
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Palladium-catalyzed asymmetric allenylic alkylation: construction of multiple chiral thiochromanone derivatives†
Li-Xia
Liu
 ab,
Yu-Qing
Bai
ab,
Xiang
Li
a,
Chang-Bin
Yu
*a and
Yong-Gui
Zhou
*a
ab,
Yu-Qing
Bai
ab,
Xiang
Li
a,
Chang-Bin
Yu
*a and
Yong-Gui
Zhou
*a
aState Key Laboratory of Catalysis, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, Dalian 116023, China. E-mail: cbyu@dicp.ac.cn; ygzhou@dicp.ac.cn
bUniversity of Chinese Academy of Sciences, Beijing 100049, China
First published on 25th April 2023
Abstract
The development of a new strategy for the construction of chiral cyclic sulfide-containing multiple stereogenic centers is highly desirable. Herein, by the combination of base-promoted retro-sulfa-Michael addition and palladium-catalyzed asymmetric allenylic alkylation, the streamlined synthesis of chiral thiochromanones containing two central chiralities (including a quaternary stereogenic center) and an axial chirality (allene unit) was successfully realized with up to 98% yield, 49.0![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 dr and >99% ee.
:1 dr and >99% ee.
Introduction
Organic sulfur compounds play important roles in pharmaceuticals, natural products and other fields, in which molecules containing chiral cyclic sulfide skeletons generally have special biological activity.1 For example, diltiazem (A),2a a calcium channel blocking agent, is used for treating supraventricular tachycardia, a rhythm disturbance of the heart. Additionally, the rivastigmine analogue (B)2b is synthesized as an acetylcholine-esterase (AChE) inhibitor for the treatment of Alzheimer's disease, and 7-thia-DCK (C)2c is an anti-AIDS agent (Scheme 1). Therefore, developing simple and efficient methods for the synthesis of chiral cyclic sulfide derivatives has become an intriguing area. Among the developed methods, organocatalytic sulfa-Michael-initiated cascade reactions are particularly appealing, which construct chiral cyclic sulfides containing one or more stereocenters.3 In contrast, metal-catalyzed asymmetric reactions4 are underexplored owing to the poisoning of metal catalysts by sulfur,5 especially sulfur anions. Considering thiochromanes are important components of cyclic sulfide derivatives,2b,c,6 it is vital to build chiral thiochromane derivatives containing multiple stereogenic centers.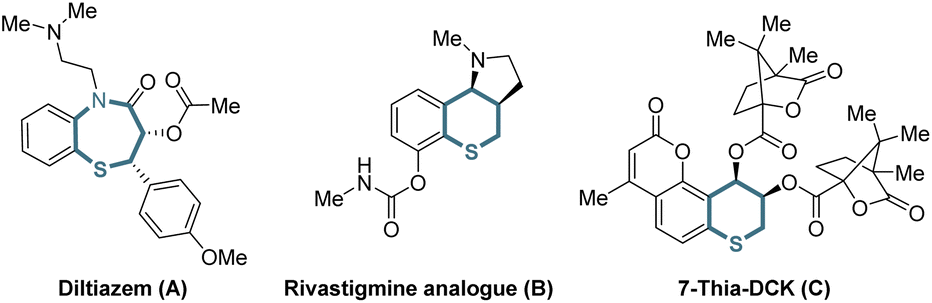 | ||
| Scheme 1 Selected bioactive cyclic sulfide derivatives. | ||
The allene moiety is present widely in natural products and bioactive compounds,7 and is an extremely versatile functional group in organic synthesis as well.8 Recently, Pd-catalyzed asymmetric allenylic alkylations9 have been developed as an efficient approach to construct chiral allenes. Previously, metal-catalyzed asymmetric reactions to build multiple chiral compounds, utilizing retro-oxa-Michael addition to simultaneously racemize two stereocenters, have been developed.10 However, these asymmetric reactions involving a retro-sulfa-Michael addition process were less developed, in which only organocatalytic examples were reported.3d,h,11 Combining palladium-catalyzed asymmetric allenylic alkylation and retro-sulfa-Michael addition to construct multiple chiral thiochromanone derivatives, some issues would need to be considered. (1) The rate of the racemization process via retro-sulfa-Michael addition should be faster than the rate of the next allenylic alkylation. (2) The sulfur is also nucleophilic,12 which would compete with the carbon nucleophile, leading to different chemoselective products. (3) Regioselective products would be observed with an electrophilic π-allylpalladium intermediate (allene partner).13 (4) The precise control of multiple chiralities including axial and central chirality is also a challenge. Hence, multiple possible products might be produced (Scheme 2). In this article, we reported the synthesis of enantioenriched multiple substituted thiochromanone derivatives containing two central chiralities and an axial chirality through the combination of retro-sulfa-Michael addition and palladium-catalyzed allenylic alkylation under basic conditions with up to 49.0:1 dr and >99% ee.
