
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePalladium mono-N-protected amino acid complexes: experimental validation of the ligand cooperation model in C–H activation†
Sara
Fernández-Moyano
 ,
Vanesa
Salamanca
and
Ana C.
Albéniz
*
,
Vanesa
Salamanca
and
Ana C.
Albéniz
*
IU CINQUIMA/Química Inorgánica, Universidad de Valladolid, 47071-Valladolid, Spain. E-mail: albeniz@uva.es
First published on 26th May 2023
Abstract
Mechanistic proposals for the C–H activation reaction enabled by mono-N-protected amino acid ligands (MPAAs) have been supported by DFT calculations. The direct experimental observation of the ligand-assisted C–H activation has not yet been reported due to the lack of well-defined isolated palladium complexes with MPAAs that can serve as models. In this work, palladium complexes bearing chelating MPAAs (NBu4)[Pd(κ2-N,O–AcN–CHR–COO)(C6F5)py] (Ac = MeC(O); R = H, Me) and [Pd(κ2-N,O–MeNH–CH2–COO)(C6F5)py] have been isolated and characterized. Their evolution in a solution containing toluene leads to the C–H activation of the arene and the formation of the C6F5–C6H4Me coupling products. This process takes place only for the ligands with an acyl protecting group, showing the cooperating role of this group in a complex with a chelating MPAA, therefore experimentally validating this working model. The carboxylate group is inefficient in this C–H activation.
Introduction
Mono-N-protected amino acids (MPAAs) are extremely important ligands in palladium catalyzed C–H functionalization reactions.1 Yu's group introduced them in 2008 and they showed that these ligands strongly accelerated coupling reactions that involved a C–H activation.2 They also demonstrated the utility of these ligands in enantioselective C–H functionalization.2–4 Since then, MPAAs have been used in a myriad of transformations of C(sp3)–H and C(sp2)–H bonds and they are one of the most important types of cooperating ligands in these reactions.5,6MPAAs usually conform to the formulation PGNH-backbone-COOH where PG = acyl group. The presence of several donor atoms of moderate coordination ability makes MPAAs potential polydentate ligands that can act in a mono and dianionic forms. Therefore, different coordination modes to the metal are possible, which complicate the study of these systems, but this is important in the metal speciation in a catalytic process. Possible binding modes are: monodentate (κ1-O)− (X ligand), chelating (κ2-N,O)− (XL ligand), chelating (κ2-N,O)2− (X2 ligand) and bridging (μ2-O,O)− (μ-XL ligand), the latter leading to polynuclear complexes (Scheme 1).
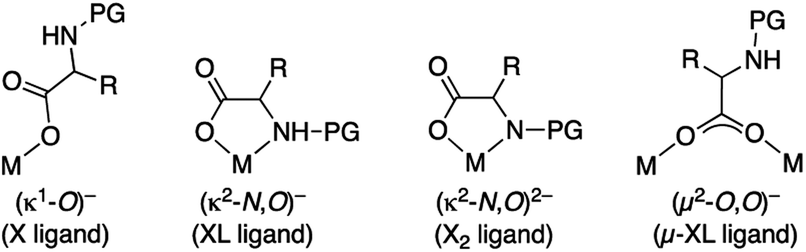 | ||
| Scheme 1 Coordination modes of mono-N-protected amino acids. | ||
The role of MPAAs in catalytic C–H functionalization can be manifold and these ligands can be involved in the stabilization of intermediates or in other steps of the catalysis different from the C–H cleavage.7,8 However their main role is to facilitate the C–H activation step and the importance of the presence of the acyl monoprotection to enable this process was early recognized.2,5b This protecting group exerts a cooperating role in the C–H cleavage via the involvement of the acyl oxygen in a concerted metalation deprotonation (CMD) mechanism. It has been proposed that the MPAA is coordinated to palladium in a chelating dianionic (κ2-N,O)2− form,1c,9 and the transfer of the C–H hydrogen to the acyl oxygen occurs in the transition state, transforming the dianionic MPAA into a monoanionic ligand (Scheme 2(a)).10 This working model also explains the asymmetric induction observed in enantioselective C–H functionalization,11 and it is commonly proposed with the support of computational studies.12 However, the situation can be more complex and it is worth noting that calculations have also supported that the C–H activation in a MPAA–Pd mononuclear complex can be assisted by an external base such as acetate or carbonate (outer sphere CMD) and, in fact, the energy difference between both pathways can be small.13 In addition, just the carboxylate moiety in the MPAA can assist the C–H activation with no involvement of the protecting group as found in some computational studies (Scheme 2(b)).14 Dinuclear rather than mononuclear palladium complexes have been proposed as relevant species in the C–H activation step and, in the reported example, calculations show that a bridging (μ2-O,O)− MPAA is present but the amino acid is not directly involved in the C–H cleavage, which is assisted by an additional coordinated acetate.15 For C–H functionalizations that use silver bases, the formation of heterometallic Pd–Ag species has been suggested based on DFT calculations. In this case a CMD process is assisted by the MPAA carboxylate moiety as shown in Scheme 2(b).14
