DOI:
10.1039/D3SC00874F
(Edge Article)
Chem. Sci., 2023,
14, 5705-5711
Copper-catalyzed [1,3]-nitrogen rearrangement of O-aryl ketoximes via oxidative addition of N–O bond in inverse electron flow†
Received
16th February 2023
, Accepted 28th April 2023
First published on 1st May 2023
Abstract
The [1,3]-nitrogen rearrangement reactions of O-aryl ketoximes were promoted by N-heterocyclic carbene (NHC)-copper catalysts and BF3·OEt2 as an additive, affording ortho-aminophenol derivatives in good yields. The reaction of substrates with electron-withdrawing substituents on the phenol moiety are accelerated by adding silver salt and modifying the substituent at the nitrogen atom. Density functional theory calculations suggest that the rate-determining step of this reaction is the oxidative addition of the N–O bond of the substrate to the copper catalyst. The negative ρ values of the substituent at both the oxime carbon and phenoxy group indicate that the donation of electrons by the oxygen and nitrogen atoms accelerates the oxidative addition.
Introduction
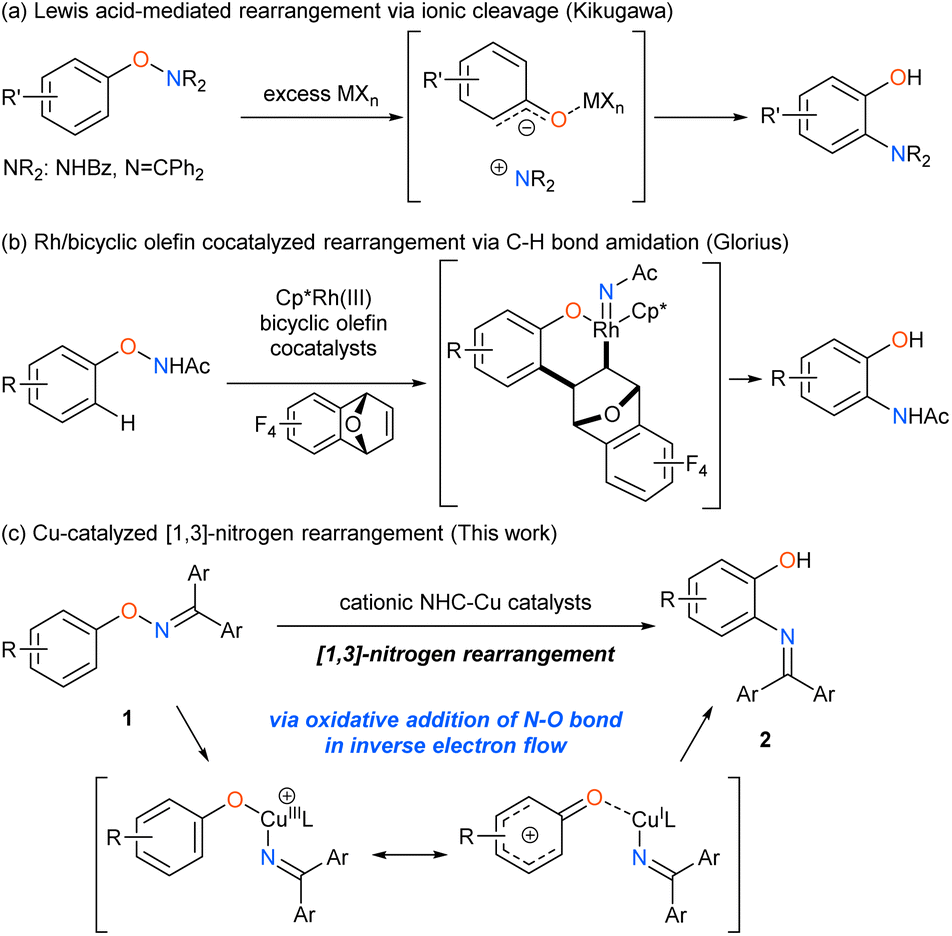
Catalytic transformations involving the cleavage of an N–O bond have recently gathered much attention as a unique method to synthesize nitrogenous compounds,1 because the starting materials are readily accessible and storable despite the weak N–O bond (bond dissociation energy: 55–65 kcal mol−1).2 The N–O bond cleavage, which is the driving force of the catalytic cycle, occurs via various mechanisms, including the oxidative addition to low-valent metal catalysts, homolytic cleavage, single electron transfer, and ionic cleavage, to generate unique reactive intermediates, such as aminyl radicals,3 nitrenes,4 and nitreniums.5 In contrast to the catalytic transformations in which the oxygen atom of the N–O bond is typically cleaved off as a leaving group, rearrangement reactions yield molecules with both nitrogen and oxygen functional groups in an atom-efficient manner,6,7 by using metal catalysts8 and organocatalysts9 under mild reaction conditions. However, [1,3]-nitrogen rearrangement reactions of O-arylhydroxylamine derivatives still remain unexplored to date although the fundamental rearrangement process can directly synthesize ortho-aminophenol derivatives10,11 (Scheme 1). In early times, Kikugawa reported that excess amounts of Lewis acids, such as AlCl3 and ZrCl4 promoted the [1,3]-nitrogen rearrangement reaction of O-arylhydroxylamine derivatives via ionic cleavage of N–O bond (Scheme 1a),12 although the process was applied to only a limited number of substrates due to the harsh reaction conditions. Glorius has recently developed the catalytic version of the [1,3]-nitrogen rearrangement reaction of N-phenoxyacetamides by using rhodium and bicyclic olefin co-catalysts via C–H activation (Scheme 1b).13 However, the state-of-the-art transformation still suffers from low efficiency in the reaction of substrates having the electron-withdrawing group on the phenoxy ring due to a side reaction derived from the nature of the nitrene intermediate. In this context, we envisioned that cationic N-heterocyclic carbene (NHC)-copper catalysts, which efficiently promoted [1,3]-oxygen rearrangement of N-alkoxyanilines,5b,d would be effective for the challenging rearrangement reaction. Herein, we report that the Cu-catalyzed [1,3]-nitrogen rearrangement reactions of O-aryl ketoximes 1 produced the corresponding ortho-aminophenol derivatives 2 in good to high yields with excellent regioselectivities and high functional group compatibility (Scheme 1c). Our mechanistic investigations revealed that the present reaction proceeds via unusual oxidative addition which is accelerated by donation of electrons from N–O bond to Cu catalyst in inverse electron flow.
