
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
De novo glycan sequencing by electronic excitation dissociation MS2-guided MS3 analysis on an Omnitrap-Orbitrap hybrid instrument†
Juan
Wei
 *ab,
Dimitris
Papanastasiou
c,
Mariangela
Kosmopoulou
c,
Athanasios
Smyrnakis
c,
Pengyu
Hong
d,
Nafisa
Tursumamat
a,
Joshua A.
Klein
b,
Chaoshuang
Xia
b,
Yang
Tang
be,
Joseph
Zaia
b,
Catherine E.
Costello
be and
Cheng
Lin
*b
*ab,
Dimitris
Papanastasiou
c,
Mariangela
Kosmopoulou
c,
Athanasios
Smyrnakis
c,
Pengyu
Hong
d,
Nafisa
Tursumamat
a,
Joshua A.
Klein
b,
Chaoshuang
Xia
b,
Yang
Tang
be,
Joseph
Zaia
b,
Catherine E.
Costello
be and
Cheng
Lin
*b
aShanghai Jiao Tong University, 800 Dongchuan Road, Shanghai, 200240, China
bCenter for Biomedical Mass Spectrometry, Boston University Chobanian & Avedisian School of Medicine, Boston, MA 02118, USA. E-mail: chenglin@bu.edu
cFasmatech Science and Technology, 15310 Athens, Greece
dDepartment of Computer Science, Brandeis University, Waltham, MA 02454, USA
eDepartment of Chemistry, Boston University, Boston, MA 02215, USA
First published on 2nd June 2023
Abstract
Comprehensive de novo glycan sequencing remains an elusive goal due to the structural diversity and complexity of glycans. Present strategies employing collision-induced dissociation (CID) and higher energy collisional dissociation (HCD)-based multi-stage tandem mass spectrometry (MSn) or MS/MS combined with sequential exoglycosidase digestions are inherently low-throughput and difficult to automate. Compared to CID and HCD, electron transfer dissociation (ETD) and electron capture dissociation (ECD) each generate more cross-ring cleavages informative about linkage positions, but electronic excitation dissociation (EED) exceeds the information content of all other methods and is also applicable to analysis of singly charged precursors. Although EED can provide extensive glycan structural information in a single stage of MS/MS, its performance has largely been limited to FTICR MS, and thus it has not been widely adopted by the glycoscience research community. Here, the effective performance of EED MS/MS was demonstrated on a hybrid Orbitrap-Omnitrap QE-HF instrument, with high sensitivity, fragmentation efficiency, and analysis speed. In addition, a novel EED MS2-guided MS3 approach was developed for detailed glycan structural analysis. Automated topology reconstruction from MS2 and MS3 spectra could be achieved with a modified GlycoDeNovo software. We showed that the topology and linkage configurations of the Man9GlcNAc2 glycan can be accurately determined from first principles based on one EED MS2 and two CID-EED MS3 analyses, without reliance on biological knowledge, a structure database or a spectral library. The presented approach holds great promise for autonomous, comprehensive and de novo glycan sequencing.
Introduction
Glycosylation, a diverse and complex post-translational modification, plays vital roles in many biological processes.1–5 Elucidation of glycan structures presents considerable challenges due to their structural complexity and heterogeneity, requiring analytical tools that can provide detailed information on their branching patterns, linkages, and stereochemical configurations. Database searching methods are limited to the identification of previously characterized glycans as there can be no genome-predicted glycan database because of the non-template-driven nature of glycan biosynthesis. Thus, discovery of novel glycan structures must be achieved by de novo sequencing. Though NMR can provide detailed structural information for glycans,6 it typically requires milligrams of purified sample, an amount that is not usually available from biological sources. Tandem mass spectrometry (MS/MS) has been effectively applied for glycan characterization owing to its high sensitivity, specificity, and ease of implementation with on-line separation methods.7–9 Although tandem MS analysis is commonly performed with collision-induced dissociation (CID) and higher energy collisional dissociation (HCD), glycan analysis by collision-based MS/MS is characterized by preferential cleavage of glycosidic bonds with few linkage-defining cross-ring fragments, and frequent losses of labile modifications. Consequently, a single stage of CID MS/MS analysis often fails to provide sufficient structural details, and sequential tandem mass spectrometry (MSn) is usually needed to determine the glycan branching pattern and linkages, and to differentiate structural isomers.10–17 However, the MSn approach is low-throughput and difficult to automate. Present strategies for automation of the MSn process are limited by the availability of glycan structure databases or tandem mass spectral libraries.18–20Major limitations of collision-based MS/MS arise from the slow-heating nature of collisional activation, and may be overcome by employing alternative, radical-driven ion activation methods, such as free radical activated glycan sequencing,21,22 ultraviolet photodissociation,23–25 charge transfer dissociation,26 and various electron-activated dissociation (ExD) methods.15,27–34 Among them, electronic excitation dissociation (EED) MS/MS has recently emerged as a powerful tool for structural glycomics. EED can produce detailed structural information in a single stage of MS/MS analysis, and has been successfully implemented with on-line LC and ion mobility separation for effective characterization of glycan mixtures.34–40 The power of an integrated approach that combines EED MS/MS with on-line chromatographic separation and software-assisted spectral interpretation was demonstrated in a recent study that employed porous graphitic carbon (PGC)-LC-EED-MS/MS.40 Candidate topologies of 18 oligomannose glycans released from RNase B were reconstructed from their EED MS2 spectra by GlycoDeNovo,41 a de novo glycan sequencing software, without reliance on any database. Putative topologies were consistently ranked as the top structures by the software.40 Many linkages could also be determined de novo, although in some cases, biological insights, such as the glycan biosynthetic rules, were needed to fully define the structure.