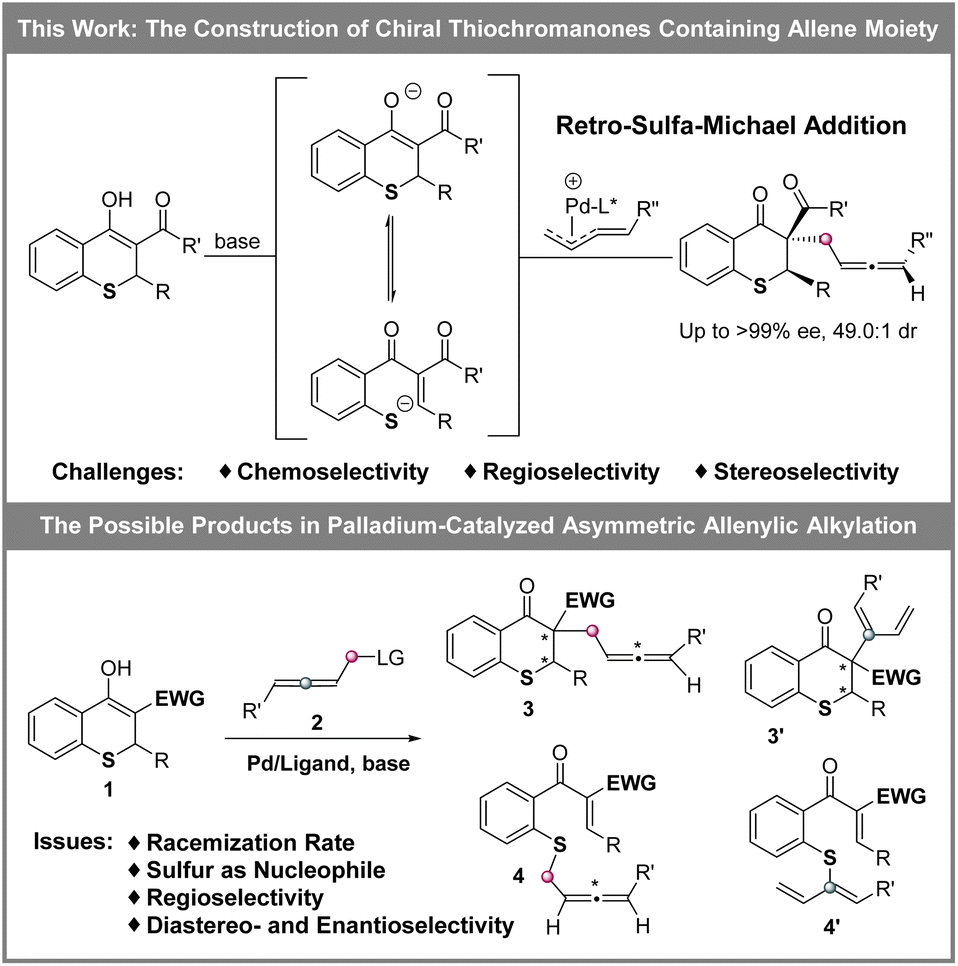 | ||
| Scheme 2 Construction of multiple chiral thiochromanone derivatives. | ||
Results and discussion
The regio- and chemoselectivity of asymmetric allenylic alkylation might be ascribed to the electron-withdrawing group (EWG) in the thiochromanone derivatives. Thus, we initially explored the effect of electron-withdrawing groups on the reaction (Table 1). Just as we speculated, different electron-withdrawing groups led to regio- and chemoselective isomers on the allenylic alkylation. When the nitro group was used as the EWG, the chemoselective isomer 4aa with sulfur as a nucleophile was observed (entry 1). Then, the desired product 3ba with carbon as a nucleophile was obtained when the acetyl group was introduced as the EWG instead of the nitro group with low diastereoselectivity (entry 2). It is delightful that 14.7:1 dr was gained using the methoxycarbonyl as the EWG (entry 3).
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | EWG | Product | Yieldb (%) | drb (%) | eec (%) | |
| Major | Minor | |||||
| a Reaction conditions: 1 (0.1 mmol), 2a (1.5 equiv.), Pd(dba)2 (10 mol%), L1 (11 mol%), DBU (1.2 equiv.), THF (1.0 mL), 5 Å MS (50 mg), 30 °C, 10–24 h. b Yield and diastereomeric ratio were measured by analysis of 1H NMR spectra using 1,3,5-trimethoxybenzene as the internal standard. c Determined by HPLC. | ||||||
| 1 | NO2 (1a) | 4aa | 74 | — | 84 | — |
| 2 | C(O)Me (1b) | 3ba | 94 | 3.5:1 |
70 | 90 |
| 3 | CO2Me (1c) | 3ca | 94 | 14.7:1 |
98 | 31 |
| 4 | C(O)N(CH2)5 (1d) | 3da | 12 | 5.0:1 |
96 | 5 |
| 4'da | 84 | — | — | — | ||
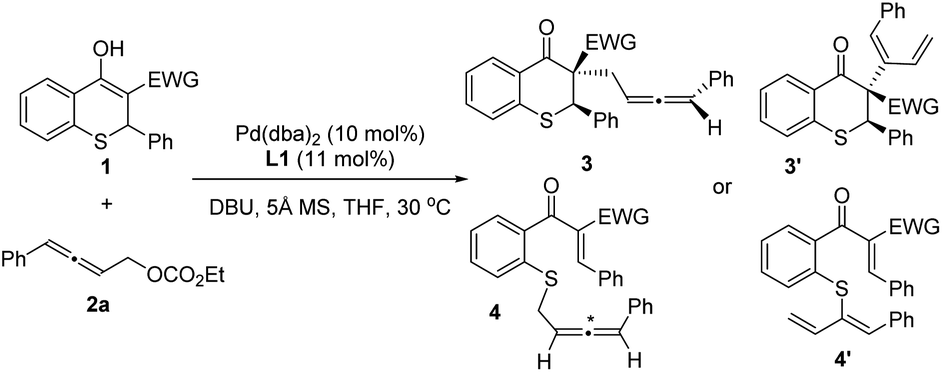
In addition, using amido as the EWG, the regio- and chemoselective isomer 4′da was discovered as the main product when nucleophilic 1d reacted with π-allylpalladium species. The yield of the desired product 3da was only 12% with 5.0:1 dr and 96% ee for the major diastereoisomer (entry 4). Besides, using 1,3-bis(diphenylphosphino)propane as the ligand, trace rac-3′da was observed, in which different isomers might be obtained owing to the effect of ligand. On the whole, methoxycarbonyl should be the optimal electron-withdrawing group, with which the allenylic alkylation could proceed smoothly to deliver the desired major isomer.