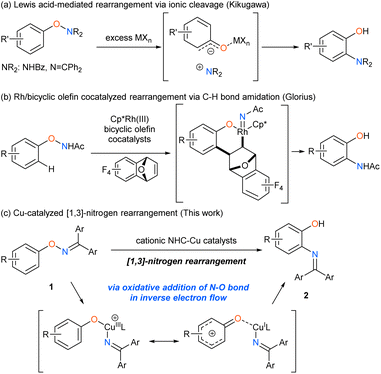 |
| | Scheme 1 [1,3]-Nitrogen rearrangement reactions of O-arylhydroxylamines. | |
Results and discussion
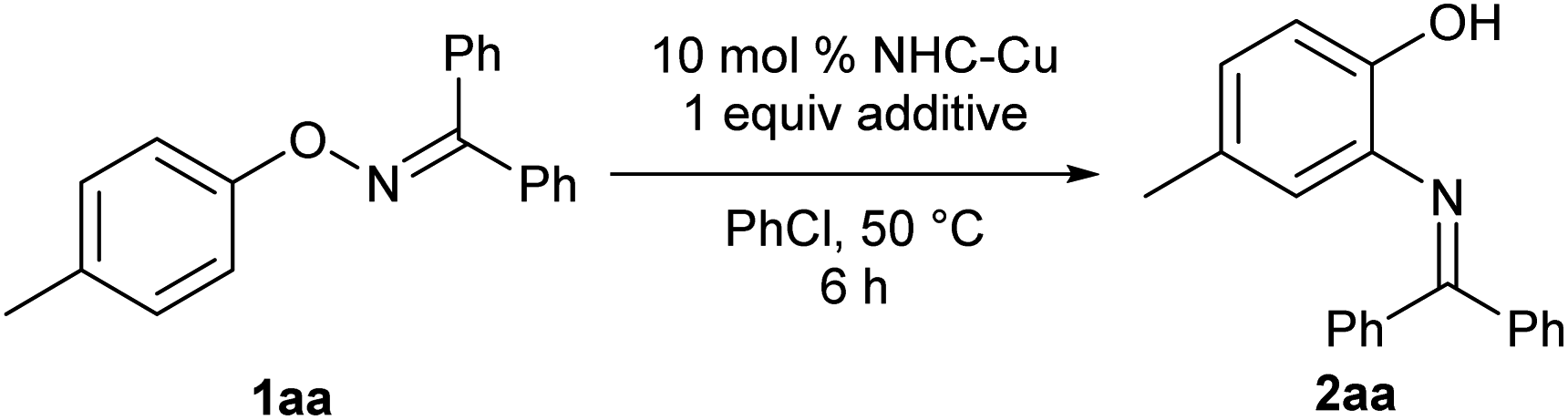
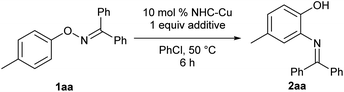
The catalytic transformation was achieved when O-p-tolyl oxime 1aa, which was derived from benzophenone, was treated with catalytic amounts of IPrCuBr [10 mol%, IPr: N,N′-bis(2,6-diisopropylphenyl)-imidazol-2-ylidene] and AgSbF6 (10 mol%) in chlorobenzene at 50 °C for 6 h. The reaction afforded corresponding ortho-aminophenol derivative 2aa in 24% yield along with a considerable amount (64%) of the recovered starting material 1aa (Table 1, entry 1). In our initial screening, other typical Lewis acids, such as Yb(OTf)3 and CoCl2/AgSbF6, did not exhibit any catalytic activities (see ESI†). Preliminary screening of NHC ligands suggested that IPr was the most appropriate ligand for the present reaction (entries 1–3, see also ESI†). Because extending the reaction time did not improve the chemical yield in the reaction that used IPrCuBr and AgSbF6, we presumed that the Cu catalyst was deactivated by complexation with product 2aa, which interfered with the catalyst turnover. Therefore, we examined additives to help regeneration of the copper catalyst through decomplexation from the product. To our delight, the use of 1 equivalent of BF3·OEt2 improved the catalyst turnover, affording desired product 2aa in 84% yield after aqueous workup with saturated ammonium chloride solution (entry 4). The reaction of 1aa proceeded smoothly even without AgSbF6 to afford desired product 2aa in almost the same yield as that in entry 4, albeit with a slightly longer reaction time (entry 5), indicating that BF3·OEt2 functions in not only the regeneration of the copper catalyst, but also the generation of the cationic copper species. Noteworthy, the reaction of 1aa in the presence of BF3·OEt2 without IPrCuBr resulted in the low chemical yield of desired product 2aa with recovery of starting material 1aa (entry 6), indicating that the copper catalyst is essential for the completion of this reaction. The reaction at lower temperature (30 °C) did not improve the chemical yield (see ESI†).
Table 1 Optimization of reaction conditionsa
|
|
| Entry |
NHC–Cu catalyst (mol%) |
Additive |
2aa (%)b |
1aa (%)b |
|
The reaction of 1aa (0.1 mmol) was carried out in the presence of the catalyst and the additive in chlorobenzene (0.7 mL) at 50 °C.
The yield was deterimined by NMR spectroscopy using CH2Br2 as the internal standard.
Hydrolysis using NH4Cl aq. was conducted after the reaction.
For 15 min.
For 1 h.
|
| 1 |
IPrCuBr (10), AgSbF6 (10) |
None |
24 |
64 |
| 2 |
SIPrCuBr (10), AgSbF6 (10) |
None |
25 |
32 |
| 3 |
IMesCuBr (10), AgSbF6 (10) |
None |
<1 |
76 |
| 4cd |
IPrCuBr (10), AgSbF6 (10) |
BF3·OEt2 |
84 |
2 |
| 5ce |
IPrCuBr (10) |
BF3·OEt2 |
85 |
4 |
| 6ce |
None |
BF3·OEt2 |
18 |
68 |
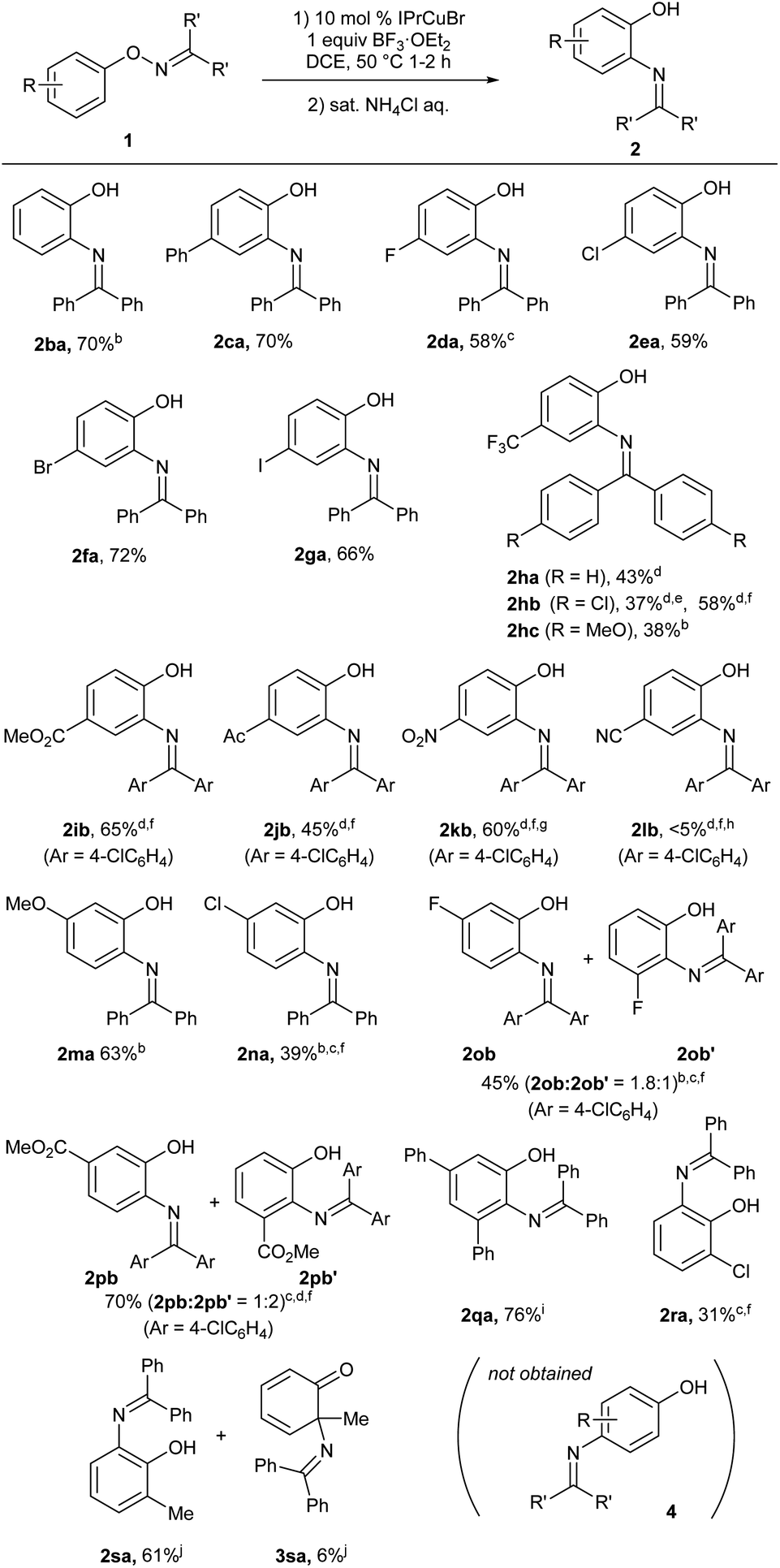
