
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEnantioselective total syntheses of six natural and two proposed meroterpenoids from Psoralea corylifolia†
Xiao-Wei
Chen
a,
Zi-Chao
Hou
a,
Chi
Chen
a,
Ling-Hui
Zhang
a,
Meng-En
Chen
a and
Fu-Min
Zhang
 *ab
*ab
aState Key Laboratory of Applied Organic Chemistry, College of Chemistry and Chemical Engineering, Lanzhou University, Lanzhou 730000, China. E-mail: zhangfm@lzu.edu.cn
bShanghai Engineering Research Center of Molecular Therapeutics and New Drug Development, School of Chemistry and Molecular Engineering, East China Normal University, Shanghai 200062, China
First published on 2nd May 2023
Abstract
The first enantioselective total syntheses of six natural and two proposed meroterpenoids isolated from Psoralea corylifolia have been achieved in 7–9 steps from 2-methylcyclohexanone. The current synthetic approaches feature a high level of synthetic flexibility, stereodivergent fashion and short synthetic route, thereby providing a potential platform for the preparation of numerous this-type meroterpenoids and their pseudo-natural products.
Synthetic strategies play a central role in the total syntheses of natural products; therefore, numerous synthetic strategies have been developed, such as diversity-oriented synthesis, function-oriented synthesis, two-phase synthesis, convergent synthesis, modular synthesis, C–H bond functionalization, and so on.1 Although considerable advances in these regards have been made, the pursuit of “ideal synthesis”2 has been attracting attention from synthetic chemists to continually explore new synthetic strategies, including the innovative applications of existing synthetic methodologies.
The carbonyl group is one of the most fundamental functionalities in organic chemistry,3 either as a crucial scaffold in countless organic molecules or as a universal and valuable reactive linchpin in synthetic chemistries. Due to its polarized C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond and the resultant acidic proton at the α-position of the carbonyl and/or the activation of the β-position through the formation of enolization-related reactive species, carbonyl-based chemical conversions have been an essential topic in organic synthesis, spawning numerous venerable organic reactions, particularly at the ipso-, α-, and β-positions of the carbonyl group.4 These excellent achievements of carbonyl conversions serve as a great reservoir for designing and developing novel transformations in the total syntheses of natural products.5 To add “a drop water” in “the ocean” of carbonyl-based conversion synthetic strategies, in the current work we pursued the collective total syntheses of natural meroterpenes using such a strategy.
O bond and the resultant acidic proton at the α-position of the carbonyl and/or the activation of the β-position through the formation of enolization-related reactive species, carbonyl-based chemical conversions have been an essential topic in organic synthesis, spawning numerous venerable organic reactions, particularly at the ipso-, α-, and β-positions of the carbonyl group.4 These excellent achievements of carbonyl conversions serve as a great reservoir for designing and developing novel transformations in the total syntheses of natural products.5 To add “a drop water” in “the ocean” of carbonyl-based conversion synthetic strategies, in the current work we pursued the collective total syntheses of natural meroterpenes using such a strategy.
Meroterpenoids of the monoterpene phenol family were isolated from Psoralea corylifolia, a well-known medicinal plant that has been widely used for the treatment of various diseases in China, India, and other countries.6 Some representative natural meroterpenes are shown in Scheme 1, including (+)-psoracorylifol F (1a),7a (−)-7α,8β-hydroxy-12β-cyclobakuchiol C (1b),7c corypsoriol J (1c),7b (−)-7β,8α-hydroxy-12β-psoracorylifol F (1d),7c (−)-corypsoriol H (1e),7b (−)-psoracorylifol G (1f),7f (−)-8α-hydroxy-cyclobakuchiol C (1g),7c corypsoriol I (1h),7b and other analogues.7 Structurally, these meroterpenes possess a densely functionalized cyclohexane containing four contiguous chiral centers, with one of which being all-carbon stereocenter. Besides the distinct stereochemistry of the substituents at C-7, C-8, and C-12 positions, they also feature a diversity of oxidation states for the sidechain at C-12 position as well as the aryl ring at C-7 position. Therefore, they may serve as an ideal arsenal to interpret the magic of the stereodivergent synthesis.1n To the best of our knowledge, the total syntheses of these meroterpenes bearing four contiguous stereogenic centers have not been achieved to date.8 Inspired by the unique structural features of the abovementioned meroterpenes and our research interest in carbonyl-related synthetic chemistry,9 we designed a carbonyl-based conversion synthetic strategy and anticipated that these target molecules could be collectively synthesized through sequential transformations directed by the carbonyl group of synthetic intermediates in each crucial step (Scheme 1). Guided by this strategy, we speculated that the structural simplification of all target meroterpenes 1a–1h to the key common intermediate 2 or its diastereomer 2′ would be feasible. For the 7α-aryl-12β-alkyl series, the hydroxy group at the C-13 position of 7α,8β-hydroxy-12β-cyclobakuchiol C (1b) or the C-3 position of corypsoriol J (1c) would be introduced via site-selective oxidation from psoracorylifol F (1a) or its C-8 methylether (not shown), respectively. Psoracorylifol F (1a), in turn, might be obtained via stereoselective reduction from the intermediate 2. For 7β-aryl-12β-alkyl meroterpenes, such as 7β,8α-hydroxy-12β-psoracorylifol F (1d), the stereochemistry of the aryl ring could be reversed through an in situ carbonyl-mediated enolization/protonation process from the intermediate 2; the β-hydroxyl group at the C-8 position would be acquired by the subsequent stereoselective reduction of the carbonyl group. Comparably, the 7β-aryl-12α-alkyl series, such as corypsoriol H (1e), psoracorylifol G (1f), 8α-hydroxy-cyclobakuchiol C (1g), and corypsoriol I (1h), could be achieved as well via similar transformations employing compound 2′, a diastereomer of intermediate 2, as the key precursor. The aryl ring at the C-7 position and transformable isopropenyl substituent at the C-12 position of the desired intermediates 2 and 2′ would be simultaneously assembled, in a stereochemistry-tunable fashion, using our developed Cu-mediated one-pot Michael addition/α-arylation reaction from enone 3.9a Inspired by the excellent work of Stoltz,10 the vinyl group of enone 3 would be generated through the Pd-catalyzed decarbonylative dehydration of acid 4. The conjugated double bond in the acid 4 would be introduced by the carbonyl-based regioselective dehydrogenation of the δ-keto ester 5. The all-carbon quaternary center at the C-9 position of δ-keto ester 5 would be constructed by the catalytic asymmetric Michael addition of the enolate of 2-methylcyclohexanone 6 to methyl acrylate 7.
