
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of biolabile thioalkyl-protected phosphates from an easily accessible phosphotriester precursor†
Lloyd D.
Murphy
 a,
Kathryn E.
Huxley
a,
Ava
Wilding
b,
Cyane
Robinson
b,
Quentin P. O.
Foucart
a and
Lianne I.
Willems
*a
a,
Kathryn E.
Huxley
a,
Ava
Wilding
b,
Cyane
Robinson
b,
Quentin P. O.
Foucart
a and
Lianne I.
Willems
*a
aYork Structural Biology Laboratory and York Biomedical Research Institute, Department of Chemistry, University of York, York YO10 5DD, UK. E-mail: lianne.willems@york.ac.uk
bDepartment of Chemistry, University of York, York YO10 5DD, UK
First published on 19th April 2023
Abstract
Robust methods for the synthesis of mixed phosphotriesters are essential to accelerate the development of novel phosphate-containing bioactive molecules. To enable efficient cellular uptake, phosphate groups are commonly masked with biolabile protecting groups, such as S-acyl-2-thioethyl (SATE) esters, that are removed once the molecule is inside the cell. Typically, bis-SATE-protected phosphates are synthesised through phosphoramidite chemistry. This approach, however, suffers from issues with hazardous reagents and can give unreliable yields, especially when applied to the synthesis of sugar-1-phosphate derivatives as tools for metabolic oligosaccharide engineering. Here, we report the development of an alternative approach that gives access to bis-SATE phosphotriesters in two steps from an easy to synthesise tri(2-bromoethyl)phosphotriester precursor. We demonstrate the viability of this strategy using glucose as a model substrate, onto which a bis-SATE-protected phosphate is introduced either at the anomeric position or at C6. We show compability with various protecting groups and further explore the scope and limitations of the methodology on different substrates, including N-acetylhexosamine and amino acid derivatives. The new approach facilitates the synthesis of bis-SATE-protected phosphoprobes and prodrugs and provides a platform that can boost further studies aimed at exploring the unique potential of sugar phosphates as research tools.
Introduction
Glycans are a structurally diverse class of biomolecules that play essential roles in diverse biological and pathological processes. Metabolic oligosaccharide engineering (MOE) has emerged as a powerful approach to characterise and manipulate glycan function in a native context.1 In MOE, chemical tags or other modifications are introduced into glycan structures by treating living cells or organisms with unnatural carbohydrate derivatives, which are metabolised by the cell's natural biosynthetic machinery into the activated sugar nucleotides required for glycosylation. A prerequisite for the success of this approach is that the unnatural carbohydrates are efficiently taken up inside the cell.2–5 With a few exceptions,6,7 the cellular entry mechanism of MOE probes is generally assumed to involve passive diffusion. Therefore, these probes are typically designed as caged precursors which are able to cross the cell membrane more efficiently and carry biolabile protecting groups that can be cleaved by endogenous intracellular enzyme activity to release the bioactive molecules.2,8Most MOE approaches make use of unnatural analogues of monosaccharides, such as azide- or alkyne-tagged derivatives of N-acetylglucosamine (GlcNAc). Several phosphorylated monosaccharide derivatives have, however, shown significantly improved MOE efficiency by bypassing problematic steps in the cellular biosynthetic pathways towards the corresponding sugar nucleotide donors.8–12 To enable efficient cellular uptake, the phosphates on these probes are typically masked with two S-acyl-2-thioethyl (SATE) protecting groups, as shown for a tagged GlcNAc-1-phosphate derivative in Fig. 1.
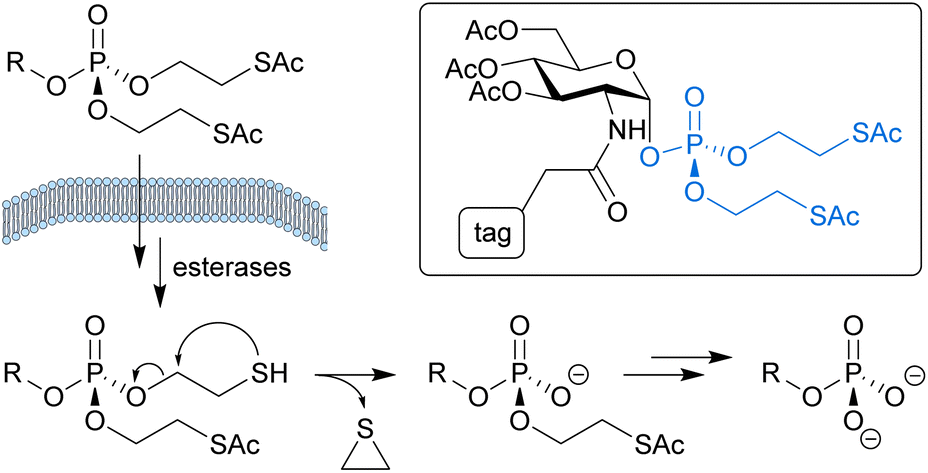 | ||
| Fig. 1 Biolabile S-acyl-2-thioethyl protecting groups enable intracellular delivery of phosphate-containing bioactive molecules from masked precursors, such as the tagged GlcNAc-1-phosphate derivative shown (with ‘tag’ representing a photoaffinity label8 or fluorophore,9 for example). Once inside the cell, the free phosphate is released through esterase-mediated hydrolysis of the thioesters followed by spontaneous decomposition of the resulting mercaptoethyl groups. | ||
First described for the intracellular delivery of nucleoside 5′-monophosphate derivatives of an anti-HIV drug,13 SATE protection is still commonly used as a prodrug strategy for nucleoside monophosphates and phosphonates.14,15 The biological instability of SATE groups is attributed to hydrolysis of the thioesters by carboxy- or thioesterase activity naturally present in mammalian cells.13,16 The resulting mercaptoethyl groups are presumed to spontaneously decompose by an intramolecular nucleophilic displacement mechanism that generates ethylene disulfide and releases the desired phosphate monoester (Fig. 1).