To date, glycan structural elucidation by EED MS/MS has only been demonstrated on Fourier transform-ion cyclotron resonance (FTICR) MS instruments. Broad adoption of EED MS/MS by the glycoscience research community has been hindered by the limited accessibility and high operating cost of FTICR MS. Recently, electromagnetostatic cells that may be installed in non-ICR instruments have become commercially available, allowing protein sequencing by electron-based dissociation on Orbitrap, time-of-flight (TOF), and ion trap mass spectrometers.42–48 However, effective ExD characterization of glycans on these instruments has yet to be fully realized, in part because ion-electron interaction occurs during analyte transfer through the ExD cell without trapping, and this limits the ExD efficiency, particularly for higher-energy ExD processes. Improved electron activated dissociation (EAD) efficiency may be achieved by trapping the precursor ions for more extensive interactions with electrons.49
In this study, we took advantage of a new ExD cell design on a prototype Omnitrap instrument.50Fig. 1a shows the schematic of an Omnitrap platform, consisting of a system of linear ion trap segments, connected to the rear of the HCD cell of a QE-HF Orbitrap instrument via a transfer hexapole. The segmented ion trap offers flexibilities for ion storage and manipulation, enabling high performance MSn analysis by utilizing a wide range of fragmentation methods, including CID and ExD. Interfacing the Omnitrap with an Orbitrap allows subsequent detection of fragment ions with high mass accuracy and resolving power, and this is beneficial for MS/MS-based glycan structural analysis as isobaric fragments are frequently produced by glycans. The principle of operation for ExD experiments on a QE-Omnitrap instrument is illustrated in Fig. 1b. ExD analysis is carried out in Q5 with an orthogonally mounted electron source consisting of a tantalum disc cathode and electrostatic focusing lenses. Electrons are injected into Q5 when a positive potential is applied to the X-electrodes and blocked when a negative potential is applied. The E2 split electrode provides a deflection pulse to ensure electron injection only during the correct RF phase. Unlike conventional ion traps that employ sinusoidal RF waveforms, rectangular waveforms are applied to the Omnitrap electrodes. The rectangular waveform produces a “static’ trapping field, allowing precise control of the electron energy. Time-controlled ion-electron interaction is possible due to analyte trapping in Q5 during the entire ExD event, and this should lead to improved ExD efficiencies.
 | ||
| Fig. 1 (a) Schematic of the hybrid Omnitrap-Orbitrap platform; (b) principle of operations for ExD analysis performed on the Omnitrap system. The illustration of the ExD cell in (b) is rotated 90 degrees compared to (a). | ||
Here, we report, for the first time, efficient detailed glycan structural characterization by EED MS/MS on a hybrid QE-Omnitrap instrument. To address the challenges in de novo glycan sequencing by MS2, we developed a novel EED MS2-guided MS3 strategy to differentiate iso-topologies and to eliminate ambiguities in linkage assignment among different antennae. Additionally, the GlycoDeNovo software was modified to facilitate automated interpretation of both MS2 and MS3 spectra. The potential of this approach for de novo glycan sequencing is demonstrated for characterization of the canonical Man9 chitobiose structure (Man9GlcNAc2) and the biantennary G2 glycan.
Experimental
Materials
The Man9GlcNAc2 and G2 glycan standards were purchased from ProZyme-Agilent (Hayward, MA). HPLC grade water, acetonitrile (CAN), chloroform, and sodium acetate were obtained from Fisher Scientific (Pittsburgh, PA). Micro bio-spin columns (0.8 mL bed volume) were acquired from Bio-Rad Laboratories (Hercules, CA). Methyl iodide, dimethyl sulfoxide (DMSO), sodium hydroxide beads (20–40 mesh), sodium borodeuteride (NaBD4), and acetic acid were purchased from Sigma-Aldrich (St. Louis, MO).Sample preparation
All glycans were deutero-reduced and permethylated according to protocols described in detail elsewhere.36,51 Briefly, 2 μg of glycan was dissolved in 200 μL of NaBD4 (250 mM) in 100 mM NH4OH solution. Following a 2 h incubation at room temperature, the reaction was quenched by gradual addition of 10% acetic acid until bubbling ceased. Excess NaBD4 was removed with addition of methanol. For solid-phase permethylation, an empty spin column was filled with suspended NaOH beads and conditioned with 400 μL of DMSO. Deutero-reduced glycans were dissolved in 120 μL of DMSO plus 5 μL of H2O and loaded onto the NaOH column. An aliquot of 100 μL of methyl iodide was added to the column, and the spin-column was settled for 1.5 h, with additions of 100 μL methyl iodide every 30 min. Permethylated glycans were extracted by chloroform and dried with a SpeedVac system (ThermoFisher Scientific, Waltham, MA).Mass spectrometry analysis
Deutero-reduced and permethylated glycans were dissolved in 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50 H2O:ACN solution containing 100 μM sodium acetate, to a concentration of 1 or 5 pmol μL−1, for MS analysis on QE-Omnitrap or FTICR MS, respectively. All QE-Omnitrap experiments were performed on a Q Exactive HF instrument (Thermo Scientific, Bremen, Germany) modified with an Omnitrap (Fasmatech, Athens, Greece). The Omnitrap platform is equipped with an ExD source consisting of a tantalum disc (1.6 mm diameter) and a series of electrostatic focusing lenses to guide electrons into the Q5 segment (Fig. 1). A TriVersa NanoMate nanoESI source (Advion, Ithaca, NY) was used for sample introduction into the mass spectrometer, with an injection time of 5 ms for MS2 and up to 300 ms for MS3 analyses. For EED MS/MS, precursors were selected by the QE quadrupole, and sent to Q5 in the Omnitrap where they were irradiated with 17–20 eV electrons for 50 ms. For CID-EED MS3 analysis, CID was conducted in the Omnitrap Q2 with pulsed argon gas (10 ms dipolar excitation). The CID fragment of interest was isolated in Q2 and sent to Q5 for EED fragmentation, followed by the transfer of product ions back to the Orbitrap for mass analysis. HCD (50 eV) experiments were performed in the QE HCD cell. All spectra were acquired with 5 microscans and a fixed resolving power of 60 K at m/z 200.
:50 H2O:ACN solution containing 100 μM sodium acetate, to a concentration of 1 or 5 pmol μL−1, for MS analysis on QE-Omnitrap or FTICR MS, respectively. All QE-Omnitrap experiments were performed on a Q Exactive HF instrument (Thermo Scientific, Bremen, Germany) modified with an Omnitrap (Fasmatech, Athens, Greece). The Omnitrap platform is equipped with an ExD source consisting of a tantalum disc (1.6 mm diameter) and a series of electrostatic focusing lenses to guide electrons into the Q5 segment (Fig. 1). A TriVersa NanoMate nanoESI source (Advion, Ithaca, NY) was used for sample introduction into the mass spectrometer, with an injection time of 5 ms for MS2 and up to 300 ms for MS3 analyses. For EED MS/MS, precursors were selected by the QE quadrupole, and sent to Q5 in the Omnitrap where they were irradiated with 17–20 eV electrons for 50 ms. For CID-EED MS3 analysis, CID was conducted in the Omnitrap Q2 with pulsed argon gas (10 ms dipolar excitation). The CID fragment of interest was isolated in Q2 and sent to Q5 for EED fragmentation, followed by the transfer of product ions back to the Orbitrap for mass analysis. HCD (50 eV) experiments were performed in the QE HCD cell. All spectra were acquired with 5 microscans and a fixed resolving power of 60 K at m/z 200.
