
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceVisible-light-induced [3+2] cycloadditions of donor/donor diazo intermediates with alkenes to achieve (spiro)-pyrazolines and pyrazoles†
Yu
Zhang‡
 a,
Yanchuan
Li‡
bc,
Shao-Fei
Ni‡
d,
Jin-Peng
Li
d,
Dingding
Xia
a,
Xinyu
Han
ac,
Jingchuan
Lin
ac,
Jinxin
Wang
*c,
Shoubhik
Das
*ef and
Wei-Dong
Zhang
*abcg
a,
Yanchuan
Li‡
bc,
Shao-Fei
Ni‡
d,
Jin-Peng
Li
d,
Dingding
Xia
a,
Xinyu
Han
ac,
Jingchuan
Lin
ac,
Jinxin
Wang
*c,
Shoubhik
Das
*ef and
Wei-Dong
Zhang
*abcg
aShanghai Frontiers Science Center for Chinese Medicine Chemical Biology, Institute of Interdisciplinary Integrative Medicine Research, Shanghai University of Traditional Chinese Medicine, No. 1200, Cailun Road, Shanghai 201203, China. E-mail: wdzhangy@hotmail.com
bSchool of Pharmaceutical Sciences, Zhejiang Chinese Medical University, Hangzhou 310053, China
cSchool of Pharmacy, Second Military Medical University, Shanghai 200433, China. E-mail: jxwang2013@126.com
dDepartment of Chemistry, Key Laboratory for Preparation and Application of Ordered Structural Materials of Guangdong Province, Shantou University, Shantou 515063, China
eDepartment of Chemistry, University of Antwerp, Antwerp, Belgium. E-mail: Shoubhik.Das@uni-bayreuth.de
fDepartment of Chemistry, University of Bayreuth, Bayreuth, Germany
gInstitute of Medicinal Plant Development, Chinese Academy of Medical Sciences & Peking Union Medical College, Beijing 100193, China
First published on 8th September 2023
Abstract
To date, [3 + 2] cycloadditions of diazo esters with alkynes or alkenes have been a robust tool to generate pyrazoles and pyrazolines. However, methods capable of generating donor/donor diazo species from readily available N-tosylhydrazones to furnish [3 + 2] cycloadditions, remain elusive. Herein, we describe the first visible-light-induced [3 + 2] cycloadditions of donor/donor diazo precursors with alkenes to afford pyrazoles and novel (spiro)pyrazolines bearing a quaternary center. This protocol shows a tolerable substrate scope covering versatile carbonyl compounds and alkenes. Late-stage functionalization of bioactive molecules, one-pot approach, and gram-scale synthesis have also been introduced successfully to prove the practicability. At last, mechanistic experiments and DFT studies suggested the formation of non-covalent interactions enabling the activation of N-tosylhydrazones and the formation of the donor/donor diazo intermediates.
Introduction
Pyrazoles and their partially saturated counterparts pyrazolines have been frequently used in modern drug discovery for anti-bacterial, antineoplastic, anti-inflammatory, and anticonvulsant activities.1,2 In particular, pyrazoles appear in 14 of the top 200 highest-grossing pharmaceutical products in 2021 such as Eliquis ($16.73 billion) and Imbruvica ($9.78 billion) (Scheme 1a).3 Generally, pyrazolines serve as potent cannabinoid CB1 receptor antagonists that are used for the treatment of obesity and related metabolic diseases.4 Pyrazolines are also frequently used for the therapy of cancer and inflammatory diseases.5 Moreover, both of them are ubiquitous skeletons in natural products such as alkaloids, pigments, and vitamins.6 Consequently, the development of facile and sustainable strategies enabling the construction of pyrazolines and pyrazoles is in high demand.7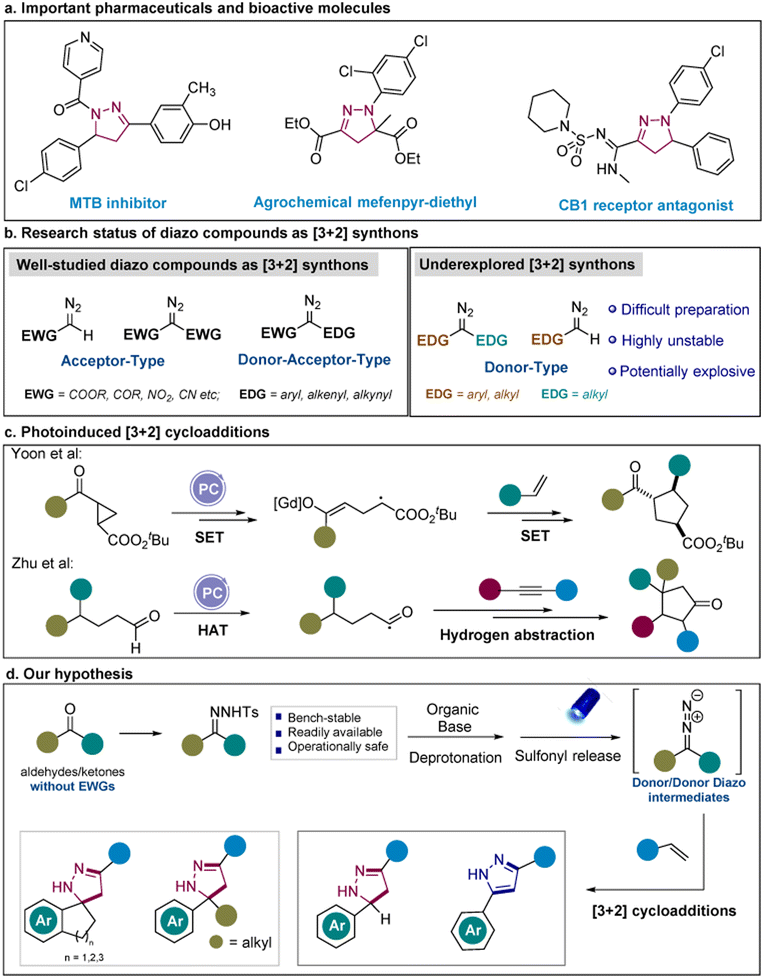 | ||
| Scheme 1 Visible-light mediated [3 + 2] cycloadditions and our design. | ||
To date, [3 + 2] cycloadditions have been the most general approaches to construct 5-membered heterocycles.8 Among them, [3 + 2] cycloadditions of diazo esters or other α-stabilized diazo compounds with alkynes or alkenes provided robust tools for the generation of pyrazoles and pyrazolines.9–13 Nevertheless, the application of diazo compounds has been hampered significantly by their abnormal reactivities and hazardous properties14 since diazo compounds in most of the studies require at least one electron-withdrawing group (typically an ester group) to stabilize (Scheme 1b).11,15,16 To extend the scope of diazo compounds and achieve structural diversity, precursors of diazo compounds such as N-tosylhydrazones and N-sulfonyl-1,2,3-triazoles have been introduced to form corresponding diazo intermediates.17–20 For instance, the Bamford–Stevens reaction, the Shapiro reaction and other approaches that rely on deprotonation of N-tosylhydrazones to generate diazo compounds have been reported,21–24 which generally require thermal or oxidative conditions. Moreover, cycloadditions of arynes and N-tosylhydrazones have been disclosed by the group of Shi.25 When compared with the use of limited diazo compounds or hydrazonoyl chlorides,26,27 this work features the application of in situ formed donor-type diazo compounds to afford indazoles with structural diversity.