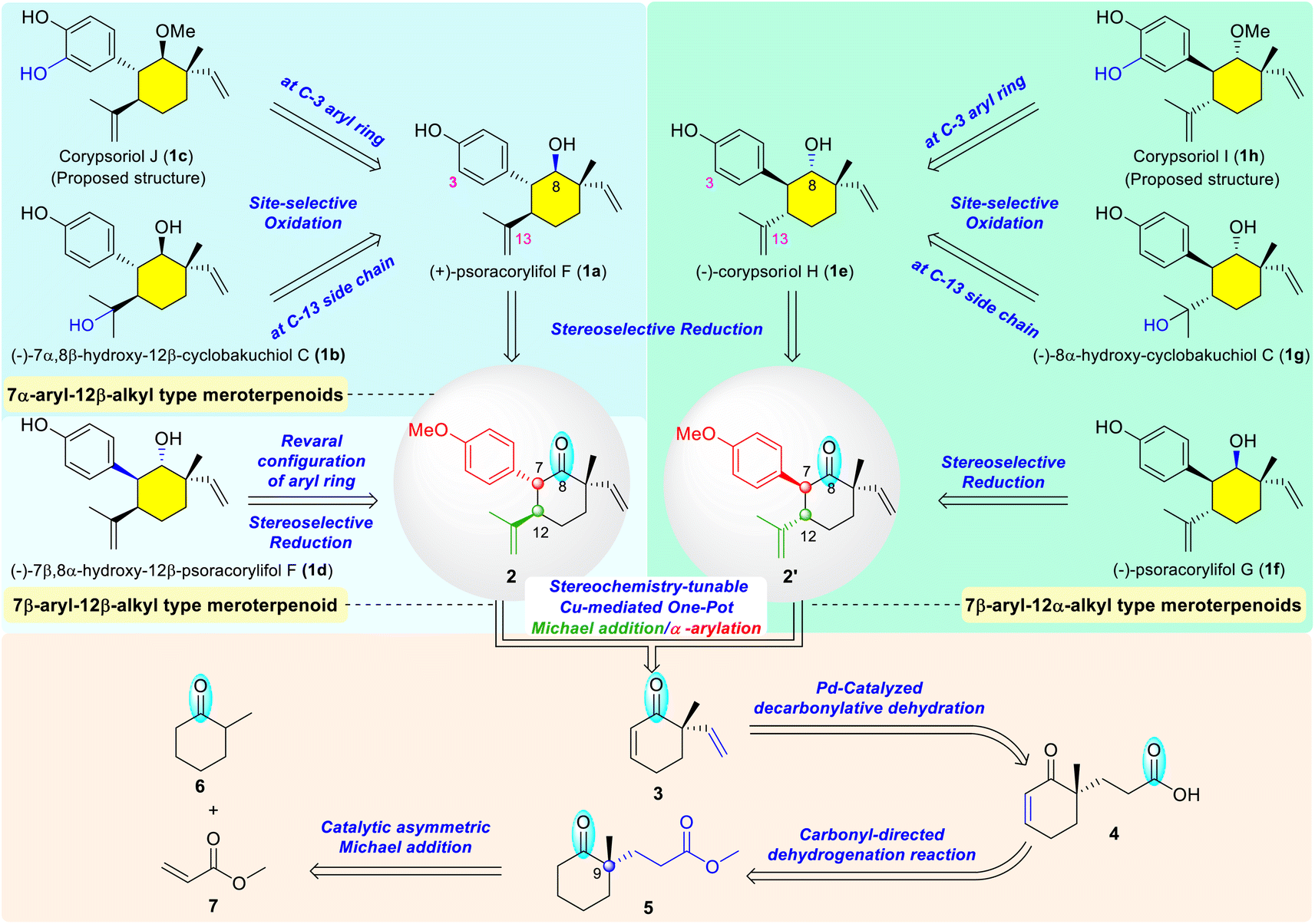 | ||
| Scheme 1 The retrosynthetic analysis of meroterpenes using carbonyl-based conversions. | ||
Following our retrosynthetic considerations, we commenced our total synthesis with the installation of the quaternary stereogenic centers of the target molecules. Using a modification of the method described by Cheong and Carter,11 an intermolecular Michael addition between commercially available 2-methylcyclohexanone (6) and methyl acrylate (7) catalyzed by a chiral thiourea catalyst (Cat. 1) afforded the expected adduct 5 with excellent enantiomeric excess (92% ee) and yield (80%) (Scheme 2). The conjugated double bond was introduced via carbonyl-directed dehydrogenation.12 Although the classical enolization and subsequent oxidation developed by Saegusa13 led to the desired desaturated enone 5′ (not shown, 65% yield),14 1.0 equivalent amount of Pd(OAc)2 was necessary to achieve the complete conversion of the starting material 5. Hence, a one-step approach using a stoichiometric amount of 2-iodoxybenzoic acid (IBX) as an oxidant15 was examined. To our delight, δ-keto ester 5 was smoothly converted to the corresponding conjugated enone 5′ (86% yield), which was then hydrolyzed to release the acid 4 in 97% yield.14 To further improve the synthetic efficiency, a two-step/one-column protocol was applied, affording the acid 4 in 83% yield. Subsequently, the Pd-catalyzed decarbonylative dehydration conditions developed by Stoltz10 were employed to introduce a vinyl group at the C-9 position, and the expected enone 3 was smoothly obtained in 70% yield. After the introduction of a vinyl group at the quaternary carbon center on the cyclohexenone skeleton, we turned our attention to the assembly of vicinal difunctionalities of the precursors 2 and 2′ from enone 3. Our developed Cu-mediated one-pot Michael addition/α-arylation sequence was extensively investigated by varying the reaction temperature, the amount of the diaryliodonium salt, copper salts, additives, and reaction solvent.14 Under the optimal reaction conditions, the expected product 2 along with its isomer 2′ was isolated in good stereoselectivity (8![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 dr) with moderate yield (40%, two-steps in one-pot). At this stage, the expected functionalities, including three chiral centers and one transformable carbonyl group, were efficiently assembled in the cyclohexane skeleton through five-step asymmetric transformations from two chemical feedstocks. With the key intermediate 2 in hand, we continued our efforts to achieve the collective total syntheses of representative target meroterpenes.
:1 dr) with moderate yield (40%, two-steps in one-pot). At this stage, the expected functionalities, including three chiral centers and one transformable carbonyl group, were efficiently assembled in the cyclohexane skeleton through five-step asymmetric transformations from two chemical feedstocks. With the key intermediate 2 in hand, we continued our efforts to achieve the collective total syntheses of representative target meroterpenes.