SATE-protected sugar phosphates are typically synthesised from a bis-SATE phosphoramidite intermediate (1), which in turn is prepared from the highly reactive reagent diisopropylphosphoramidous dichloride (Scheme 1A, top panel).13 Reaction of 1 with a substrate alcohol in the presence of 1H-tetrazole gives a phosphite, which is further oxidised to obtain the bis-SATE-protected phosphoester. Aside from the issue that diisopropylphosphoramidous dichloride is not commercially available in some countries including the UK, necessitating synthesis and isolation of the hazardous reagent, this methodology also suffers from low and inconsistent yields, particularly when used in the context of sugar-1-phosphates.9,10,17–19 Recently, a modification to the phosphoramidite approach, starting from a commercially available bis(diisopropylamino)chlorophosphine reagent, was shown to improve the synthesis of a SATE-protected GlcNAc-1-phosphate derivative.19
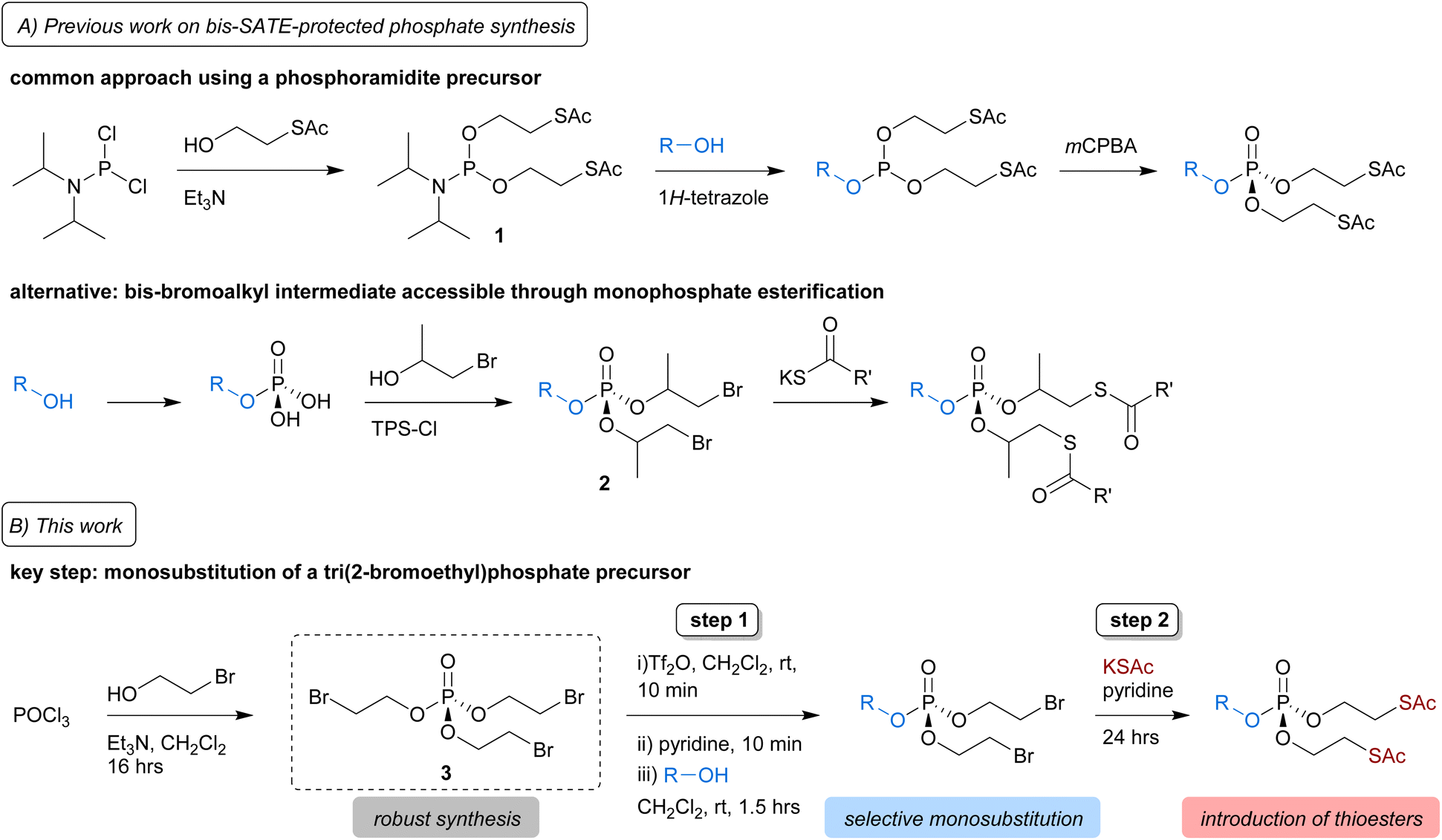 | ||
| Scheme 1 Synthesis of bis-SATE protected phosphates. (A) The common approach for bis-SATE phosphotriester synthesis involves reaction of a substrate (R–OH) with phosphoramidite 1, followed by oxidation. Alternatively, after introduction of a monophosphate onto a substrate, a bis-bromoalkyl phospotriester intermediate 2 can be formed through esterification and the bromides subsequently substituted for thioacyl groups.20 (B) In this work, we describe a two-step approach involving triflic anhydride/pyridine-mediated selective monosubstitution of tri(2-bromoethyl)phosphate precursor 3 by a substrate, followed by substitution of the bromides for thioacetates. | ||
A different strategy was reported to solve issues with the preparation of an S-pivaloyl-2-thioisopropyl phosphoramidite reagent needed for the synthesis of caged mononucleoside derivatives.20 Starting from a substrate monophosphate, the required thioalkyl-protected phosphotriester was generated via esterification either directly with the required thioalkyl groups or via bromoalkyl intermediate 2 (Scheme 1A, bottom panel). In this work, we develop an alternative method for the synthesis of SATE-protected mixed phosphotriesters that does not require the initial installation of a phosphomonoester onto the substrate, while avoiding the use of reactive phosphoramidite intermediates.