Synthetic methods mediated by visible-light activation such as relying on photocatalysts (photosensitizers)28–34 and EDA complex35–45 have enabled untapped transformations and diverted the previously harsh reaction conditions. Inspired by these strategies, we focused on designing of visible-light mediated [3 + 2] cycloaddition reactions to synthesize heterocyclic compounds via the formation of new C–N bonds. While [2 + 2] and [4 + 2] cycloaddition reactions have been widely applied in photoredox catalysis, the application of [3 + 2] cycloadditions to build five-membered rings has been scarcely investigated.46–48 To the best of our knowledge, only a few research articles have reported the use of photocatalytic [3 + 2] cycloadditions to synthesize five-membered cyclopentane rings via the generation of new C–C bonds.49–54 Up to now, the presence of an expensive transition metal-based photocatalyst has been essential for achieving these transformations (Scheme 1c). Notably, the group of König disclosed the formation of donor/donor diazo compounds from N-tosylhydrazones to facilitate the homologation of carbonyl compounds under UV light.55 Very recently, our group also reported the formation of a donor/donor carbene from the N-tosylhydrazones which was induced by the photosensitizer.56 In this case, we particularly wanted to focus on the in situ generation of underexplored donor/donor diazo compounds under visible-light irradiation to demonstrate the wide application of our strategy (Scheme 1d).
Results and discussion
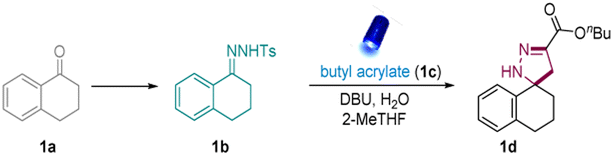
At the beginning of our investigation, a readily available N-tosylhydrazone (1b), conveniently obtained from ketone 1a, was reacted with butyl acrylate (1c) in the presence of different organic bases and inorganic bases such as 5-diazabicyclo(4.3.0)non-5-ene (DBN), 1,5,7-triazabicyclo-[4,4,0]dec-5-ene (TBD), and 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) (Table S2†) (Table 1, entries 1–4). To our delight, after the careful investigation and evaluation of bases, solvents, and quantities of 1c, the desired spiropyrazoline (1d) was obtained in a 90% isolated yield in the presence of 3.0 equivalents of water (Table 1, entry 1). Moreover, an investigation of different solvents demonstrated that 2-MeTHF, a biomass-derived solvent that is widely used in organic chemistry,57 was the most suitable solvent (Table 1, entries 5 and 6). The reaction was also performed in the absence of water. Unsurprisingly, less product was obtained without water (Table 1, entry 7). To investigate the role of water in the system, we replace it with phase transfer catalyst (PTC) and it showed better reactivity compared the reaction without the addition of water (Table 1, entry 9). Therefore, we postulated that 2-Me-THF has a good miscibility with water and this facilitates water probably facilitated the dissolution of N-tosylhydrazone anion, which enhanced the [3 + 2] cycloaddition. Finally, the control experiments exhibited no product formation in the absence of light or base, emphasizing the required presence of light and base in this system (Table 1, entries 9 and 10).|
|
||
|---|---|---|
| Entry | Change from standard conditions | Yieldb [%] |
| a Reaction conditions: 1b (0.2 mmol), butyl acrylate (0.3 mmol, 1.5 equiv.), base (0.6 mmol, 3.0 equiv.), H2O (0.6 mmol, 3.0 equiv.), solvent (1.0 mL), irradiation with 40 W blue Kessil lamp (λ = 456 nm), r.t., argon atmosphere, 16 h., n.d. = not detected. b Yields were determined by 1H NMR analysis of the crude reaction mixture using 1,3,5-trimethoxybenzene as the internal standard. c PTC: tetrabutylammonium bromide. | ||
| 1 | None | 90 |
| 2 | DBN instead of DBU | 76 |
| 3 | Cs2CO3 instead of DBU | n.d. |
| 4 | Et3N instead of DBU | n.d. |
| 5 | THF instead of 2-MeTHF | 89 |
| 6 | MeCN instead of 2-MeTHF | 50 |
| 7 | Without H2O | 76 |
| 8 | 3.0 equiv. PTC instead of H2O | 82c |
| 9 | In the dark | n.d. |
| 10 | Without base | n.d. |
With the best reaction conditions in hand, we investigated the scope and generality of [3 + 2] cycloaddition reactions between various N-tosylhydrazones and alkenes. First, diverse N-tosylhydrazones from substituted benzocyclohexanones were evaluated. Gratifyingly, both the EWG-substituted and EDG-substituted N-tosylhydrazones exhibited good to excellent reactivities to provide functionalized spiropyrazolines (Scheme 2, 2d–7d). Next, heteroaromatic cyclic ketones containing 6,7-dihydro-4-benzothiophenone (8a), boc-protected 1,5,6,7-tetrahydro-4H-indol-4-one (9a), and 7,8-dihydroquinolin-5(6H)-one (10a) also successfully underwent [3 + 2] cycloadditions to provide the desired spiropyrazolines (8d–10d). Moreover, 4-chromanone (11a) and 2-methyl-1-tetralone (12a) were also applied in this transformation. After the successful exploration of heteroaromatic cyclic ketones with six-member rings, we aimed to extend the scope to other heteroaromatic cyclic ketones, including five and seven-member rings. Thus, N-tosylhydrazones from 1-indanone and its derivatives were successfully converted into spiropyrazolines in medium to good yields (13d–17d). Moreover, 1-acenaphthenone also worked well to form the corresponding pyrazoline in excellent yield (18d). Inspired by these results, N-tosylhydrazone from benzocycloheptanone was investigated and exhibited good reactivity under the standard conditions (19d). Acyclic ketones were also investigated and to our delight, a diverse array of substituted acetophenones with methoxy-, dimethylamino-, phenoxy-, ester-, methylsulfonyl-, chloro-, dichloro-, and phenyl groups were feasibly synthesized with good to excellent yields (20d–35d). Significantly, diverse heteroaryl alkyl ketones containing benzofuran, indole, benzothiophene, benzo[d]oxazole, pyridine, and thiophene moieties were successfully converted into the corresponding pyrazoline derivatives (36d–44d). In addition to acetophenones, the use of other aryl alkyl ketones with different chain lengths or branched chains also proceeded smoothly (45d–52d). Furthermore, we aimed to extend the scope of ketones bearing diverse groups instead of general aryl or alkyl groups. Among them, the ketones with an ester group located at the β- and γ-site of the ketone or phenoxy group located at the β-site proceeded excellently to generate the desired pyrazolines with quaternary centers (52d–54d). Moreover, N-tosylhydrazones derived from aldehydes (55d, 56d) or butyl pyruvate (57d) were also appropriate substrates.