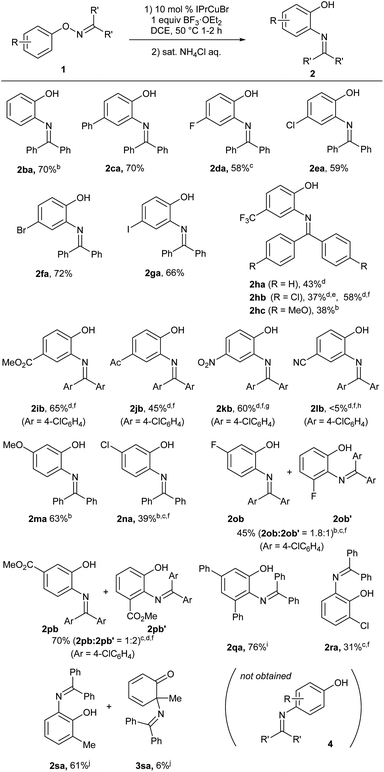
The scope of O-aryl ketoximes for the present catalytic [1,3]-nitrogen rearrangement under the optimized reaction conditions (Table 1, entry 5) is summarized in Scheme 2. The reaction of benzophenone oximes 1aa–1ga having various substituents at the para position proceeded at 50–60 °C, affording corresponding ortho-aminophenol derivatives 2aa–2ga in good yields, respectively. In particular, bromo (2fa) and iodo (2ga) groups were tolerated under the reaction conditions. In contrast, substrate 1ha with a strong electron-withdrawing trifluoromethyl group required an elevated temperature (80 °C) to afford desired product 2ha in moderate yield. Thus, we examined the electronic effect of the oxime carbon substituents on the reaction efficiency and found that the electron-deficient p-chlorophenyl groups (1hb) at the oxime carbon significantly improved the mass balance at 80 °C. Furthermore, the chemical yield was improved by adding 10 mol% AgSbF6. In contrast, 1hc, which possesses p-anisyl groups at the oxime carbon was rapidly decomposed even at 60 °C, affording 2hc in a low yield (see ESI†). The use of the chlorophenyl group as a substituent at the oxime carbon was effective for substrates possessing other electron-withdrawing groups, such as methoxycarbonyl (1ib), acetyl (1jb), and nitro (1kb) groups, affording desired products 2ib, 2jb, and 2kb, respectively, in good to acceptable yields, whereas substrate 1lb with a cyano group was unreactive under the present reaction conditions. Substrate 1ma and 1na, which have a methoxy group and a chloro group, respectively, at the position meta to the oxime moiety, selectively afforded 2ma and 2na, respectively, through the migration of the diphenylimino group to the less hindered ortho position, whereas the reaction of 1pb, which has a strongly electron-withdrawing methoxycarbonyl group at the meta position, preferentially afforded 2pb′, which was derived from the migration to the more hindered ortho position adjacent to the methoxycarbonyl group. The substrate 1qa having bulky phenyl groups at two meta positions of the phenoxy group afforded 2qa in good yield. In the case of substrates 1ra and 1sa, which have a chloro and a methyl group, respectively, at the position ortho to oxime moiety, the corresponding 6-substituted 2-aminophenol derivatives 2ra and 2sa were selectively obtained in good to moderate yields. It is noteworthy that a trace amount of 2-aminocyclohexadienone derivative 3sa, which was formed through the rearrangement of the nitrogen functional group to the methyl-bound ortho position, was observed in the Cu-catalyzed reaction of the ortho-methyl-substituted substrate 1sa. It should be noted that para-aminophenol derivatives 4 were not formed in the copper-catalyzed reactions of substrates 1ba, 1ma, 1na, 1ob, 1pb, 1qa, 1ra, and 1sa, all of which do not possess any substituents at the position para to the oxime moiety.