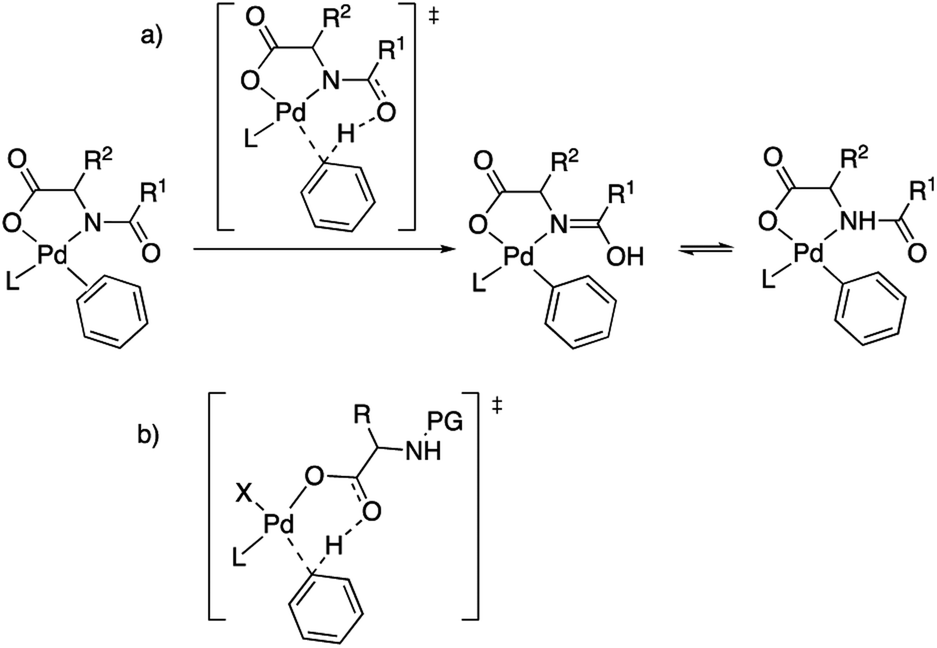 | ||
| Scheme 2 Models for MPAA cooperation in C–H activation: (a) chelating model with assistance of the acyl protecting group; (b) carboxylate assistance with no involvement of the protecting group. | ||
Mechanistic studies on some C–H functionalization reactions enabled by MPAAs, usually alkenylation reactions of arenes, have inferred the formation of mononuclear MPAA–palladium species during catalysis by kinetic experiments,16 mass spectrometry studies,10,11a,12b,17 and diffusion NMR spectroscopy.12b However, the actual C–H activation step has never been modeled experimentally. The analysis of the optimal Pd/MPAA ratio for the acceleration of a C–H functionalization reaction has been analyzed as an indication of the nuclearity of the catalytic species in the C–H cleavage step. This analysis can be complicated by the fact that, because MPAAs can significantly lower the barrier for C–H activation, other steps in the catalytic cycle can become turnover limiting. In some cases, MPAAs can have a detrimental effect on other steps in the catalytic cycle and this has been shown by Stahl et al.18 Thus, the optimal Pd/MPAA ratio can be the result of a compromise between opposing effects of the ligand during the catalytic cycle and it may not reflect the actual composition of the C–H activation transition state. The experimental validation of the proposed model for C–H activation enabled by MPAAs requires the observation of this step in a system where the coordination mode of the amino acid is well-stablished and the role of the MPAA in the C–H cleavage can be separated from the influence of other additives (bases, OAc−, etc.). We describe here the synthesis of well-defined palladium complexes with chelating MPAAs and we show that they react with arenes via C–H activation. By comparing acyl and methyl protecting groups, it is possible to unequivocally show the ability of acyl-MPAAs to induce the C–H cleavage via metal–ligand cooperation.
Results and discussion
Palladium complexes with a chelating N-acetylglycine (2) and N-acetylvaline (3) were prepared by reaction of (NBu4)[PdBr2(C6F5)py] (1) and the corresponding amino acid ligand in the presence of silver carbonate as shown in eqn (1). Both complexes bear a pentafluorophenyl group that allowed us to monitor the C–H activation reactions by detecting the coupling products C6F5–aryl (see below). This strategy has proved useful in our former studies on the role of cooperating bipyridone ligands in C–H activation.19 The presence of a fluorinated tag is also very useful in the characterization of the species formed. Pyridine, a ligand of moderate coordination ability, completes the coordination sphere of palladium and imparts enough stability to the complexes without blocking further reactivity by ligand substitution.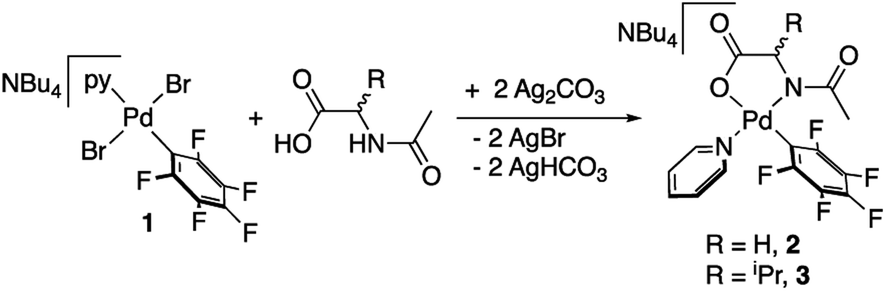 | (1) |
Complexes 2 and 3 were characterized by NMR, elemental analyses and mass spectrometry. The MPAA ligand is coordinated in a chelating dianionic (κ2-N,O)2− form. A 1H–19F HOESY NMR experiment shows a cross peak between the methyl substituent of the acetyl group and the Fortho of the perfluoroaryl indicating, therefore, that they are close to each other in space (see Fig. S30 and S35, in the ESI†). This experiment confirms the coordination of the nitrogen of the amino acid to palladium and unequivocally determines the stereochemistry of the complexes. In addition, no dissociation of the pyridine ligand was observed in solution and the complexes are monomeric as shown by 19F NMR DOSY experiments (see Section 1.6, ESI†). We could not obtain crystals suitable for X-ray diffraction and attempts at crystallization of complex 2 only led to the ligand reorganization and the formation of the bis MPAA complex 4 and the bis-aryl 5 that crystallized out (eqn (2)). The molecular structures of these complexes are shown in Fig. 1.20 This reorganization is slow at temperatures equal or below 298 K and the formation of 4 and 5 occurs after keeping layered solutions of 2 for long periods of time.