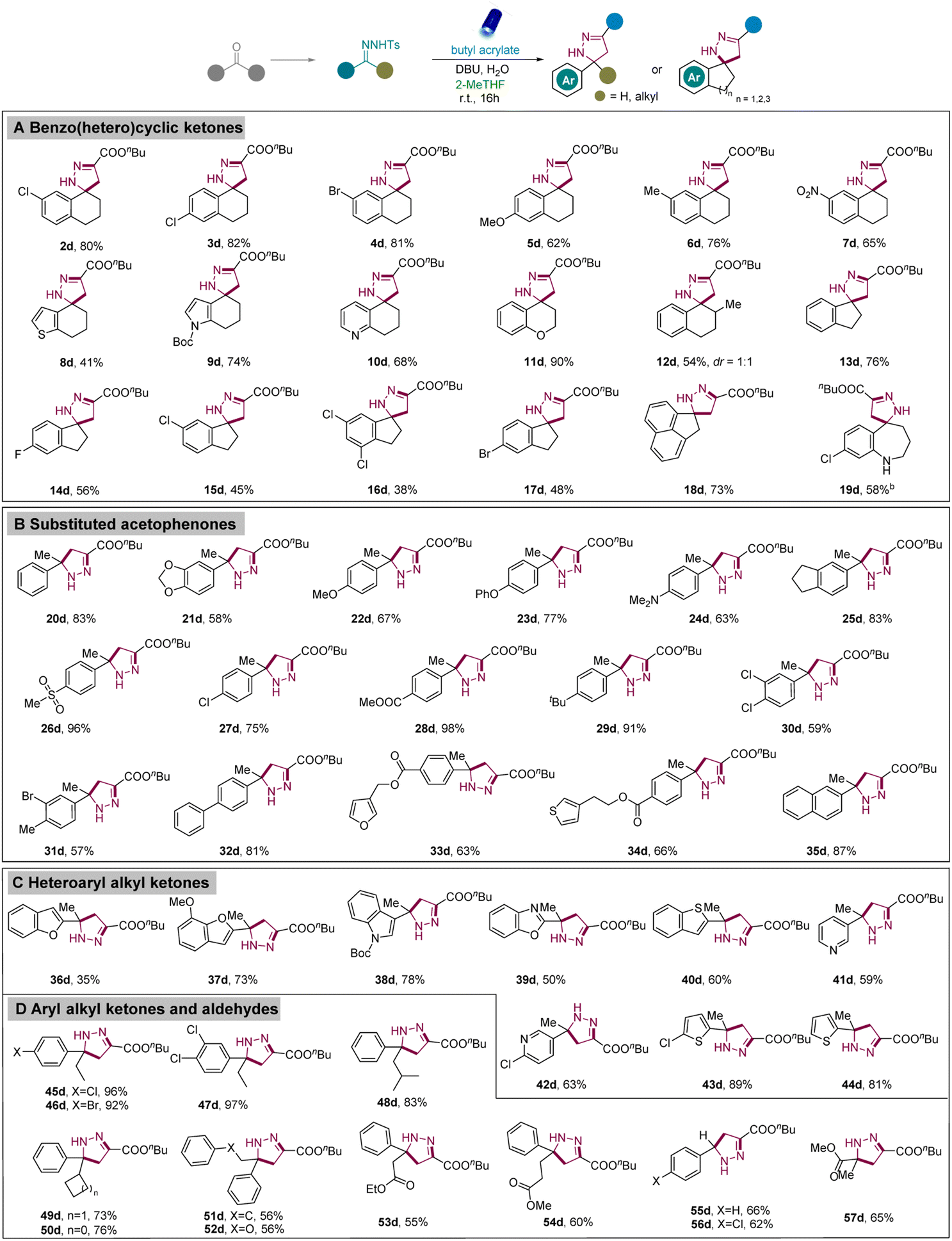 | ||
| Scheme 2 Scope of N-tosylhydrazones derived from diverse ketones to obtain pyrazolines.a,b Reaction conditions: aN-tosylhydrazones (0.2 mmol), butyl acrylate (0.3 mmol, 1.5 equiv.), base (0.6 mmol, 3.0 equiv.), H2O (0.6 mmol, 3.0 equiv.), 2-MeTHF (1.0 mL), irradiation with 40 W Kessil lamp (λ = 456 nm), r.t., argon atmosphere, 16 h; b40 h. | ||
Next, the amenability of this mild strategy toward alkenes was assessed (Scheme 3). The reaction with various acrylates including diverse and potentially reactive functional groups such as tertbutyl (58d), allyl (59d), alkynyl (60d), 1-methyl-cyclopentyl (61d), 2-methoxyethyl (62d), and methyl (63d) groups furnished the corresponding products in good to high yields. Additional functional groups such as furyl (64d), benzoate (65d), and pyridyl (66d) were also well-tolerated. Moreover, diverse acryl amides bearing different functional groups (67d–72d) such as pyridyl, benzyl, morpholine, piperidyl, and phenyl groups were also successfully converted into the desired products. Remarkably, α,β-unsaturated-enone (73d) was compatible with our system to afford the corresponding pyrazoline, albeit in a lower yield. The generality of alkenes was further evaluated by using vinylphosphonate (74d), which exhibited excellent reactivity. Nevertheless, lower reactivity was revealed when acrylonitrile (75d) was used, and only a trace amount of product was observed when methyl vinyl sulfone (75d) was applied.
