DOI:
10.1039/D3SC00658A
(Edge Article)
Chem. Sci., 2023,
14, 10420-10428
Buckybowl and its chiral hybrids featuring eight-membered rings and helicene units†
Received
6th February 2023
, Accepted 7th March 2023
First published on 14th March 2023
Abstract
Here we report the synthesis of a novel buckybowl (7) with a high bowl-to-bowl inversion barrier (ΔG‡ = 38 kcal mol−1), which renders the rate of inversion slow enough at room temperature to establish two chiral polycyclic aromatic hydrocarbons (PAHs). By strategic fusion of eight-membered rings to the rim of 7, the chiral hybrids 8 and 9 are synthesized and display helicity and positive and negative curvature, allowing the enantiomers to be configurationally stable and their chiroptical properties are thoroughly examined. Computational and experimental studies reveal the enantiomerization mechanisms for the chiral hybrids and demonstrate that the eight-membered ring strongly affects the conformational stability. Because of its static and doubly curved conformation, 9 shows a high binding affinity towards C60. The OFET performance of 7–9 could be tuned and the hybrids show ambipolar characteristics. Notably, the 9·C60 cocrystal exhibits well-balanced ambipolar performance with electron and hole mobilities of up to 0.19 and 0.11 cm2 V−1 s−1, respectively. This is the first demonstration of a chiral curved PAH and its complex with C60 for organic devices. Our work presents new insight into buckybowl-based design of PAHs with configurational stability and intriguing optoelectronic properties.
Introduction
Incorporating nonhexagonal rings into sp2 hexagonal nanographenes has emerged as a promising strategy to construct nonplanar chiral polycyclic aromatic hydrocarbons (PAHs).1 The introduction of five-membered rings into a hexagonal network typically causes positive Gaussian curvature, leading to bowl-shaped aromatic hydrocarbons, so-called buckybowls or π-bowls,2 with a variety of distinctive optoelectronic and supramolecular properties.3 Recently, buckybowls have drawn increasing attention as attractive platforms for late-stage functionalization due to their diverse reactivity and rich, well-established transmetalation chemistry, allowing the preparation of newly chiral PAHs, so-called hybridized buckybowls.4 For example, corannulene has been extensively utilized to construct well-defined chiral PAHs (Fig. 1a),5 which exhibit unique dynamic behaviors that emerge from their chirality and nonplanarity.6 These notable examples affirm the competency of buckybowls for chemical tailoring to extend molecular dimensionality, achieving topologically distinct PAHs with promising physical properties. For example, Zhang and coworkers reported the synthesis of a helicene-fused buckybowl as a near-infrared fluorescence probe in biological applications.7 Hybridized corannulenes suffer generally from configurational lability associated with low-energy bowl-to-bowl inversion, from a stereochemical view; however, these molecules are non-rigid and thus achiral.1a
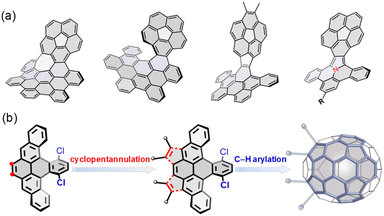 |
| | Fig. 1 (a) Representative of hybridized buckybowls; (b) structural design and synthetic strategy to a new C70 subunit for late-stage hybridization in this work. | |
Very recently, a growing number of sp2 hexagonal networks containing eight-membered rings considered to be subunits of elusive Mackay crystals,8 have been synthesized and characterized.9 It has been found that the eight-membered ring deeply impacts the overall geometry and the selective fusion of an aromatic ring with an eight-membered ring gives rise to twist structures, which possess exceptionally high enantiomerization free energy barriers (ΔG‡).10 On the other hand, fusion of eight-membered rings to buckybowls is largely underexplored. It is hypothesized that, in this hybrid system, the merged positive and negative regions open the possibility of controlling the electronic properties and frontier orbital profiles across the eight-membered rings, which are expected to tune barriers for interconversion and rates for bowl inversion by an intricate network of twisted eight-membered rings, buckybowls, and helical superstructures. As such, one can expect that such unusual structural features offer interesting design opportunities for the development of attractive chiral hybrids, which are fundamentally different from other merged systems.
Herein, we report the synthesis of a novel buckybowl 7 and its chiral hybrids (8 and 9) shown in Scheme 1. The molecular design is expanded to π-conjugated hybridized buckybowl systems, which are composed of eight-membered rings fused in different fashions (Fig. 1b). The buckybowl 7, as a new C70 subunit, contains four five-membered rings and has a high energy barrier for bowl-to-bowl conversion, a feature that allows for the configurational stability of chiral materials. Furthermore, the eight-membered rings and buckybowl units are merged in a unique manner and raise the possibility of creating chiral PAHs, which show topologically distinct helical geometries and novel physical properties. This model system would help elucidate the effect of structural curvature on configurational stability and molecular properties.
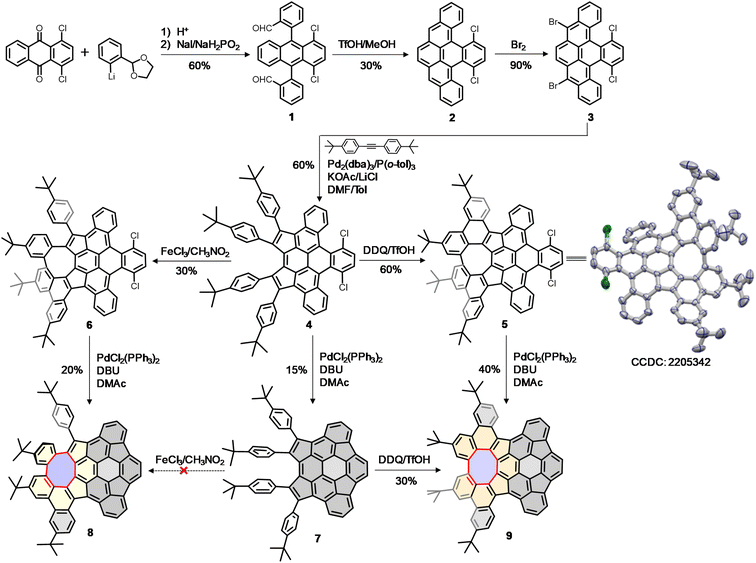 |
| | Scheme 1 Synthetic steps to buckybowl 7 and its chiral hybrids 8 and 9. | |
Results and discussion
Design and synthesis
Our synthesis began with the nucleophilic addition of lithiated 2-(2-bromophenyl)-1,3-dioxolane to 1,4-dichloroanthraquinone followed by reductive aromatization with NaI/NaH2PO2 in acetic acid, yielding a mixture of stereoisomers of dialdehyde 1. The subsequent reductive Friedel–Crafts cyclization of the isomeric 1 in the presence of trifluoromethanesulfonic acid (TfOH) and methanol afforded the fully aromatic compound 2 in 30% yield, which underwent a smooth bromination to afford 3 in 90% yield. The palladium-catalyzed intermolecular cyclopentannulation of 3 with diarylacetylene provided 4 as a sole major product in 60% yield. The Scholl-type oxidation of 4 using 2,3-dichloro-5,6-dicyanobenzoquinone (DDQ) in the presence of TfOH yielded 5 in 60% yield, which was further confirmed by single-crystal X-ray analysis. However, when 4 was treated with FeCl3, the formation of 5 was suppressed, giving 6 as the sole product in 30% yield. Finally, the palladium-catalyzed intramolecular C–H arylation of 4–6 gave the desired products 7–9 in yields of 15–40%. Interestingly, the Scholl-type oxidation of buckybowl 7 using DDQ/TfOH was also successful, to selectively generate 9 in 30% yield. However, our attempts to convert 7 into 8 using FeCl3 were unsuccessful.