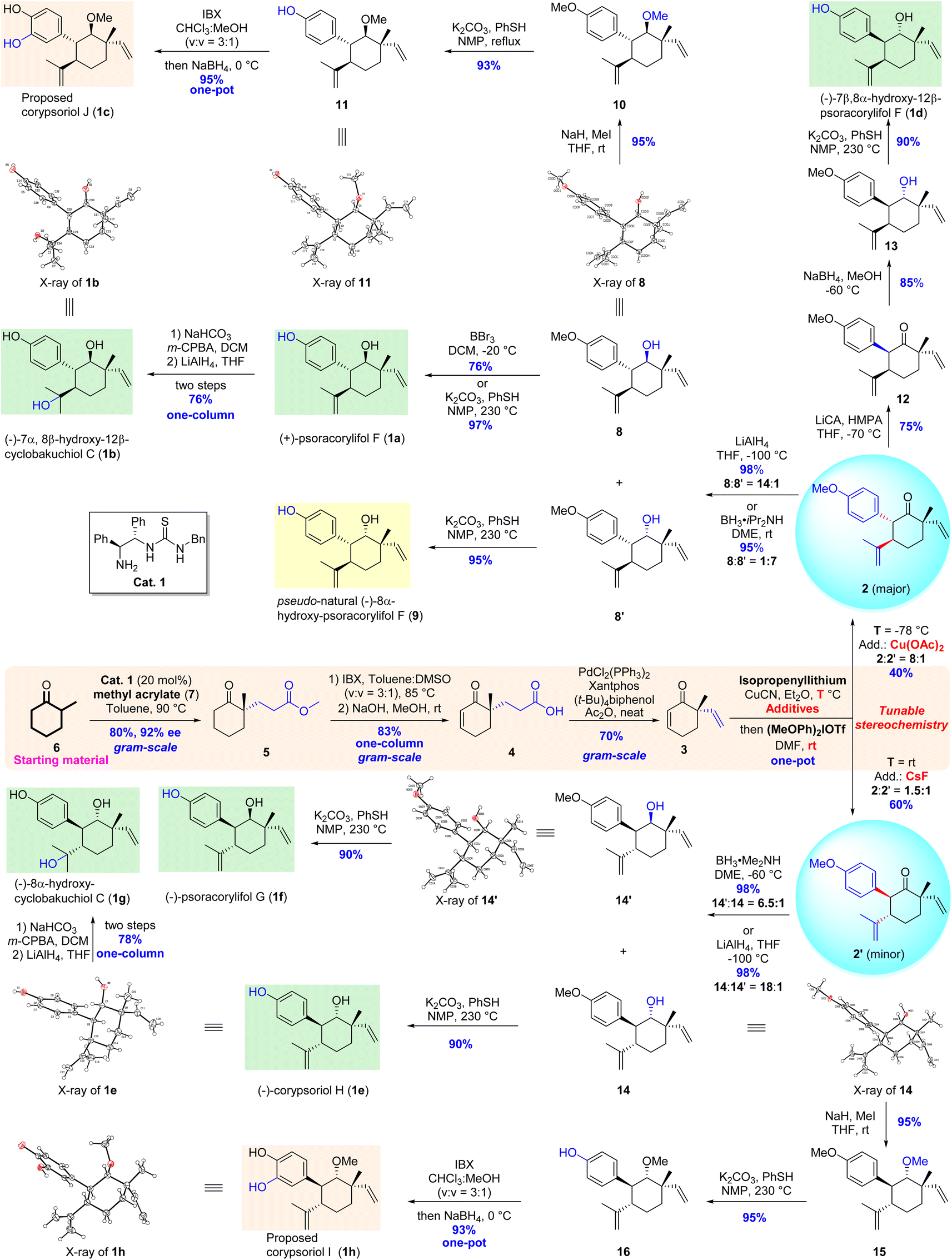 | ||
| Scheme 2 Collective total syntheses of meroterpenes from Psoralea corylifolia. | ||
The oxidation state at the C-8 position of precursor 2 was adjusted by reduction with LiAlH4 in tetrahydrofuran (THF) at −100 °C to produce the major 8β-OH alcohol 8 and its minor isomer 8′ with excellent levels of selectivity (14:1 dr) and yield (98%), while the selectivity was reversed (1:7 dr with 95% yield) using BH3·iPr2NH as a reductant in 1,2-dimethoxyethane (DME) at room temperature. The structure of alcohol 8 was further confirmed by X-ray crystallographic analysis, in which the absolute configurations of the four stereogenic centers were unambiguously verified. Subsequently, the treatment of alcohol 8 with BBr3 in CH2Cl2 removed the methyl ether at the aryl ring to afford (+)-psoracorylifol F (1a) in 76% yield, and an improved yield (97%) was observed when a combination of PhSH and K2CO3 in N-methylpyrrolidone (NMP) at 230 °C was applied.16 Pleasingly, the spectral data of 1a were consistent with the reported literature.7a Hence, the first total synthesis of (+)-psoracorylifols F was achieved in just 7 steps (Scheme 2). A similar deprotection of the methylether of the product 8′ yielded pseudo-natural (−)-8α-hydroxy-psoracorylifol F (9).
The site-selective epoxidation of the disubstituted double bond of (+)-psoracorylifol F with m-chloroperoxybenzoic acid (m-CPBA) and the subsequent reduction of the resulting epoxide with LiAlH4 produced natural (−)-7α,8β-hydroxy-12β-cyclobakuchiol C (1b) in 76% yield (two steps), and its structure was also confirmed by X-ray crystallographic analysis (Scheme 2).
To achieve the total synthesis of corypsoriol J (1c), alcohol 8 was treated with MeI to produce methylether 10 in 95% yield. The selective removal of methyl phenol ether was then examined, and the expected phenol 11 was obtained in excellent yield (93%) using the PhSH/K2CO3/NMP reaction system.16 To install an OH group at the C-3 position of the aryl ring, a two-step one-pot transformation17 involving IBX-mediated oxidation and subsequent reduction of the resultant ortho diketone with NaBH4 yielded the proposed corypsoriol J (1c) (95% yield) (Scheme 2). Unfortunately, the spectral data of synthetic corypsoriol J were not consistent with the reported ones of the natural product.7b Efforts to obtain the X-ray crystallographic structures of the synthetic corypsoriol J and its three derivatives were not successful.14 However, subsequent synthetic endeavors further confirmed that the structure of the proposed corypsoriol J needs to be revised.