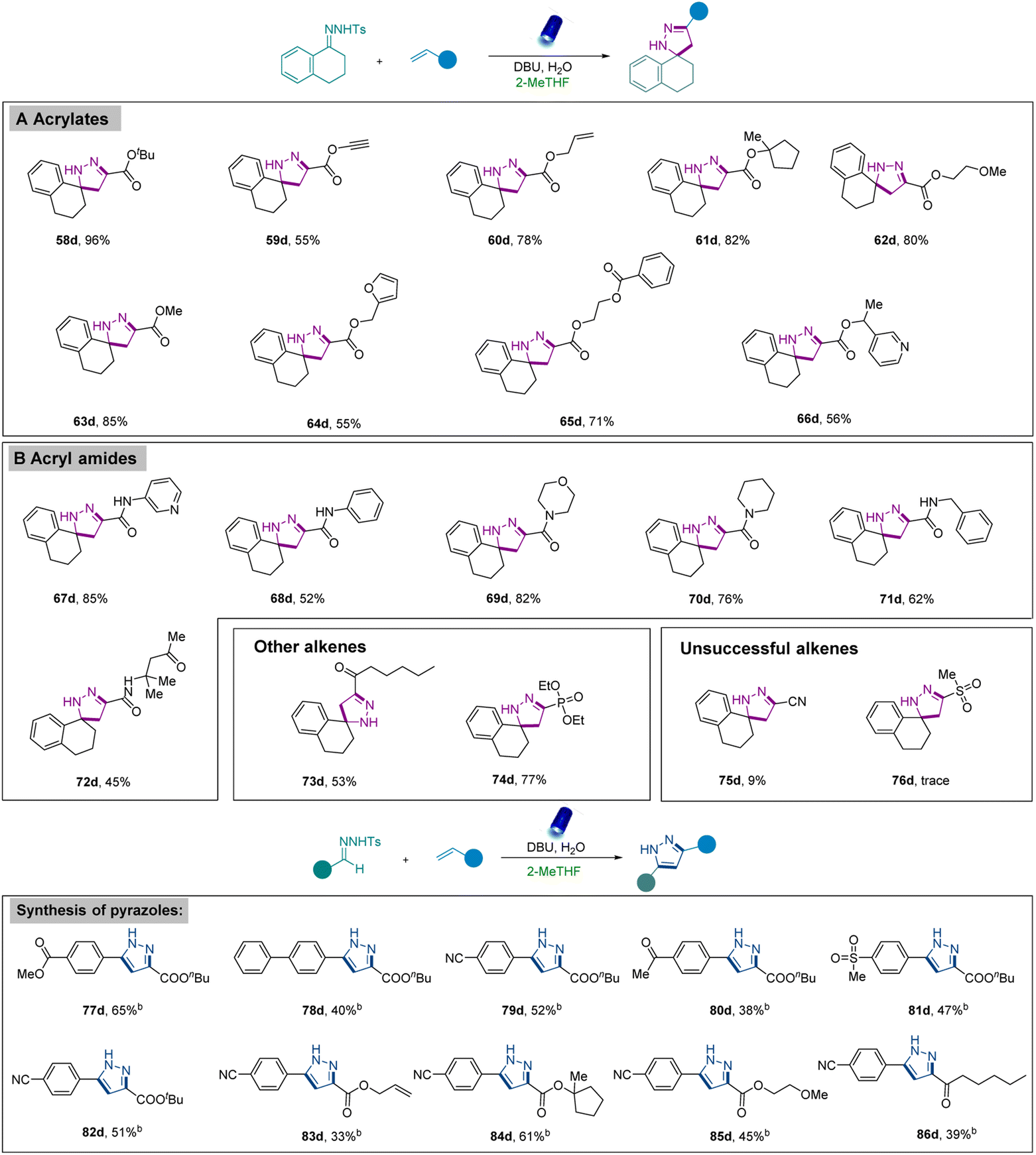 | ||
| Scheme 3 Scope of different acrylates to obtain pyrazolines and synthesis of pyrazoles using different N-tosylhydrazones and acrylates.a,b Reaction conditions: aN-tosylhydrazones (0.2 mmol), alkenes (0.3 mmol, 1.5 equiv.), base (0.6 mmol, 3.0 equiv.), H2O (0.6 mmol, 3.0 equiv.), 2-MeTHF (1.0 mL), irradiation with 40 W Kessil lamp (λ = 456 nm), r.t., argon atmosphere, 16 h; bbase (1.0 mmol, 5.0 equiv.). | ||
Inspired by these results, the synthesis of pyrazoles was also investigated using our strategy. It was observed that the N-tosylhydrazones obtained from aldehydes provided a mixture of pyrazoles and pyrazolines, and we argued that the elimination reaction from the in situ formed pyrazolines could generate pyrazoles at the end. However, only limited product was observed under standard reaction conditions (Scheme 2, 55d, 56d). Therefore, reaction conditions were slightly modified by adding additional bases or additional oxidants to facilitate further elimination of pyrazolines and afford the desired pyrazoles (Table S5 in the ESI†). Encouragingly, careful investigations identified that higher amounts of bases (5 equivalents) enabled the formation of pyrazoles in good yields. Furthermore, this revised protocol was also employed to synthesize pyrazoles using diverse N-tosylhydrazones. Among them, N-tosylhydrazones bearing -ester, -phenyl, -cyano, -acetyl, and -methyl sulfonyl groups (77d–82d) at the para position were converted to desirable pyrazoles in medium to good yields. Furthermore, in addition to butyl acrylate, acrylates bearing various functional groups such as allyl (83d) and 1-methyl-cyclopentyl (84d) were also suitably converted into the corresponding pyrazoles. The [3 + 2] cycloaddition reaction was also successfully performed using α,β-unsaturated enone (86d).