A comparative study was performed on a 12T solariX FTICR MS equipped with a hollow cathode dispenser (Bruker Daltonics, Bremen, Germany). Samples were loaded into a pulled fused silica capillary and directly infused into the mass spectrometer by nano-electrospray. Targeted ions were isolated by the front-end quadrupole and accumulated in an external hexapole collision cell for up to 400 ms. For EED MS/MS analysis, precursor ions were irradiated by 18–20 eV electrons for 100–300 ms. For MS3 analysis, quadrupole-selected precursors were accumulated and fragmented by CID in the collision cell, and fragments were sent to the ICR cell and selected by an in-cell sweep isolation for EED MS3 analysis. Mass spectra were acquired with a low-mass cutoff of m/z 200, and a 512k or 1 M data size, resulting in a transient length of 288 ms or 577 ms, and an estimated resolving power of 66 K or 130 K at m/z 400, respectively. Five and forty spectra were averaged for EED MS/MS and CID-EED MS3 analyses, respectively.
Data analysis
The QE-Omnitrap mass spectral data were processed by Xcalibur and FreeStyle (Thermo Scientific, Bremen, Germany). Peak lists exported from FreeStyle were deconvoluted using ms_deisotope and subsequently interpreted by GlycoDeNovo. FTICR spectra were processed by DataAnalysis 4.4 (Bruker, Bremen, Germany), and peak picking was performed using the SNAP algorithm. Fragments were annotated according to the Domon and Costello nomenclature.52 Manual spectra interpretation was assisted by GlycoWorkbench 2.53 Candidate topologies were automatically reconstructed by GlycoDeNovo and ranked by IonClassifier.41Results and discussion
We first compared the performance of EED MS2 on two instrument platforms, FTICR MS and QE-Omnitrap MS. For FTICR MS analysis, the reduced and permethylated Man9GlcNAc2 glycan solution (5 pmol μL−1) was loaded into a pulled glass capillary, and directly infused into the mass spectrometer using nano-electrospray ionization. For QE-Omnitrap analysis, the same batch of the derivatized Man9GlcNAc2 glycan was diluted to a concentration of 1 pmol μL−1, and directly infused using an Advion TriVersa NanoMate ESI source. Instrumental parameters were optimized to achieve the highest EED efficiency.Fig. 2 shows the EED MS2 spectra of the doubly-sodiated Man9GlcNAc2 precursor (m/z 1218.1042) acquired on the QE-Omnitrap and on the FTICR MS, with all assigned fragments listed in ESI Tables S1 and S2,† respectively. EED produced very similar fragmentation patterns on these two instruments, while the EED efficiency achieved on QE-Omnitrap was significantly higher than on FTICR MS, producing on average 5–10 times higher relative fragment abundance and signal-to-noise ratio. With a higher S/N ratio, 31 more peaks were assigned in the QE-Omnitrap EED MS2 spectrum than in the corresponding FTICR spectrum, representing a 25% increase in the number of assigned peaks. Notably, the Omnitrap spectrum of the five-fold diluted sample solution was acquired with a shorter ion injection time (5 ms vs. 400 ms) and electron irradiation time (50 ms vs. 200 ms) than the ICR spectrum. The shorter spectral acquisition time and higher EED efficiency achieved on QE-Omnitrap make it far more suitable for high-throughput LC-MS/MS analysis, allowing characterization of a higher number of glycoforms, including those present in lower abundance. The higher EED efficiency achieved on the QE-Omnitrap instrument may have benefited from collisional cooling and focusing in the higher-pressure Q5 that improves the electron–ion interaction, whereas in the ultra-high vacuum of the ICR cell, axial excitation and magnetron expansion can lead to poorer ion-electron overlap.54
 | ||
| Fig. 2 EED MS2 spectra and cleavage maps of reduced- and permethylated Man9GlcNAc2 ([M + 2Na]2+ at m/z 1218.1042) acquired on (a) QE-Omnitrap MS and (b) FTICR MS, respectively. Each EED MS2 spectrum was the summed result of 5 scans. A complete series of C-ions and Y/Z/1,5X triplets are labeled in red and blue, respectively. Linkage-diagnostic cross-ring and internal fragments are labeled in green. Asterisk indicates singly charged fragments with two sodium atoms. A complete list of assigned fragments can be found in ESI Tables S1 and S2.† | ||
On either instrument platform, EED of the Man9GlcNAc2 glycan produced a complete series of C-type ions (C1/C1‡, C2/C2‡, C3β/C3β‡, C3α/C3α‡, C4‡, C5‡, where the symbol ‡ indicates loss of two hydrogens), and Y/Z/1,5X triplets. The presence of these sequence ions allows de novo reconstruction of the Man9GlcNAc2 candidate topologies by GlycoDeNovo. The canonical structure, listed in bold in Fig. 3a with its topology encircled in Fig. 3b, is one of the five candidates with the highest IonClassifier score. The IonClassifier was established by a machine learning approach that identifies common spectral context of a certain type of fragment in a training data set, defined as a collection of its related neighboring peaks, represented by their respective mass shifts and relative abundances. The IonClassifier can be used to evaluate the accuracy of an assignment by examining the spectral context of the fragment.41 The IonClassifier score of a candidate topology is the sum of the IonClassifier scores of all its supporting peaks, namely, glycosidic fragments consistent with the topology. Here, the five top-ranked topologies are indistinguishable at the MS2 level, as they produce the same set of supporting peaks, including non-reducing-end glycosidic fragments with compositions of Hex, Hex2, Hex3, Hex5, Hex9, and Hex9HexNAc. Previously, the correct Man9GlcNAc2 topology was assigned based on prior glycan biosynthetic knowledge.40 Among the five top-ranked candidates, only topology 2, with a linear tri-hexose branch and a branched penta-hexose branch, can be derived from the structure of the tetradecasaccharide N-linked glycan precursor (Fig. 3c).