Static structures
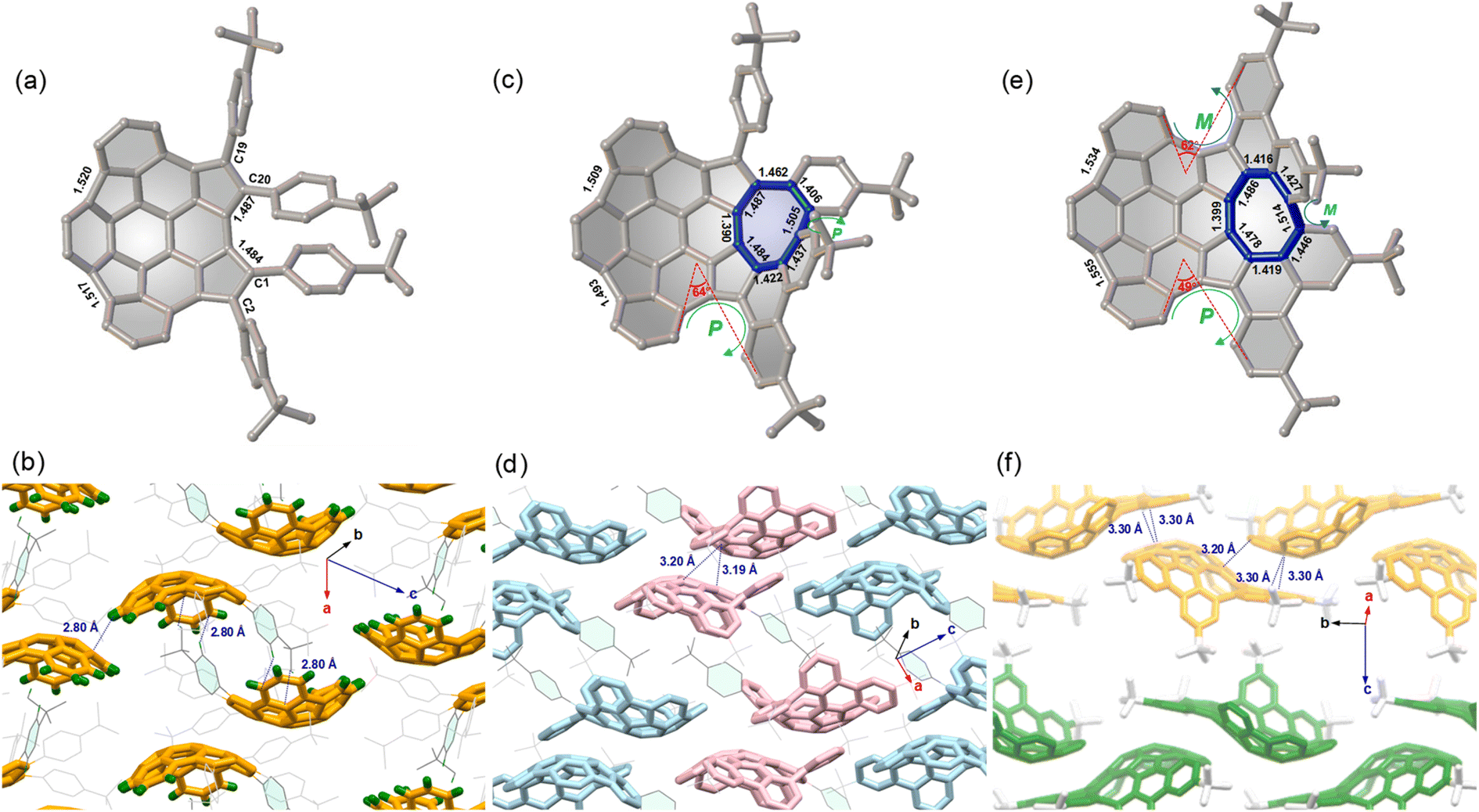
The molecular structures 7–9 were determined by single crystal X-ray diffraction analysis. Compound 7 displays a bowl-shaped geometry (Fig. 2a), whereas compounds 8 and 9 display helicity and positive and negative curvatures (Fig. 2c and e). Obviously, the degree of bowl curvature and helicene folding are the most characteristic structural features of 7–9. The bowl depth of 7, here defined as the average perpendicular displacement of four rim carbons (C1, C2, C19, and C20) from the least-squares plane of the central six-membered ring, is 1.88 Å and is observed to decrease to 1.77 Å for 8 and 1.70 Å for 9. For the [5]helicene unit in 8, the dihedral angle between the mean planes of the two outer benzene rings is 64°, which is larger than that for pristine carbo-[5]helicene (46°).10a However, the two dihedral angles of [5]helicene units in 9 are 49° and 62°, respectively. Notably, the eight-membered ring exhibits tub-shaped conformation with twist angles (angle between the planes of the terminal benzene rings) of 69° for 8 and 60° for 9. The larger twist angle of 8 is presumably due to the additional steric environment presented by the pendant phenyl group. The C–C bond lengths of the eight-membered rings in 8 and 9 are relatively long and are arranged alternately. In addition, there is significant bond-length elongation (1.484–1.555 Å) in the five-membered rings in 7–9, which minimizes the aromatic character of the rings.
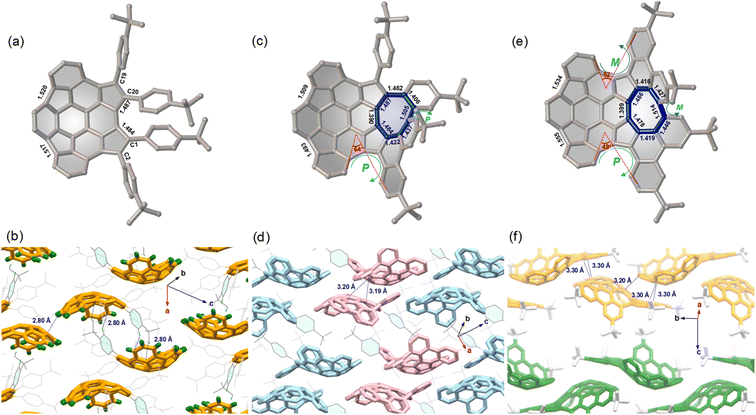 |
| | Fig. 2 Crystal structures and molecular packing of 7 (a and b), 8 (c and d), and 9 (e and f) with selected bond lengths (Å) and close contact distances. (M,PP)-8 is colored in pink and (P,MM)-8 is colored in light blue in (d); (M,MMP)-9 is colored in yellow and (P,PPM)-9 is colored in green in (f); partial hydrogen atoms and solvent molecules are omitted for clarity. | |
For compound 7, the compounds pack in a “substituent-in-bowl” motif, as the pendant phenyl groups partially occupy volume between the adjacent bowls to prevent columnar stacking (Fig. 2b). As a result, two such buckybowls are linked together to form a dimer structure by multiple C–H⋯π contacts (2.80 Å) between pendant phenyl groups and convex π-surfaces in the crystal. For compound 8, the crystal is a racemate composed of a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of enantiomers. To distinguish between the possible conformers, chirality arising from the curvature of each buckybowl is denoted as P or M, according to the nomenclature suggested by Scott and Petrukhina.11 Accordingly, the pair of enantiomers in the crystal of 8 are assigned as (M,PP)-8 (herein the bowl chirality is M and the helical chirality is PP) and (P,MM)-8 (herein the bowl chirality is P and the helical chirality is MM) (Fig. 2d). The racemic 8 also crystallizes with a “substituent-in-bowl” motif similar to that of 7 described above. The dimers engage in concave–convex contacts of 3.19 Å and 3.20 Å. In this way, compound 8 is more densely packed when compared to 7. Unlike the structures of 7 and 8, 9 has a 2D layered structure (Fig. 2f). In each layer, the molecules display the same helicity and are arranged in wavy stacks through the interactions between the positive and negative π-surfaces of the adjacent enantiomers (M,MMP)-9 or (P,PPM)-9, thus maintaining substantial spatial overlap between the adjacent molecules.