Due to 7β,8α-hydroxy-12β-psoracorylifol F (1d) featuring a cis-configuration of the aryl ring at the C-7 position and an alkyl substituent at the C-12 position, a one-pot protocol involving the enolization/protonation of the key precursor 2 was carried out to reverse the configuration of the aryl ring. The desired product 12 was obtained in 75% yield using lithium isopropylcyclohexylamide (LiCA) as a base in THF at −70 °C, along with a recovery of the precursor 2 (10% yield). The subsequent stereoselective reduction of the carbonyl group of compound 12 generated alcohol 13 as a single isomer (85% yield), without the isolation of another diastereoisomer. The demethylation of 13 completed the total synthesis of (−)-7β,8α-hydroxy-12β-psoracorylifol F (1d) in 90% yield, and its spectral data were consistent with those of the corresponding natural product7c (Scheme 2).
Having accomplished the total syntheses of two natural 7α-aryl-12β-alkyl meroterpenoids, a proposed one, a pseudo-natural product, along with a 7β-aryl-12β-alkyl meroterpenoid, we focused on a third set of meroterpenoids, namely, 7β-aryl-12α-alkyl substituted natural products 1e–1h (Scheme 2), whose C-7 and C-12 stereocenters are reversal compared with the synthetic compounds 1a–1c. Fortunately, these natural meroterpenoids possess the same configuration of the minor product 2′ obtained from the previous key one-pot Michael addition/α-arylation reaction. Therefore, the tuning of the selectivity of the one-pot reaction is important. Various reaction conditions were again investigated to optimize the selectivity; the best results were obtained with an acceptable amount of product 2′ and its isomer 2 (ratio = 1:1.5, 60% combined yield for two steps).14 With the desired precursor 2′ in hand, chemical transformations similar to those for the syntheses of target molecules 1a–1c were performed to complete the total syntheses of four 7β-aryl-12α-alkyl substituted meroterpenoids. The stereoselective reduction of the carbonyl group at the C-8 position with LiAlH4 in THF at −100 °C provided two isomers 14 and 14′ with an excellent level (18:1 dr); in contrast, the treatment of 2′ with BH3·Me2NH in DME at −60 °C resulted in reverse selectivity, affording isomers 14′ and 14 (6.5:1 dr). The demethylation of compounds 14 and 14′ yielded (−)-corypsoriol H (1e) and (−)-psoracorylifol G (1f), respectively. The site-selective epoxidation of (−)-corypsoriol H and subsequent reductive ring-opening of the resultant epoxide completed the total synthesis of (−)-8α-hydroxy-cyclobakuchiol C (1g) in 78% yield (two steps). The spectroscopic data of these three synthesized natural products were fully consistent with those reported in the literature, respectively.7b,c,f Finally, using an approach similar to that for the proposed corypsoriol J, corypsoriol I (1h) was also synthesized in 4 steps with 66% overall yield from the precursor 2′. Its chemical structure was unambiguously confirmed by X-ray crystallographic analysis. Unfortunately, the spectral data of synthetic corypsoriol I were also not consistent with the reported literature7b (Scheme 2). Therefore, the chemical structures of natural corypsoriols I and J need to be reassigned.