 | ||
| Fig. 3 (a) Top-ranked candidate topologies reconstructed by GlycoDeNovo from the EED MS2 spectrum of deutero-reduced and permethylated Man9GlcNAc2 in Fig. 2; (b) graphic representations of the five top-ranked isotopolomers of Man9GlcNAc2; (c) SNFG representation of the N-glycan precursor. | ||
Once a topology is defined, most linkages can be independently determined based on linkage-diagnostic fragments without further reference to the precursor structure. For example, the presence of 3,5A4 (m/z 1145.5566), 0,4A4 (m/z 1117.5256), and C4/Z3β (m/z 1247.5889) ions not only places the pentasaccharide branch to the 6-antenna, but also localizes the tri-hexose branch to the 3-antenna, since a high-abundance C/Z ion is associated with the loss of the C3-substituent from a Z ion. The observation of 1,3A4 (m/z 723.3408) and 0,2X2 (m/z 618.3319) ions also corroborates with the assignment of the tri-hexose to the 3-antenna. Within the 6-antenna, the presence of 0,4A3α (m/z 505.2253), 3,5A3α (m/z 533.2568), C3α/Z4α′′ (m/z 635.2886), and 0,2X3α (m/z 728.8606) ions is consistent with a penta-hexose structure consisting of two di-hexose branches, at the 6- and 3-positions of the branching mannose, respectively, although the 1→6 linkage assignment is not conclusive, as the observed 0,4A3α and 3,5A3α ions can theoretically be generated from the linear tri-hexose 3-antenna as well. Similarly, the 1→2 linkage between the second and third mannose residues on the 3-antenna cannot be assigned definitively, since its diagnostic 1,3A3β (m/z 519.2411) ion is isomeric to the 1,3A3α′′ at the 6-antenna. Finally, while the presence of a 1,3A2 ion (m/z 315.1410) and the absence of 0,2X4, 3,5A2, and 0,4A2 ions strongly suggest the existence of at least one 1→2-linked non-reducing-end mannose residue, its location (or their locations) cannot be unambiguously determined. ESI Scheme S1b† shows a hypothetical structure that can produce the same group of cross-ring and internal fragments as the true structure (ESI Scheme S1a†). Detailed discussions on the general EED mechanism and formation of specific diagnostic fragments have been presented elsewhere.36,37,40,55
The analysis above clearly illustrates the challenges of MS2-based de novo glycan sequencing. Even with complete series of glycosidic fragments and cross-ring fragments, it is sometimes not possible to differentiate iso-topologies or accurately determine linkages among different branches. Additional structural details may be revealed by performing sequential tandem MS analysis on MS2 fragment(s) of interest. Presently, there are several ways to choose MS2 fragments as precursors for later stages of MSn analysis, each with its merits and limitations. The most straightforward method is to select product ion(s) of the highest abundance. Although such an approach allows autonomous precursor selection, the highest-abundance fragments do not always produce the most structurally informative MS3 spectra. Alternatively, precursors may be selected by expert users, aided by existing knowledge of the glycan fragmentation behavior and/or biological insights.56–58 This is typically performed during direct infusion analyses on ion trap instruments, whose low mass resolving power can negatively impact the accuracy of the fragment assignment. Further, without chromatographic separation, each MS2 precursor may consist of multiple isomeric structures, and each MS2 fragment chosen for further MSn analysis can be a mixture of isobaric structures. Consequently, confident structural assignment requires investigation of many gas-phase disassembly pathways, often involving deeper level of MSn. This process is difficult to automate, and the need to perform MSn>3 limits both the sensitivity and throughput. Recently, Sun and coworkers developed a glycan intelligent precursor selection (GIPS) strategy to guide MSn experiments.59,60 GIPS utilizes a statistical model to calculate the distinguishing power of fragments, which dictates the selection of precursor(s) for next stage of tandem MS analyses. The GIPS approach has only been demonstrated for the identification of the glycan branching patterns, but not for linkage determination. Moreover, when calculating the distinguishing power, it only considers structures present in the existing glycan database, and thus it cannot identify new structures.
The MSn method may also be combined with spectral library search for glycan structural elucidation, as utilized in logically derived sequence (LODES)/MSn.20,61,62 The LODES/MSn approach employs a built-in logical procedure to successively break down an oligosaccharide sequence into its monosaccharide and disaccharide components. The CID spectra of these smaller fragments can then be searched against a spectral database for structural assignment. The LODES/MSn approach can provide detailed structural information, provided that the corresponding spectral library of disaccharide standards is available. However, it still requires investigation of many MSn pathways, and is not readily compatible with on-line LC-MS analysis. Multiple LC injections were needed to fully define the structure of glycans as small as trisaccharides.62 For larger oligosaccharides, such as released N-linked glycans, knowledge on their biosynthesis was utilized to reduce the number of MSn pathways that need to be examined. For complex mixtures, it was necessary to perform multi-dimensional LC fractionation off-line before LODES/MSn analysis.20
Here, precursor(s) for MS3 analyses were selected based on their projected differentiating power on top-ranked topology candidates identified by GlycoDeNovo from the EED MS2 spectrum. For Man9GlcNAc2, the top five topologies (Fig. 3b) can be uniquely differentiated by performing MS3 analysis on the CID-generated Hex3 and Hex5 fragments (Fig. 4a). EED of a Hex5 fragment, 0,4A4 at m/z 1117.5259, produced glycosidic fragments with either one or two Hex units (C1 ion at m/z 259.1162, B2 at m/z 445.2045, and C2 at m/z 463.2153), but not with three or four Hex units (Fig. 3b), hence eliminating topologies 1, 3, and 5 from consideration. The remaining two topologies, 2 and 4, can be distinguished by EED analysis of a Hex3 fragment, B3β at m/z 649.3046. The presence of two Hex2 fragments (B2 at m/z 445.2048 and C2 at m/z 463.2162) in the EED spectrum of B3β (Fig. 3c) clearly established topology 2 as the only structure consistent with the MS3 results. The same conclusion could be reached by applying a modified GlycoDeNovo algorithm for analysis of the MS3 spectra. In this case, the software treated the mass difference between the intact glycan and the MS2 fragment as a reducing-end modification. GlycoDeNovo successfully identified a penta-saccharide with two di-hexose branches and a linear tri-hexose structure as the top-ranked topologies for the Hex5 and Hex3 substructures, respectively. Thus, we showed that the correct topology of the Man9GlcNAc2 glycan can be defined by just two MS3 analyses, autonomously from the first principles, with no need for reference spectral libraries, or biological knowledge. Here, the applicability of EED MS/MS to analysis of singly charged precursor is essential for the CID-EED MS3 workflow, as both the 0,4A4 and B3β fragments selected for MS3 analysis were singly charged, and therefore could not be characterized by charge-reducing MS/MS methods, such as ETD and ECD.
 | ||
| Fig. 4 (a) CID MS2 spectra of deutero-reduced- and permethylated Man9GlcNAc2 ([M + 2Na]2+ at m/z 1218.1042) acquired on the QE-Omnitrap system. Fragments selected for EED MS3 are labeled. (b) and (c) CID-EED MS3 spectra and cleavage maps of the 0,4A4 (m/z 1117.5259) and B3β (m/z 649.3046) ions, respectively, acquired on QE-Omnitrap. Linkage-diagnostic cross-ring and internal fragments are labeled in green. Peaks marked by asterisks are background peaks with mass defects that are not expected from glycan fragments. Complete lists of assigned fragments can be found in ESI Tables S3 and S4.† | ||
It is worth noting that a given substructure may be present in several fragments. For MS2 fragments with the same differentiating power, the choice is generally made based on the fragment ion abundance and the ability to achieve a clean isolation. Sometimes, more than one MS2 fragment can generate similarly informative MS3 spectra. For example, the Y3β2+ ion at m/z 904.9469 also retained the Man5 substructure without interference from the Man3 branch. EED of Y3β2+ (ESI Fig. S1 and Table S5†) similarly produced non-reducing-end glycosidic fragments with compositions of Hex1 or Hex2, but not Hex3 or Hex4, consistent with the branched Hex5 substructure assignment based on the EED spectrum of the 0,4A4 ion.