:1 mixture of enantiomers. To distinguish between the possible conformers, chirality arising from the curvature of each buckybowl is denoted as P or M, according to the nomenclature suggested by Scott and Petrukhina.11 Accordingly, the pair of enantiomers in the crystal of 8 are assigned as (M,PP)-8 (herein the bowl chirality is M and the helical chirality is PP) and (P,MM)-8 (herein the bowl chirality is P and the helical chirality is MM) (Fig. 2d). The racemic 8 also crystallizes with a “substituent-in-bowl” motif similar to that of 7 described above. The dimers engage in concave–convex contacts of 3.19 Å and 3.20 Å. In this way, compound 8 is more densely packed when compared to 7. Unlike the structures of 7 and 8, 9 has a 2D layered structure (Fig. 2f). In each layer, the molecules display the same helicity and are arranged in wavy stacks through the interactions between the positive and negative π-surfaces of the adjacent enantiomers (M,MMP)-9 or (P,PPM)-9, thus maintaining substantial spatial overlap between the adjacent molecules.
Conformational dynamics
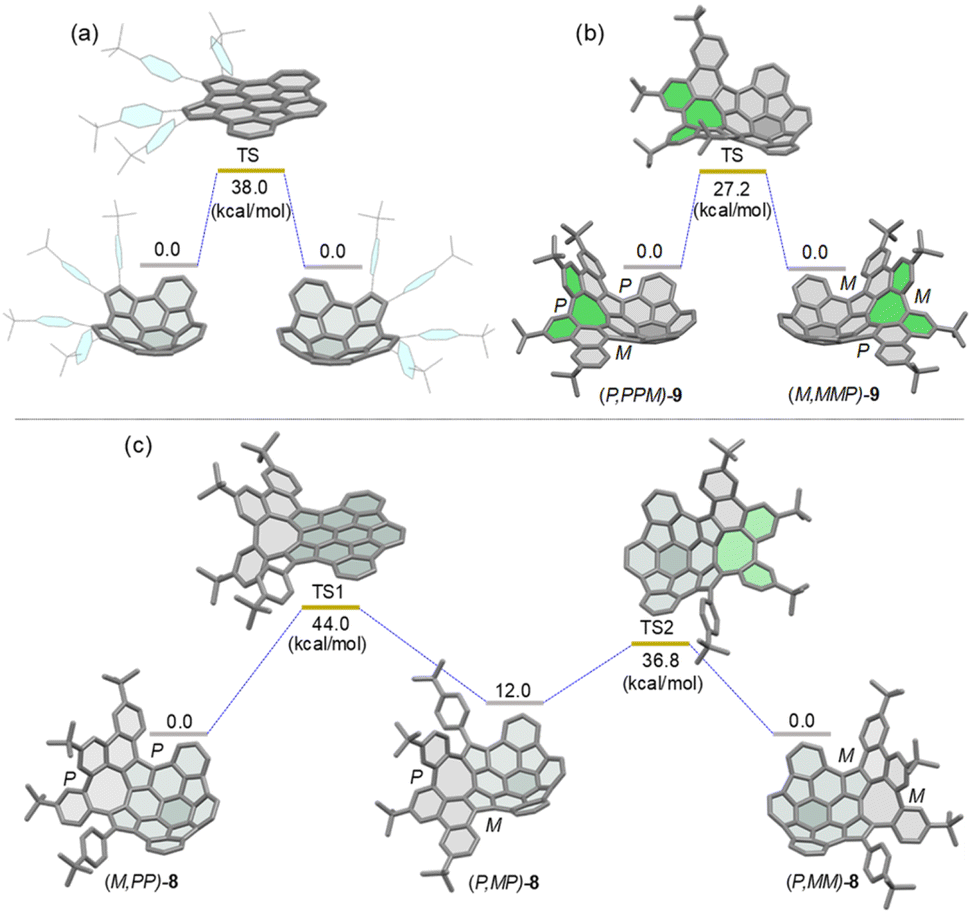
Density functional theory (DFT) calculations at the B3LYP/6-311G(d,p) level of theory revealed that the bowl-to-bowl inversion of 7 proceeds via a planar transition state with an inversion barrier of 38 kcal mol−1 (Fig. 3a), which is comparable with the calculated bowl inversion energy for triazasumanene, a chiral buckybowl that has a half-life time of 54 billion years at 20 °C.12 This barrier value is much higher than that of corannulene (∼10 kcal mol−1)13 and sumanene (∼20 kcal mol−1).14 For 9, DFT calculations indicated that its enantiomerization in fact proceeds by a single-step mechanism in which only inversion of the helicity of the twisted eight-membered ring moiety (highlighted in green in Fig. 3b) takes place to arrive at the enantiomer, with an activation barrier of 27.2 kcal mol−1, which is higher than that of carbo-[5]helicene (ΔG‡ = 24.1 kcal mol−1 at 298 K).15 On the other hand, DFT calculations predicted the stepwise pathway for enantiomerization of 8 (Fig. 3c). In the first step, (M,PP)-8 converts to a local minimum (P,MP)-8, through a transition state (TS1) with a planar buckybowl unit, accompanied by a synchronous inversion of the [5]helicene unit. The activation barrier for TS1 is 44.0 kcal mol−1. The second process occurs via a transition state (TS2) with a tub-shaped eight-membered ring to give the other enantiomer (P,MM)-8. The activation barrier for TS2 is 36.8 kcal mol−1, which is higher than the activation barrier for the transition state for 9. The emergence of steric interactions between the pendant phenyl group and the neighboring benzene rings could be responsible for an increase in its activation energy.