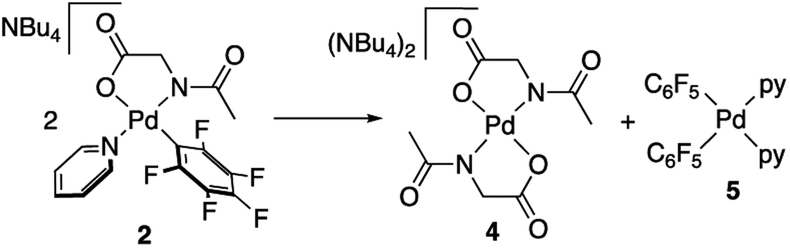 | (2) |
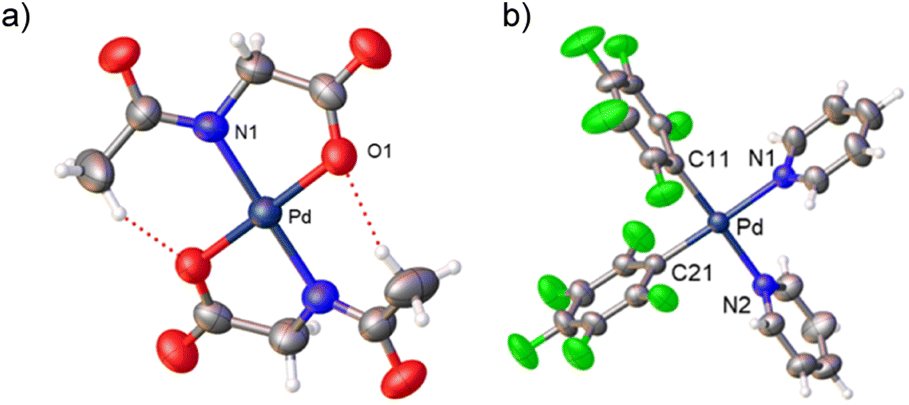 | ||
| Fig. 1 X-ray molecular structure of complexes 4 ((a) NBu4+ omitted for clarity) and 5 (b) (ORTEP, 40% probability). Selected bond lengths (Å): 4, Pd–N(1) = 2.018(3), Pd–O(1) = 2.002(3); 5, Pd–N(1) = 2.104(3); Pd–N(2) = 2.104(3); Pd–C(11) = 1.994(3); Pd–C(21) = 1.994(3). | ||
Derivatives of N-methylglycine (sarcosine) were also prepared. In this case the alternative synthetic route depicted in Scheme 3 was used. The amino acid is acting as a chelating monoanionic (κ2-N,O)− ligand in complexes 7 and 8. The less acidic NH moiety in sarcosine does not deprotonate even when carbonate is used as a base. In fact, the synthetic route depicted in eqn (1) only gives complex 8 when sarcosine is used albeit in lower yield than that in the procedure shown in Scheme 3. Complexes 7 and 8 were completely characterized and Fig. 2 shows the molecular structure of complex 8 (see Fig. S16† for complex 7).20 It is noteworthy that both derivatives are monomeric showing the higher preference of the monoanionic amino acid ligand to bind in a chelating fashion rather than a dimeric form with a bridging (μ2-O,O)− coordination mode.
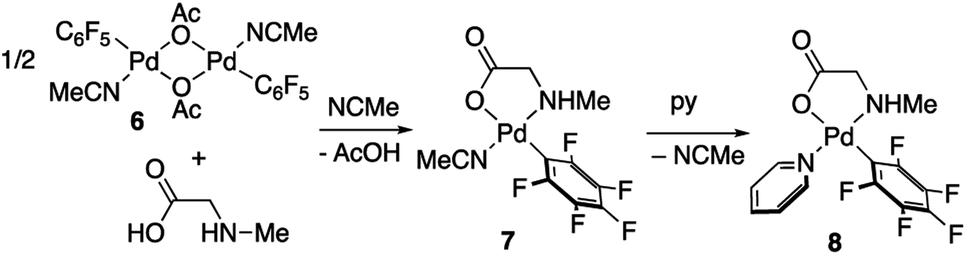 | ||
| Scheme 3 Synthesis of complexes 7 and 8. | ||
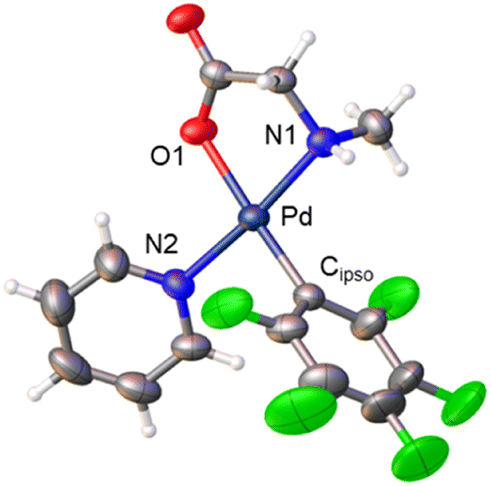 | ||
| Fig. 2 X-ray molecular structure of complex 8 (ORTEP, 40% probability). Selected bond lengths (Å): Pd–O(1) = 2.065(3); Pd–N(1) = 2.045(3); Pd–N(2) = 2.039(3); Pd–Cipso = 1.995(5). | ||
With these complexes at hand we tested their ability to enable the C–H activation process by heating them up in the presence of an arene and in the absence of other reagents (Table 1). When the N-acetylglycine derivative 2 was heated either in toluene or in ethylbenzoate at 130 °C the C–H functionalization of the arene was observed and the biaryl coupling products C6F5–C6H4R (R = Me, COOEt) were formed as major decomposition products in 30 min (entries 1 and 2, Table 1).
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | Complex | Solvent | Time | Conversion, % | C6F5–arene, % | C6F5H, % | “Pd(C6F5)2”,b % |
| a Reaction temperature: 130 °C. The percentages shown correspond to the mol% of fluorinated compounds and they were determined by integration of 19F NMR signals. b Pd-species formed in parenthesis. c Other unidentified Pd–C6F5 complexes account for the remaining 12% in entry 1 and 37% in entry 9. d Reaction temperature: 90 °C due to the low boiling point of HFIP. | |||||||
| 1 | 2 | Toluene | 30 min | 82 | 53 | 17 | —c |
| 2 | 2 | PhCOOEt | 30 min | 100 | 83 | 17 | — |
| 3 | 2 | Pyridine | 30 min | 10 | 6 | — | 4 ([Pd(C6F5)2py2]) |
| 4 | 2 | Toluene/DMA (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) :1) |
30 min | 100 | 75 | 12 | 13 (9) |
| 5 | 2 | Toluene/DMA (1:1) |
60 min | 100 | 79 | 21 | — |
| 6 | 3 | Toluene/DMA (1:1) |
60 min | 100 | 77 | 23 | — |
| 7 | 8 | Toluene/DMA (1:1) |
60 min | 38 | 10 | 5 | 23 ([Pd(C6F5)2py2]) |
| 8 | 9 | Toluene/DMA (1:1) |
60 min | 42 | 16 | 26 | — |
| 9d | 2 | Toluene/HFIP (1:1) |
60 min | 100 | 31 | 27 | 5 (C6F5–C6F5)c |
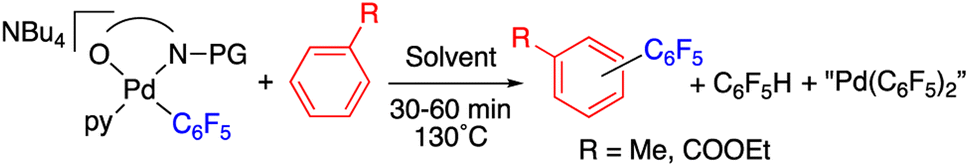
Complex 2 is very stable in pyridine and the product of the C–H arylation of this arene is only observed as a minor product (entry 3, Table 1). The C–H functionalization of toluene occurs when the arene is used as reactant and solvent as well as in a mixture of the arene and a cosolvent such as DMA (entries 1 and 4, Table 1). HFIP was also tested and the coupling products were observed, albeit in lower amount (entry 9). The C–H activation products were obtained as a mixture of regioisomers, the meta substitution being the predominant one (Table S1, ESI†). Pentafluorobenzene and aryl reorganization complexes bearing the “Pd(C6F5)2” moiety were also observed as byproducts. The formation of bispentafluorophenyl complexes most probably occurs by transmetalation of C6F5 groups from one Pd to another as observed and studied before.21
The C–H functionalization occurs for the acyl protected amino acid complexes 2 and 3, whereas it is inefficient for the methyl protected sarcosine derivative 8 (cf. entries 5–7, Table 1). This experimentally shows the feasibility of the chelating model of assistance of the C–H activation with the direct involvement of the acyl group. It also shows that the presence of the amino acid carboxylate moiety is not enough for an efficient C–H cleavage (entry 7, Table 1).