In summary, the first enantioselective total syntheses of six meroterpenoids, i.e. (+)-psoracorylifol F, (−)-7α, 8β-hydroxy-12β-cyclobakuchiol C, (−)-7β, 8α-hydroxy-12β-psoracorylifol F, (−)-corypsoriol H, (−)-psoracorylifol G, and (−)-8α-hydroxy-cyclobakuchiol C, and two proposed corypsoriols I and J, along with pseudo-natural (−)-8α-hydroxyl-psoracorylifol F, have been accomplished using a carbonyl-based conversion synthetic strategy to install all stereogenic centers and the corresponding functionalities. The key transformations include organocatalytic Michael addition to construct a quaternary carbon center, Nicolaou oxidation to yield an enone, Pd-catalyzed decarbonylative dehydration to introduce a vinyl group, one-pot Michael addition/α-arylation to assemble two vicinal stereocenters, epimerization to reverse the configuration of the aryl group, and stereoselective reduction of ketone to acquire hydroxyl groups with different C-8 configurations. We envision that our stereodivergent synthetic route for the meroterpenoids reported here will facilitate further investigation of the biological activities of natural products, pseudo-natural ones, and their non-natural analogues. Further extension of this carbonyl-based conversion synthetic strategy for the total syntheses of natural products is being carried out in our laboratory.
Data availability
The datasets and spectra supporting this article have been uploaded as part of the ESI material.† Crystallographic data for compounds 1b, 1e, 1h, 8, 11, 14 and 14′ have been deposited in the Cambridge Crystallographic Data Center under accession numbers CCDC 2226760 (1b), CCDC 2226749 (1e), CCDC 2226751 (1h), CCDC 2226753 (8), CCDC 2226754 (11), CCDC 2226756 (14), and CCDC 2226759 (14′).†Author contributions
F.-M. Z. designed the project and wrote the manuscript. X.-W. C., Z.-C. H., C. C., and L.-H. Z. performed all the experiments. X.-W. C. prepared the ESI.† L.-H. Z. and M.-E. C. prepared some starting material and part of the ESI.† All of the authors discussed the results.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful to the NSFC (No. 22278200, 21971095 and 91956203) for financially supporting this work.Notes and references
- For recently selected books, reviews, and examples, see: (a) P.-Q. Huang, Z.-J. Yao and R. P. Hsung, Efficiency in natural product total synthesis, Wiley, Hoboken, 2018 CrossRef; (b) K. C. Nicolaou, S. Rigol and R. Yu, CCS Chem., 2019, 1, 3–37 CAS; (c) B. Mikulak-Klucznik, P. Gołębiowska, A. A. Bayly, O. Popik, T. Klucznik, S. Szymkuć, E. P. Gajewska, P. Dittwald, O. Staszewska-Krajewska, W. Beker, T. Badowski, K. A. Scheidt, K. Molga, J. Mlynarski, M. Mrksich and B. A. Grzybowski, Nature, 2020, 588, 83–88 CrossRef CAS PubMed; (d) P. A. Wender, V. A. Verma, T. J. Paxton and T. H. Pillow, Acc. Chem. Res., 2008, 41, 40–49 CrossRef CAS PubMed; (e) C. J. Gerry and S. L. Schreiber, Curr. Opin. Chem. Biol., 2020, 56, 1–9 CrossRef CAS PubMed; (f) E. Lenci and A. Trabocchi, Eur. J. Org. Chem., 2022, 2022, e202200575 CrossRef CAS; (g) R. Chen, Y. Shen, S. Yang and Y. Zhang, Angew. Chem., Int. Ed., 2020, 59, 14198–14210 CrossRef CAS PubMed; (h) Y. Kanda, Y. Ishihara, N. C. Wilde and P. S. Baran, J. Org. Chem., 2020, 85, 10293–10320 CrossRef CAS PubMed; (i) C. K. G. Gerlinger and T. Gaich, Chem.