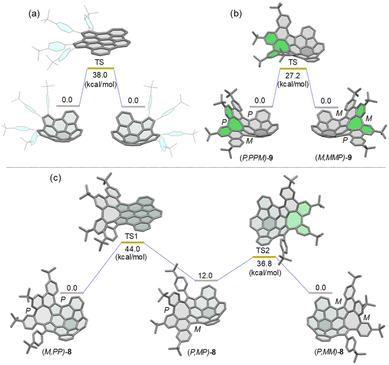 |
| | Fig. 3 Calculated diagrams for the bowl inversion process of 7 (a) and enantiomerization of (P,PPM)-9 (b) and (M,PP)-8 (c) at the B3LYP/6-311G(d,p) level. | |
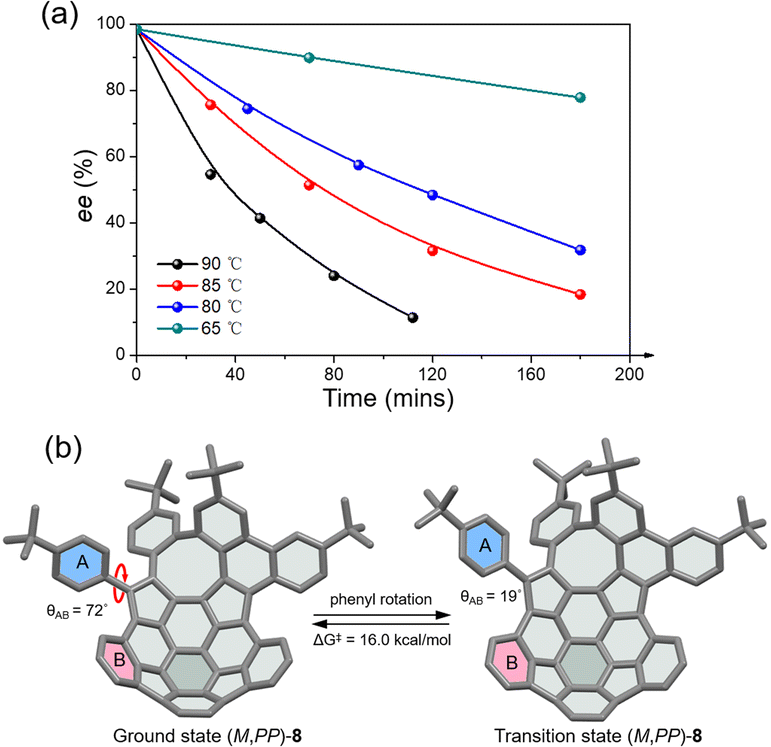
The racemic 8 and 9 were readily resolved into their corresponding enantiomers by chiral high-performance liquid chromatography (HPLC). The racemization barrier for 9 was experimentally determined by measurement of the decrease in HPLC of the enantiomeric excess of 9 (ee, 98.4%) in toluene at various temperatures (Fig. 4a). Fitting the data by using the Arrhenius and Eyring plots gave the thermodynamic parameters ΔH‡ of 27.6 kcal mol−1, ΔS‡ of −1.05 J (mol−1 K), and ΔG‡ of 27.7 kcal mol−1 at 298 K (see details in the ESI†). This value is in perfect agreement with the calculated ΔG‡ for enantiomerization. As expected, compound 8 exhibits a higher racemization barrier than 9. In fact, upon heating diphenyl ether solution for 5 h at 453 K, no racemization of 8 was observed (Fig. S20†). Additionally, variable-temperature 1H NMR (VT-NMR) of 8 revealed a process (Fig. S15†), which is ascribed to the pendant phenyl group rotation. The coalescence temperature for the ortho hydrogens of the pendant phenyl group is 60 °C, corresponding to a rotational barrier of 15.4 kcal mol−1. The rotation barrier was further investigated at the B3LYP/6-311G(d,p) level (Fig. 4b), which revealed that the angles between the planes of rings A and B in the ground state and transition state are 72° and 19°, giving a rotation barrier of 16.0 kcal mol−1, in reasonable agreement with the measured value.
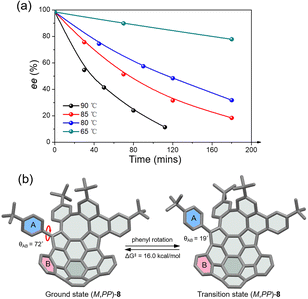 |
| | Fig. 4 (a) Racemization kinetic plots of enantioenriched 9 in toluene at various temperatures; (b) the rotation process and rotation barrier of the pendant phenyl ring in (M,PP)-8 at the B3LYP/6-311G(d,p) level. | |
Optical and electrochemical properties and aromaticity
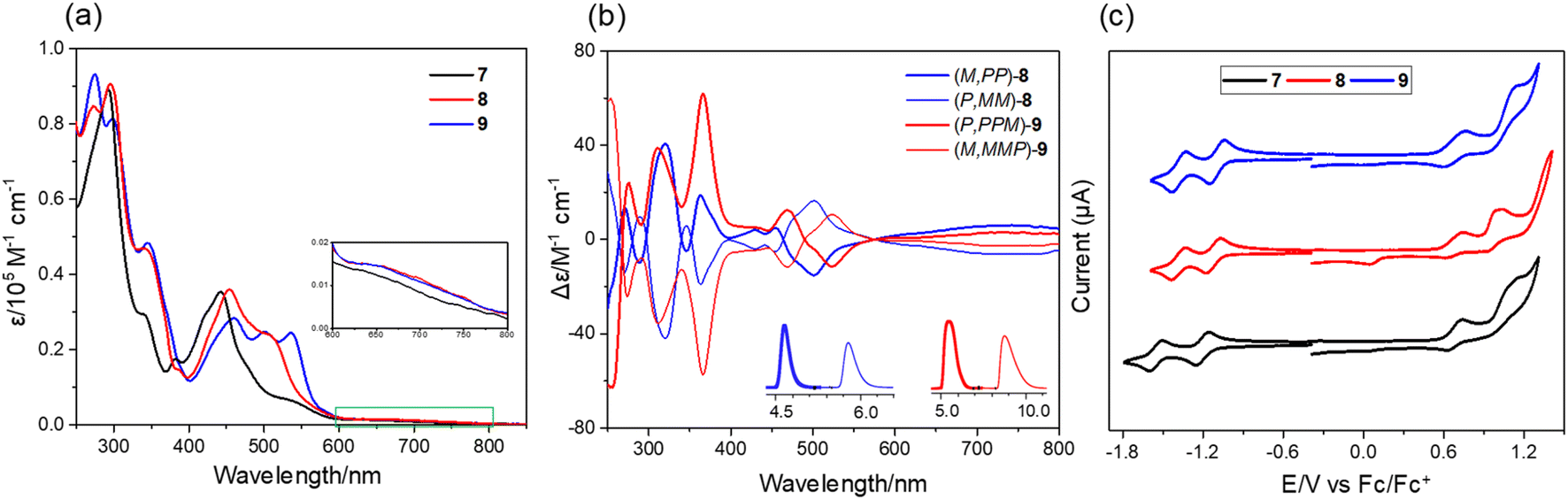
The UV-vis spectra of 7–9 show strong absorption in the visible region (400–600 nm) of the light spectrum, with a broad, low energy absorbance tail that extends into 800 nm (Fig. 5a). For compound 7, it possesses a prominent band centered at 442 nm together with a wide shoulder at ∼527 nm (ε = 6900 M−1 cm−1). The π-extended 8 shows a bathochromic shift (10 nm) of maximum absorption with a relatively more intense shoulder absorption (λ = 506 nm, ε = 24000 M−1 cm−1) as compared to 7. The absorption maximum of 9 is further red-shifted as a result of its more extended π-system. The spectral profiles of 7–9 well agree with the simulated spectra using time dependent density functional theory (TD-DFT) calculations at the CAM-B3LYP/6-311G(d,p) level of theory, which predicted that the low-energy absorbance tails correspond to the forbidden HOMO-to-LUMO with a low oscillator strength (for 7: λcalc = 637 nm, f = 0.007; for 8: λcalc = 645 nm, f = 0.005; for 9: λcalc = 657 nm, and f = 0.005) (Fig. S7†). The chiroptical properties of the enantiomers of 8 and 9 were investigated by circular dichroism (CD) measurement (Fig. 5b). The absolute configuration of the enantiomers was assigned by comparison of the experimental CD spectra with the TD-DFT simulated CD spectra (Fig. S8†). The corresponding enantiomers show perfect mirror-image CD spectra with opposite cotton effects. For 8, the first fraction (M,PP)-8 shows four positive and five negative cotton effects in the range of 250–550 nm and the most intense CD dis-symmetry factor (|gabs|) is 2 × 10−3 at 367 nm. For 9, the first fraction of (P,PPM)-9 shows four positive effects and one negative cotton effect, with the most intense |gabs| of 6 × 10−3 at 370 nm, whose value is comparable to those of (P)-(+)-[5]helicene.16 Similar to other buckybowls17 and fullerenes,18 none of the three compounds produce easily measured fluorescence. The CV of 7 shows that its first-wave reduction potential (−1.20 V vs. Fc/Fc+) occurs about 100 meV higher than that of 8 and 9 (−1.12 and −1.10 eV, respectively), indicating that incorporation of an eight-membered ring can increase the electron-accepting character of these systems (Fig. 5c). The difference in the first oxidation peak of 7–9 is quite small, decreasing from 0.68 V for 7 to 0.67 V for 8 to 0.66 V for 9. The DFT calculations reveal that fusion of the eight-membered ring to buckybowl 7 leads to a more delocalized wave function, which results in a slight stabilization of LUMOs in 8 and 9 when compared to 7 (Fig. S6†). However, the HOMOs of 7–9 are found to be mainly localized in the bowl region, which may account for the essentially same oxidation potential for 7–9.