The reaction of complex 2 in a mixture of toluene/DMA was monitored by 19F NMR at 90 °C. Fig. 3 shows the course of the reaction. In addition to the toluene C–H functionalization products and a small amount of C6F5H, a new organometallic product (9) was formed (eqn (3)). This complex was also observed when the decomposition was carried out at 130 °C for short reaction times (Fig. 4 and entry 4, Table 1).
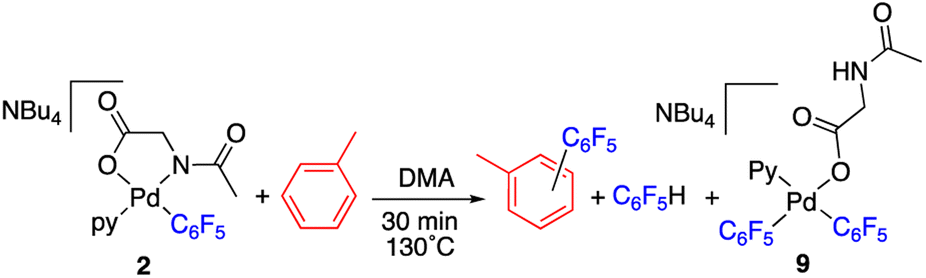 | (3) |
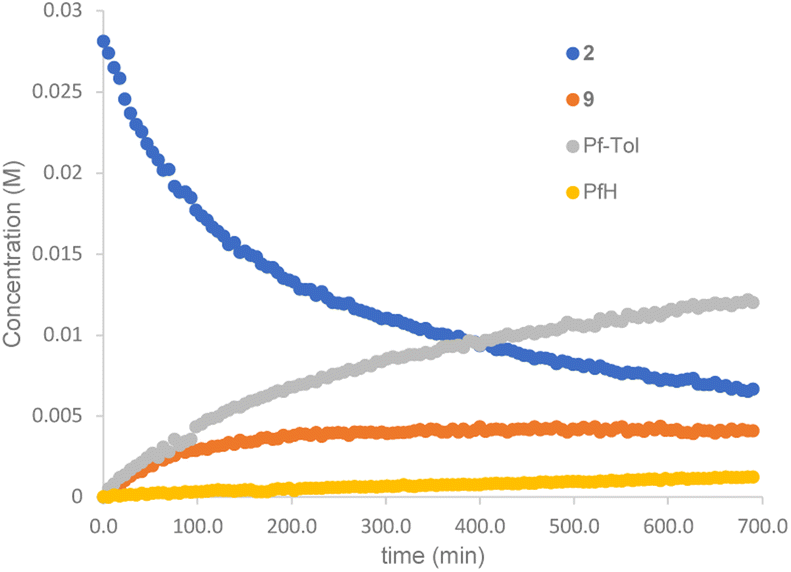 | ||
| Fig. 3 19F NMR follow up of the decomposition of complex 2 in a mixture of toluene/DMA at 90 °C (Pf = C6F5). | ||
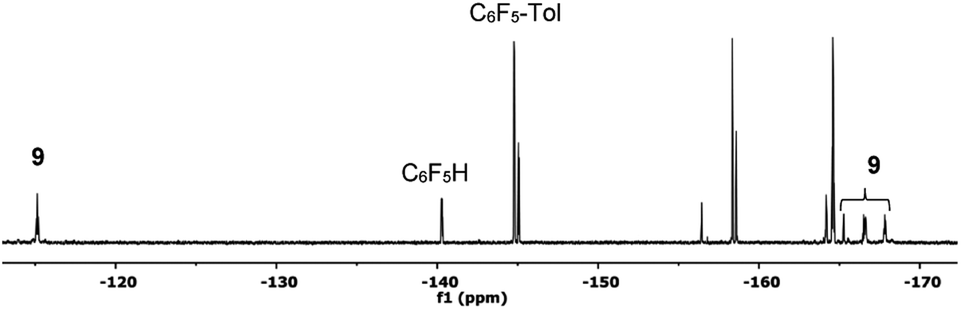 | ||
| Fig. 4 19F NMR spectrum (470.17 MHz) of the product mixture obtained after heating complex 2 at 130 °C in toluene/DMA (1/1) for 30 min (entry 4, Table 1). Only the Fortho region for the organic products has been labelled for clarity. | ||
Complex 9 shows the characteristic NMR pattern of a “Pd(C6F5)2” moiety where both aryls are chemically inequivalent. After thorough experimentation, 9 could be identified as a palladium derivative with a monodentate (κ1-O)− amino acid ligand. This derivative was independently synthesized as shown in Scheme 4 and it is a rare example of this coordination mode for a MPAA ligand.22 Complex 9 was characterized by NMR and its formulation and monomeric nature was confirmed by DOSY experiments (Section 1.6, ESI†).