Considering the prevalence and importance of pyrazolines and pyrazoles in the pharmaceutical industry, we aimed to demonstrate the application of our protocol via the functionalization of existing natural products and drug-like molecules (Scheme 4). Flavanone and related derivatives (87d, 88d) are highly valuable natural products with several significant pharmacological properties such as anticancer, antioxidant, and antiviral activities. These compounds were successfully converted into the corresponding spiropyrazoline analogues. Importantly, this transformation has not yet been reported, indicating the significance of our approach in the field of superior drug development and/or discovery. Next, our attention turned to the investigation of an array of N-tosylhydrazones derived from a wide range of bioactive natural products such as dehydroabietic acid, celestolide, and L-menthol (89d–91d). These products were also converted into the desired (spiro)pyrazolines successfully. Moreover, various complex acrylates derived from abiraterone, a drug used for treating prostate cancer, and several natural products such as diacetonefructose and nerol were also suitable substrates for the preparation of pyrazolines (92d–97d). Pyrazole-substituted complex molecules were also generated with our approach (98d–100d). To further explore the practicality of this protocol, gram-scale synthesis was smoothly performed to obtain the desired pyrazolines. Meanwhile, a one-pot approach starting from the respective ketone directly and furnished the pyrazoline, clearly demonstrating that the reaction could proceed even from the aldehydes/ketones rather than the preformation of N-tosylhydrazones.
 | ||
| Scheme 4 Modification of complex molecules, gram-scale synthesis, and one-pot approach.a–c Reaction conditions: aN-tosylhydrazones (0.2 mmol), alkenes (0.3 mmol, 1.5 equiv.), base (0.6 mmol, 3.0 equiv.), H2O (0.6 mmol, 3.0 equiv.), 2-MeTHF (1.0 mL), irradiation with 40 W Kessil lamp (λ = 456 nm), r.t., argon atmosphere, 16 h; bbase (1.0 mmol, 5.0 equiv.); cfurther details of the gram-scale synthesis and one-pot synthesis procedures are provided in the ESI.† | ||
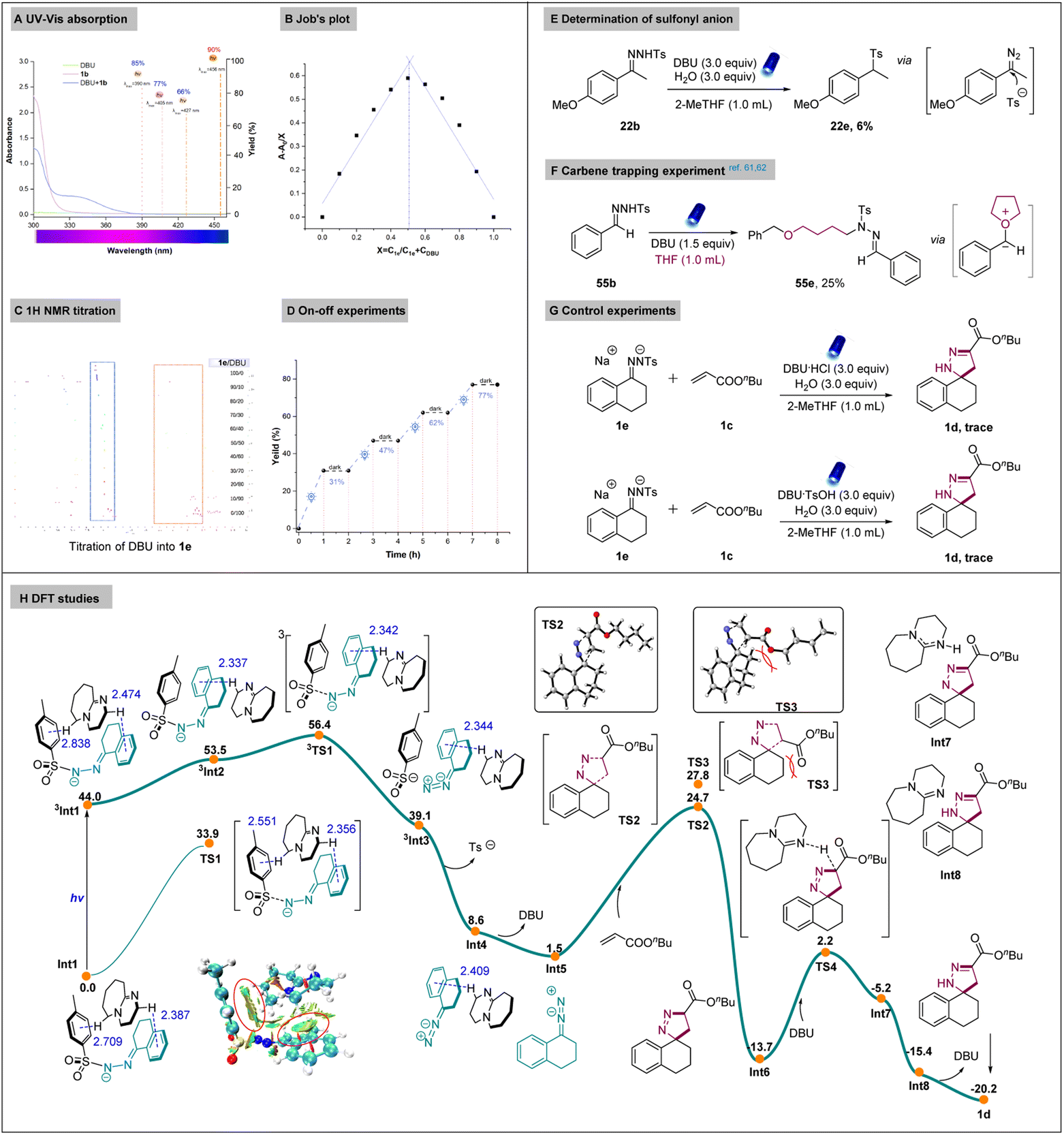
We further aimed to gather more mechanistic information of our reaction (Scheme 5). First, ultraviolet/visible (UV/Vis) absorption spectra were obtained and a clear red (bathochromic) shift was observed between DBU and the N-tosylhydrazone anion, with a visible-light absorption tailing at 400–460 nm. This implied the formation of the non-covalent interaction. Examining the influence of different wavelengths revealed that the best reactivity was obtained under the irradiation of light with a wavelength of 456 nm (Scheme 5A). Furthermore, the Job's plot of the complex was investigated by the UV-vis absorption spectrometry (details in the ESI†). The maximal absorption was observed when the ratio of N-tosylhydrazone anion and DBU in THF was 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, indicating the existence of the non-covalent interaction from N-tosylhydrazone anion 1e and DBU (Scheme 5B). We further performed NMR titration experiments to investigate the interactions of N-tosylhydrazone anion 1e and DBU. An upfield shift of 1e was observed with increasing DBU concentration, providing further evidence that the complexation occurred between 1e and DBU (Scheme 5C). On-off experiments were also carried out to demonstrate the necessity of the light in our system and the stability of the system under darkness, demonstrating its potential for practical use (Scheme 5D). Later, we performed the reaction under standard conditions without alkenes and sulfonylation of N-tosylhydrazone was observed, it is known that diazo intermediate enable the nucleophilic addition with diverse nucleophiles such as alcohols, boronic acids and sodium azides.58–60 Therefore, we assumed that sulfonyl anion is existed in our system (Scheme 5E). It is also known that diazo compounds can generate oxonium ylide with oxetane and 2,5-dihydrofurans via a carbene intermediate.61,62 To verify this, a carbene-trapping experiment was carried out with THF, and product 55e was obtained via the formation of an oxonium ylide intermediate (Scheme 5F). Afterward, we evaluated the reactivity of 1e and ammonium salts of DBU under standard conditions. However, no products were formed, indicating the necessity of the base (Scheme 5G).