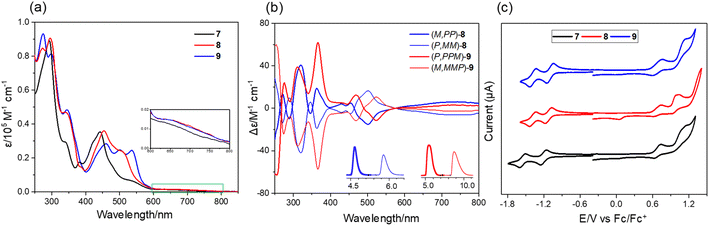 |
| | Fig. 5 (a) UV-vis absorption of 7–9 in chloroform (∼10−5 M); (b) CD spectra of the enantiomers of 8 and 9 in chloroform. (inset: resolution of the enantiomers of 8 and 9 by chiral analytical HPLC.); (c) cyclic voltammograms of 7–9 in nitrogen-purged dichloromethane with tetrabutylammonium hexafluorophosphate (TBAPF6, 0.1 M) as the supporting electrolyte and a scan rate of 100 mV s−1. | |
Nucleus-independent chemical shift (NICS), 3D isochemical shielding surface (3D ICSS), and anisotropy of the induced current density (ACID) calculations were performed to evaluate the aromatic character of 7–9. For 7–9, the NICS(1)zz values of five-membered rings are between 13.5 and 26.7 ppm, indicating the antiaromatic character (Fig. 6a–c). The values of the two central six-membered rings in 7 are −3.2 to 6.6 ppm, indicating the nonaromatic character. The values of the six-membered rings next to the eight-membered rings in 8 and 9 are slightly more positive than the corresponding ring in 7, suggesting stronger dearomatization. This is attributed to the antiaromatic eight-membered ring that these rings in 8 and 9 are fused with. Remarkably, the NICS(1)zz values on concave sides (NICS(1)zz-concave) of the six-membered rings in buckybowl units of 7–9 are more negative than the values on concave sides (NICS(1)zz-convex), while the NICS(1) values on concave/convex sides of the five-membered rings are nearly identical. From 8 to 9, the NICS(1)zz value of the eight-membered ring is increased, indicating an enhancement in antiaromaticity upon planarization. These values are significantly lower than that of parent planar cyclooctatetraene with D4h symmetry (62.5).19 The 3D ICSS map revealed several independent deshielding areas of 7 above the planes of the five-membered rings on the concave side with the shielding area distributed over the six-membered rings in the structure (Fig. 6d). In going from 7 to 8 and finally 9, additional deshielding areas are observed at the eight-membered rings, which suggests that five- and eight-membered rings are locally antiaromatic and the local aromaticity is nearly exclusively distributed over some of the six-membered rings in the structures (Fig. 6e and f). As expected, the ACID plots of 7–9 confirm that the counterclockwise induced currents appear at five-membered rings, while the six-membered rings sustain clockwise currents (Fig. S9†). Note that nondirectional currents are observed at eight-membered rings or two central six-membered rings.
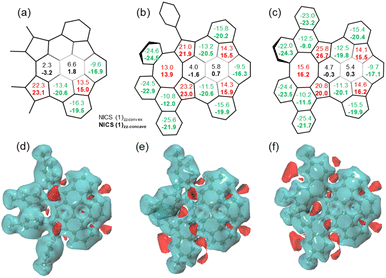 |
| | Fig. 6 NICS(1)zz values (ppm) of 7 (a), 8 (b), and 9 (c) calculated at the B3LYP/6-311G(d,p) level of theory; 3D ICCS maps of 7 (d), 8 (e), and 9 (f) showing the shielding area (light blue) and deshielding area (red) (isovalue = 4). | |
Host–guest complexes with C60
Due to their geometrically and electronically complementary nature, 7–9 are proposed as prospective components to complex with fullerenes.20 Upon addition of C60, the systematic shifts of the protons in the 1H NMR spectra are observed (Fig. 7a), which suggest that the expected complexes are formed. The titration data versus C60 for 7–9 were fitted successfully to a 1:1 binding mode,21 yielding binding constants of 660 M−1 for 7 (Fig. S21†), 5900 M−1 for 8 (Fig. S22†), and 7450 M−1 for 9 (Fig. 7b). The relatively low binding constant for 7 is likely due to the rotation of pendant phenyl groups. Furthermore, crystallographic analysis of crystals obtained from the mixture of 9 and C60 in toluene confirms the 1:1 complex (Fig. 7c). In the cocrystal, each molecule of C60 interacts directly with a total of three molecules of 9. Two enantiomeric 9 wrap around C60 in its “polar” regions by the positive and negative π-surfaces, respectively, but allow close C⋯C contacts between the “equatorial” regions of two C60 molecules with a centroid-to-centroid distance of 10.15 Å. In addition, each C60 also forms multiple C–H⋯π interactions (2.80 Å) with the third molecule of 9. By this way, compounds 9 form wavy chains around the polar regions of the C60 molecules with close contacts between positive and negative π-surfaces, and the voids between the chains accommodate C60 molecules to create one-dimensional zig-zag arrays along the b-axis, and thus 9 and C60 ensemble separately in segregated stack cocrystals. In the cocrystal, the measured distances between the centroid of C60 and the centroids of the aromatic rings of 9 are in the range of 6.65–6.84 Å, indicating the strong π–π interactions and that the cocrystallization is favored by the combined effect of topological and electronic complementarities. Notably, in contrast to the several reported structures of fullerene cocrystals,22 no solvent molecules or C60 disorder is observed in the crystal, which suggests that the static conformation of 9 significantly increases its ability to bind C60 and results in a tight packing complex.