–Eur. J., 2019, 25, 10782–10791 CrossRef CAS PubMed; (j) M. Elkin and T. R. Newhouse, Chem. Soc. Rev., 2018, 47, 7830–7844 RSC; (k) C. W. Coley, D. A. Thomas, J. A. M. Lummiss, J. N. Jaworski, C. P. Breen, V. Schultz, T. Hart, J. S. Fishman, L. Rogers, H. Gao, R. W. Hicklin, P. P. Plehiers, J. Byington, J. S. Piotti, W. H. Green, A. J. Hart, T. F. Jamison and K. F. Jensen, Science, 2019, 365, eaax1566 CrossRef CAS PubMed; (l) M. Sc. L. Koser, M. Sc. V. M. Lechner and T. Bach, Angew. Chem., In. Ed., 2021, 60, 20269–20273 CrossRef PubMed; (m) F. Schneider, K. Samarin, S. Zanella and T. Gaich, Science, 2020, 367, 676–681 CrossRef CAS PubMed; (n) L. Li, Z. Chen, X. Zhang and Y. Jia, Chem. Rev., 2018, 118, 3752–3832 CrossRef CAS PubMed; (o) R. Roddan, E. M. Carter, B. Thair and H. C. Hailes, Nat. Prod. Rep., 2022, 39, 1375–1382 RSC; (p) M. Munda, S. Niyogi, K. Shaw, S. Kundu, R. Nandia and A. Bisai, Org. Biomol. Chem., 2022, 20, 727–748 RSC; (q) S. P. Pitre and L. E. Overman, Chem. Rev., 2022, 122, 1717–1751 CrossRef CAS PubMed; (r) O. Baudoin, Angew. Chem., Int. Ed., 2020, 59, 17798–17809 CrossRef CAS PubMed; (s) H. Yan and F.-E. Chen, Adv. Synth. Catal., 2022, 364, 1934–1961 CrossRef CAS; (t) L. Cai, I. B. Seiple and Q. Li, Acc. Chem. Res., 2021, 54, 1891–1908 CrossRef CAS PubMed; (u) Y. Jiao and T. Luo, Nat. Sci. Rev., 2022, nwac029 CrossRef PubMed; (v) Y. Zhang, A. A. Vinogradov, J. S. Chang, Y. Goto and H. Suga, Org. Lett., 2022, 24, 7894–7899 CrossRef CAS PubMed; (w) J. Li, F. Chen and H. Renata, J. Am. Chem. Soc., 2022, 144, 19238–19242 CrossRef CAS PubMed.
- For recently selected references, see: (a) R. Noyori, Nat. Chem., 2009, 1, 5–6 CrossRef CAS PubMed; (b) P. A. Wender, et al., Organic synthesis: Theory and applications, ed. T. Hudlicky, JAI Press, Greenwich, 1993, vol. 2, pp. 27–66 Search PubMed; (c) J. B. Hendrickson, J. Am. Chem. Soc., 1975, 97, 5784–5800 CrossRef CAS; (d) B. M. Trost, Science, 1991, 254, 1471–1477 CrossRef CAS PubMed; (e) N. Z. Burns, P. S. Baran and R. W. Hoffmann, Angew. Chem., Int. Ed., 2009, 48, 2854–2867 CrossRef CAS PubMed.
- For the selected book and reviews, see: (a) Modern carbonyl chemistry, ed. J. Otera, Wiley-VCH, Weinheim. 2000 Search PubMed; (b) Y. Xue and G. Dong, Acc. Chem. Res., 2022, 55, 2341–2354 CrossRef CAS PubMed; (c) D. J. Foley and H. Waldmann, Chem. Soc. Rev., 2022, 51, 4094–4120 RSC; (d) H.-M. Huang, P. Bellotti and F. Gloriu, Acc. Chem. Res., 2022, 55, 1135–1147 CrossRef CAS PubMed; (e) X.-J. Dai, C.-C. Li and C.-J. Li, Chem. Soc. Rev., 2021, 50, 10733–10742 RSC; (f) H. Albright, A. J. Davis, J. L. Gomez-Lopez, H. L. Vonesh, P. K. Quach, T. H. Lambert and C. S. Schindler, Chem. Rev., 2021, 121, 9359–9406 CrossRef CAS PubMed; (g) Á. Péter, S. Agasti, O. Knowles, E. Pye and D. J. Procter, Chem. Soc. Rev., 2021, 50, 5349–5365 RSC; (h) S. Wang and B. König, Angew. Chem., Int. Ed., 2021, 60, 21624–21634 CrossRef CAS PubMed; (i) Z. Yang, Acc. Chem. Res., 2021, 54, 556–568 CrossRef CAS PubMed; (j) B. Delayre, Q. Wang and J. Zhu, ACS Cent. Sci., 2021, 7, 559–569 CrossRef CAS PubMed.
- (a) P.-Q. Huang, Organic named reactions, reagents and rules, Chemical Industry Press, 2nd edn, Beijing, 2019 Search PubMed; (b) L. L. Jack, Named reactions, Springer-Verlag Berlin Heidelberg, 2009 Search PubMed; (c) L. Kürti and B. Czakó, Strategic applications of named reactions in organic synthesis: background and detailed mechanisms, Elsevier Academic Press, San Diego, CA, 2005 Search PubMed.