Our approach is based on recent reports showing that mixed phosphates and phosphonates can be accessed through triflic anhydride-mediated activation of a trialkylphosph(on)ate.21,22 Key to these strategies was the selective monosubstitution of one of the phosphoester groups. We hypothesised that we could use a similar strategy for the synthesis of bis-SATE-protected phosphates from the precursor tri(2-bromoethyl)phosphate (3) (Scheme 1B). Here, we describe the development of this approach, which involves triflic anhydride/pyridine-mediated selective monosubstitution of phosphotriester 3 by the free hydroxyl group of a substrate, followed by substitution of the bromides for thioacetate groups. We demonstrate the feasibility of this strategy for the synthesis of bis-SATE-protected sugar phosphates and further explore the scope and compatibility of the methodology on a range of substrates with diverse protecting groups.
Results and discussion
Prompted by the reported difficulties with phosphoramidite chemistry for SATE-protected sugar phosphate synthesis, we decided to perform the validation and optimisation of our new methodology on glucose as a model substrate. C1- and C6-phosphorylated pyranose sugars occur naturally within cellular metabolic pathways towards the biosynthesis of glycosylation donors. Of these two positions, the primary hydroxyl group at C6 is most nucleophilic and sterically accessible. Moreover, SATE-protected glucose-6-phosphate derivatives have been described as pro-inhibitors for intracellular carbonic anhydrase activity.23We thus began our method development with the installation of a bis-SATE-protected phosphate at the C6 position of 1,2,3,4-tetra-O-benzylglucopyranoside 5 (Scheme 2). Initially, we aimed to install the thioalkyl-protected phosphate directly onto 5 in one step by using a phosphotriester precursor already equipped with the required SATE groups (4). We were able to synthesise this precursor (4) via a straightforward two-step procedure from the commercially available reagents POCl3, 2-bromoethanol and potassium thioacetate. Both reagent 4 and the bromoalkyl intermediate 3 were readily produced with good yields on a gram scale. Subsequent reaction of glucose substrate 5 with precursor 4 in the presence of triflic anhydride and pyridine was, however, not successful (Scheme 2). Despite being able to detect the desired bis-SATE-protected phosphate 6 by mass spectrometry, we could not isolate any product.
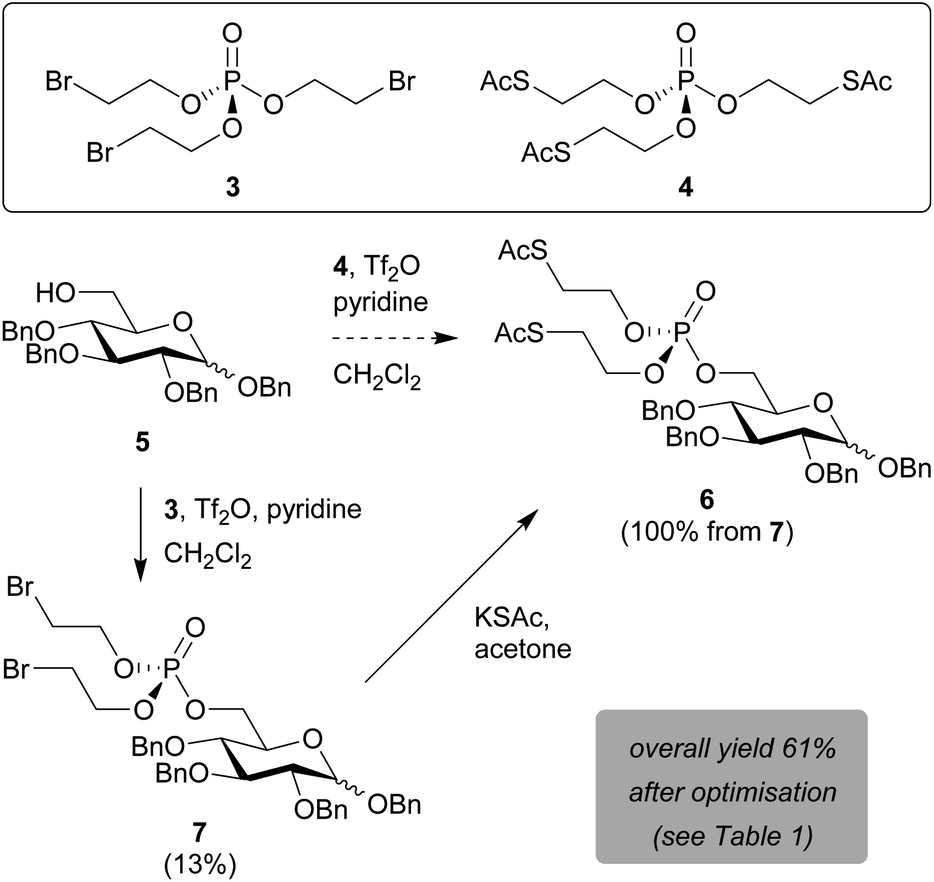 | ||
| Scheme 2 Installation of a bis-SATE-protected phosphate at C6 of 1,2,3,4-tetra-O-benzylglucopyranoside. Initial yields are shown prior to optimisation of reaction conditions. | ||
As an alternative, in line with the approach shown in Scheme 1A,20 we attempted to install a phosphate masked with two bromoalkyl groups onto the glucose substrate first, prior to substitution of the bromides for thioacetates. The triflic anhydride/pyridine-mediated reaction of 5 with tri(2-bromoethyl)phosphate 3 successfully yielded intermediate 7 in 13% yield (Scheme 2). The bis-SATE-protected phosphate 6 was then quantitatively obtained through a substitution reaction with potassium thioacetate.