As discussed earlier, once topology 2 is established as the correct structure, the Hex5 and Hex3 branches can be accurately located to the 6- and 3-antennae, respectively, on the basis of the characteristic cross-ring and abundant C/Z internal fragments observed in the EED MS2 spectrum. Within the 6-antenna, one of the di-hexose branches can be assigned to the 3-position based on the presence of a high-abundance C3/Z4α′′ ion at m/z 635.2886, but the location of the other di-hexose branch cannot be confidently assigned due to potential interference by fragments from the linear tri-hexose 3-antenna. With MS3, the Hex5 substructure can be isolated for accurate linkage analysis. EED MS3 of the 0,4A4 ion generated both the 0,4A3 ion at m/z 505.2262 and the 3,5A3 ion at m/z 533.2575, thus unambiguously assigning the second di-hexose branch to the 6-position. The presence of the 1,3A2 ion at m/z 315.1416 and a C‡/Y (Hex) ion at m/z 243.0844, together with the lack of a 0,2X4β/0,4A4 ion at m/z 751.3359, placed the terminal mannose residues to the 2-position. Similarly, the linear tri-hexose branch at the 3-antenna was isolated in the B3β fragment, whose EED MS3 analysis generated 1,3A2 at m/z 315.1415, 1,3A3 at m/z 519.2409, C‡/Y (Hex) ion at m/z 243.0842, but 0,2X4β at m/z 283.1149, supporting the 1→2 linkages between the mannose residues. Collectively, the linkages within both antennae could be determined de novo by the CID-EED MS3 analysis.
With the Omnitrap-Orbitrap system, the MS3 experiment can be performed in several different configurations. The CID-EED sequence was chosen here as CID typically generates a smaller number of fragments in higher abundance than EED. Thus, the CID spectrum is usually not as congested as the EED spectrum, making it easier to achieve a clean isolation of the high-abundance MS2 fragment of interest. For Man9GlcNAc2, isolation of the 0,4A4 fragment at m/z 1117.5256 from EED MS2 can be complicated by co-isolation of the doubly charged 3,5A6 ion at m/z 1115.0348 (Fig. 2a and Table S1†), whereas the interfering 3,5A6 ion was absent in the CID spectrum of Man9GlcNAc2 (Fig. 4a). On the other hand, EED is the preferred final-stage dissociation method, producing MS3 spectra with much higher structural information content than CID. The EED and CID MS3 cleavage maps of the 0,4A4 ion are shown in Fig. 4b and ESI Fig. S2,† respectively. It is evident that many linkage-diagnostic fragments, such as 1,3A and C‡/Y (Hex) ions, were absent in the CID spectrum (Fig. S2†). Finally, although it is possible to perform CID-EED MS3 analysis on a hybrid Qh-FTICR MS instrument, selection of the MS3 precursor can only be achieved with in-cell isolation, resulting in a much lower ion count for the precursor of interest. Consequently, even with more than 10 times the ion injection time, the CID-EED MS3 spectrum of the 0,4A4 ion on FTICR MS (ESI Fig. S3†) was of much lower quality than that obtained on the Omnitrap-Orbitrap system (Fig. 4b).
While the discussion thus far has been focused on the Man9GlcNAc2 structure, the workflow presented here can be effectively applied to characterize other glycan structures, including complex-type glycans. Note that the Man9GlcNAc2 structure was chosen as the primary example not because of its apparent structural simplicity, but rather, because it represents one of the hardest cases for isotopolomer differentiation. With only one type of monosaccharide residue (hexose) present in all branches, many structures can produce the same set of glycosidic fragments and are therefore impossible to differentiate at the MS2 level, even with the IonClassifier. Two additional MS3 analyses were required to unambiguously define the canonical structure. On the other hand, complex-type glycans consist of a larger variety of monosaccharide building blocks that have different masses and are less likely to produce isotopologies. True topologies of many complex-type glycans can be identified at the MS2 level, though there are cases where MS3 is necessary. Fig. 5 shows one such case for characterization of the biantennary G2 glycan. At the MS2 level, topologies 1 and 2 (T1 and T2) would produce the same set of glycosidic fragments (Hex, HexHexNAc, Hex2HexNAc, etc.), and were co-ranked as the top candidate by the IonClassifier based on the EED MS/MS spectrum of the deutero-reduced and permethylated G2 glycan (ESI Fig. S4†). These two topologies could be differentiated based on the HCD-EED MS3 spectrum of the Y4 ion (ESI Fig. S5†), where the presence of HexHexNAc fragments and their complementary ions supported T1 as the correct topology. Lists of assigned peaks for the EED spectra of the G2 glycan and its Y4 fragment can be found in ESI Tables S6 and S7.†
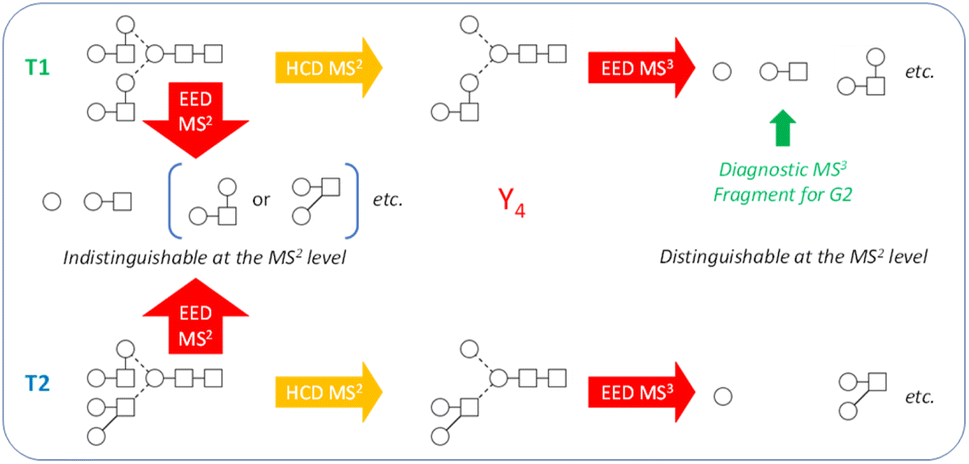 | ||
| Fig. 5 Isotopologies T1 and T2 were indistinguishable by MS2 of the G2 glycan, whereas diagnostic HexHexNAc fragments generated by HCD-EED MS3 of the Y4 ion can be used to define T1 as the correct topology. | ||
Conclusions
The recently introduced hybrid Omnitrap-Orbitrap system allows EED MS2 analysis of glycans to be performed with high sensitivity, fragmentation efficiency, and spectral acquisition rate that are crucial for high-throughput analysis. Candidate topology structures can be automatically reconstructed and ranked by the GlycoDeNovo software based on the EED MS2 spectrum. Top-ranked topology candidates identified by EED MS2 can then be used to guide the selection of MS2 fragments for MS3 analysis. With one EED MS2 and two CID-EED MS3 spectra, the complete structure of even the particularly challenging example, Man9GlcNAc2, including its topology and linkages, could be confidently assigned de novo without the need for biological knowledge or a spectral library. With high sensitivity and analysis speed, the novel EED MS2-guided-MS3 approach presented here should be fully compatible with on-line LC separation, and holds great promise for automated, de novo, and comprehensive glycan structural elucidation.Data availability
All mass spectral data are included in the ESI.†Author contributions
JW and CL conceptualized the experimental design. JW and CX prepared the glycan samples for analysis. DP, MK, and AS performed the initial analysis on the Omnitrap system. JW performed the comparative study on FTICR MS. CX and CL performed additional analysis on the Omnitrap-Orbitrap system. JW, NT, and CL did the data interpretation. JAK assisted with spectral deconvolution by ms_deisotope. PH performed automated spectral interpretation by GlycoDeNovo. JW and CL co-wrote the manuscript. All authors contributed to the editing and revision of the manuscript.Conflicts of interest
DT, MK, and AS are affiliated with Fasmatech, the inventor and manufacturer of the Omnitrap instrument.Acknowledgements
This work was supported by NIH grants R01 GM132675, R01 GM133963, R24 GM134210, S10 RR025082, and NSFC grant 82204330. The content is solely the responsibility of the authors and does not represent the official views of the funding agency.References
- K. Ohtsubo and J. D. Marth, Glycosylation in cellular mechanisms of health and disease, Cell, 2006, 126, 855–867 CrossRef CAS PubMed.