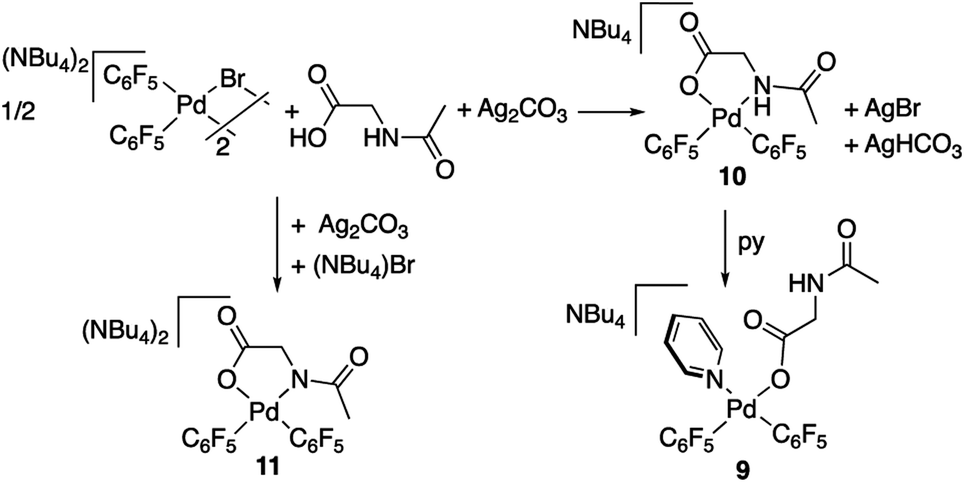 | ||
| Scheme 4 Synthesis of bis-pentafluorophenyl palladium complexes with the N-acetylglycine ligand. | ||
The decomposition of complex 9 occurs slowly in toluene/DMA at 130 °C to give the biaryl coupling product and C6F5H, the latter in a higher relative molar amount than that observed for complex 2 (entry 8, Table 1 and Fig. S4†). This product distribution is consistent with the reaction pathway shown in Scheme 5. First, the protonation of one of the pentafluorophenyl groups of the complex takes place and this could occur by the amino acid acting as a hydrogen donor. This process leads to the formation of complex 2 that is responsible for the C–H activation of toluene.
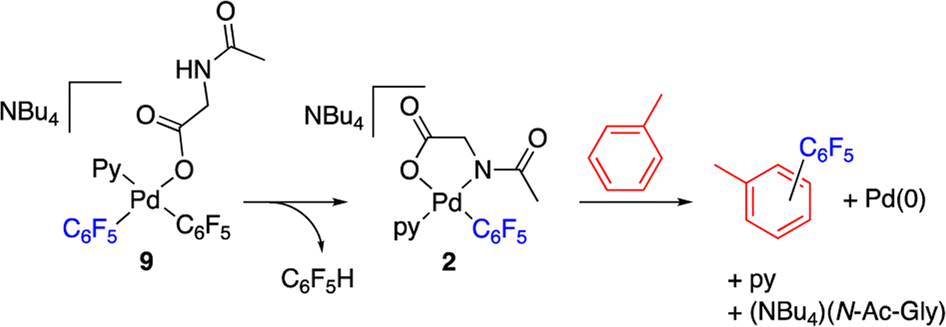 | ||
| Scheme 5 Plausible decomposition route of complex 9via C–H activation. | ||
The reaction of complex 2 in toluene was modelled by DFT calculations using the M06 functional and including solvation in the optimizations through the SMD implicit solvent method (see the ESI† for details). The reaction profile for the formation of C6F5–C6H4–m-Me is represented in Fig. 5 and the resulting energy barriers are consistent with the experimental conditions needed for the decomposition of 2. The C–H activation of toluene assisted by the MPAA requires the cis arrangement of the coordinated arene and amido fragment of the amino acid ligand (intermediate c1). In the case of complex 2 this requires a cis–trans isomerization and a rotation of the acyl group around the C–N bond. The energy difference between complex 2 and the isomer closest to the required stereochemistry (2brot) is not high (less than 5 kcal mol−1) and the calculated barrier for rotation about the C(acyl)–N bond of the amino acid is 18.6 kcal mol−1, much lower than the barrier for C–H activation (see Fig. S56, ESI†). Therefore, at the temperature needed for the decomposition of the complex, both processes are expected to be faster than the subsequent C–H cleavage. The overall barrier for the C–H activation from 2 is 29.2 kcal mol−1 and it is determined by a CMD transition state with the assistance of the acyl oxygen. The formation of the coupling product follows, by reductive elimination of the pentafluorophenyl and the tolyl groups. This process is very energy demanding as expected for the perfluorinated aryl group and, in fact, the reductive elimination step shows the higher energy barrier in the calculated reaction profile. The measured kinetic isotope effect, determined in separate experiments, has a low value, KIE = 1.4 ± 0.1, and it is consistent with a scenario where the C–H cleavage is not controlling the reaction rate and there is a subsequent step of higher energy barrier. Since the C–H activation precedes this step a measurable but small KIE may be observed derived from the different equilibrium concentrations of c2.23 The barriers for reductive elimination are much lower for non-polyfluorinated aryls,19b,24 and, therefore, in these cases the C–H activation step is expected to be the one with highest energy barrier.‡ It has to be noted that, by itself, the low KIE value observed does not rule out other possibilities such as the existence of a previous step close in energy to the C–H activation, or an electrophilic metalation pathway for the C–H activation (secondary KIE by rehybridization). Nonetheless, the latter is less likely for the anionic electron rich complexes involved in these reactions.