:1, indicating the existence of the non-covalent interaction from N-tosylhydrazone anion 1e and DBU (Scheme 5B). We further performed NMR titration experiments to investigate the interactions of N-tosylhydrazone anion 1e and DBU. An upfield shift of 1e was observed with increasing DBU concentration, providing further evidence that the complexation occurred between 1e and DBU (Scheme 5C). On-off experiments were also carried out to demonstrate the necessity of the light in our system and the stability of the system under darkness, demonstrating its potential for practical use (Scheme 5D). Later, we performed the reaction under standard conditions without alkenes and sulfonylation of N-tosylhydrazone was observed, it is known that diazo intermediate enable the nucleophilic addition with diverse nucleophiles such as alcohols, boronic acids and sodium azides.58–60 Therefore, we assumed that sulfonyl anion is existed in our system (Scheme 5E). It is also known that diazo compounds can generate oxonium ylide with oxetane and 2,5-dihydrofurans via a carbene intermediate.61,62 To verify this, a carbene-trapping experiment was carried out with THF, and product 55e was obtained via the formation of an oxonium ylide intermediate (Scheme 5F). Afterward, we evaluated the reactivity of 1e and ammonium salts of DBU under standard conditions. However, no products were formed, indicating the necessity of the base (Scheme 5G).
 | ||
| Scheme 5 Mechanistic investigations of visible light-mediated [3 + 2] cycloadditions. | ||
To further exclude whether the non-covalent interaction was formed from 1e and different ammonium salts. We performed the UV-Vis spectra measurement and no red shift was observed in the mixture of the two species (Fig. S3†). However, the obvious red shift was observed from the combination of 1e and DBU (Fig. S5†). DFT calculation was carried out to provide deep understanding of the reaction mechanism. As shown in Scheme 5H, non-covalent adduct Int1 between deprotonated N-tosylhydrazone anion and DBU could be formed. In Int1, two C–H⋯π interactions are observed, which is confirmed by the NCI (non-covalent interaction) analysis. Under irradiation, the excited state 3Int1 is formed, from where the homolytic N–S bond cleavage takes place via transition state 3TS1 to afford intermediate 3Int3. The Gibbs free energy needs for this process is 12.4 kcal mol−1, lower than the 33.9 kcal mol−1 for the transition state TS1 in singlet state. With the release of sulfonyl anion and DBU, Int5 is generated, from which the [3 + 2] cycloaddition with alkene take place via two transition states with regioselectivity. Structure analysis indicated big steric hindrance between the substituent of the alkene and Int5 in the unfavourable transition state TS3, which is 3.1 kcal mol−1 higher than TS2. Energy decomposition analysis also indicates that the regioselectivity mainly come from the structure distortion of the Int5. The linear C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) NN structure in Int5 bended to 143.9° in TS3 due to the large steric hindrance, compared to 146.5° in TS4 (see Fig. S8 in ESI†). From Int6, the stepwise proton transfer with the help of DBU will drive the reaction to the final spiropyrazoline product. Other mechanism for the proton transfer process were also considered, all of them have higher reaction barriers (see Fig. S9†), which are not competitive compared with the stepwise proton transfer process.
NN structure in Int5 bended to 143.9° in TS3 due to the large steric hindrance, compared to 146.5° in TS4 (see Fig. S8 in ESI†). From Int6, the stepwise proton transfer with the help of DBU will drive the reaction to the final spiropyrazoline product. Other mechanism for the proton transfer process were also considered, all of them have higher reaction barriers (see Fig. S9†), which are not competitive compared with the stepwise proton transfer process.
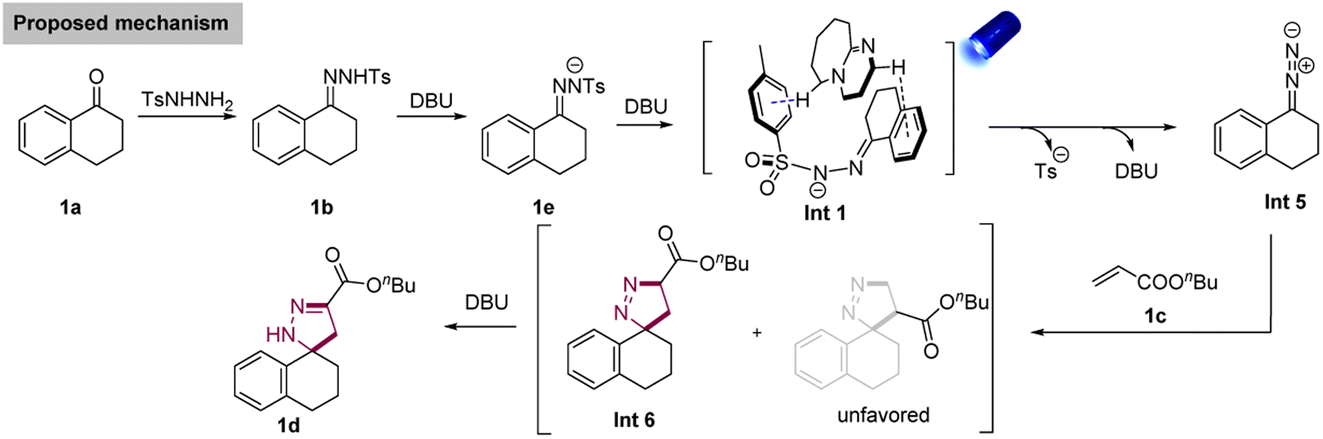
Based on these mechanistic investigations, a proposed mechanism is provided in Scheme 6. First, N-tosylhydrazone 1b was afforded by the condensation reaction between a ketone (1a) and 4-methylbenzenesulfonohydrazide. Next, 1b was deprotonated to afford the corresponding N-tosylhydrazone anion (1e). A non-covalent interaction Int 1 generated between 1e and DBU, and Int 1 was irradiated by the blue light (λ = 456 nm) to afford the Int 5via the homolytic N–S bond cleavage and the release of DBU. Subsequently, the in situ formed donor/donor diazo species further underwent [3 + 2] cycloaddition with alkene 1c, affording the desired spiropyrazoline 1d.