- For recently selected examples, see: (a) Z. Wu, X. Xu, J. Wang and G. Dong, Science, 2021, 374, 734–740 CrossRef CAS PubMed; (b) J. Xie, X. Liu, N. Zhang, S. Choi and G. Dong, J. Am. Chem. Soc., 2021, 143, 19311–19316 CrossRef CAS PubMed; (c) Z. Wu and G. Dong, Angew. Chem., Int. Ed., 2022, 61, e202201239 CAS; (d) S. Jin, X. Zhao and D. Ma, J. Am. Chem. Soc., 2022, 144, 15355–15362 CrossRef CAS PubMed; (e) G. Sennari, K. E. Gardner, S. Wiesler, M. Haider, A. Eggert and R. Sarpong, J. Am. Chem. Soc., 2022, 144, 19173–19185 CrossRef CAS PubMed; (f) W. M. Amberg and E. M. Carreira, J. Am. Chem. Soc., 2022, 144, 15475–15479 CrossRef CAS PubMed; (g) T. Ma, H. Cheng, M. Pitchakuntla, W. Ma and Y. Jia, J. Am. Chem. Soc., 2022, 144, 20196–20200 CrossRef CAS PubMed; (h) B. Yang, J. Dai, Y. Luo, K. K. Lau, Y. Lan, Z. Shao and Y. Zhao, J. Am. Chem. Soc., 2021, 143, 4179–4186 CrossRef CAS PubMed; (i) Z. Xin, H. Wang, H. He, X. Zhao and S. Gao, Angew. Chem., Int. Ed., 2021, 60, 12807–12812 CrossRef CAS PubMed; (j) P. Yang, Y.-Y. Li, H. Tian, G.-L. Qian, Y. Wang, X. Hong and J. Gui, J. Am. Chem. Soc., 2022, 144, 17769–17775 CrossRef CAS PubMed; (k) Y. Zhao, J. Hu, R. Chen, F. Xiong, H. Xie and H. Ding, J. Am. Chem. Soc., 2022, 144, 2495–2500 CrossRef CAS PubMed; (l) Y.-J. Hu, C.-C. Gu, X.-F. Wang, L. Min and C.-C. Li, J. Am. Chem. Soc., 2021, 143, 17862–17870 CrossRef CAS PubMed; (m) R. Andres, Q. Wang and J. Zhu, Angew. Chem., Int. Ed., 2022, 61, e202201788 CrossRef CAS PubMed.
- For the recently selected reviews, see: (a) B. Z. Amirzakariya and A. Shakeri, Phytochemistry, 2022, 197, 113130–113166 CrossRef CAS PubMed; (b) N. Mahajan, B. Koul, P. Gupta, B. A. Shahd and J. Singh, S. Afr. J. Bot., 2022, 149, 963–993 CrossRef CAS; (c) F. Alam, G. N. Khan and M. H. H. B. Asad, Phytother. Res., 2018, 32, 597–615 CrossRef PubMed; (d) X. Zhang, W. Zhao, Y. Wang, J. Lu and X. Chen, Am. J. Chin. Med., 2016, 44, 35–60 CrossRef CAS PubMed; (e) L. Chen, S. Chen, P. Sun, X. Liu, Z. Zhan and J. Wang, Chin. Med., 2023, 18, 4 CrossRef CAS PubMed; (f) A. R. Kangane, N. V. Gawali and S. D. Lokhande, World J. Pharm. Sci., 2023, 12, 1486–1498 CAS.
- (a) G. Xiao, X. Li, T. Wu, Z. Cheng, Q. Tang and T. Zhang, Fitoterapia, 2012, 83, 1553–1557 CrossRef CAS PubMed; (b) Q.-X. Xu, W. Xu and X.-W. Yang, Tetrahedron, 2020, 76, 131343–131348 CrossRef CAS; (c) M. X. Xiu, Y. M. Zhao, Y. Zhang, D. X. Xiong, D. Wang, H. S. Lee and L. Cui, Fitoterapia, 2021, 151, 104881–104885 CrossRef CAS PubMed; (d) H.-T.-Y. Gao, G.-Z. Lang, Y.-D. Zang, J. Ma, J.-Z. Yang, F. Ye, J.-Y. Tian, P.-P. Gao, C.-J. Li and D.-M. Zhang, Bioorg. Chem., 2021, 112, 104924–104936 CrossRef CAS PubMed; (e) Q. Xu, Q. lv, L. Liu, Y. Zhang and X. Yang, Chin. Med., 2021, 16, 98–112 CrossRef CAS PubMed; (f) X.-W. Yang, Q. Lü, Q.-X. Xu, W. Xu and Y.-T. Zhang, Chin. Tradit. Herb. Drugs, 2022, 53, 3269–3277 Search PubMed.
- Two relative skeleton rearrangement natural products have been synthesized by Tong and coworkers, see: (a) J. Ren, Y. Liu, L. Song and R. Tong, Org. Lett., 2014, 16, 2986–2989 CrossRef CAS PubMed ; for the total syntheses of three relative natural products bearing three stereocentres, see:; (b) H. Kawashima, Y. Kaneko, M. Sakai and Y. Kobayashi, Chem.–Eur. J., 2014, 20, 272–278 CrossRef CAS PubMed; (c) H. Kawashima, M. Sakai, Y. Kaneko and Y. Kobayashi, Tetrahedron, 2015, 71, 2387–2392 CrossRef CAS; (d) V. Maurya and C. Appayee, Org. Lett., 2018, 20, 4111–4115 CrossRef CAS PubMed; (e) M. Shoji, T. Esumi, N. Tanaka, M. Takeuchi, S. Yamaji, M. Watanabe, E. Takahashi, H. Kido, M. Yamamoto and T. Kuzuhara, PLoS One, 2021, 16, e0248960 CrossRef CAS PubMed.