- A. Varki, R. D. Cummings, J. D. Esko, H. H. Freeze, P. Stanley, C. R. Bertozzi, G. W. Hart and M. E. Etzler, Essentials of Glycobiology, Cold Spring Harbor Laboratory Press, 2009 Search PubMed.
- K. W. Moremen, M. Tiemeyer and A. V. Nairn, Vertebrate protein glycosylation: diversity, synthesis and function, Nat. Rev. Mol. Cell Biol., 2012, 13, 448–462 CrossRef CAS PubMed.
- C. Reily, T. J. Stewart, M. B. Renfrow and J. Novak, Glycosylation in health and disease, Nat. Rev. Nephrol., 2019, 15, 346–366 CrossRef PubMed.
- K. T. Schjoldager, Y. Narimatsu, H. J. Joshi and H. Clausen, Global view of human protein glycosylation pathways and functions, Nat. Rev. Mol. Cell Biol., 2020, 21, 729–749 CrossRef CAS PubMed.
- M. D. Battistel, H. F. Azurmendi, B. Yu and D. I. Freedberg, NMR of glycans: shedding new light on old problems, Prog. Nucl. Magn. Reson. Spectrosc., 2014, 79, 48–68 CrossRef CAS PubMed.
- J. Zaia, Mass spectrometry and the emerging field of glycomics, Chem. Biol., 2008, 15, 881–892 CrossRef CAS PubMed.
- S. J. North, P. G. Hitchen, S. M. Haslam and A. Dell, Mass spectrometry in the analysis of N-linked and O-linked glycans, Curr. Opin. Struct. Biol., 2009, 19, 498–506 CrossRef CAS PubMed.
- X. Dong, Y. Huang, B. G. Cho, J. Zhong, S. Gautam, W. Peng, S. D. Williamson, A. Banazadeh, K. Y. Torres-Ulloa and Y. Mechref, Advances in mass spectrometry-based glycomics, Electrophoresis, 2018, 39, 3063–3081 CrossRef CAS PubMed.
- V. Reinhold, H. Zhang, A. Hanneman and D. Ashline, Toward a platform for comprehensive glycan sequencing, Mol. Cell. Proteomics, 2013, 12, 866–873 CrossRef CAS PubMed.
- D. J. Ashline, Y. Yu, Y. Lasanajak, X. Song, L. Hu, S. Ramani, V. Prasad, M. K. Estes, R. D. Cummings and D. F. Smith, Structural characterization by multistage mass spectrometry (MSn) of human milk glycans recognized by human rotaviruses, Mol. Cell. Proteomics, 2014, 13, 2961–2974 CrossRef CAS PubMed.
- D. J. Harvey, Negative ion mass spectrometry for the analysis of N-linked glycans, Mass Spectrom. Rev., 2020, 39, 586–679 CrossRef CAS PubMed.
- S. Yan, L. Brecker, C. Jin, A. Titz, M. Dragosits, N. G. Karlsson, V. Jantsch, I. B. Wilson and K. Paschinger, Bisecting galactose as a feature of N-glycans of wild-type and mutant Caenorhabditis elegans, Mol. Cell. Proteomics, 2015, 14, 2111–2125 CrossRef CAS PubMed.
- J. Zhao, S. Li, C. Li, S.-L. Wu, W. Xu, Y. Chen, M. Shameem, D. Richardson and H. Li, Identification of low abundant isomeric N-glycan structures in biological therapeutics by LC/MS, Anal. Chem., 2016, 88, 7049–7059 CrossRef CAS PubMed.
- J. Jiao, H. Zhang and V. N. Reinhold, High performance IT-MS sequencing of glycans (spatial resolution of ovalbumin isomers), Int. J. Mass Spectrom., 2011, 303, 109–117 CrossRef CAS PubMed.
- J. M. Prien, D. J. Ashline, A. J. Lapadula, H. Zhang and V. N. Reinhold, The high mannose glycans from bovine ribonuclease B isomer characterization by ion trap MS, J. Am. Soc. Mass Spectrom., 2009, 20, 539–556 CrossRef CAS PubMed.
- D. J. Ashline, H. Zhang and V. N. Reinhold, Isomeric complexity of glycosylation documented by MS n, Anal. Bioanal. Chem., 2017, 409, 439–451 CrossRef CAS PubMed.
- S. T. Tsai, C. Y. Liew, C. Hsu, S. P. Huang, W. C. Weng, Y. H. Kuo and C. K. Ni, Automatic full glycan structural determination through logically derived sequence tandem mass spectrometry, ChemBioChem, 2019, 20, 2272 CrossRef CAS.
- S. Sun, C. Huang, Y. Wang, Y. Liu, J. Zhang, J. Zhou, F. Gao, F. Yang, R. Chen, B. Mulloy, W. Chai, Y. Li and D. Bu, Toward automated identification of glycan branching patterns using multistage mass spectrometry with intelligent precursor selection, Anal. Chem., 2018, 90, 14412–14422 CrossRef CAS PubMed.