Having demonstrated the viability of our new two-step approach, we set out to optimise the reaction conditions using the more readily available substrate 3-phenylpropanol (Table S1 in the ESI†). We found that the introduction of a 10 minute preactivation time, during which the phosphate precursor 3 is reacted with triflic anhydride (Scheme 1B, step 1i) before the addition of pyridine (step 1ii), greatly increased yields. It is also important to note that the use of freshly distilled triflic anhydride and fresh anhydrous pyridine is important to achieve reproducible results. Overall, we were able to increase the yield of the incorporated bis-(2-bromoethyl)phosphate from 17% to 51% after optimisation (Table S1,† entry 2). On increasing the preactivation time further, a reduction in yield was observed, suggesting instability of the intermediate phospho-triflate species in solution. Similarly, we observed a drop in yield when the reaction mixture was left for more than 10 minutes between the addition of pyridine (Scheme 1B, step 1ii) and the substrate (step 1iii).
Moving back to our model carbohydrate substrate, we applied the optimised reaction conditions to the reaction of partially protected glucose derivative 5 with phosphotriester 3. Pleasingly, we found the yield of bis-(2-bromoethyl)-protected phosphate 7 to be improved from 13% to 31% (Table S2,† entry 1). We then tested the effect of increasing the reaction time after addition of the sugar substrate to the preactivated phosphotriester/triflic anhydride/pyridine mixture (Scheme 1B, step 1iii). The results (Table S2†) show that further increases in yield were achieved with increasing reaction time, doubling from 31% after 30 minutes to 62% at an optimal reaction time of 1.5 hours (entry 3). The main carbohydrate by-product we observed was identified as a triflated species that can be hydrolysed back to starting material (see ESI†).
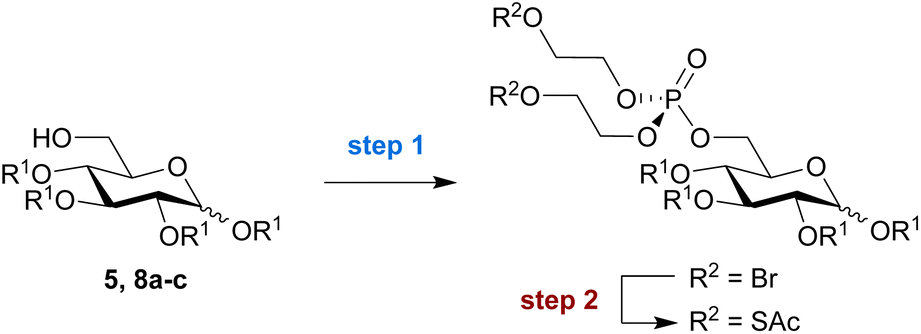
Next, we aimed to explore the compatibility and efficiency of the reaction on a small panel of differently protected glucose derivatives. In addition to the benzyl ether-protected substrate used above (5), we also tested glucose derivatives protected with allyl ethers (8a), acetyl esters (8b), and benzoyl esters (8c) under the same reaction conditions (Table 1). Installation of the bis-SATE-protected phosphate at the C6 position was successful for all tested substrates, with each reaction producing reproducible results across at least two independent replicates. Yields for the phosphoester substitution reaction (step 1) were somewhat lower for the allyl- and benzoyl-protected substrates 8a and 8c, respectively (32–37%) while the acetylated (8b) and benzylated (5) substrates reacted more efficiently (56–61%). The subsequent thioacetate substitution (step 2) proceeded quantitatively for all substrates except the acetyl-protected derivative, which surprisingly yielded only 55% of purified product. Overall, the reaction remained most successful on benzylated substrate 5, which provides access to bis-SATE-protected glucose-6-phosphate derivative 6 in a reproducible yield of around 60% over both steps.
|
|
|||
|---|---|---|---|
| Substrate | Yields (%)b | ||
| Step 1 | Step 2 | Overall | |
| a Reaction conditions: step (1) (i) 3, Tf2O, CH2Cl2, RT, 10 min., (ii) pyridine, RT, 10 min., (iii) 5, 8a, 8b or 8c, RT, 1.5 h; step (2) KSAc, pyridine, RT, 18 h. b Yields are reported as averages of duplicate reactions. Overall yield represents the combined yield over steps 1 and 2. c The product could not be fully separated from unreacted reagent 3. Yields were calculated from relative integration of characteristic signals on 1H NMR. | |||
| 5 R1 = Bn | 61% | Quant | 61% |
| 8a R1 = allylc | 32% | Quant | 32% |
| 8b R1 = Ac | 56% | 55% | 31% |
| 8c R1 = Bz | 37% | Quant | 37% |
Encouraged by these promising results, we extended our strategy to the installation of a bis-SATE-protected phosphate at the anomeric position of a set of appropriately protected glucose derivatives (Table 2). Initially, step 2 was performed with KSAc in acetone as before (Scheme 2). This resulted in low conversion to thioalkyl product, while the lack of recoverable starting material suggested that degradation was taking place. Therefore, we explored alternative solvents for the substitution step, which led to significantly improved results (Table S3†). The use of pyridine provided the highest conversion into thioester product and enabled the successful isolation of bis-SATE-protected glucose-1-phosphate derivatives of 9a–d in overall yields ranging from 14% to 30% over both steps (Table 2).