 | ||
| Scheme 6 Proposed mechanism of visible light-mediated [3 + 2] cycloadditions. | ||
Conclusions
In summary, we described the visible light-mediated [3 + 2] cycloaddition reactions to afford pyrazoles and (spiro)pyrazolines bearing a quaternary carbon center. This strategy provides a novel and benign approach to access donor/donor diazo species from the corresponding N-tosylhydrazones in situ and strongly extends the scope of visible-light mediated [3 + 2] cycloadditions. This protocol demonstrates tolerance for a broad range of functional groups and can be used to transform a wide range of bioactive compounds into functionalized pyrazoles and pyrazolines.Data availability
The data supporting this manuscript is available in the ESI† of and available on request.Author contributions
Y. Z. and S. D. conceived the project. Y. Z. and Y. L. performed the experiments and analysed the data. D. X. reproduced part of substrate scope. S. N. and J. L. performed the DFT calculation. Y. L., X. H. and C. L. prepared the synthesized starting materials and ESI.† Y. Z., S. D., J. W. and W.-D. Z. wrote the manuscript. All authors have given approval to the final version of the manuscript.Conflicts of interest
A Chinese Patent on this work has been applied with the number (2023101291361) on 17 February 2023. The remaining authors declare no competing interests.Acknowledgements
This work was supported by the Chenguang Program of Shanghai Education Development Foundation and Shanghai Municipal Education Commission (22CGA51, Y. Z.), National Natural Science Foundation of China (82141203, 82003624, W. Z.), the National Key Research and Development Program of China (2022YFC3502000, W. Z.), Shanghai Municipal Science and Technology Major Project (ZD2021CY001, W. Z.), Three-year Action Plan for Shanghai TCM Development and Inheritance Program [ZY(2021–2023)-0401, W. Z. ], Innovation Team and Talents Cultivation Program of National Administration of Traditional Chinese Medicine (ZYYCXTDD-202004, W. Z.). We are also grateful to Dr Lu Lu for carrying out the NMR experiments. S.-F. N. acknowledge funding from the STU Scientific Research Foundation for Talents (NTF20022).Notes and references
- E. Vitaku, D. T. Smith and J. T. Njardarson, J. Med. Chem., 2014, 57, 10257–10274 CrossRef CAS PubMed.
- A. P. Taylor, R. P. Robinson, Y. M. Fobian, D. C. Blakemore, L. H. Jones and O. Fadeyi, Org. Biomol. Chem., 2016, 14, 6611–6637 RSC.
- N. A. McGrath, M. Brichacek and J. T. Njardarson, J. Chem. Educ., 2010, 87, 1348–1349 CrossRef CAS.
- M. R. Shaaban, A. S. Mayhoub and A. M. Farag, Expert Opin. Ther. Pat., 2012, 22, 253–291 CrossRef CAS PubMed.
- J. H. Lange, M. A. van der Neut, A. P. den Hartog, H. C. Wals, J. Hoogendoorn, H. H. van Stuivenberg and C. G. Kruse, Bioorg. Med. Chem. Lett., 2010, 20, 1752–1757 CrossRef CAS PubMed.
- V. Kumar, K. Kaur, G. K. Gupta and A. K. Sharma, Eur. J. Med. Chem., 2013, 69, 735–753 CrossRef CAS PubMed.
- M. A. Ali, M. S. Yar, M. Kumar and G. S. Pandian, Nat. Prod. Res., 2007, 21, 575–579 CrossRef CAS PubMed.
- M. Breugst and H. U. Reissig, Angew. Chem., Int. Ed., 2020, 59, 12293–12307 CrossRef CAS PubMed.
- F. Zhang, L. Xin, Y. Yu, S. Liao and X. Huang, Synthesis, 2021, 53, 238–254 CrossRef CAS.
- X. Li, C. Golz and M. Alcarazo, Angew. Chem., Int. Ed., 2021, 60, 6943–6948 CrossRef CAS PubMed.
- Y. L. Su, K. Dong, H. Zheng and M. P. Doyle, Angew. Chem., Int. Ed., 2021, 60, 18484–18488 CrossRef CAS PubMed.
- Z. Chen, Y. Zheng and J. A. Ma, Angew. Chem., Int. Ed., 2017, 56, 4569–4574 CrossRef CAS PubMed.
- K. J. Hock, L. Mertens, F. K. Metze, C. Schmittmann and R. M. Koenigs, Green Chem., 2017, 19, 905–909 RSC.
- J. R. Fulton, V. K. Aggarwal and J. de Vicente, Eur. J. Org Chem., 2005, 2005, 1479–1492 CrossRef.
- Z. Yang, M. L. Stivanin, I. D. Jurberg and R. M. Koenigs, Chem. Soc. Rev., 2020, 49, 6833–6847 RSC.
- P. Zhao, Z. Li, J. He, X. Liu and X. Feng, Sci. China: Chem., 2021, 64, 1355–1360 CrossRef CAS.
- M. Akter, K. Rupa and P. Anbarasan, Chem. Rev., 2022, 122, 13108–13205 CrossRef CAS PubMed.
- Y. Xia and J. Wang, Chem. Soc. Rev., 2017, 46, 2306–2362 RSC.
- Y. He, Z. Huang, K. Wu, J. Ma, Y. G. Zhou and Z. Yu, Chem. Soc. Rev., 2022, 51, 2759–2852 RSC.
- M. Linden, S. Hofmann, A. Herman, N. Ehler, R. M. Bär and S. R. Waldvogel, Angew. Chem., Int. Ed., 2023, 62, e202214820 CrossRef CAS PubMed.
- M. Ghavre, Asian J. Org. Chem., 2020, 9, 1901–1923 CrossRef CAS.
- Q. Xiao, Y. Zhang and J. Wang, Acc. Chem. Res., 2013, 46, 236–247 CrossRef CAS PubMed.
- D. Arunprasath, B. Devi Bala and G. Sekar, Adv. Synth. Catal., 2019, 361, 1172–1207 CrossRef CAS.
- M. Tang, W. Zhang and Y. Kong, Org. Biomol. Chem., 2013, 11, 6250–6254 RSC.
- P. Li, J. Zhao, C. Wu, R. C. Larock and F. Shi, Org. Lett., 2011, 13, 3340–3343 CrossRef CAS PubMed.