- C. Y. Liew, C.-C. Yen, J.-L. Chen, S.-T. Tsai, S. Pawar, C.-Y. Wu and C.-K. Ni, Structural identification of N-glycan isomers using logically derived sequence tandem mass spectrometry, Commun. Chem., 2021, 4, 92 CrossRef CAS PubMed.
- J. Gao, D. A. Thomas, C. H. Sohn and J. L. Beauchamp, Biomimetic reagents for the selective free radical and acid–base chemistry of glycans: application to glycan structure determination by mass spectrometry, J. Am. Chem. Soc., 2013, 135, 10684–10692 CrossRef CAS PubMed.
- N. Desai, D. A. Thomas, J. Lee, J. Gao and J. L. Beauchamp, Eradicating mass spectrometric glycan rearrangement by utilizing free radicals, Chem. Sci., 2016, 7, 5390–5397 RSC.
- A. Devakumar, Y. Mechref, P. Kang, M. V. Novotny and J. P. Reilly, Identification of isomeric N-glycan structures by mass spectrometry with 157 nm laser-induced photofragmentation, J. Am. Soc. Mass Spectrom., 2008, 19, 1027–1040 CrossRef CAS PubMed.
- B. J. Ko and J. S. Brodbelt, 193 nm ultraviolet photodissociation of deprotonated sialylated oligosaccharides, Anal. Chem., 2011, 83, 8192–8200 CrossRef CAS PubMed.
- L. E. Pepi, F. E. Leach III, D. R. Klein, J. S. Brodbelt and I. J. Amster, Investigation of the experimental parameters of ultraviolet photodissociation for the structural characterization of chondroitin sulfate glycosaminoglycan isomers, J. Am. Soc. Mass Spectrom., 2021, 32, 1759–1770 CrossRef CAS PubMed.
- D. Ropartz, P. Li, M. Fanuel, A. Giuliani, H. Rogniaux and G. P. Jackson, Charge transfer dissociation of complex oligosaccharides: comparison with collision-induced dissociation and extreme ultraviolet dissociative photoionization, J. Am. Soc. Mass Spectrom., 2016, 27, 1614–1619 CrossRef CAS PubMed.
- B. Budnik, K. Haselmann, Y. N. Elkin, V. Gorbach and R. Zubarev, Applications of electron-ion dissociation reactions for analysis of polycationic chitooligosaccharides in Fourier transform mass spectrometry, Anal. Chem., 2003, 75, 5994–6001 CrossRef CAS PubMed.
- J. T. Adamson and K. Hakansson, Electron capture dissociation of oligosaccharides ionized with alkali, alkaline earth, and transition metals, Anal. Chem., 2007, 79, 2901–2910 CrossRef CAS PubMed.
- J. J. Wolff, I. J. Amster, L. L. Chi and R. J. Linhardt, Electron detachment dissociation of glycosaminoglycan tetrasaccharides, J. Am. Soc. Mass Spectrom., 2007, 18, 234–244 CrossRef CAS PubMed.
- J. J. Wolff, T. N. Laremore, H. Aslam, R. J. Linhardt and I. J. Amster, Electron-induced dissociation of glycosaminoglycan tetrasaccharides, J. Am. Soc. Mass Spectrom., 2008, 19, 1449–1458 CrossRef CAS PubMed.
- C. Zhao, B. Xie, S. Y. Chan, C. E. Costello and P. B. O'Connor, Collisionally activated dissociation and electron capture dissociation provide complementary structural information for branched permethylated oligosaccharides, J. Am. Soc. Mass Spectrom., 2008, 19, 138–150 CrossRef CAS PubMed.
- J. J. Wolff, F. E. Leach, T. N. Laremore, D. A. Kaplan, M. L. Easterling, R. J. Linhardt and I. J. Amster, Negative electron transfer dissociation of glycosaminoglycans, Anal. Chem., 2010, 82, 3460–3466 CrossRef CAS PubMed.
- X. Yu, Y. Huang, C. Lin and C. E. Costello, Energy-dependent electron activated dissociation of metal-adducted permethylated oligosaccharides, Anal. Chem., 2012, 84, 7487–7494 CrossRef CAS PubMed.
- X. Yu, Y. Jiang, Y. Chen, Y. Huang, C. E. Costello and C. Lin, Detailed glycan structural characterization by electronic excitation dissociation, Anal. Chem., 2013, 85, 10017–10021 CrossRef CAS PubMed.
- Y. Tang, Y. Pu, J. Gao, P. Hong, C. E. Costello and C. Lin, De novo glycan sequencing by electronic excitation dissociation and fixed-charge derivatization, Anal. Chem., 2018, 90, 3793–3801 CrossRef CAS PubMed.
- Y. Tang, J. Wei, C. E. Costello and C. Lin, Characterization of Isomeric Glycans by Reversed Phase Liquid Chromatography-Electronic Excitation Dissociation Tandem Mass Spectrometry, J. Am. Soc. Mass Spectrom., 2018, 29, 1295–1307 CrossRef CAS PubMed.
- J. Wei, Y. Tang, M. E. Ridgeway, M. A. Park, C. E. Costello and C. Lin, Accurate identification of isomeric glycans by trapped ion mobility spectrometry-electronic excitation dissociation tandem mass spectrometry, Anal. Chem., 2020, 92, 13211–13220 CrossRef CAS PubMed.
- H.-T. K. Wong, X. Chen, R. Wu, Y.-L. E. Wong, Y.-L. W. Hung and T.-W. D. Chan, Dissociation of mannose-rich glycans using collision-based and electron-based ion activation methods, J. Am. Soc. Mass Spectrom., 2022, 33, 803–812 CrossRef CAS PubMed.
- Y. Pu, M. E. Ridgeway, R. S. Glaskin, M. A. Park, C. E. Costello and C. Lin, Separation and identification of isomeric glycans by selected accumulation-trapped ion mobility spectrometry-electron activated dissociation tandem mass spectrometry, Anal. Chem., 2016, 88, 3440–3443 CrossRef CAS PubMed.
- J. Wei, Y. Tang, Y. Bai, J. Zaia, C. E. Costello, P. Hong and C. Lin, Toward automatic and comprehensive glycan characterization by online PGC-LC-EED MS/MS, Anal. Chem., 2020, 92, 782–791 CrossRef CAS PubMed.
- P. Hong, H. Sun, L. Sha, Y. Pu, K. Khatri, X. Yu, Y. Tang and C. Lin, GlycoDeNovo–an efficient algorithm for accurate de novo glycan topology reconstruction from tandem mass spectra, J. Am. Soc. Mass Spectrom., 2017, 28, 2288–2301 CrossRef CAS PubMed.