 |
| | Scheme 2 Copper-catalyzed reactions of 1.a a The reaction of 1 (0.1 mmol) was carried out in the presence of IPrCuBr (0.01 mmol) and BF3·OEt2 (0.1 mmol) in 1,2-DCE (0.7 mL) at 50 °C for 1–2 h and the reaction workup was performed by adding saturated ammonium chloride solution. b At 60 °C. c 0.05 mmol scale. d At 80 °C. e 40% of 1hb was recovered. f The reaction was carried out with 10 mol% of AgSbF6. g Yield based on recovered starting material (brsm); 27% of 1kb was recovered. h 84% of 1lb was recovered. i At 70 °C. j 0.5 h. | |
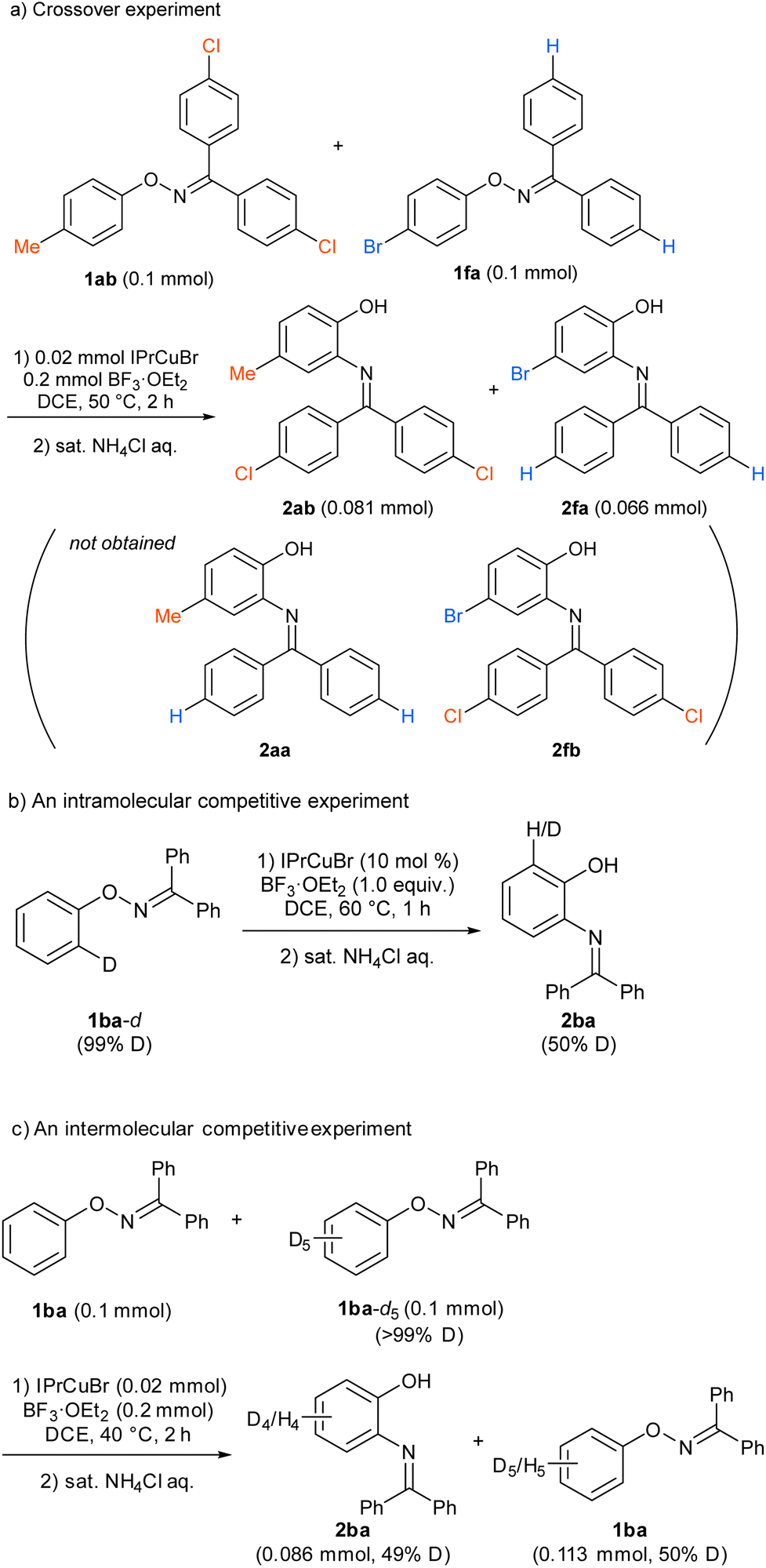
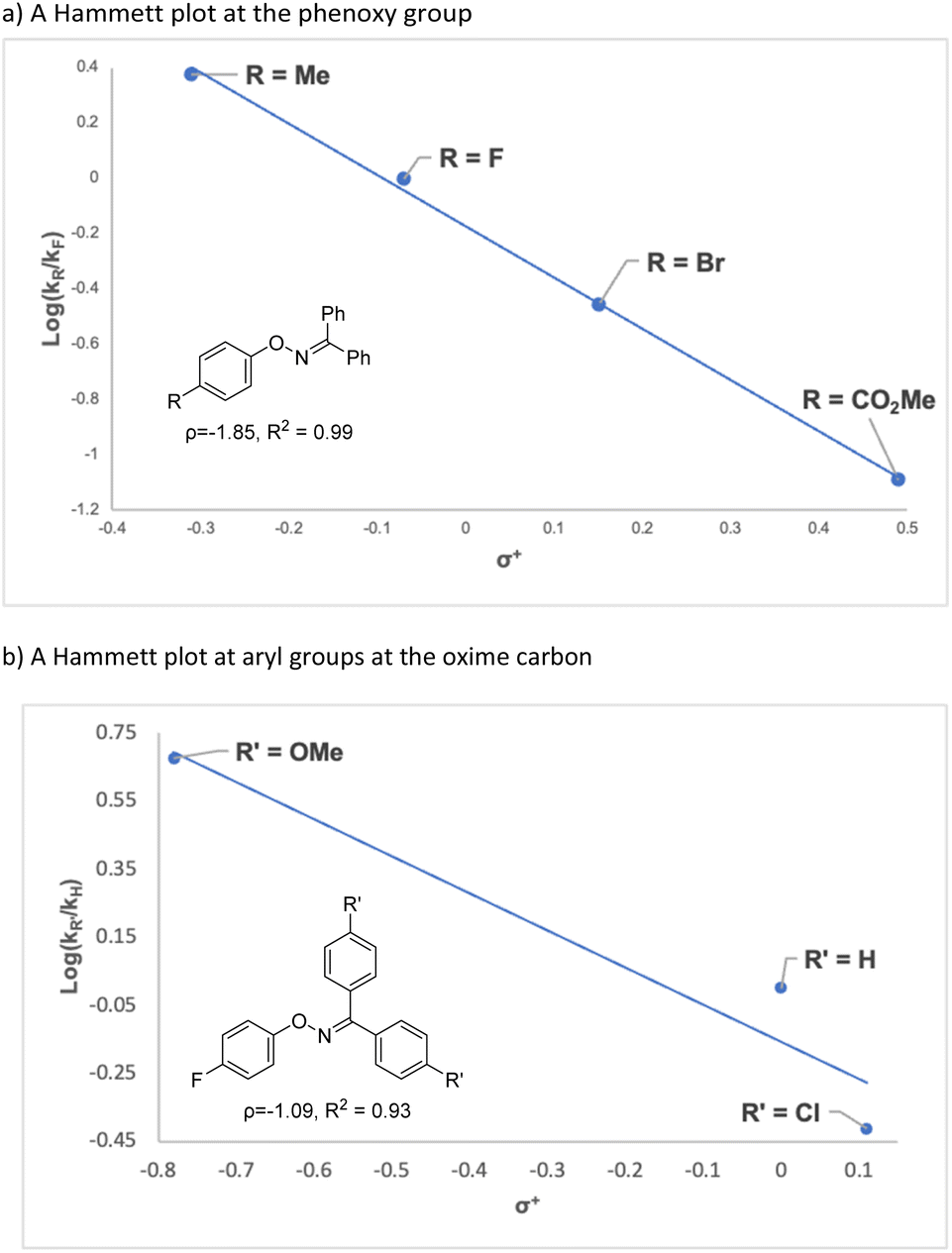
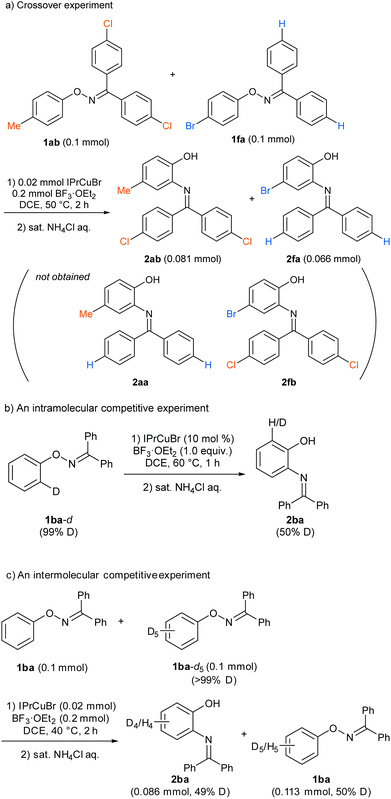
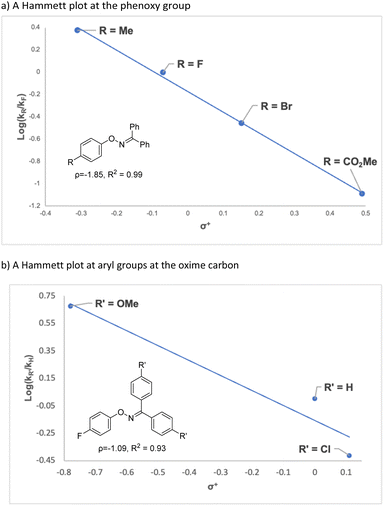
To gain an insight into the reaction mechanism, the reaction of a mixture of 1ab and 1fa, which showed similar reactivity, was conducted under the standard reaction conditions (Scheme 3a). The reaction afforded 2ab and 2fa, which were derived from starting materials 1ab and 1fa, respectively; crossover products, such as 2aa and 2fb, were not observed by high-resolution mass spectrometry. The result clearly indicates that the nitrogen rearrangement proceeds intramolecularly. In addition, no significant kinetic isotope effect was observed in intra- and intermolecular competitive experiments that used deuterated substrates 1ba-d and 1ba/1ba-d5, respectively (Schemes 3b and 3c). The results suggest that cleavage of the C–H bond at the ortho position is not involved in the rate-determining step (RDS) of the present rearrangement reaction. Then, a Hammett type analysis was conducted for both the phenoxy group and the aryl groups at the oxime carbon to understand electronic nature of a rate-determining step of the present rearrangement reaction. The rearrangement reaction was conducted under the conditions using IPrCuBr (10 mol%) and BF3·OEt2 (1.0 equiv) at 50 °C, and consumption of starting material 1 was monitored by 1H NMR spectroscopy and normalized to an internal standard (CH2Br2). The reaction rate constant kR of each reaction was determined from a first-order plot of −ln[SM] versus time (see ESI†). In terms of the slope of the approximate formula, the Hammett correlation at both the phenoxy group (ρ = −1.85, R2 = 0.99, Fig. 1a) and the aryl groups at the oxime carbon (ρ = −1.09, R2 = 0.93, Fig. 1b) versus σ+ values showed a good linear correlation with a negative slope.‡ These results indicate that the rate determining step of the present reaction is accelerated by electron-donating groups at both the phenoxy group and the aryl groups at the oxime carbon.
 |
| | Scheme 3 Mechanistic studies. | |
 |
| | Fig. 1 Hammett plot analysis. | |
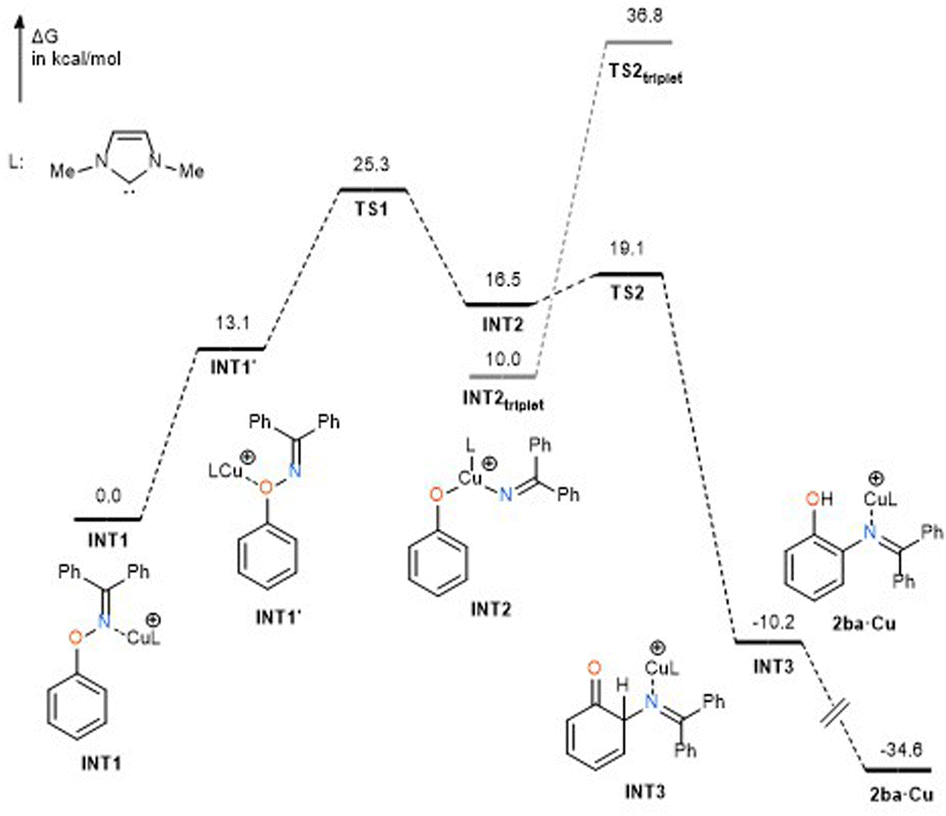