|
|
||||
|---|---|---|---|---|
| Substrate | Yields (%) | Anomeric ratioc | ||
| Step 1 | Step 2 | Overallb |
α![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :β (s.m.) :β (s.m.) |
|
| a Reaction conditions: step (1) (i) 3, Tf2O, CH2Cl2, RT, 10 min., (ii) pyridine, RT, 10 min., (iii) 9a–d, RT, 1.5 h; step (2) KSAc, pyridine, RT, 18 h. b Overall yield represents the combined yield over steps 1 and 2. c Anomeric ratios were determined from the relative integration of anomeric proton peaks in the 1H NMR spectra of the products after step 2. Numbers in parentheses represent the anomeric ratio of the starting material (s.m.). | ||||
| 9a R1 = Bn | 26% | 78% | 20% | 1:0 (2:1) |
| 9b R1 = allyl | 20% | 72% | 14% | 3:1 (4:1) |
| 9c R1 = Ac | 33% | 72% | 24% | 1.2:1 (4:1) |
| 9d R1 = Bz | 41% | 74% | 30% | 5:1 (4:1) |
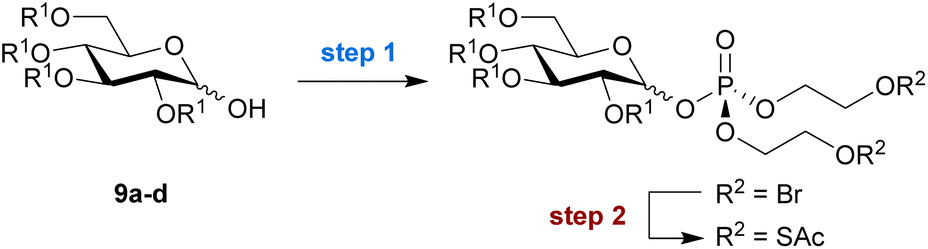
Analysis of the anomeric product ratios after the two-step procedure revealed that most reactions favoured formation of the α-anomeric phosphate, the anomer that generally acts as a direct metabolic precursor for sugar nucleotide biosynthesis and would thus be desired in the context of MOE. An exception is the acetylated substrate 9c, which showed a slight preference for the β-anomer, leading to an equimolar mixture of anomeric products. Benzyl-protected substrate 9a gave full α selectivity. Finally, we performed the reaction with substrate 9c on a gram scale and obtained essentially the same yield (25% over two steps), demonstrating the excellent scalability of the approach.
Aiming to push the boundaries of our new synthetic strategy further, we shifted our focus towards N-acetylhexosamines, substrates that provide additional challenges due to the presence of the N-acyl group capable of forming a 1,2-oxazoline through neighbouring group participation. Several SATE-protected N-acetylhexosamine-1-phosphate derivatives have been developed as MOE reporters.8–12 Therefore, we attempted to perform our phosphorylation strategy on 3,4,6-tri-O-acetylated GlcNAc 10 and its tribenzylated analogue 11 (Scheme 3). Unfortunately, these reactions were unsuccessful and only starting material could be isolated. We were also unable to detect any product from the reaction on 6-azido-tagged GlcNAc derivative 12.
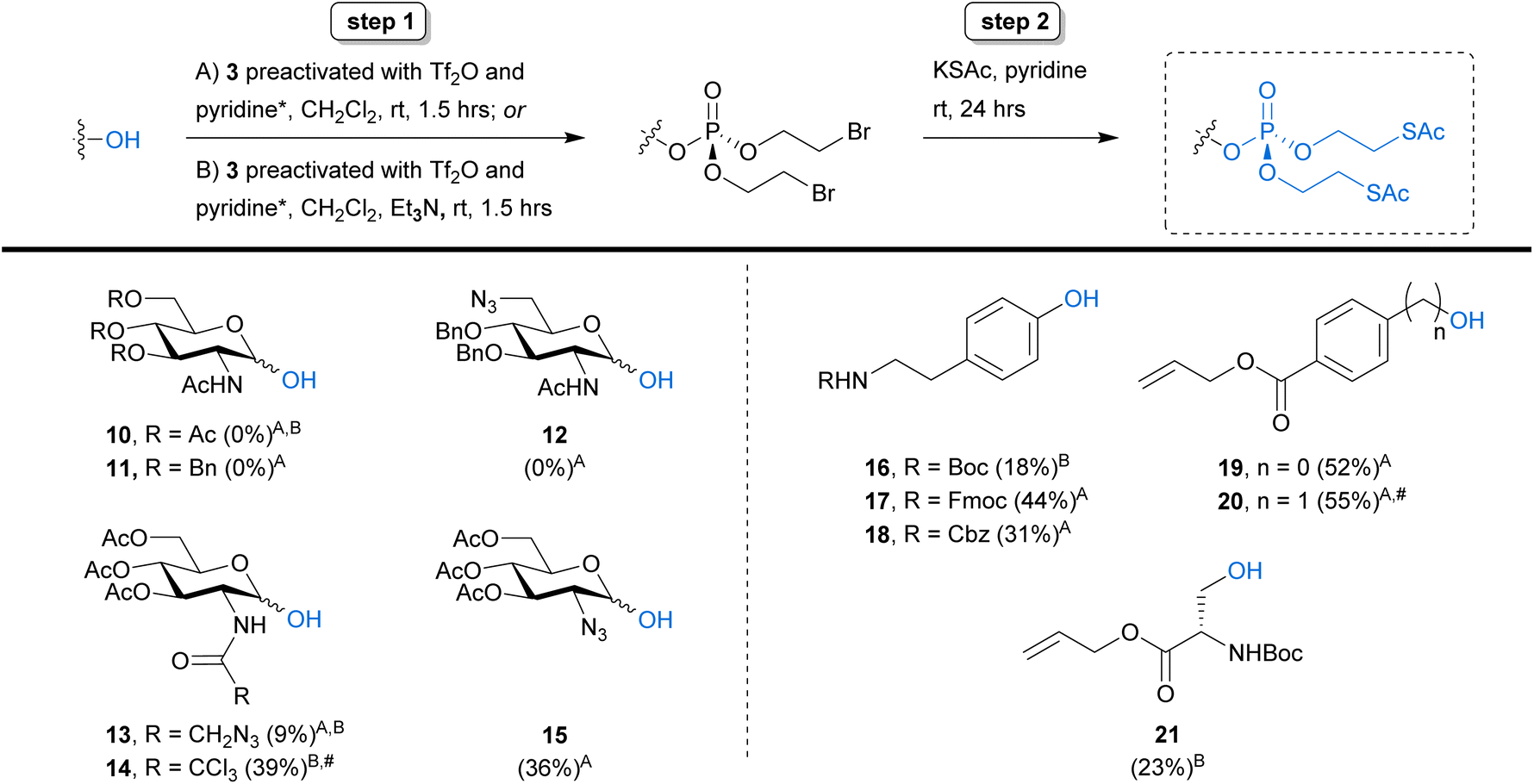 | ||
| Scheme 3 Extending the scope of the bis-SATE-protected phosphate synthesis methodology. The two-step reaction procedure was applied to various GlcNAc derivatives (10–15) and amino acid analogues (16–21). Yields are shown following either the optimised procedure (A) or the adapted protocol with the addition of triethylamine (B) and represent overall yields over both steps of the reaction unless indicated otherwise. *Preactivation conditions: (i) 3, Tf2O, CH2Cl2, RT, 10 min., (ii) pyridine, RT, 10 min. #: Yield of bis-(2-bromoethyl)-phosphate intermediate after step 1. | ||
Because GlcNAc donors are notorious for forming oxazolines or oxazolinium ions during glycosylation reactions,24 we hypothesised that oxazoline formation might be hindering reaction progress. We therefore monitored the reaction with substrate 10 by 1H NMR and indeed observed formation of an oxazolinium ion (Fig. S1†) as soon as 5 minutes after the start of the reaction. This observation was supported by high resolution mass spectrometry data, confirming the presence of an oxazoline (Fig. S2†) alongside unreacted 10. Interestingly, when we tested the N-azido-acetylated analogue 13 we observed low but detectable levels of product formation and were able to isolate the bis-SATE-protected phosphate in 9% yield (Scheme 3). We speculated that electronic effects might cause the higher reactivity of 13 as compared to 10, with the azide in 13 deactivating the N-acyl group and thereby reducing the probability of oxazolinium ion formation. To test this possibility, we prepared the electron-withdrawing trichloroacetyl derivative 14. Surprisingly, we observed no reaction with this substrate under the optimised reaction conditions (Scheme 3, conditions A).