- T. Baba, P. Ryumin, E. Duchoslav, K. Chen, A. Chelur, B. Loyd and I. Chernushevich, Dissociation of biomolecules by an intense low-energy electron beam in a high sensitivity time-of-flight mass spectrometer, J. Am. Soc. Mass Spectrom., 2021, 32, 1964–1975 CrossRef CAS PubMed.
- L. H. Di Stefano, D. Papanastasiou and R. A. Zubarev, Size-dependent hydrogen atom attachment to gas-phase hydrogen-deficient polypeptide radical cations, J. Am. Chem. Soc., 2018, 140, 531–533 CrossRef CAS PubMed.
- J. B. Shaw, N. Malhan, Y. V. Vasil’ev, N. I. Lopez, A. Makarov, J. S. Beckman and V. G. Voinov, Sequencing grade tandem mass spectrometry for top-down proteomics using hybrid electron capture dissociation methods in a benchtop orbitrap mass spectrometer, Anal. Chem., 2018, 90, 10819–10827 CrossRef CAS PubMed.
- K. L. Fort, C. N. Cramer, V. G. Voinov, Y. V. Vasil’ev, N. I. Lopez, J. S. Beckman and A. J. R. Heck, Exploring ECD on a benchtop Q Exactive orbitrap mass spectrometer, J. Proteome Res., 2018, 17, 926–933 CrossRef CAS PubMed.
- K. Jeanne Dit Fouque, D. Kaplan, V. G. Voinov, F. H. V. Holck, O. N. Jensen and F. Fernandez-Lima, Proteoform differentiation using tandem trapped ion mobility, electron capture dissociation, and ToF mass spectrometry, Anal. Chem., 2021, 93, 9575–9582 CrossRef CAS PubMed.
- J. B. Shaw, W. Liu, Y. V. Vasil’ev, C. C. Bracken, N. Malhan, A. Guthals, J. S. Beckman and V. G. Voinov, Direct determination of antibody chain pairing by top-down and middle-down mass spectrometry using electron capture dissociation and ultraviolet photodissociation, Anal. Chem., 2020, 92, 766–773 CrossRef CAS PubMed.
- J. B. Shaw, D. A. Cooper-Shepherd, D. Hewitt, J. L. Wildgoose, J. S. Beckman, J. I. Langridge and V. G. Voinov, Enhanced top-down protein characterization with electron capture dissociation and cyclic ion mobility spectrometry, Anal. Chem., 2022, 94, 3888–3896 CrossRef CAS PubMed.
- T. Baba, K. Rajabi, S. Liu, P. Ryumin, Z. Zhang, K. Pohl, J. Causon, J. Y. Le Blanc and M. Kurogochi, Electron impact excitation of ions from organics on singly protonated peptides with and without post-translational modifications, J. Am. Soc. Mass Spectrom., 2022, 33, 1723–1732 CrossRef CAS PubMed.
- D. Papanastasiou, D. Kounadis, A. Lekkas, I. Orfanopoulos, A. Mpozatzidis, A. Smyrnakis, E. Panagiotopoulos, M. Kosmopoulou, M. Reinhardt-Szyba, K. Fort, A. Makarov and R. A. Zubarev, The omnitrap platform: a versatile segmented linear ion trap for multidimensional multiple-stage tandem mass spectrometry, J. Am. Soc. Mass Spectrom., 2022, 33, 1990–2007 CrossRef CAS PubMed.
- P. Kang, Y. Mechref, I. Klouckova and M. V. Novotny, Solid-phase permethylation of glycans for mass spectrometric analysis, Rapid Commun. Mass Spectrom., 2005, 19, 3421–3428 CrossRef CAS PubMed.
- B. Domon and C. E. Costello, A systematic nomenclature for carbohydrate fragmentations in FAB-MS/MS spectra of glycoconjugates, Glycoconjugate J., 1988, 5, 397–409 CrossRef CAS.
- A. Ceroni, K. Maass, H. Geyer, R. Geyer, A. Dell and S. M. Haslam, GlycoWorkbench: a tool for the computer-assisted annotation of mass spectra of glycans, J. Proteome Res., 2008, 7, 1650–1659 CrossRef CAS PubMed.
- Y. E. Fung, C. M. Adams and R. A. Zubarev, Electron ionization dissociation of singly and multiply charged peptides, J. Am. Chem. Soc., 2009, 131, 9977–9985 CrossRef CAS PubMed.
- Y. Huang, Y. Pu, X. Yu, C. E. Costello and C. Lin, Mechanistic study on electronic excitation dissociation of the cellobiose-Na+ complex, J. Am. Soc. Mass Spectrom., 2015, 27, 319–328 CrossRef PubMed.
- D. J. Ashline, A. J. Lapadula, Y.-H. Liu, M. Lin, M. Grace, B. Pramanik and V. N. Reinhold, Carbohydrate structural isomers analyzed by sequential mass spectrometry, Anal. Chem., 2007, 79, 3830–3842 CrossRef CAS PubMed.
- J. M. Prien, D. J. Ashline, A. J. Lapadula, H. Zhang and V. N. Reinhold, The high mannose glycans from bovine ribonuclease B isomer characterization by ion trap MS, J. Am. Soc. Mass Spectrom., 2008, 20, 539–556 CrossRef PubMed.
- J. Jiao, H. Zhang and V. N. Reinhold, High performance IT-MSn sequencing of glycans: Spatial resolution of ovalbumin isomers, Int. J. Mass Spectrom., 2011, 303, 109–117 CrossRef CAS PubMed.
- S. Sun, C. Huang, Y. Wang, Y. Liu, J. Zhang, J. Zhou, F. Gao, F. Yang, R. Chen and B. Mulloy, Toward automated identification of glycan branching patterns using multistage mass spectrometry with intelligent precursor selection, Anal. Chem., 2018, 90, 14412–14422 CrossRef CAS PubMed.
- C. Huang, H. Wang, D. Bu, J. Zhou, J. Dong, J. Zhang, H. Gao, Y. Wang, W. Chai and S. Sun, Multistage mass spectrometry with intelligent precursor selection for N-glycan branching pattern analysis, Carbohydr. Polym., 2020, 237, 116122 CrossRef CAS PubMed.
- H. C. Hsu, C. Y. Liew, S.-P. Huang, S.-T. Tsai and C.-K. Ni, Simple method for de novo structural determination of underivatised glucose oligosaccharides, Sci. Rep., 2018, 8, 1–12 Search PubMed.
- S.-P. Huang, H. C. Hsu, C. Y. Liew, S.-T. Tsai and C.-K. Ni, Logically derived sequence tandem mass spectrometry for structural determination of galactose oligosaccharides, Glycoconjugate J., 2021, 38, 177–189 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sc00870c |
| This journal is © The Royal Society of Chemistry 2023 |