Afterwards, the optimization of the asymmetric allenylic alkylation was examined using thiochromanone 1c and allenylic carbonates 2 as substrates. First, different allenylic carbonates were screened (Table 2, entries 1–3) and ethyl (4-phenylbuta-2,3-dien-1-yl) carbonate 2a might be better (entry 1). We next investigated the effect of solvent on this alkylation reaction (entries 4–6). The reactions performed smoothly in a variety of solvents, offering good stereoselectivities and reactivities. Then, tetrahydrofuran was chosen as the optimal solvent. Next, organic and inorganic bases were investigated, and 1,8-diazabicyclo-[5.4.0]undec-7-ene (DBU) still performed better (entries 7–9).
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Solvent | Base/L | [Pd] | Yieldb (%) | drb | eec (%) |
| Major/minor | ||||||
| a Reaction conditions: 1c (0.1 mmol), 2a (1.5 equiv.), [Pd] (10 mol%), L (11 mol%), base (1.2 equiv.), solvent (1.0 mL), 5 Å MS (50 mg), 30 °C, 10–72 h. b Yield and diastereomeric ratio were measured by analysis of 1H NMR spectra using 1,3,5-trimethoxybenzene as the internal standard. c Determined by chiral HPLC. d 2a′ instead of 2a. e 2a′′ instead of 2a. f 0 °C instead of 30 °C. g −20 °C instead of 30 °C. h Isolated yield for the reaction at the 0.2 mmol scale, 72 h. | ||||||
| 1 | THF | DBU/L1 | Pd(dba)2 | 94 | 14.7:1 |
98/31 |
| 2d | THF | DBU/L1 | Pd(dba)2 | >95 | 12.7:1 |
97/59 |
| 3e | THF | DBU/L1 | Pd(dba)2 | >95 | 12.7:1 |
97/30 |
| 4 | MeCN | DBU/L1 | Pd(dba)2 | 91 | 14.2:1 |
98/42 |
| 5 | DMF | DBU/L1 | Pd(dba)2 | 90 | 14.0:1 |
97/11 |
| 6 | Toluene | DBU/L1 | Pd(dba)2 | >95 | 11.4:1 |
96/69 |
| 7 | THF | Et3N/L1 | Pd(dba)2 | 92 | 5.1:1 |
90/83 |
| 8 | THF | TMG/L1 | Pd(dba)2 | >95 | 8.7:1 |
95/84 |
| 9 | THF | KOtBu/L1 | Pd(dba)2 | >95 | 11.0:1 |
97/55 |
| 10 | THF | DBU/L2 | Pd(dba)2 | >95 | 4.0:1 |
79/31 |
| 11 | THF | DBU/L3 | Pd(dba)2 | >95 | 9.8:1 |
98/77 |
| 12 | THF | DBU/L4 | Pd(dba)2 | >95 | 18.6:1 |
95/30 |
| 13 | THF | DBU/L5 | Pd(dba)2 | >95 | 15.0:1 |
98/65 |
| 14 | THF | DBU/L4 | Pd2(dba)3 | >95 | 15.3:1 |
98/54 |
| 15 | THF | DBU/L4 | [Pd(C3H5)Cl]2 | 86 | 13.3:1 |
98/16 |
| 16 | THF | DBU/L4 | Pd(OAc)2 | >95 | 15.0:1 |
98/9 |
| 17f | THF | DBU/L4 | Pd(dba)2 | >95 | 31.0:1 |
98/11 |
| 18g | THF | DBU/L4 | Pd(dba)2 | 92h | 32.3:1 |
99/52 |
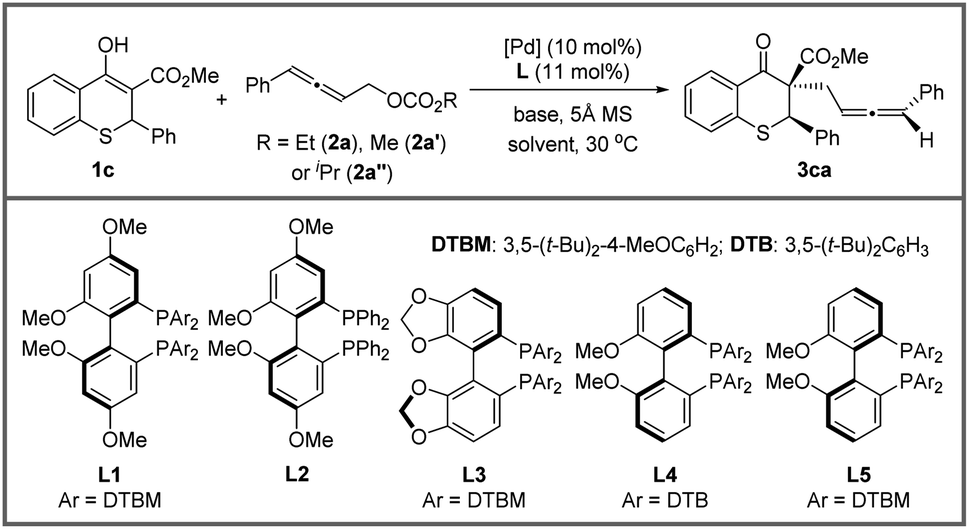
A chiral ligand, as we all know, is the key to stereoselectivity in the asymmetric reaction. Different axially chiral bisphosphine ligands with large steric hindrance were screened in the asymmetric allenylic alkylation (entries 11–13), and the large steric hindrance was necessary (entries 1 and 10). Surprisingly, the reaction delivered the desired product in 18.6:1 dr with L4. Subsequently, different catalyst precursors were examined with L4, including Pd2(dba)3, [Pd(C3H5)Cl]2 and Pd(OAc)2, with which all reactions resulted in lower diastereoselectivities (entries 14–16). Lastly, to further improve the diastereoselectivity and enantioselectivity, the effect of temperature was tested (entries 17–18). When the temperature decreased, higher stereoselectivity could be achieved under −20 °C. Finally, optimized conditions were established in entry 18 of Table 2.