While these data support the hypothesis that oxazolininium ion formation is hindering efficient anomeric phosphorylation of GlcNAc substrates under the triflic anhydride/pyridine-promoted reaction conditions, further experimentation is required to fully understand the lack of reaction with these substrates and the mechanism leading to formation of an oxazoline or oxazolinium species remains elusive. The activation of anomeric glycosyl phosphotriesters using triflic acid (a by-product of our tri(2-bromoethyl)phosphate activation procedure) as a promoter has been reported previously.25,26 However, we have not been able to observe the formation of any phosphotriester product that could act as a potential intermediate towards an oxazoline. Furthermore, we detected oxazoline formation by 1H NMR upon treatment of 10 with triflic anhydride and pyridine alone, in the absence of phosphotriester 3, supporting a potential mechanism independent of anomeric phosphate installation (Fig. S1†). Nevertheless, when trialling the addition of triethylamine into the phosphorylation reaction to neutralise any triflic acid that was being formed (Scheme 3, conditions B), the trichloroacetyl derivative 14 was converted successfully into the bis-(2-bromoethyl)phosphate intermediate in 39% yield. Unfortunately, the same conditions did not result in notable changes to the reaction outcome for substrates 10 and 13. We also attempted to vary the concentration of the reactants and the molar equivalents of triflic anhydride and pyridine in the reaction with 10, but this did not lead to successful phosphotriester formation.
Synthesis of GlcNAc-containing glycoconjugates often involves installation of an azide at C2 as a temporary masking group. This strategy has previously been used to synthesise a fluorescent, GlcNAc-1-phosphate-derived MOE probe: after installation of a bis-SATE-protected anomeric phosphate onto 2-deoxy-2-azido GlcNAc 15 (Scheme 3), the amide functionality was regained through selective reduction of the azide with concomitant N-acylation.9 To explore if the same strategy could also give access to bis-SATE-protected GlcNAc-1-phosphate derivatives using our new methodology, we applied our optimised two-step reaction sequence to compound 15. This reaction was successful and yielded a satisfying 36% of bis-SATE-protected phosphotriester with the α anomer being the major product (7:1 ratio to β anomer), comparing well with the 25% yield previously reported for its synthesis via phosphoramidite chemistry.9 The resulting bis-SATE-protected 2-azidosugar phosphate can be converted into GlcNAc-1-phosphate derivatives as reported.9
Finally, we turned our attention to the phosphorylation of amino acids. We selected a small set of phospho-serine and -tyrosine analogues to test our new methodology. This also enabled us to explore compatibility with additional protecting groups, including Boc, Fmoc and Cbz. The Fmoc, CBz and allyl protecting groups in tyrosine mimics 17, 18 and 19 were well tolerated, giving yields between 31% and 52% (Scheme 3). Initial attempts with the Boc-protected substrate 16 resulted in cleavage of the Boc group, likely due to triflic acid formation during the reaction. When repeated in the presence of triethylamine (conditions B), however, we successfully obtained the desired product in 18% yield. For allyl-protected derivative 20, installation of the bis-(2-bromoethyl)phosphate was successful (55%) but we detected loss of the phosphate group during thioacetate treatment. This might be caused by bromide substitution at the benzylic carbon in a manner analogous to the reported chloride-mediated cleavage of benzyl groups during phosphotriester synthesis with POCl3.27 Lastly, installation of the protected phosphate onto Boc-protected serine derivative 21 was successful in the presence of base. These results demonstrate the viability of our new approach for the preparation of bis-SATE-protected phospho-amino acids.