In order to specify the rate-determining step, DFT calculations were preliminarily performed for the reaction mechanism at the level of B3LYP/SDD (Cu) 6-31+(d,p) (for others) using substrate 1ba and N,N′-dimethylimidazol-2-ylidene (IMe) as the model ligand (Fig. 2).§ Although the coordination of the copper catalyst to the nitrogen atom of 1ba to form complex INT1 is 13.1 kcal mol−1 more stable in energy than the coordination to the oxygen atom (INT1′), N–O bond cleavage was found to occur from the oxygen-coordinated copper complex INT1′, when the reaction pathway from TS1 was traced using the intrinsic reaction coordinate (IRC) method (see ESI†). More importantly, transition state TS1 of the N–O bond cleavage process has 6.2 kcal mol−1 higher energy than that of the C–N bond forming process (TS2), suggesting that the rate-determining step of the present rearrangement reaction is N–O bond cleavage. It should be noted that the triplet state of the intermediate INT2triplet, which was formed after N–O bond cleavage, was calculated to be more stable than the singlet state INT2. However, the transition state TS2triplet for the triplet state was calculated to be much higher than that for the singlet, suggesting that the C–N bond formation occurs in a singlet state. Considering the results of Hammett analysis, we conclude that the present rearrangement reaction proceeds via an oxidative addition of the N–O bond to the copper atom,¶ and the key process is accelerated by donation of electrons from both nitrogen and oxygen atoms. It should be pointed out that the transition metal-catalyzed reactions of oxime esters, which involve oxidative addition of the oxime N–O bond, are generally accelerated by electron-withdrawing substituents at both oxygen and carbon atoms.14 In fact, Okamoto and Ohe reported that the ρ value for the oxidative addition of the oxime ester N–O bond to ruthenium is +0.19 at the oxime carbon.15 Thus, the N–O bond cleavage process in the present rearrangement reaction can be regarded as an oxidative addition with inverse electron flow. On the other hand, the inverse electron flow in oxidative addition observed in the present reaction is in good agreement with the report by Amgoune and Bourissou; the ρ value for the oxidative addition of para-substituted iodoarenes to cationic Au(I) complex is −1.09.16 Concerning the C–N bond forming process, natural bond order (NBO) analysis of transition state TS2 indicates that donor–acceptor interactions from the nitrogen moiety to the phenoxy benzene ring prevail. Indeed, the donation by the nitrogen lone pair to the vacant p orbital at the ortho position is calculated as ca. 58 kcal mol−1, whereas opposite interactions are not significant (less than 2 kcal mol−1, see ESI†). Thus, we conclude that the C–N bond forming process is mainly governed by the nucleophilic attack of the nitrogen atom on the phenyloxenium. The angle consisting of ortho carbon, nitrogen, and oxime-derived sp2 carbon atoms was calculated as 127.3° (see ESI†), which corresponds to the nucleophilic attack of the lone pair of the nitrogen atom.