Under the optimized reaction conditions, thiochromanone derivatives 1b–1p were reacted with 2a to explore the generality of the substrates (Scheme 3). Fortunately, thiochromanone derivatives with various aryl substituted R1 could react smoothly in this palladium-catalyzed asymmetric allenylic alkylation to furnish the desirable products in good yields (82–96%), diastereoselectivities (24.0:1–49.0:1) and enantioselectivities (98–99%), which displayed valuable functional group tolerance in aromatic rings including methyl, fluoro, chloro, bromo and methoxyl. Among them, the substrate with an electron-rich substituent is relatively more active than that with an electron-deficient substituent (for details about reaction time, please see the ESI†). The reaction was slightly sensitive to the steric bulk of R1. When o-tolyl (1e) was introduced, the reactivity, diastereoselectivity and enantioselectivity of this alkylation were a little lower. Notably, the halogens in the para-position of the aromatic ring (1h–1j) could slimly improve the diastereoselectivity to 49.0:1. However, alkyl (methyl) had a negative effect on the diastereoselectivity (1l), which was 9.0:1. Furthermore, various R2 groups were well tolerated, such as the strong electron-withdrawing nitro group (1m), electron-donating methoxy group (1n) and methyl group (1o). Increasing the steric hindrance of the R3 group from methoxyl to tert-butoxyl, the reaction delivered the desired product 3pa in 95% yield, 32.3:1 dr and 99% ee for the major diastereoisomer. We also investigated the thiochromanone derivative bearing the heterocyclic aromatic substituted group (2-benzofuranyl) and found that it gave the corresponding product 3qa in 85% yield, albeit in lower diastereoselectivity (5.7:1 dr) and 97% ee for the major diastereoisomer. When 1b with acetyl instead of methoxycarbonyl reacted with 2a, the strong impact of ligands was observed after screening a series of chiral ligands, and (1R,1′R,2S,2′S)-DuanPhos was selected as the optimal ligand, with which the reaction delivered 3ba at 30 °C in 61% yield, 24.0:1 dr and 74% ee for the major diastereoisomer. When the reaction temperature was decreased to 0 °C, the reaction could not perform. Besides, thiochromanone 1d with the amide group reacted with 2a under the optimized conditions to deliver the desired product 3da with low 34% yield, in which there was the side product 4′da with 48% yield and a little recovered 1d. To assign the absolute configuration, 3mk was synthesized and its absolute configuration was assigned as (2R,3S,Ra) by X-ray diffraction analysis (for details, please see the ESI†).