Conclusions
We have developed a novel approach for the synthesis of bis-SATE-protected phosphates that avoids the use of unstable phosphoramidite precursors. The reaction involves triflic anhydride/pyridine-mediated activation of a tri(bromoalkyl) phosphotriester precursor that can be easily prepared from phosphoryl chloride on a large scale. With this strategy, we facilitate straightforward access to biologically relevant phosphotriesters with biolabile thioalkyl protecting groups. We successfully applied the new methodology to the synthesis of SATE-protected sugar phosphates and phospho-amino acid derivatives and demonstrated it to be compatible with a range of commonly used protecting groups. In addition to its potential impact on the development of SATE-protected prodrugs,14,15 the approach offers a valuable extension to the available toolkit for the synthesis of phosphorylated saccharides.28 By tuning the ester groups on the phosphotriester precursor, different types of mixed phosphates should also be within reach. In particular, our new synthetic route towards SATE-protected sugar-1-phosphates has great potential to boost the development of novel phosphorylated metabolic precursors as MOE probes for the study of glycan structure and function.Experimental procedures
Compounds 9a,299b,309c,319d,3210,3313,914,3416,2717,2718,2719,2720,27 and 2135 were synthesised as described previously and characterisation data were found to be in accordance with the literature. Compound 15 was synthesised through an adapted literature procedure9 with the azido transfer reagent made in situ.36 Characterisation data matched those described previously.9 Experimental procedures and NMR spectra for all new compounds are provided in the ESI.†
Data availability
All data associated with this publication are provided in the ESI.†Author contributions
This study was conceptualised by LIW and KEH. LDM and LIW co-wrote the manuscript with input from KEH. Compounds were synthesised and characterised by LDM, KEH, AW, CR and QPOF under supervision of LIW. KEH performed initial testing and optimisation of the methodology. LDM explored the substrate scope and validated the main findings. LIW acquired and managed the funding to support the work.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was funded by the European Research Council (ERC) under the European Union's Horizon 2020 research and innovation programme [Grant agreement No 851448]. The authors would like to thank Gideon Davies and Martin Fascione for valuable input and Natasha Hatton for providing 3,4,6-tri-O-acetyl-2-deoxy-2-trichloroacetamido-α/β-D-glucopyranose.References
- For recent reviews, see: (a) N. Nischan and J. J. Kohler, Glycobiology, 2016, 26, 789 CrossRef PubMed; (b) T. J. Sminia, H. Zuilhof and T. Wennekes, Carbohydr. Res., 2016, 435, 121 CrossRef CAS PubMed; (c) P. A. Gilormini, A. R. Batt, W. R. Pratt and C. Biot, Chem. Sci., 2018, 9, 7585 RSC; (d) M. Kufleitner, L. M. Haiber and V. Wittmann, Chem. Soc. Rev., 2023, 52, 510 RSC; (e) K. E. Huxley and L. I. Willems, Biochem. Soc. Trans., 2021, 49, 903 CrossRef CAS PubMed.
- A. K. Sarkar, T. A. Fritz, W. H. Taylor and J. D. Esko, Proc. Natl. Acad. Sci. U. S. A., 1995, 92, 3323 CrossRef CAS PubMed.
- B. E. Collins, T. J. Fralich, S. Itonori, Y. Ichikawa and R. L. Schnaar, Glycobiology, 2000, 10, 11 CrossRef CAS PubMed.
- M. B. Jones, H. Teng, J. K. Rhee, N. Lahar, G. Baskaran and K. J. Yarema, Biotechnol. Bioeng., 2004, 85, 394 CrossRef CAS PubMed.
- N. D. Pham, C. S. Fermaintt, A. C. Rodriguez, J. E. McCombs, N. Nischan and J. J. Kohler, Glycoconjugate J., 2015, 32, 515 CrossRef CAS PubMed.
- P. A. Gilormini, C. Lion, D. Vicogne, T. Levade, S. Potelle, C. Mariller, Y. Guérardel, C. Biot and F. Foulquier, Chem. Commun., 2016, 52, 2318 RSC.
- M. Bardor, D. H. Nguyen, S. Diaz and A. Varki, J. Biol. Chem., 2005, 280, 4228 CrossRef CAS PubMed.
- S. H. Yu, M. Boyce, A. M. Wands, M. R. Bond, C. R. Bertozzi and J. J. Kohler, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 4834 CrossRef CAS PubMed.
- H. Y. Tan, R. Eskandari, D. Shen, Y. Zhu, T.-W. Liu, L. I. Willems, M. G. Alteen, Z. Madden and D. J. Vocadlo, J. Am. Chem. Soc., 2018, 140, 15300 CrossRef CAS PubMed.
- B. Schumann, et al. , Mol. Cell, 2020, 78, 824 CrossRef CAS PubMed.