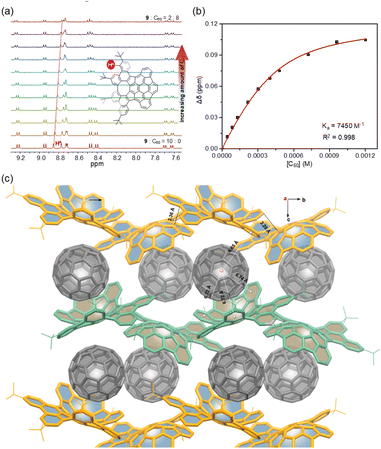 |
| | Fig. 7 (a) Selected regions of 1H NMR spectra during titration of 9 with C60 (the total concentration [9] + [C60] = 6.0 × 10−4 M, toluene-d8, 298 K); (b) fitting curves for the proton HA in 9vs. [C60]; (c) crystal packing of 9·C60 with selected centroid–centroid distances and close contact distances (Å). | |
Charge-carrier transport properties
The solution-processed organic thin film transistors (OTFTs) were fabricated with the ‘‘bottom-gate bottom-contact’’ geometry (see details in the ESI†). Devices based on 7 exhibit exclusive hole transporting characteristics with a mobility of 1.1 × 10−5 cm2 V−1 s−1. However, the devices based on 8 show ambipolar characteristics with hole and electron mobilities of 1.8 × 10−5 and 2.9 × 10−6 cm2 V−1 s−1. The increase in electron mobility is due to the presence of an eight-membered ring, which leads to energetic lowering of the LUMO levels. For 9, it also exhibits ambipolar characteristics but with slightly higher mobilities for holes and electrons of 7.6 × 10−5 and 3.8 × 10−6 cm2 V−1 s−1 as compared to 8. Finally, the intrinsic charge-carrier transport of 9 was evaluated through single-crystal organic field-effect transistors (SC-OFETs), which were fabricated by a “gold strips” technique23 (see details in the ESI†). The single crystals of 9 and 9·C60 were prepared by slow diffusion of methanol into toluene solution, forming microbelts with several μm width and hundreds of μm length (Fig. 8a and c). According to the X-ray diffraction peaks (Fig. S29†) and corresponding selected area electron diffraction (SAED) patterns (Fig. 8b and d), the growth directions of the crystals 9 and 9·C60 are determined to be along the [010] direction. For 9, the microbelts show well-balanced ambipolar behavior with averaged hole and electron mobilities of 0.024 and 0.016 cm2 V−1 s−1, along the [010] direction (Fig. 8e). Among these results, the maximum hole and electron mobilities are 0.036 and 0.030 cm2 V−1 s−1, respectively. The intermolecular electronic couplings based on the bulk crystal structure were investigated by using GGA:PW91/TZP, as implemented in the Amsterdam Density Functional (ADF) program. The calculations predicted nearly equal values for the effective transfer integrals for holes and electrons (9.8 vs. 9.7 meV) (Fig. S10†), explaining why both types of carriers in 9 possess comparable values. For 9·C60, the cocrystals also exhibit ambipolar characteristics with averaged hole and electron mobilities of 0.06 and 0.10 cm2 V−1 s−1, along the [010] direction (Fig. 8f), and the maximum hole and electron mobilities are measured to be 0.11 and 0.19 cm2 V−1 s−1, which is the highest value previously reported for ambipolar OFETs of stoichiometric host–guest cocrystals.24 The ADF calculations indicated that the segregated zigzag arrays of C60 in the cocrystal can significantly increase the transfer integrals for electrons in the 9·C60 cocrystal (Fig. S11†), which can be explained by the fact that the electron mobility in 9·C60 is larger than that in 9. We note that this is the first OFET with conformationally rigid chiral PAHs featuring merged positive and negative segments to demonstrate their application as active materials in organic electronic devices.
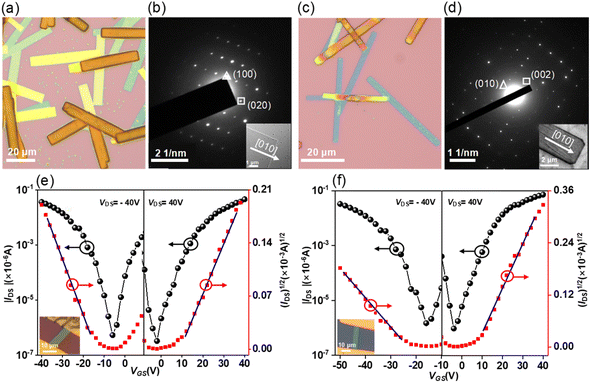 |
| | Fig. 8 Optical microscopy images of microbelts 9 (a) and 9·C60 (c); the selected-area electron diffraction (SAED) patterns of microbelts 9 (b) and 9·C60 (d) (inset: the TEM image of a single-crystalline microbelt); the transfer characteristics of an OFET based on microbelts 9 (W/L = 7.8/22.1) (e) and 9·C60 (W/L = 4.8/23.3) (f). | |
Conclusions
In summary, we have presented novel approaches to a new C70 subunit (7) and its hybrids featuring the fusion of eight-membered rings (8 and 9). The structural variations could allow for deeper understanding and fine-tuning of molecular properties of these fascinating compounds. The resulting PAHs 7–9 are thoroughly characterized by spectroscopic and X-ray analysis and DFT calculations. The chiral hybrids 8 and 9 show lowered LUMO levels by ∼0.1 eV compared to 7. Benefiting from the intricate network of a twisted eight-membered ring, static buckybowls, and helical super-structures, 8 and 9 are configurationally stable chiral PAHs. The activation energy for the racemization of 9 is found to be 27.7 kcal mol−1, whereas 8 displayed a substantially higher racemization barrier than 9. Our results provided crucial insight on the interplay between the molecular structure and configurational stability, thus providing guiding design principles for chiral materials with high enantiomerization free energy barriers. It also appears that the OFET characteristics of 7–9 can be tuned and the cocrystal 9·C60 shows promising ambipolar transport properties. We are exploring the applications of these topologically interesting, curved PAHs for chiral organic devices and shape-selective molecular recognition.
Data availability
The details of experimental procedures and supporting experimental data are available from ESI.†
Author contributions
L. Z received and designed the project. Y. D synthesized and characterized the compounds. X. L, H. H, and H. Y performed DFT calculations. M. C made and characterized the devices. All authors discussed the results and contributed to the manuscript preparation.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (NSFC) (No. 21871022, 22005018, and 22175013). L. Z. thanks Dr Sean Parkin (University of Kentucky) and Dr Maxime A. Siegler (Johns Hopkins University) for crystal structure refinement. The authors acknowledge Prof. Huanli Dong from ICCAS for assistance with TEM measurements.