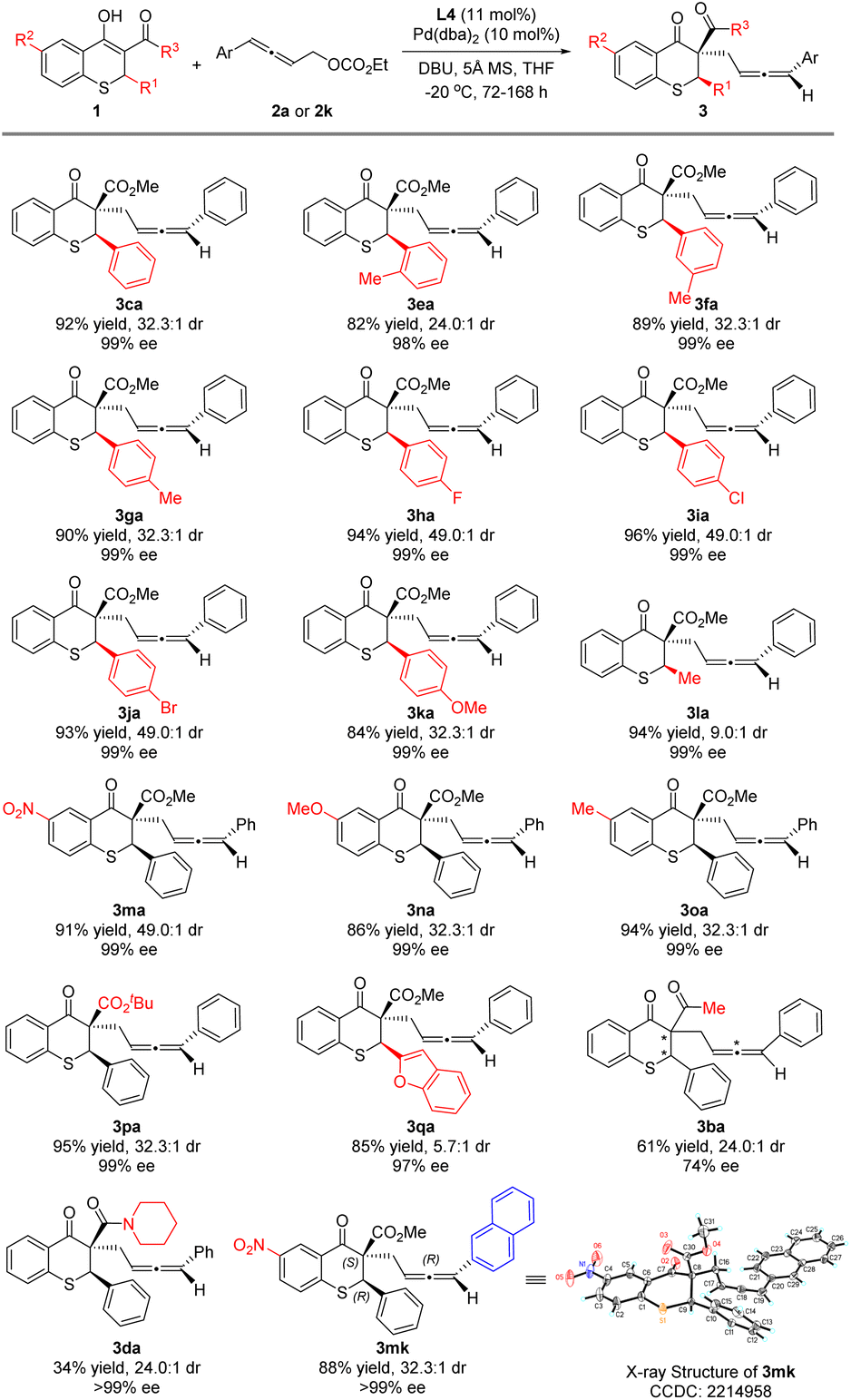 | ||
| Scheme 3 Substrate scope for the thiochromanone derivatives 1. | ||
Reactions of 1c with various allenylic carbonates 2b–2m were next investigated (Scheme 4). The methyl substituent at the ortho, meta or para-position on the benzene ring of the allenylic carbonates (2b–2d) almost did not affect the stereoselectivity of the reaction, and better diastereoselectivity was obtained with the meta-position substituent. Subsequently, when the fluoro, chloro, bromo or methoxy group was introduced at the meta-position, the alkylation reaction underwent smoothly to deliver 3ce, 3cf, 3cg or 3ch in satisfactory yield, excellent ee and dr. Similarly, the reaction was conducted with 3,5-dimethylphenyl or 4-phenylphenyl as the R, furnishing the product 3ci or 3cj in high yield and stereoselectivity. Moreover, allenylic carbonates bearing aromatic substituents, such as naphthyl (2k) and thienyl (2l), were also suitable substrates. The alkyl substituted allenylic carbonate 2m also produced the product with a 97% ee value, albeit in 54% yield and 7.3:1 dr. In addition, when the trisubstituted allene partner (2n) was used, the alkylation reaction underwent smoothly to deliver product 3cn in 72% yield with 6.7:1 dr and 90% ee for the major diastereoisomer at 30 °C, while no reaction was observed at 0 °C.