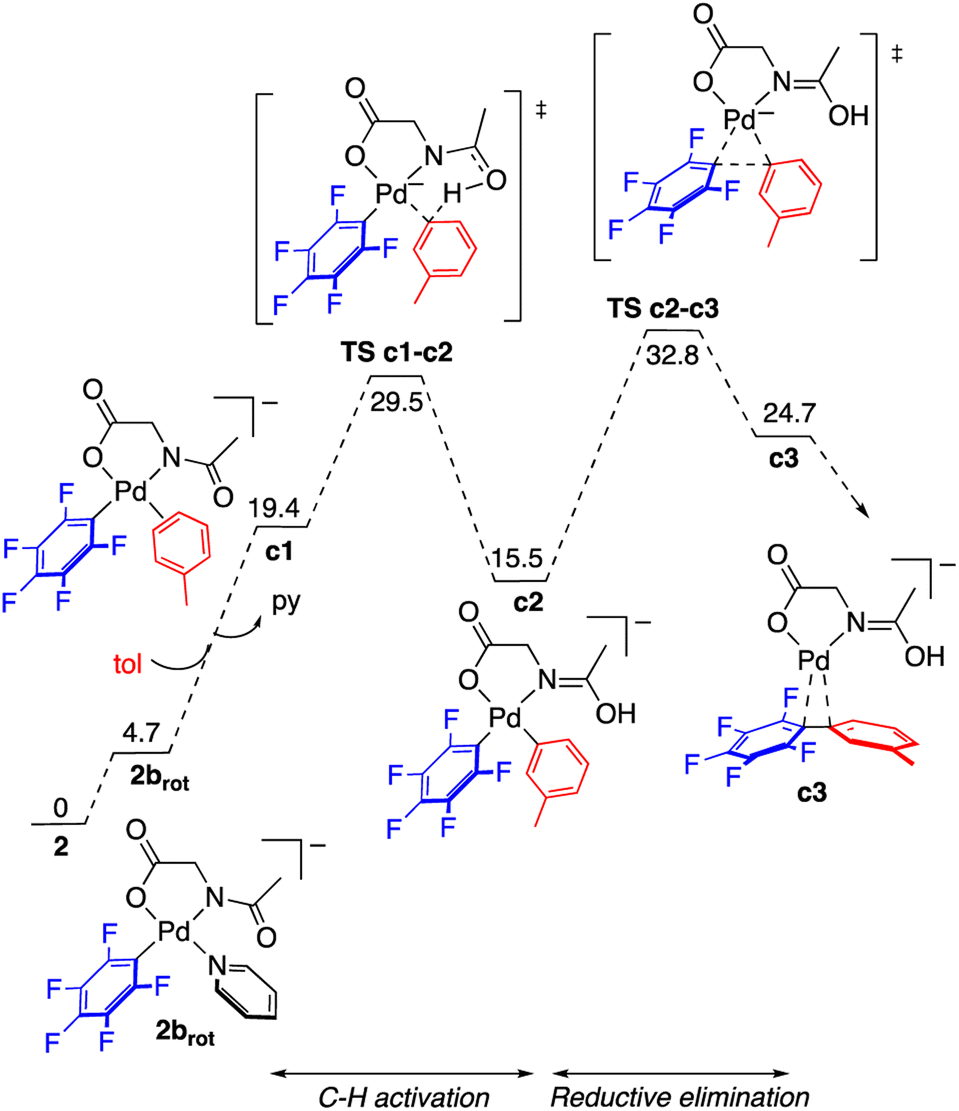 | ||
| Fig. 5 Gibbs energy profile for the formation of the C–H functionalization product C6F5–C6H4–m-Me from complex 2 in toluene at 130 °C (energies in kcal mol−1). | ||
The palladium complexes with coordinated N-acetylglycinates 9–11 depicted in Scheme 4 show some interesting features regarding the coordination modes of this ligand. The protonated nitrogen atom of the monoanionic N-acetylglycinate is a weak donor and complex 10 shows a fluxional behavior in solution consistent with the fast decoordination–recoordination of this atom. The 19F NMR spectrum of 10 shows broad signals at room temperature and equivalent pentafluorophenyl groups, which indicate a fast cis–trans isomerization and nitrogen inversion. This process could occur: (i) via dissociation of the NHAc group, topomerization and recoordination or (ii) via an associative NHAc substitution by the carboxylate and formation of intermediate carboxylato-bridged dimeric species (Scheme 6). The mass spectrum of 10 shows the monomeric complex depicted in Schemes 4 and 6 as the predominant species ([Pd(C6F5)2(AcNH–CH2COO)]−, m/z 555.9168) but it also shows a minor fragment of molecular weight corresponding to [{Pd(C6F5)2}2(AcNH–CH2COO)]− (m/z 995.8060) indicating that the formation of dimeric species is also possible. Unfortunately, complex 10 is not stable in solution long enough to perform additional DOSY experiments. Therefore, the occurrence of dimeric species as reaction intermediates for the protonated monoanionic N-acetylglycinato cannot be ruled out. Musaev, Lewis et al. have reported the synthesis of dimeric species for analogous monoanionic MPAAs, where the complex bears a metalated benzylamine fragment.25 This precedent and the results here show that the (κ2-N,O)− and bridging (μ2-O,O)− coordination modes for monoanionic MPAA ligands are close in energy and the adoption or one or the other can be determined by the additional ligands completing the coordination sphere of palladium. In our case, the monomeric species 10 are more stable than the dimeric situation by 2.7 kcal mol−1 as shown by DFT calculations. The presence of equimolar amounts of pyridine leads to complex 9 (Scheme 4) showing that the use of an additional ligand in C–H functionalization reactions (dual-ligand enabled reactions),6e,26 may ensure the formation of monomeric species throughout the C–H activation process: dianionic (κ2-N,O)2− before C–H activation (exemplified by complex 2) and monoanionic (κ1-O)− after C–H activation (as in complex 9). Complex 11 (Scheme 4) was also synthesized and it shows a static behavior with well-defined 19F NMR signals corresponding to the two inequivalent pentafluorophenyl rings. The much better donor amido AcN moiety leads to the preferred chelating coordination mode of the dianionic N-acetylglycinate.