- M. F. Debets, et al. , Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 25293 CrossRef CAS PubMed.
- A. Cioce, et al. , ACS Chem. Biol., 2021, 16, 1961 CrossRef CAS PubMed.
- (a) C. Périgaud, G. Gosselin, I. Lefebvre, J.-L. Girardet, S. Benzaria, I. Barber and J.-L. Imbach, Bioorg. Med. Chem. Lett., 1993, 3, 2521 CrossRef; (b) I. Lefebvre, C. Périgaud, A. Pompon, A. M. Aubertin, J. L. Girardet, A. Kirn, G. Gosselin and J. L. Imbach, J. Med. Chem., 1995, 38, 3941 CrossRef CAS PubMed.
- U. Pradere, E. C. Garnier-Amblard, S. J. Coats, F. Amblard and R. F. Schinazi, Chem. Rev., 2014, 114, 9154 CrossRef CAS PubMed.
- Z. Xie, L. Lu, Z. Wang, Q. Luo, Y. Yang, T. Fang, Z. Chen, D. Ma, J. Quan and Z. Xi, Eur. J. Med. Chem., 2022, 243, 114796 CrossRef CAS PubMed.
- B. R. Meade, et al. , Nat. Biotechnol., 2014, 32, 1256 CrossRef CAS PubMed.
- J. F. A. Pijnenborg, E. A. Visser, M. Noga, E. Rossing, R. Veizaj, D. J. Lefeber, C. Büll and T. J. Boltje, Chemistry, 2021, 27, 4022 CrossRef CAS PubMed.
- J. F. A. Pijnenborg, E. Rossing, J. Merx, M. J. Noga, W. H. C. Titulaer, N. Eerden, R. Veizaj, P. B. White, D. J. Lefeber and T. J. Boltje, Nat. Commun., 2021, 12, 7024 CrossRef CAS PubMed.
- B. N. Kakde, E. Capota, J. J. Kohler and U. K. Tambar, J. Org. Chem., 2021, 86, 18257 CrossRef CAS PubMed.
- S. Peyrottes, C. Périgaud, A.-M. Aubertin, G. Gosselin and J.-L. Imbach, Antiviral Chem. Chemother., 2001, 12, 223 CrossRef CAS PubMed.
- H. Huang, J. Ash and J. Y. Kang, Org. Lett., 2018, 20, 4938 CrossRef CAS PubMed.
- P. Adler, A. Pons, J. Li, J. Heider, B. R. Brutiu and N. Maulide, Angew. Chem., Int. Ed., 2018, 57, 13330 CrossRef CAS PubMed.
- G. M. Rankin, D. Vullo, C. T. Supuran and S.-A. Poulsen, J. Med. Chem., 2015, 58, 7580 CrossRef CAS PubMed.
- M. H. S. Marqvorsen, M. J. Pedersen, M. R. Rasmussen, S. K. Kristensen, R. Dahl-Lassen and H. H. Jensen, J. Org. Chem., 2017, 82, 143 CrossRef CAS PubMed.
- O. J. Plante, E. R. Palmacci, R. B. Andrade and P. H. Seeberger, J. Am. Chem. Soc., 2001, 123, 9545 CrossRef CAS PubMed.
- R. Arihara, K. Kakita, N. Suzuki, S. Nakamura and S. Hashimoto, J. Org. Chem., 2015, 80, 4259 CrossRef CAS PubMed.
- C. D. Spicer, M. Pujari-Palmer, H. ne Autefage, G. Insley, P. Procter, H. Engqvist and M. M. Stevens, ACS Cent. Sci., 2020, 6, 226 CrossRef CAS PubMed.
- N. Wang, Y. Kong, J. Li, Y. Hu, X. Li, S. Jiang and C. Dong, Bioorg. Med. Chem., 2022, 68, 116806 CrossRef CAS PubMed.
- M. A. Fernández-Herrera, S. Mohan, H. López-Muñoz, J. M. V. Hernández-Vázquez, E. Pérez-Cervantes, M. L. Escobar-Sánchez, L. Sánchez-Sánchez, I. Regla, B. M. Pinto and J. Sandoval-Ramírez, Eur. J. Med. Chem., 2010, 45, 4827 CrossRef PubMed.
- P. Wei, D. Zhang, Z. Gao, W. Cai, W. Xu, L. Tang and G. Zhao, Synth. Commun., 2015, 45, 1457 CrossRef CAS.
- S. Kim and P. Nagorny, Org. Lett., 2022, 24, 2294 CrossRef CAS PubMed.
- S. Yin, L. Li, L. Su, H. Li, Y. Zhao, Y. Wu, R. Liu, F. Zou and G. Ni, Carbohydr. Res., 2022, 517, 108575 CrossRef CAS PubMed.
- C. A. Kondor, J. N. Gorantla, G. D. Leonard and C. Fehl, Bioorg. Med. Chem., 2022, 70, 11691 CrossRef PubMed.
- N. Barroca-Aubry, A. Pernet-Poil-Chevrier, A. Domard and S. Trombotto, Carbohydr. Res., 2010, 345, 1685 CrossRef CAS PubMed.
- S. Bregant and A. B. Taylor, J. Org. Chem., 2005, 70, 2430 CrossRef CAS PubMed.
- H. Ye, R. Liu, D. Li, Y. Liu, H. Yuan, W. Guo, L. Zhou, X. Cao, H. Tian, J. Shen and P. G. Wang, Org. Lett., 2013, 15, 18 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Optimisation data, experimental procedures and NMR spectra of all new compounds. See DOI: https://doi.org/10.1039/d3sc00693j |
| This journal is © The Royal Society of Chemistry 2023 |