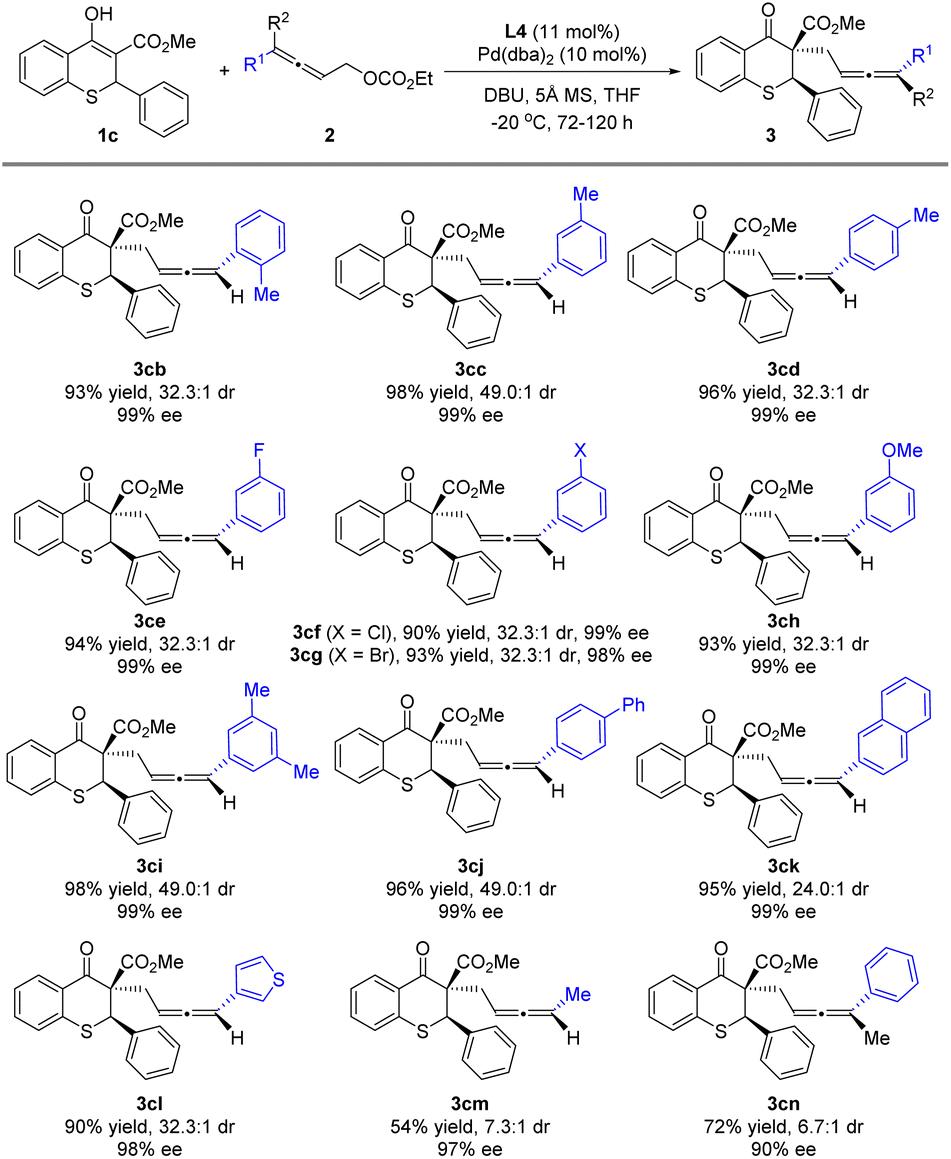 | ||
| Scheme 4 Substrate scope for the allenylic carbonates 2 | ||
Based on the above experimental results and the putative mechanism on palladium-catalyzed allenylic alkylation,9a,e,k a plausible mechanism was proposed in Scheme 5. First, oxidative addition of racemic allene 2a formed the π-allylpalladium complexes A and A′, which were in rapid equilibrium. Subsequently, the nucleophile would attack from the back side of the terminal carbon atom to give the chiral product 3ca, and regenerate the active Pd(0) species.
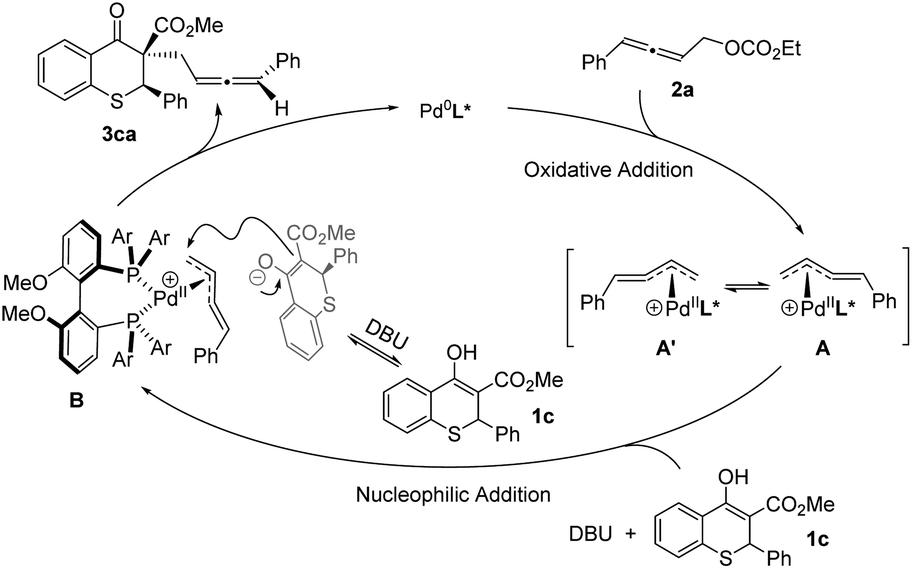 | ||
| Scheme 5 Plausible mechanism. | ||
To illustrate the practicality of this asymmetric allenylic alkylation reaction, a scale-up synthesis of 3ca was carried out (Scheme 6a). The product was isolated in 89% yield with 24.0:1 dr and 99% ee for the major diastereoisomer under the standard conditions without loss of activity and enantioselectivity. Next, the elaboration of the product was proceeded. The chiral 3ca could be converted to the corresponding chiral sulfoxide 5 or sulfone 6 using 3-chloroperoxybenzoic acid as the oxidant at different temperatures. Furthermore, the reductions of 3ca were concentrated on carbonyl and allenyl groups, respectively. The allenyl group could be easily hydrogenated with 10% Pd/C, affording the single reductive isomer 7 in 95% yield and 97% ee, showing that the diastereoselectivity of 3ca was ascribed to the allene unit. A selective reduction of the carbonyl group of 3ca with lithium aluminum hydride (LiAlH4) at −78 °C proceeded smoothly, providing the chiral alcohol 8 in 69% yield, 13.3:1 dr and 99% ee for the major diastereoisomer (Scheme 6b). The relative configuration of the hydroxyl and phenyl in compound 8 was assigned as cis-8 by the NOE spectrum (for details, please see the ESI†).