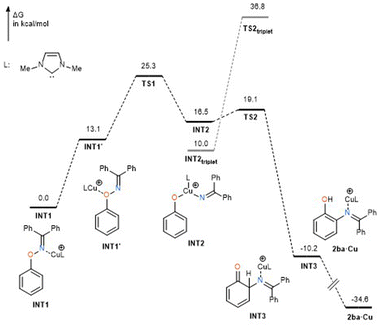 |
| | Fig. 2 DFT calculations. | |
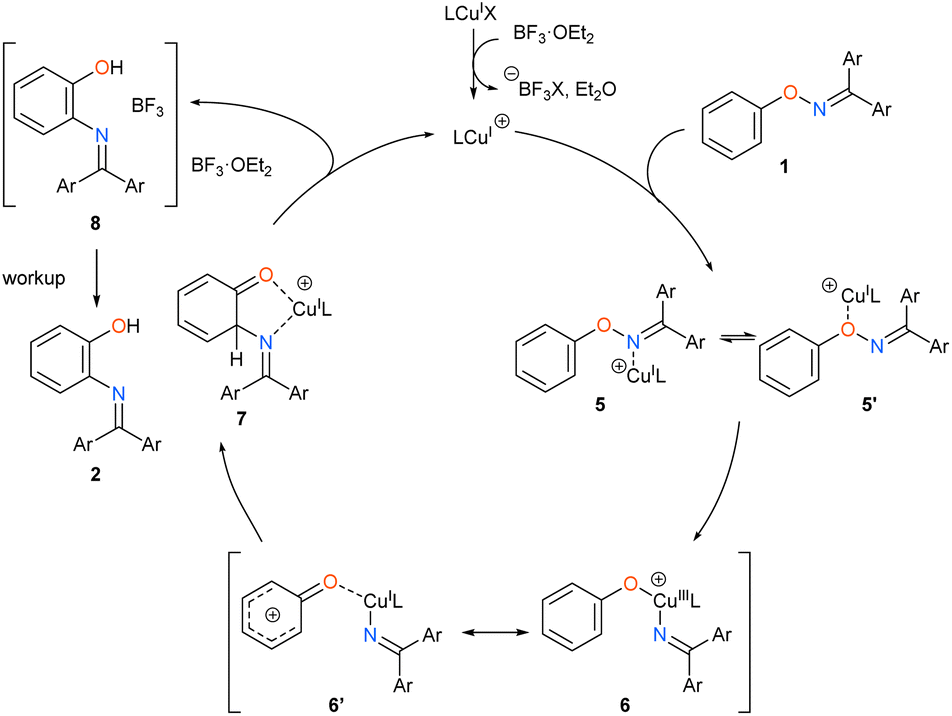
On the basis of the above results, we propose a reaction mechanism of the catalytic [1,3]-nitrogen rearrangement as shown in Scheme 4. First, the cationic NHC-copper catalyst is generated by abstracting the halogen atom on the copper complex by either BF3·OEt2 or silver salt. Then, the cationic NHC-copper catalyst coordinates to the substrate 1. Next, the oxidative addition of the N–O bond to Cu(I) catalyst from the oxygen-coordinated σ complex 5′ yields Cu(III) complex 6. Owing to the contribution of the canonical form 6′, which has a phenyloxenium structure, a C–N bond is formed at the proximal ortho position via the nucleophilic attack of the nitrogen atom. Resulting intermediate 7 undergoes complexation with BF3·OEt2, regenerating the cationic NHC-copper catalyst. Finally, the product-BF3 complex 8, of which the exact structure remains unknown, is transformed into desired product 2 by a workup process. We observed intriguing substituent effects that mesomerically electron donating groups, such as methoxy (1ma), chloro (1na), and fluoro (1ob), at the meta of phenoxy group preferentially led to the product derived from the migration of the imino group to the less hindered ortho position while an electron-withdrawing methoxycarbonyl group (1pb) led to the migration to the ortho position next to the electron-withdrawing group (Scheme 2). This is presumably because the nucleophilic attack of the nitrogen atom occurs more electron-deficient ortho position, of which the electron density is affected by the meta substituent, in the phenyloxenium 6′, although steric repulsion between the meta substituent and the nitrogenous migrating group also affects the selectivity to some extent (see ESI†).||.
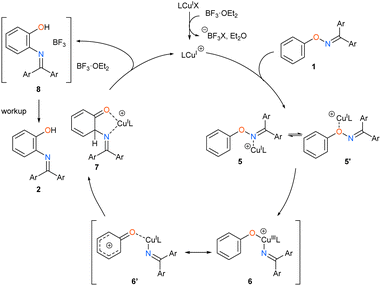 |
| | Scheme 4 Proposed mechanism. | |
Conclusion
In conclusion, we have developed a catalytic [1,3]-nitrogen rearrangement reaction of O-aryl ketoximes by the use of cationic NHC–Cu(I) catalyst. The present rearrangement reaction is applicable to a wide range of functional groups and useful for the synthesis of functionalized ortho-aminophenol derivatives under mild reaction conditions. Inverse electron flow in the oxidative addition of the N–O bond is expected to be the key to realize atom-efficient transformations via N–O bond cleavage.
Data availability
All experimental procedures, characterization, and computational data for this study can be found in the ESI.†
Author contributions
Conceptualization: IN; methodology: MS and IN; investigation MS; writing: MS, MT, and IN; supervision: MT. All authors have given approval to the final version of the manuscript.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by JSPS KAKENHI Grant Number JP17H06447, a Grant-in-Aid for Scientific Research on Innovative Areas “Hybrid Catalysis for Enabling Molecular Synthesis on Demand” and JP20H02731, Grant-in-Aid for Scientific Research (B) from MEXT, Japan. The computation was performed using Research Center for Computational Science, Okazaki, Japan (Project: 22-IMS-C127).
Notes and references
-
(a) K. Narasaka and M. Kitamura, Eur. J. Org. Chem., 2005, 4505–4519 CrossRef CAS;
(b) F. W. Patureau and F. Glorius, Angew. Chem., Int. Ed., 2011, 50, 1977–1979 CrossRef CAS PubMed;
(c) H. Huang, X. Ji, W. Wu and H. Jiang, Chem. Soc. Rev., 2015, 44, 1155–1171 RSC;
(d) H. Huang, J. Cai and G.-J. Deng, Org. Biomol. Chem., 2016, 14, 1519–1530 RSC;
(e) K. A. Rykaczewski, E. R. Wearing, D. E. Blackmun and C. S. Schindler, Nat. Synth., 2022, 1, 24–36 CrossRef;
(f) K. Hirano and M. Miura, J. Am. Chem. Soc., 2022, 144, 648–661 CrossRef CAS PubMed.