Notes and references
-
(a) M. Rickhaus, M. Mayor and M. Juríček, Chem. Soc. Rev., 2017, 46, 1643 RSC;
(b) S. H. Pun and Q. Miao, Acc. Chem. Res., 2018, 51, 1630 CrossRef CAS PubMed;
(c) M. A. Majewski and M. Stępień, Angew. Chem., Int. Ed., 2019, 58, 86 CrossRef CAS PubMed;
(d) A. Narita, X.-Y. Wang, X. Feng and K. Müllen, Chem. Soc. Rev., 2015, 44, 6616 RSC;
(e) C. Chaolumen, I. A. Stepek, K. E. Yamada, H. Ito and K. Itami, Angew. Chem., Int. Ed., 2021, 60, 23508 CrossRef PubMed;
(f) Y. Fei and J. Liu, Adv. Sci., 2022, 9, 2201000 CrossRef CAS PubMed.
-
(a) Y.-T. Wu and J. S. Siegel, Chem. Rev., 2006, 106, 4843 CrossRef CAS PubMed;
(b) V. M. Tsefrikas and L. T. Scott, Chem. Rev., 2006, 106, 4868 CrossRef CAS PubMed;
(c) M. Rickhaus, M. Mayor and M. Juríček, Chem. Soc. Rev., 2017, 46, 1643 RSC;
(d) M. A. Majewski and M. Stępień, Angew. Chem., Int. Ed., 2019, 58, 86 CrossRef CAS PubMed;
(e) T.-C. Wu, H.-J. Hsin, M.-Y. Kuo, C.-H. Li and Y.-T. Wu, J. Am. Chem. Soc., 2011, 133, 16319 CrossRef CAS PubMed;
(f) J. Liu, S. Osella, J. Ma, R. Berger, D. Beljonne, D. Schollmeyer, X. Feng and K. Müllen, J. Am. Chem. Soc., 2016, 138, 8364 CrossRef CAS PubMed;
(g) Y. Zou, W. Zeng, T. Y. Gopalakrishna, Y. Han, Q. Jiang and J. Wu, J. Am. Chem. Soc., 2019, 141, 7266 CrossRef CAS PubMed.
-
(a) M. Saito, H. Shinokubo and H. Sakurai, Mater. Chem. Front., 2018, 2, 635 RSC;
(b) T. Amaya, S. Seki, T. Moriuchi, K. Nakamoto, T. Nakata, H. Sakane, A. Saeki, S. Tagawa and T. Hirao, J. Am. Chem. Soc., 2009, 131, 408 CrossRef CAS PubMed;
(c) H. Yokoi, Y. Hiraoka, S. Hiroto, D. Sakamaki, S. Seki and H. Shinokubo, Nat. Commun., 2015, 6, 8215 CrossRef PubMed;
(d) Y. Wang, Y. Li, W. Zhu, J. Liu, X. Zhang, R. Li, Y. Zhen, H. Dong and W. Hu, Nanoscale, 2016, 8, 14920 RSC;
(e) T.-C. Wu, M.-K. Chen, Y.-W. Lee, M.-Y. Kuo and Y.-T. Wu, Angew. Chem., Int. Ed., 2013, 52, 1289 CrossRef CAS PubMed;
(f) Y. Tanaka, N. Fukui and H. Shinokubo, Nat. Commun., 2020, 11, 3873 CrossRef CAS PubMed;
(g) S. M. Elbert, A. Haidisch, T. Kirschbaum, F. Rominger, U. Zschieschang, H. Klauk and M. Mastalerz, Chem. - Eur. J., 2020, 26, 10585 CrossRef CAS PubMed.
-
(a) A. Haupt and D. Lentz, Chem. - Eur. J., 2019, 25, 3440 CrossRef CAS PubMed;
(b) X. Fu, Y. Zhen, Z. Ni, Y. Li, H. Dong, J. S. Siegel and W. Hu, Angew. Chem., Int. Ed., 2020, 59, 14024 CrossRef CAS PubMed;
(c) G. Gao, M. Chen, J. Roberts, M. Feng, C. Xiao, G. Zhang, S. Parkin, C. Risko and L. Zhang, J. Am. Chem. Soc., 2020, 142, 2460 CrossRef CAS PubMed;
(d) K. Shi, T. Lei, X. Wang, J. Wang and J. Pei, Chem. Sci., 2014, 5, 1041 RSC.
-
(a) K. Kawasumi, Q. Zhang, Y. Segawa, L. T. Scott and K. Itami, Nat. Chem., 2013, 5, 739 CrossRef CAS PubMed;
(b) K. Kato, Y. Segawa, L. T. Scott and K. Itami, Angew. Chem., Int. Ed., 2018, 57, 1337 CrossRef CAS PubMed;
(c) H.-A. Lin, K. Kato, Y. Segawa, L. T. Scott and K. Itami, Chem. Sci., 2019, 10, 2326 RSC;
(d) T. Fujikawa, D. V. Preda, Y. Segawa, K. Itami and L. T. Scott, Org. Lett., 2016, 18, 3992 CrossRef CAS PubMed;
(e) T. Guo, A. Li, J. Xu, K. K. Baldridge and J. S. Siegel, Angew. Chem., Int. Ed., 2021, 60, 25809 CrossRef CAS PubMed;
(f) S. Lampart, L. M. Roch, A. K. Dutta, Y. Wang, R. Warshamanage, A. D. Finke, A. Linden, K. K. Baldridge and J. S. Siegel, Angew. Chem., Int. Ed., 2016, 55, 14648 CrossRef CAS PubMed;
(g) A. K. Dutta, A. Linden, L. Zoppi, K. K. Baldridge and J. S. Siegel, Angew. Chem., Int. Ed., 2015, 54, 10792 CrossRef CAS PubMed;
(h) J. M. Fernández-García, P. J. Evans, S. M. Rivero, I. Fernández, D. García-Fresnadillo, J. Perles, J. Casado and N. Martín, J. Am. Chem. Soc., 2018, 140, 17188 CrossRef PubMed;
(i) S. Zank, J. M. Fernández-García, A. J. Stasyuk, A. A. Voityuk, M. Krug, M. Solá, D. M. Guldi and N. Martín, Angew. Chem., Int. Ed., 2022, 61, e202112834 CrossRef CAS PubMed.
-
(a) Y. Tokimaru, S. Ito and K. Nozaki, Angew. Chem., Int. Ed., 2017, 56, 15560 CrossRef CAS PubMed;
(b) K. Kise, S. Ooi, H. Saito, H. Yorimitsu, A. Osuka and T. Tanaka, Angew. Chem., Int. Ed., 2022, 61, e202112589 CrossRef CAS PubMed;
(c) G. Báti, D. Csókás, T. Yong, S. M. Tam, R. R. S. Shi, R. D. Webster, I. Pápai, F. García and M. C. Stuparu, Angew. Chem., Int. Ed., 2020, 59, 21620 CrossRef PubMed;
(d) Q. Xu, C. Wang, Y. Zhao, D. Zheng, C. Shao, W. Guo, X. Deng, Y. Wang, X. Chen, J. Zhu and H. Jiang, Org. Lett., 2020, 22, 7397 CrossRef CAS PubMed;
(e) Y. Tokimaru, S. Ito and K. Nozaki, Angew. Chem., Int. Ed., 2018, 57, 9818 CrossRef CAS PubMed.
- Y. Wu, S. Ying, L. Su, J. Du, L. Zhang, B. Chen, H. Tian, H. Xu, M. Zhang, X. Yan, Q. Zhang, S. Xie and L. Zheng, J. Am. Chem. Soc., 2022, 144, 10736 CrossRef CAS PubMed.