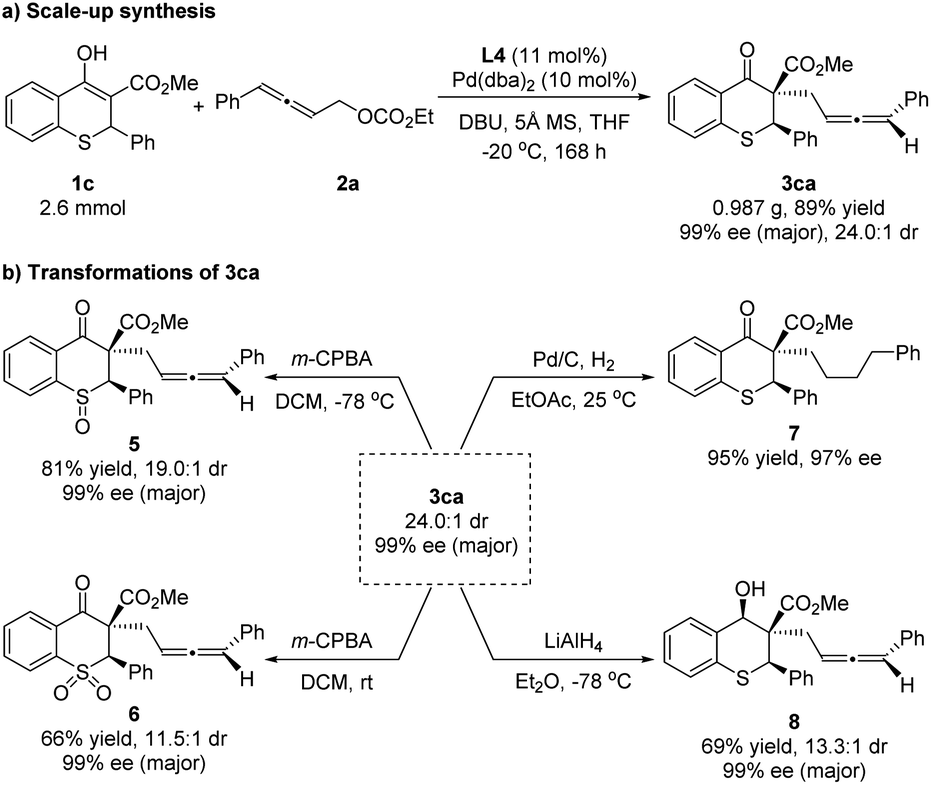 | ||
| Scheme 6 Scale-up synthesis and transformations of 3ca. | ||
Conclusions
In summary, we have realized the synthesis of enantioenriched multiple substituted thiochromanone derivatives containing two central chiralities and an axial chirality (allene unit) based on the combination of retro-sulfa-Michael addition and palladium-catalyzed asymmetric allenylic alkylation under basic conditions, overcoming the challenges of chemo-, regio- and stereoselectivities. The reaction showed great functional group tolerance, and a broad range of highly enantioenriched products could be conveniently prepared with up to 98% yield, 49.0:1 dr and >99% ee. Further investigations utilizing the racemization strategy through retro-hetero-Michael addition on other reactions are being actively pursued in our laboratory.
Data availability
Experimental data has been uploaded as part of the ESI.†Author contributions
L.-X. L. performed the experiments and prepared the ESI.† Y.-Q. B. and X. L. checked the data. Prof. C.-B. Y. and Y.-G. Z. conceived and directed the project. L.-X. L. and C.-B. Y. prepared the draft and Y.-G. Z. revised the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is dedicated to Prof. Li-Xin Dai on the occasion of his 100th birthday. Financial support from the National Natural Science Foundation of China (21871255) and K. C. Wong Education Foundation of CAS (GJTD-202008) is acknowledged.Notes and references
- (a) A. Nudelman, The Chemistry of Optically Active Sulfur Compounds, New York, 1984 Search PubMed; (b) L. A. Damani, Sulphur-Containing Drugs and Related Organic Compounds: Chemistry, Biochemistry and Toxicology, Wiley, New York, 1989 Search PubMed; (c) E. A. Ilardi, E. Vitaku and J. T. Njardarson, J. Med. Chem., 2014, 57, 2832–2842 CrossRef CAS PubMed; (d) M. E. Cinar and T. Ozturk, Chem. Rev., 2015, 115, 3036–3140 CrossRef CAS PubMed; (e) M. Feng, B. Tang, S. H. Liang and X. Jiang, Curr. Top. Med. Chem., 2016, 16, 1200–1216 CrossRef CAS PubMed; (f) P. Devendar and G. F. Yang, Top. Curr. Chem., 2017, 375, 82 CrossRef PubMed; (g) G. Turkoglu, M. E. Cinar and T. Ozturk, Top. Curr. Chem., 2017, 375, 84 CrossRef PubMed; (h) N. Wang, P. Saidhareddy and X. Jiang, Nat. Prod. Rep., 2020, 37, 246–275 RSC.