- T. Jin and Y. Yamamoto, Angew. Chem., Int. Ed., 2007, 46, 3323–3325 CrossRef CAS PubMed.
- C. Spiteri, S. Keeling and J. E. Moses, Org. Lett., 2010, 12, 3368–3371 CrossRef CAS PubMed.
- G. Tan, M. Das, R. Kleinmans, F. Katzenburg, C. Daniliuc and F. Glorius, Nat. Catal., 2022, 5, 1120–1130 CrossRef CAS.
- A. Dewanji, L. van Dalsen, J. A. Rossi-Ashton, E. Gasson, G. E. Crisenza and D. J. Procter, Nat. Catal., 2023, 15, 43–52 CAS.
- J. Xuan and W. J. Xiao, Angew. Chem., Int. Ed., 2012, 51, 6828–6838 CrossRef CAS PubMed.
- K. L. Skubi, J. B. Kidd, H. Jung, I. A. Guzei, M. H. Baik and T. P. Yoon, J. Am. Chem. Soc., 2017, 139, 17186–17192 CrossRef CAS PubMed.
- C. K. Prier, D. A. Rankic and D. W. MacMillan, Chem. Rev., 2013, 113, 5322–5363 CrossRef CAS PubMed.
- N. L. Reed and T. P. Yoon, Chem. Soc. Rev., 2021, 50, 2954–2967 RSC.
- N. A. Romero and D. A. Nicewicz, Chem. Rev., 2016, 116, 10075–10166 CrossRef CAS PubMed.
- Z. Y. Cao, T. Ghosh and P. Melchiorre, Nat. Chem., 2018, 9, 3274 Search PubMed.
- G. E. Crisenza, D. Mazzarella and P. Melchiorre, J. Am. Chem. Soc., 2020, 142, 5461–5476 CrossRef CAS PubMed.
- T. P. Yoon, M. A. Ischay and J. Du, Nat. Chem., 2010, 2, 527–532 CrossRef CAS PubMed.
- I. Bosque and T. Bach, ACS Catal., 2019, 9, 9103−9109 Search PubMed.
- J. D. Lasso, D. J. Castillo-Pazos, M. Sim, J. Barroso-Flores and C. J. Li, Chem. Sci., 2023, 14, 525–532 RSC.
- M. J. Cabrera-Afonso, A. Granados and G. A. Molander, Angew. Chem., Int. Ed., 2022, 61, e202202706 CrossRef CAS PubMed.
- J. Mateos, F. Rigodanza, P. Costa, M. Natali, A. Vega-Peñaloza, E. Fresch and L. Dell'Amico, Nat. Synth., 2023, 2, 26–36 CrossRef PubMed.
- A. Vega-Penaloza, S. Paria, M. Bonchio, L. Dell'Amico and X. Companyo, ACS Catal., 2023, 9, 6058–6072 CrossRef.
- F. Glaser, C. Kerzig and O. S. Wenger, Angew. Chem., Int. Ed., 2020, 59, 10266–10284 CrossRef CAS PubMed.
- K. Sun, A. Shi, Y. Liu, X. Chen, P. Xiang, X. Wang and B. Yu, Chem. Sci., 2022, 13, 5659–5666 RSC.
- A. Dewanji, L. van Dalsen, J. A. Rossi-Ashton, E. Gasson, G. E. Crisenza and D. J. Procter, Nat. Chem., 2023, 15, 43–52 CrossRef CAS PubMed.
- A. S. Harmata, T. E. Spiller, M. J. Sowden and C. R. Stephenson, J. Am. Chem. Soc., 2021, 143, 21223–21228 CrossRef CAS PubMed.
- D. Sarkar, N. Bera and S. Ghosh, Eur. J. Org Chem., 2020, 2020, 1310–1326 CrossRef CAS.
- M. Rueping, D. Leonori and T. Poisson, Chem. Commun., 2011, 47, 9615–9617 RSC.
- A. Luque, J. Groß, T. J. Zähringer, C. Kerzig and T. Opatz, Chem.–Eur. J., 2022, 28, e202104329 CrossRef CAS PubMed.
- S. Maity, M. Zhu, R. S. Shinabery and N. Zheng, Angew. Chem., Int. Ed., 2012, 51, 222–226 CrossRef CAS PubMed.
- X. Huang, J. Lin, T. Shen, K. Harms, M. Marchini, P. Ceroni and E. Meggers, Angew. Chem., Int. Ed., 2018, 57, 5454–5458 CrossRef CAS PubMed.
- S. Das, C. G. Daniliuc and A. Studer, Org. Lett., 2016, 18, 5576–5579 CrossRef CAS PubMed.
- S. Le, J. Li, J. Feng, Z. Zhang, Y. Bai, Z. Yuan and G. Zhu, Nat. Commun., 2022, 13, 4734 CrossRef CAS PubMed.
- Z. Lu, M. Shen and T. P. Yoon, J. Am. Chem. Soc., 2011, 133, 1162–1164 CrossRef CAS PubMed.
- H. Wang, S. Wang, V. George, G. Llorente and B. König, Angew. Chem., Int. Ed., 2022, 61, e202211578 CrossRef CAS PubMed.
- D. Xia, R. Wu, J. Wang, X. Han, Y. Li, Q. Li, X. Luan, X. Hong, Y. Zhang and W. D. Zhang, ACS Catal., 2023, 13, 9806–9816 CrossRef CAS.
- V. Pace, P. Hoyos, L. Castoldi, P. Domínguez de María and A. R. Alcántara, ChemSusChem, 2012, 5, 1369–1379 CrossRef CAS PubMed.
- J. Barluenga, M. Tomás-Gamasa, F. Aznar and C. Valdés, Angew. Chem., Int. Ed., 2010, 49, 4993–4996 CrossRef CAS PubMed.
- M. C. Pérez-Aguilar and C. Valdés, Angew. Chem., Int. Ed., 2012, 24, 5953–5957 CrossRef PubMed.
- J. Barluenga, M. Tomás-Gamasa and C. Valdés, Angew. Chem., Int. Ed., 2012, 24, 5950–5952 CrossRef PubMed.
- D. Qi, J. Bai, H. Zhang, B. Li, Z. Song, N. Ma and W. Xia, Green Chem., 2022, 24, 5046–5051 RSC.
- S. Jana, Z. Yang, C. Pei, X. Xu and R. M. Koenigs, Chem. Sci., 2019, 10, 10129–10134 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: experimental procedures and details, mechanistic and optimization results and NMR spectra. See DOI: https://doi.org/10.1039/d3sc04188c |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2023 |