- The [TS4]2−-IRC scan indicates a bifurcation at the C–H activation step because the energy profile shows a plateau between [Int3]2− and [TS4]2− (see Fig. S55†). At the end of this plateau, the molecular geometry is sufficiently close to that of [TS3]2− for [TS3]2− to be optimized without significant energy and geometry changes. Consequently, a shortcut [Int2]2− → [TS3]2− → [TS4]2− → [10]2− is also likely to occur: D. H. Ess, S. E. Wheeler, R. G. Iafe, L. Xu, N. Çelebi-Ölçüm and K. N. Houk, Angew. Chem., Int. Ed., 2008, 47, 7592–7601 CrossRef CAS PubMed.
- We also located a transition state directly connecting [Int2]2− and [10]2− ([TS4′]2−, ΔG‡ = 40.3 kcal mol−1, see Fig. S54†). However, since the energy required for the latter is significantly higher than for the above-mentioned path via [TS4]2−, the contribution of [TS4′]2− should be negligible..
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic procedures, NMR spectra, X-ray crystallographic data and computational details. CCDC 2238019–2238023. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3sc00555k |
| This journal is © The Royal Society of Chemistry 2023 |