- A. L. Mackay and H. Terrones, Nature, 1991, 352, 762 CrossRef.
-
(a) I. R. Márquez, S. Castro-Fernández, A. Millán and A. G. Campaña, Chem. Commun., 2018, 54, 6705 RSC;
(b) S. Matsubara, Y. Koga, Y. Segawa, K. Murakami and K. Itami, Nat. Catal., 2020, 3, 710 CrossRef CAS;
(c) K. Y. Cheung, C. K. Chan, Z. Liu and Q. Miao, Angew. Chem., Int. Ed., 2017, 56, 9003 CrossRef CAS PubMed;
(d) G. G. Miera, S. Matsubara, H. Kono, K. Murakami and K. Itami, Chem. Sci., 2022, 13, 1848 RSC;
(e) K. Nakamura, Q.-Q. Li, O. Krejčí, A. S. Foster, K. Sun, S. Kawai and S. Ito, J. Am. Chem. Soc., 2020, 142, 11363 CrossRef CAS PubMed;
(f) Y. Sakamoto and T. Suzuki, J. Am. Chem. Soc., 2013, 135, 14074 CrossRef CAS PubMed;
(g) S. H. Pun, Y. Wang, M. Chu, C. K. Chan, Y. Li, Z. Liu and Q. Miao, J. Am. Chem. Soc., 2019, 141, 9680 CrossRef CAS PubMed;
(h) R. W. Miller, A. K. Duncan, S. T. Schneebeli, D. L. Gray and A. C. Whalley, Chem. - Eur. J., 2014, 20, 3705 CrossRef CAS PubMed.
-
(a) M. A. Medel, R. Tapia, V. Blanco, D. Miguel, S. P. Morcillo and A. G. Campaña, Angew. Chem., Int. Ed., 2021, 60, 6094 CrossRef CAS PubMed;
(b) T. Kirschbaum, F. Rominger and M. Mastalerz, Angew. Chem., Int. Ed., 2020, 59, 270 CrossRef CAS PubMed;
(c) J. Urieta-Mora, M. Krug, W. Alex, J. Perles, I. Fernández, A. Molina-Ontoria, D. M. Guldi and N. Martín, J. Am. Chem. Soc., 2020, 142, 4162 CrossRef CAS PubMed;
(d) T. Kirschbaum, F. Rominger and M. Mastalerz, Chem. - Eur. J., 2020, 26, 14560 CrossRef CAS PubMed;
(e) A. Rajca, A. Safronov, S. Rajca and J. Wongsriratanakul, J. Am. Chem. Soc., 2000, 122, 3351 CrossRef CAS.
- M. A. Petrukhina, K. W. Andreini, L. Peng and L. T. Scott, Angew. Chem., Int. Ed., 2004, 43, 5477 CrossRef CAS PubMed.
- Q. Tan, S. Higashibayashi, S. Karanjit and H. Sakurai, Nat. Commun., 2012, 3, 891 CrossRef PubMed.
- T. J. Seiders, K. K. Baldridge, G. H. Grube and J. S. Siegel, J. Am. Chem. Soc., 2001, 123, 517 CrossRef CAS PubMed.
- T. Amaya, K. Mori, H. L. Wu, S. Ishida, J. Nakamura, K. Murata and T. Hirao, Chem. Commun., 2007, 1902 RSC.
-
(a) J. M. Fernández-García, P. Izquierdo-García, M. Buendía, S. Filippone and N. Martín, Chem. Commun., 2022, 58, 2634 RSC;
(b) R. H. Janke, G. Haufe, E.-U. Wurthwein and J. H. Borkent, J. Am. Chem. Soc., 1996, 118, 6031 CrossRef CAS.
-
(a) Y. Nakai, T. Mori and Y. Inoue, J. Phys. Chem. A, 2012, 116, 7372 CrossRef CAS PubMed;
(b) T. Hosokawa, Y. Takahashi, T. Matsushima, S. Watanabe, S. Kikkawa, I. Azumaya, A. Tsurusaki and K. Kamikawa, J. Am. Chem. Soc., 2017, 139, 18512 CrossRef CAS PubMed.
- H. Dang, M. Levitus and M. A. Garcia-Garibay, J. Am. Chem. Soc., 2002, 124, 136 CrossRef CAS PubMed.
- S.-K. Lin, L.-L. Shiu, K.-M. Chien, T.-Y. Luh and T.-I. Lin, J. Phys. Chem., 1995, 99, 105 CrossRef CAS.
- T. Ohmae, T. Nishinaga, M. Wu and M. Iyoda, J. Am. Chem. Soc., 2010, 132, 1066 CrossRef CAS PubMed.
-
(a) D. Canevet, E. M. Pérez and N. Martín, Angew. Chem., Int. Ed., 2011, 50, 9248 CrossRef CAS PubMed;
(b) L. Sun, W. Zhu, X. Zhang, L. Li, H. Dong and W. Hu, J. Am. Chem. Soc., 2021, 143, 19243 CrossRef CAS PubMed.
- P. Thordarson, Chem. Soc. Rev., 2011, 40, 1305 RSC.
-
(a) A. Sygula, F. R. Fronczek, R. Sygula, P. W. Rabideau and M. M. Olmstead, J. Am. Chem. Soc., 2007, 129, 3842 CrossRef CAS PubMed;
(b) A. S. Filatov, M. V. Ferguson, S. N. Spisak, B. Li, C. F. Campana and M. A. Petrukhina, Cryst. Growth Des., 2014, 14, 756 CrossRef CAS;
(c) L. N. Dawe, T. A. AlHujran, H.-A. Tran, J. I. Mercer, E. A. Jackson, L. T. Scott and P. E. Georghiou, Chem. Commun., 2012, 48, 5563 RSC.
- Q. Tang, Y. Tong, H. Li, Z. Ji, L. Li, W. Hu, Y. Liu and D. Zhu, Adv. Mater., 2008, 20, 1511 CrossRef CAS.
-
(a) J. Zhang, J. Tan, Z. Ma, W. Xu, G. Zhao, H. Geng, C. Di, W. Hu, Z. Shuai, K. Singh and D. Zhu, J. Am. Chem. Soc., 2013, 135, 558 CrossRef CAS PubMed;
(b) X. Xu, T. Xiao, X. Gu, X. Yang, S. V. Kershaw, N. Zhao, J. Xu and Q. Miao, ACS Appl. Mater. Interfaces, 2015, 7, 28019 CrossRef CAS PubMed;
(c) H. T. Black and D. F. Perepichka, Angew. Chem., Int. Ed., 2014, 53, 2138 CrossRef CAS PubMed;
(d) S. K. Park, S. Varghese, J. H. Kim, S.-J. Yoon, O. K. Kwon, B.-K. An, J. Gierschner and S. Y. Park, J. Am. Chem. Soc., 2013, 135, 4757 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed synthesis and characterization of the compounds; NMR spectra and HRMS spectra of the compounds; crystal data for 5, 7, 8, 9, and 9·C60; UV and CV measurements; NMR titration measurements; VT-NMR studies; chiral separation and analysis; theoretical calculation details; device fabrication and characterization. CCDC 2205342 (for 5), 2205221 (for 7), 2208030 (for 8), 2208032 (for 9), and 2234896 (9·C60). For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3sc00658a |
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence b,
Hiroko
Yamada
b,
Hiroko
Yamada