- R. D. Bach and H. B. Schlegel, J. Phys. Chem. A, 2021, 125, 5014–5021 CrossRef CAS PubMed.
-
(a) H.-B. Yang, S. R. Pathipati and N. Selander, ACS Catal., 2017, 7, 8441–8445 CrossRef CAS;
(b) X.-Y. Yu, Q.-Q. Zhao, J. Chen, J.-R. Chen and W.-J. Xiao, Angew. Chem., Int. Ed., 2018, 57, 15505–15509 CrossRef CAS PubMed;
(c) K. M. Nakahuku, Z. Zhang, E. A. Wappes, L. M. Stateman, A. D. Chen and D. A. Nagib, Nat. Chem., 2020, 12, 697–704 CrossRef PubMed;
(d) M. R. Becker, E. R. Wearing and C. S. Schindler, Nat. Chem., 2020, 12, 898–905 CrossRef CAS PubMed;
(e) F. Du, S.-J. Jiang, R. Zeng, X.-C. Pan, Y. Lan, Y.-C. Chen and Y. Wei, Angew. Chem., Int. Ed., 2020, 59, 23755–23762 CrossRef CAS PubMed;
(f) B. Górski, A.-L. Barthelemy, J. J. Douglas, F. Juliá and D. Leonori, Nat. Catal., 2021, 4, 54–61 CrossRef;
(g) W.-X. Wei, Y. Li, Y.-T. Wen, M. Li, X.-S. Li, C.-T. Wang, H.-C. Liu, Y. Xia, B.-S. Zhang, R.-Q. Jiao and Y.-M. Liang, J. Am. Chem. Soc., 2021, 143, 7868–7875 CrossRef CAS PubMed;
(h) E. R. Wearing, D. E. Blackmun, M. R. Becker and C. S. Schindler, J. Am. Chem. Soc., 2021, 143, 16235–16242 CrossRef CAS PubMed;
(i) T. J. Fazekas, J. W. Alty, E. K. Neidhart, A. S. Miller, F. A. Leibfarth and E. J. Alexanian, Science, 2022, 375, 545–550 CrossRef CAS PubMed;
(j) T. Feng, C. Liu, Z. Wu, X. Wu and C. Zhu, Chem. Sci., 2022, 13, 2669–2673 RSC.
-
(a) H. Hayashi and T. Uchida, Eur. J. Org. Chem., 2020, 909–916 CrossRef CAS , and references therein;;
(b) H. Wang, H. Jung, F. Song, S. Zhu, Z. Bai, D. Chen, G. He, S. Chang and G. Chen, Nat. Chem., 2021, 13, 378–385 CrossRef CAS PubMed;
(c) E. Lee, Y. Hwang, Y. B. Kim, D. Kim and S. Chang, J. Am. Chem. Soc., 2021, 143, 6363–6369 CrossRef CAS PubMed;
(d) B. Du, Y. Ouyang, Q. Chen and W.-Y. Yu, J. Am. Chem. Soc., 2021, 143, 14962–14968 CrossRef CAS PubMed;
(e) Y. Zhang, D. Qiao, M. Duan, Y. Wang and S. Zhu, Nat. Commun., 2022, 13, 5630 CrossRef CAS PubMed;
(f) N. W. Carlberg and T. Rovis, J. Am. Chem. Soc., 2022, 144, 22426–22432 CrossRef PubMed;
(g) N. Li, H. J. Zhao and Z. Gu, Angew. Chem., Int. Ed., 2023, 62, e202215530 CAS.
-
(a) Y. Kikugawa, Heterocycles, 2009, 78, 571–607 CrossRef CAS , and references therein;;
(b) I. Nakamura, T. Jo, H. Tashiro and M. Terada, Org. Lett., 2017, 19, 3059–3062 CrossRef CAS PubMed;
(c) J. J. Farndon, X. Ma and J. F. Bower, J. Am. Chem. Soc., 2017, 139, 14005 CrossRef CAS PubMed;
(d) Y. Ishida, I. Nakamura and M. Terada, J. Am. Chem. Soc., 2018, 140, 8629–8633 CrossRef CAS PubMed.
- A. A. Tabolin and S. L. Loffe, Chem. Rev., 2014, 114, 5426–5476 CrossRef CAS PubMed.
- Thermally induced rearrangement reactions;
(a) L. Horner and H. Stephen, Liebigs Ann. Chem., 1957, 606, 24–47 CrossRef CAS;
(b) S. Oae and T. Sakurai, Tetrahedron, 1976, 32, 2289–2294 CrossRef CAS;
(c) P. G. Gassman and J. E. Guanrud, J. Am. Chem. Soc., 1984, 106, 1498–1499 CrossRef CAS;
(d) T. Sakurai, M. Nakamura, T. Hirai and H. Inoue, Bull. Chem. Soc. Jpn., 1992, 65, 2789–2793 CrossRef CAS;
(e) T. Kaneko, K. Kubo and T. Sakurai, Tetrahedron Lett., 1997, 38, 4779–4782 CrossRef CAS;
(f) A. Porzelle, M. D. Woodrow and N. C. O. Tomkinson, Eur. J. Org. Chem., 2008, 5135–5143 CrossRef CAS;
(g) A. Porzelle, M. D. Woodrow and N. C. O. Tomkinson, Org. Lett., 2010, 12, 812–815 CrossRef CAS PubMed;
(h) K. N. Hojczyk, P. Feng, C. Zhan and M.-Y. Ngai, Angew. Chem., Int. Ed., 2014, 53, 14559–14563 CrossRef CAS PubMed;
(i) P. Feng, K. N. Lee, J. W. Lee, C. Zhan and M.-Y. Ngai, Chem. Sci., 2016, 7, 424–429 RSC;
(j) K. N. Lee, J. W. Lee, C. Zhan and M.-Y. Ngai, Synlett, 2016, 27, 313–319 CAS;
(k) K. N. Lee, Z. Lei, C. A. M. Rivera, P. Liu and M.-Y. Ngai, Org. Biomol. Chem., 2016, 14, 5599–5605 RSC;
(l) Y. Liu, S. Bai, Y. Du, X. Qi and H. Gao, Angew. Chem., Int. Ed., 2022, 61, e202115611 CrossRef CAS PubMed . Rearrangement reactions promoted by stoichiometric amounts of either basic or acidic reagents;;
(m) Y. Kikugawa and M. Shimada, J. Chem. Soc., Chem. Commun., 1989, 1450–1451 RSC;
(n) F. Xiong, L. Lu, T.-Y. Sun, Q. Wu, D. Yan, Y. Chen, X. Zhang, W. Wei, Y. Lu, W.-Y. Sun, J. J. Li and J. Zhao, Nat. Commun., 2017, 8, 15912 CrossRef PubMed;
(o) S. Shaaban, V. Tona, B. Peng and N. Maulide, Angew. Chem., Int. Ed., 2017, 56, 10938–10941 CrossRef CAS PubMed;
(p) S. Xing, Y.-Y. Zhu, Y. Liu, J. Zhang, H. Zhang, Y. Wang, S.-F. Ni and X. Sao, Org. Lett., 2022, 24, 3378–3383 CrossRef CAS PubMed.