- (a) J.-L. Pan, T. Chen, Z.-Q. Zhang, Y.-F. Li, X.-M. Zhang and F.-M. Zhang, Chem. Commun., 2016, 52, 2382–2385 RSC; (b) T. Chen, Y.-F. Li and F.-M. Zhang, Org. Lett., 2016, 18, 4754–4757 CrossRef CAS PubMed; (c) T. Chen, R. Peng, W. Hu and F.-M. Zhang, Org. Biomol. Chem., 2016, 14, 9859–9867 RSC; (d) Z.-Q. Zhang, T. Chen and F.-M. Zhang, Org. Lett., 2017, 19, 1124–1127 CrossRef CAS PubMed; (e) S.-Z. Tang, W. Zhao, T. Chen, Y. Liu, X.-M. Zhang and F.-M. Zhang, Adv. Synth. Catal., 2017, 359, 4177–4183 CrossRef CAS; (f) S.-Z. Tang, H.-L. Bian, Z.-S. Zhan, M.-E. Chen, J.-W. Lv, S. Xie and F.-M. Zhang, Chem. Commun., 2018, 54, 12377–12380 RSC; (g) Y. An, X.-M. Zhang, Z.-Y. Li, W.-H. Xiong, R.-D. Yu and F.-M. Zhang, Chem. Commun., 2019, 55, 119–122 RSC; (h) Q. Zhang, F.-M. Zhang, C.-S. Zhang, S.-Z. Liu, J.-M. Tian, S.-H. Wang, X.-M. Zhang and Y.-Q. Tu, Nat. Commun., 2019, 10, 2507–2514 CrossRef PubMed; (i) Q. Zhang, F.-M. Zhang, C.-S. Zhang, S.-Z. Liu, J.-M. Tian, S.-H. Wang, X.-M. Zhang and Y.-Q. Tu, J. Org. Chem., 2019, 84, 12664–12671 CrossRef CAS PubMed; (j) H.-L. Bian, S.-Z. Tang, M.-E. Chen, X.-M. Zhang, J.-W. Lv, X.-W. Chen, F.-M. Qi, S.-W. Chen and F.-M. Zhang, Org. Lett., 2020, 22, 5314–5317 CrossRef CAS PubMed; (k) S. Xie, Z.-J. He, L.-H. Zhang, B.-L. Huang, X.-W. Chen, Z.-S. Zhan and F.-M. Zhang, Chem. Commun., 2021, 57, 2069–2072 RSC; (l) X. Han, L.-X. Shan, J.-X. Zhu, C.-S. Zhang, X.-M. Zhang, F.-M. Zhang, H. Wang, Y.-Q. Tu, M. Yang and W.-S. Zhang, Angew. Chem., Int. Ed., 2021, 60, 22688–22692 CrossRef CAS PubMed; (m) J.-S. Yang, K. Lu, C.-X. Li, Z.-H. Zhao, X.-M. Zhang, F.-M. Zhang and Y.-Q. Tu, Angew. Chem., Int. Ed., 2022, 61, e202114129 CAS; (n) M.-E. Chen, S.-Z. Tang, Y.-H. Hu, Q.-T. Li, Z.-Y. Gan, J.-W. Lv and F.-M. Zhang, CCS Chem., 2022, 4, 3378–3390 CrossRef CAS; (o) M.-E. Chen, Z.-Y. Gan, Y.-H. Hu and F.-M. Zhang, J. Org. Chem., 2023, 88, 3954–3964 CrossRef CAS PubMed.
- Y. Liu, S. C. Virgil, R. H. Grubbs and B. M. Stoltz, Angew. Chem., Int. Ed., 2015, 54, 11800–11803 CrossRef CAS PubMed.
- (a) J. Y. Kang and R. G. Carter, Org. Lett., 2012, 14, 3178–3181 CrossRef CAS PubMed; (b) J. Y. Kang, R. C. Johnston, K. M. Snyder, P. H.-Y. Cheong and R. G. Carter, J. Org. Chem., 2016, 81, 3629–3637 CrossRef CAS PubMed.
- For the recently selected reviews, see: (a) S. Gnaim, J. C. Vantourout, F. Serpier, P.-G. Echeverria and P. S. Baran, ACS Catal., 2021, 11, 883–892 CrossRef CAS; (b) T. Hirao, J. Org. Chem., 2019, 84, 1687–1692 CrossRef CAS PubMed.
- Y. Ito, T. Hirao and T. Saegusa, J. Org. Chem., 1978, 43, 1011–1013 CrossRef CAS.
- For the details, please see the ESI.†.
- (a) K. C. Nicolaou, Y. L. Zhong and P. S. Baran, J. Am. Chem. Soc., 2000, 122, 7596–7597 CrossRef CAS; (b) K. C. Nicolaou, T. Montagnon, P. S. Baran and Y.-L. Zhong, J. Am. Chem. Soc., 2002, 124, 2245–2258 CrossRef CAS PubMed.
- L. Sharma, M. K. Nayak and A. K. Chakraborti, Tetrahedron, 1999, 55, 9595–9600 CrossRef CAS.
- (a) A. Pezzella, L. Lista, A. Napolitano and M. ďIschia, Tetrahedron Lett., 2005, 46, 3541–3544 CrossRef CAS; (b) M. L. Nóvoa, F. J. Salazara, C. Gámez, A. Y. Angarita, E. Tropper, N. Canudas and J. E. Villamizar, Nat. Prod. Commun., 2014, 9, 355–358 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2226749, 2226751, 2226753, 2226754, 2226756, 2226759 and 2226760. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3sc00582h |
| This journal is © The Royal Society of Chemistry 2023 |