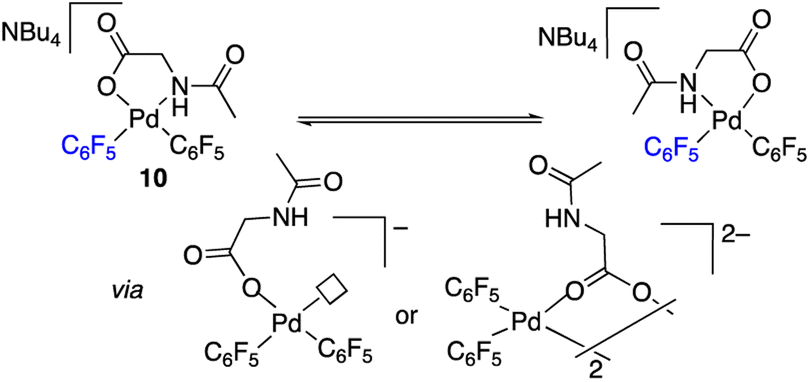 | ||
| Scheme 6 Fluxional behavior of complex 10. | ||
Conclusions
We have experimentally shown that the C–H activation of a simple arene (toluene) occurs on a palladium complex bearing a chelating dianionic acyl-monoprotected amino acid (κ2-N,O)2− in the absence of any external base, consistent with previously reported computational findings. The presence of the acyl group is necessary for the C–H activation and the acyl oxygen is involved in the CMD transition state. This gives the strongest support to the current working model for these reactions: the occurrence of an intermediate palladium complex with a chelating MPAA responsible of the C–H activation as well as the enantioselection. The carboxylato moiety in the ligand, a potential assisting group, has no significant participation in the C–H cleavage as shown by the inefficiency of the analogous methyl protected amino acid ligand (sarcosine derivative) to bring about the C–H activation of toluene. The complexes prepared show how monoprotected amino acids bind, leading to palladium monomeric complexes: dianionic chelating bidentate, monoanionic chelating bidentate and monoanionic monodentate binding modes are preferred. Our results do not support the formation of dimeric carboxylate-bridging complexes as predominant species for these ligands in the complexes studied, although in the case of monoanionic MPAA ligands they may exist as short-lived reaction intermediates. It must be noted that the nature of the ligands that complete the coordination sphere of palladium, other than the MPAA (substrate, additional auxiliary ligands, etc.), will have a strong influence in determining the preferred coordination mode of the monoanionic MPAAs during catalysis. The dianionic MPAAs bind in a chelating bidentate mode as expected for the strong deprotonated AcN donor group.Data availability
Data supporting this article have been uploaded as ESI.†Author contributions
S. F. M. and V. S. conducted the investigation under A. C. A. supervision. A. C. A. wrote the manuscript and S. F. M. prepared the ESI.† All authors contributed to the conceptualization of the project and the review and editing of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge the financial support of the Spanish MICINN (AEI, PID2019-111406GB-I00) and the Junta de Castilla y León-FEDER (VA224P20), as well as the joint support of the EU/MICINN/JCyL (C17.I01.P01.S21, H2MetAmo).Notes and references
- (a) K. M. Engle, Pure Appl. Chem., 2016, 88, 119–138 CrossRef CAS; (b) D. Wang, A. B. Weinstein, P. B. White and S. S. Stahl, Chem. Rev., 2018, 118, 2636–2679 CrossRef CAS PubMed; (c) Q. Shao, K. Wu, Z. Zhuang, S. Qian and J. Q. Yu, Acc. Chem. Res. , 2020, 53, 833–851 CrossRef CAS PubMed; (d) S. K. Sinha, S. Guin, S. Maiti, J. P. Biswas, S. Porey and D. Maiti, Chem. Rev., 2022, 122, 5682–5841 CrossRef CAS PubMed.
- B. F. Shi, N. Maugel, Y. H. Zhang and J. Q. Yu, Angew. Chem., Int. Ed., 2008, 47, 4882–4886 CrossRef CAS PubMed.
- B. F. Shi, Y. H. Zhang, J. K. Lam, D. H. Wang and J. Q. Yu, J. Am. Chem. Soc., 2010, 132, 460–461 CrossRef CAS PubMed.
- Pioneering proof of concept work that showed the ability of chiral aminoacids to induce enantioselective C–H activation: V. I. Sokolov, L. L. Troitskaya and O. A. Reutov, J. Organomet. Chem., 1979, 182, 537–546 CrossRef CAS.
- Early work: (a) D.-H. Wang, K. M. Engle, B.-F. Shi and J.-Q. Yu, Science, 2010, 327, 315–319 CrossRef CAS PubMed; (b) K. M. Engle, D. H. Wang and J. Q. Yu, J. Am. Chem. Soc., 2010, 132, 14137–14151 CrossRef CAS PubMed; (c) K. M. Engle, D. H. Wang and J. Q. Yu, Angew. Chem., Int. Ed., 2010, 49, 6169–6173 CrossRef CAS PubMed; (d) K. M. Engle, P. S. Thuy-Boun, M. Dang and J. Q. Yu, J. Am. Chem. Soc., 2011, 133, 18183–18193 CrossRef CAS PubMed.
- Some recent examples of MPAA-enabled reactions. Olefination: (a) T. K. Achar, X. Zhang, R. Mondal, M. S. Shanavas, S. Maiti, S. Maity, N. Pal, R. S. Paton and D. Maiti, Angew. Chem., Int. Ed., 2019, 58, 10353–10360 CrossRef CAS PubMed; (b) Q. J. Yao, P. P. Xie, Y. J. Wu, Y. L. Feng, M. Y. Teng, X. Hong and B. F. Shi, J. Am. Chem. Soc., 2020, 142, 18266–18276 CrossRef CAS PubMed; (c) J. Das, T. Pal, W. Ali, S. R. Sahoo and D. Maiti, ACS Catal., 2022, 12, 11169–11176 CrossRef CAS; (d) Z. Fan, X. Chen, K. Tanaka, H. S. Park, N. Y. S. Lam, J. J. Wong, K. N. Houk and J.-Q. Yu, Nature, 2022, 610, 87–93 CrossRef CAS PubMed; Cyanation: (e) H. Chen, A. Mondal, P. Wedi and M. Van Gemmeren, ACS Catal., 2019, 9, 1979–1984 CrossRef CAS; Carbonylation: (f) X. F. Bai, Q. C. Mu, Z. Xu, K. F. Yang, L. Li, Z. J. Zheng, C. G. Xia and L. W. Xu, ACS Catal., 2019, 9, 1431–1436 CrossRef CAS; Thiolation: (g) S. K. Sinha, S. Panja, J. Grover, P. S. Hazra, S. Pandit, Y. Bairagi, X. Zhang and D. Maiti, J. Am. Chem. Soc., 2022, 144, 12032–12042 CrossRef CAS PubMed, C–H/C–H cross coupling: (h) Z.-J. Cai, C.-X. Liu, Q. Gu, C. Zheng and S.-L. You, Angew. Chem., Int. Ed., 2019, 58, 2149–2153 CrossRef CAS PubMed, Arylation: (i) P. Dolui, J. Das, H. B. Chandrashekar, S. S. Anjana and D. Maiti, Angew. Chem., Int. Ed., 2019, 58, 13773–13777 CrossRef CAS PubMed; C–H addition to alkenes: (j) J. Das, P. Dolui, W. Ali, J. P. Biswas, H. B. Chandrashekar, G. Prakash and D. Maiti, Chem. Sci., 2020, 11, 9697–9702 RSC.