-
(a) K. N. Lee, D. N. Spiegowski and M.-Y. Ngai, Chem. Sci., 2017, 8, 6066–6070 RSC;
(b) Y. Wu, Z. Chen, Y. Yang, W. Zhu and B. Zhou, J. Am. Chem. Soc., 2018, 140, 42–45 CrossRef CAS PubMed;
(c) G. R. Mathi, B. Kweon, Y. Moon, Y. Jeong and S. Hong, Angew. Chem., Int. Ed., 2020, 59, 22675–22683 CrossRef CAS PubMed;
(d) Y. Wu, C. Pi, Y. Wu and X. Cui, Chem. Soc. Rev., 2021, 50, 3677–3689 RSC , and references therein;;
(e) F. Liu, M. Wang, J. Qu, H. Lu and H. Gao, Org. Biomol. Chem., 2021, 19, 7246–7251 RSC;
(f) A. Nikbakht, K. Amiri, H. Khosravi, Y. Zhou, S. Balalaie and B. Breit, Org. Lett., 2021, 23, 3343–3348 CrossRef CAS PubMed;
(g) M. Bera, H. S. Hwang, T.-W. Um, O. S. Shin and E. J. Cho, Org. Lett., 2022, 24, 1774–1779 CrossRef CAS PubMed;
(h) C. E. Raju, S. Balasubramanian and G. V. Karunakar, Org. Lett., 2022, 24, 2899–2904 CrossRef CAS PubMed;
(i) R. Mi, H. Chen, X. Zhou, N. Li, D. Ji, F. Wang, Y. Lan and X. Li, Angew. Chem., Int. Ed., 2022, 61, e202111860 CrossRef CAS PubMed.
-
(a) K. Kaur and S. Srivastava, New J. Chem., 2020, 44, 18530–18572 RSC , and references therein;;
(b) A. Y. Sukhorukov, Adv. Synth. Catal., 2020, 362, 724–754 CrossRef CAS , and references therein;;
(c) H.-Y. Chuang, M. Schupp, R. Meyrelles, B. Maryasin and N. Maulide, Angew. Chem., Int. Ed., 2021, 60, 13778–13782 CrossRef CAS PubMed.
-
(a) Y. Ogoshi, K. Shien, T. Yoshioka, H. Torigoe, H. Sato, M. Sakaguchi, S. Tomida, K. Namba, E. Kurihara, Y. Takahashi, K. Suzawa, H. Yamamoto, J. Soh and S. Toyooka, Oncol. Lett., 2019, 17, 2729–2736 CAS;
(b) C. X. Ma, J. Luo, R. A. Freedman, T. J. Pluard, J. R. Nangia, J. Lu, F. Valdez-Albini, M. Cobleigh, J. M. Jones, N. U. Lin, E. P. Winer, P. K. Marcom, S. Thomas, J. Anderson, B. Haas, L. Bucheit, R. Bryce, A. S. Lalani, L. A. Carey, M. P. Goetz, F. Gao, G. Kimmick, M. D. Pegram, M. J. Ellis and R. Bose, Clin. Cancer Res., 2022, 28, 1258–1267 CrossRef CAS PubMed;
(c) K. Mezger, S. Ebert, H. E. Muhle, U. Z. Stadt, A. Borkhardt, D. Dilloo, J. Faber, T. Feuchtinger, T. Imscweiler, N. Jorch, A. Pekrun, I. Schmid, F. Schramm, M. Zimmermann, M. A. Horstmann and G. Escherich, Blood Cancer, 2022, 69, e299997 Search PubMed.
-
(a) R. I. Tucceri, C. Barbero, J. J. Silber, L. Sereno and D. Posadas, Electrochim. Acta, 1997, 42, 919–927 CrossRef CAS;
(b) S. Menon, S. Jesny and K. G. A. Kumar, Talanta, 2018, 179, 668–675 CrossRef CAS PubMed;
(c) M. Kadivar and A. A. Aliakbar, Anal. Methods, 2021, 13, 536–551 RSC.
-
(a) E. Miyazawa, T. Sakamoto and Y. Kikugawa, J. Chem. Soc., Perkin Trans. 2, 1998, 7–12 RSC;
(b) Y. Kikugawa, C. Tsuji, E. Miyazawa and T. Sakamoto, Tetrahedron Lett., 2001, 42, 2337–2339 CrossRef CAS.
- X. Wang, T. Gensch, A. Lerchen, C. Daniliuc and F. Glorius, J. Am. Chem. Soc., 2017, 139, 6506–6512 CrossRef CAS PubMed.
-
(a) Y. Tan and J. F. Hartwig, J. Am. Chem. Soc., 2010, 132, 3676–3677 CrossRef CAS PubMed;
(b) W. P. Hong, A. V. Iosub and S. S. Stahl, J. Am. Chem. Soc., 2013, 135, 13664–13667 CrossRef CAS PubMed;
(c) D. S. Bolotin, N. A. Bokach, M. Y. Demakova and V. Y. Kukushkin, Chem. Rev., 2017, 117, 13039–13122 CrossRef CAS PubMed;
(d) Y. Takahashi, H. Tsuji and M. Kawatsura, J. Org. Chem., 2020, 85, 2654–2665 CrossRef CAS PubMed;
(e) K. Wang, H.-R. Guan, W.-L. Ren, H.-T. Yang and C.-B. Miao, J. Org. Chem., 2021, 86, 12309–12317 CrossRef CAS PubMed;
(f) C.-B. Miao, H.-R. Guan, Y. Tang, K. Wang, W.-L. Ren, X. Lyu, C. Yao and H.-T. Yang, Org. Lett., 2021, 23, 8699–8704 CrossRef CAS PubMed;
(g) Z. Xu, N. Xian, H. Chen, G.-J. Deng and H. Huang, Chin. J. Chem., 2021, 39, 1175–1180 CrossRef CAS;
(h) W.-X. Wei, X. Kong, R.-Q. Jiao, X.-S. Li, C.-T. Wang, Y. Li and Y.-M. Liang, Chem. Sci., 2022, 13, 6348–6354 RSC;
(i) Z. Qu, T. Tian, G.-J. Deng and H. Huang, Chin. Chem. Lett., 2023, 34, 107565 CrossRef CAS.
- T. Shimabayashi, K. Okamoto and K. Ohe, Chem. - Eur. J., 2017, 23, 16892 CrossRef PubMed.
- J. Rodriguez, A. Zeineddine, E. D. S. Carrizo, K. Miqueu, N. S. Merceron, A. Amgoune and D. Bourissou, Chem. Sci., 2019, 10, 7183–7192 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sc00874f |
| ‡ The ρ values of the Hammett analysis by using IPrCuBr and BF3·OEt2 were similar to that by using IPrCuBr and AgSbF6 (see ESI†). |
| § All calculations were performed using M. J. Frisch, et al., Gaussian 09, Revision D.01, Gaussian, Inc., Wallinhford CT, 2009. see ESI.† |
| ¶ The possibility that the present reaction proceeds via radical intermediates generated via either single electron transfer (SET) or homolytic N–O bond cleavage is not ruled out at the present transformation. The copper-catalyzed reaction of 1aa in the presence of a radical trapping reagent, such as TEMPO and 1,1-diphenylethylene didn't afford any adducts to evidence for such a process (see ESI† for more detail). The results are also same as Cu-catalyzed [1,3]-alkoxy rearrangement (ref. 5b). |
| || Similar substituent effects at the meta position were observed in Cu-catalyzed [1,3]-alkoxy rearrangement reactions (ref. 5b). |
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence b and
Itaru
Nakamura
b and
Itaru
Nakamura