- R. E. Plata, D. E. Hill, B. E. Haines, D. G. Musaev, L. Chu, D. P. Hickey, M. S. Sigman, J. Q. Yu and D. G. Blackmond, J. Am. Chem. Soc., 2017, 139, 9238–9245 CrossRef CAS PubMed.
- X. Cong, H. Tang, C. Wu and X. Zeng, Organometallics, 2013, 32, 6565–6575 CrossRef CAS.
- (a) D. G. Musaev, A. Kaledin, B.-F. Shi and J. Q. Yu, J. Am. Chem. Soc., 2012, 134, 1690–1698 CrossRef CAS PubMed; (b) D. G. Musaev, T. M. Figg and A. Kaledin, Chem. Soc. Rev., 2014, 43, 5009–5031 RSC.
- G. J. Cheng, Y. F. Yang, P. Liu, P. Chen, T. Y. Sun, G. Li, X. Zhang, K. N. Houk, J. Q. Yu and Y. D. Wu, J. Am. Chem. Soc., 2014, 136, 894–897 CrossRef CAS PubMed.
- (a) G. J. Cheng, P. Chen, T. Y. Sun, X. Zhang, J. Q. Yu and Y. D. Wu, Chem.–Eur. J., 2015, 21, 11180–11188 CrossRef CAS PubMed; (b) B. E. Haines, J. Q. Yu and D. G. Musaev, ACS Catal., 2017, 7, 4344–4354 CrossRef CAS; (c) T. G. Saint-Denis, R. Y. Zhu, G. Chen, Q. F. Wu and J. Q. Yu, Science, 2018, 359, eaao4798 CrossRef PubMed.
- (a) X. M. Zhong, G. J. Cheng, P. Chen, X. Zhang and Y. D. Wu, Org. Lett., 2016, 18, 5240–5243 CrossRef CAS PubMed; (b) P. Wedi, M. Farizyan, K. Bergander, C. Mück-Lichtenfeld and M. Gemmeren, Angew. Chem., Int. Ed., 2021, 60, 15641–15649 CrossRef CAS PubMed; (c) K. Mukherjee, N. Grimblat, S. Sau, K. Ghosh, M. Shankar, V. Gandon and A. K. Sahoo, Chem. Sci., 2021, 12, 14863–14870 RSC; (d) L. P. Xu, Z. Zhuang, S. Qian, J. Q. Yu and D. G. Musaev, ACS Catal., 2022, 12, 4848–4858 CrossRef CAS.
- B. E. Haines and D. G. Musaev, ACS Catal., 2015, 5, 830–840 CrossRef CAS.
- L. Fang, T. G. Saint-Denis, B. L. H. Taylor, S. Ahlquist, K. Hong, S. Liu, L. Han, K. N. Houk and J. Q. Yu, J. Am. Chem. Soc., 2017, 139, 10702–10714 CrossRef CAS PubMed.
- J. J. Gair, B. E. Haines, A. S. Filatov, D. G. Musaev and J. C. Lewis, ACS Catal., 2019, 9, 11386–11397 CrossRef CAS.
- D. E. Hill, K. L. Bay, Y. F. Yang, R. E. Plata, R. Takise, K. N. Houk, J. Q. Yu and D. G. Blackmond, J. Am. Chem. Soc., 2017, 139, 18500–18503 CrossRef CAS PubMed.
- R. Jayarajan, J. Das, S. Bag, R. Chowdhury and D. Maiti, Angew. Chem., Int. Ed., 2018, 57, 7659–7663 CrossRef CAS PubMed.
- C. A. Salazar, J. J. Gair, K. N. Flesch, I. A. Guzei, J. C. Lewis and S. S. Stahl, Angew. Chem., Int. Ed., 2020, 59, 10873–10877 CrossRef CAS PubMed.
- (a) V. Salamanca, A. Toledo and A. C. Albéniz, J. Am. Chem. Soc., 2018, 140, 17851–17856 CrossRef CAS PubMed; (b) V. Salamanca and A. C. Albéniz, Org. Chem. Front., 2021, 8, 1941–1951 RSC; (c) C. Pinilla, V. Salamanca, A. Lledós and A. C. Albéniz, ACS Catal., 2022, 12, 14527–14532 CrossRef CAS PubMed.
- Deposition numbers 2255463 (for 1), 2255464 (for 4), 2255465 (for 5), 2255468 (for 7), and 2255469 (for 8) contain the crystallographic data for this paper.†.
- (a) A. L. Casado, J. A. Casares and P. Espinet, Organometallics, 1997, 16, 5730–5736 CrossRef CAS; (b) A. C. Albéniz, P. Espinet, O. López-Cimas and B. Martín-Ruiz, Chem.–Eur. J., 2005, 11, 242–252 CrossRef PubMed.
- This coordination mode has been proposed in reactions of N-acetyl glycine in water: T. G. Appleton, D. R. Bedgood and J. R. Hall, Inorg. Chem., 1994, 33, 3834–3841 CrossRef CAS.
- E. M. Simmons and J. F. Hartwig, Angew. Chem., Int. Ed., 2012, 51, 3066–3072 CrossRef CAS PubMed.
- (a) E. Clot, C. Mégret, O. Eisenstein and R. N. Perutz, J. Am. Chem. Soc., 2009, 131, 7817–7827 CrossRef CAS PubMed; (b) E. Gioria, J. delPozo, J. M. Martínez-Ilarduya and P. Espinet, Angew. Chem., Int. Ed., 2016, 55, 13276–13280 CrossRef CAS PubMed.
- J. J. Gair, B. E. Haines, A. S. Filatov, D. G. Musaev and J. C. Lewis, Chem. Sci., 2017, 8, 5746–5756 RSC.
- (a) H. Chen, P. Wedi, T. Meuer, G. Tavakoli and M. van Gemmeren, Angew. Chem., Int. Ed., 2018, 57, 2497–2501 CrossRef CAS PubMed; (b) D. Zhao, P. Xu and T. Ritter, Chem, 2019, 5, 97–107 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details, characterization data, crystal XRD and computational details. CCDC 2255463–2255465, 2255468 and 2255469. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3sc02076b |
| ‡ Although the reductive elimination is the plausible step right after the C–H cleavage, other processes such as ligand tautomerization in the conditions herein (absence of an added base) or a change in coordination mode of the ligand to (κ1-O)− by pyridine coordination cannot be ruled out. |
| This journal is © The Royal Society of Chemistry 2023 |