- (a) C. Mordant, C. C. de Andrade, R. Touati, V. Ratovelomanana-Vidal, B. B. Hassine and J.-P. Genêt, Synthesis, 2003, 15, 2405–2409 Search PubMed; (b) M. L. Bolognesi, M. Bortolini, A. Cavalli, V. Andrisano, M. Rosini, A. Minarin and C. Melchiorre, J. Med. Chem., 2004, 47, 5945–5952 CrossRef CAS PubMed; (c) Y. Chen, Q. Zhang, B. Zhang, P. Xia, Y. Xia, Z.-Y. Yang, N. Kilgore, C. Wild, S. L. Morris-Natschke and K.-H. Lee, Bioorg. Med. Chem., 2004, 12, 6383–6387 CrossRef CAS PubMed.
- (a) W. Wang, H. Li, J. Wang and L. Zu, J. Am. Chem. Soc., 2006, 128, 10354–10355 CrossRef CAS PubMed; (b) L. Zu, J. Wang, H. Li, H. Xie, W. Jiang and W. Wang, J. Am. Chem. Soc., 2007, 129, 1036–1037 CrossRef CAS PubMed; (c) R. Dodda, J. J. Goldman, T. Mandal, C.-G. Zhao, G. A. Broker and E. R. T. Tiekink, Adv. Synth. Catal., 2008, 350, 537–541 CrossRef CAS PubMed; (d) J. Wang, H. Xie, H. Li, L. Zu and W. Wang, Angew. Chem., Int. Ed., 2008, 47, 4177–4179 CrossRef CAS PubMed; (e) Y. Gao, Q. Ren, H. Wu, M. Li and J. Wang, Chem. Commun., 2010, 46, 9232–9234 RSC; (f) X.-Q. Dong, X. Fang, H.-Y. Tao, X. Zhou and C.-J. Wang, Chem. Commun., 2012, 48, 7238–7240 RSC; (g) A. R. Choudhury and S. Mukherjee, Adv. Synth. Catal., 2013, 355, 1989–1995 CrossRef CAS; (h) S. Meninno, G. Croce and A. Lattanzi, Org. Lett., 2013, 15, 3436–3439 CrossRef CAS PubMed; (i) B.-L. Zhao and D.-M. Du, Asian J. Org. Chem., 2015, 4, 778–787 CrossRef CAS; (j) Y. Zhu, Z. Dong, X. Cheng, X. Zhong, X. Liu, L. Lin, Z. Shen, P. Yang, Y. Li, H. Wang, W. Yan, K. Wang and R. Wang, Org. Lett., 2016, 18, 3546–3549 CrossRef CAS PubMed; (k) X.-N. Ping, P.-S. Wei, X.-Q. Zhu and J.-W. Xie, J. Org. Chem., 2017, 82, 2205–2210 CrossRef CAS PubMed; (l) L. Yang, J.-Q. Zhao, Y. You, Z.-H. Wang and W.-C. Yuan, Chem. Commun., 2020, 56, 12363–12366 RSC; (m) Y. Mo, X. Zhang, Y. Yao, C. Duan, L. Ye, Z. Shi, Z. Zhao and X. Li, J. Org. Chem., 2021, 86, 4448–4456 CrossRef CAS PubMed.
- (a) T. Arai and Y. Yamamoto, Org. Lett., 2014, 16, 1700–1703 CrossRef CAS PubMed; (b) T. Arai, T. Miyazaki, H. Ogawa and H. Masu, Org. Lett., 2016, 18, 5824–5827 CrossRef CAS PubMed.
- (a) L. L. Hegedus and R. W. McCabe, Catalyst Poisoning, Marcel Dekker, New York, 1984 Search PubMed; (b) A. T. Hutton, In Comprehensive Coordination Chemistry, ed. G. Wilkinson, R. D. Gillard and J. A. McCleverty, Pergamon, Oxford, U.K, 1984, vol. 5, p. 1151 Search PubMed.
- (a) S. W. Schneller, Adv. Heterocycl. Chem., 1975, 18, 59–97 CrossRef CAS; (b) L. A. van Vliet, N. Rodenhuis, D. Dijkstra, H. Wikström, T. A. Pugsley, K. A. Serpa, L. T. Meltzer, T. G. Heffner, L. D. Wise, M. E. Lajiness, R. M. Huff, K. Svensson, S. Sundell and M. Lundmark, J. Med. Chem., 2000, 43, 2871–2882 CrossRef CAS PubMed; (c) Y. Kanbe, M.-H. Kim, M. Nishimoto, Y. Ohtake, T. Tsunenari, K. Taniguchi, I. Ohizumi, S.-I. Kaiho, Y. Nabuchi, S. Kawata, K. Morikawa, J.-C. Jo, H.-A. Kwon, H.-S. Lim and H.-Y. Kim, Bioorg. Med. Chem. Lett., 2006, 16, 4090–4094 CrossRef CAS PubMed; (d) Y.-L. Song, Y.-F. Dong, T. Yang, C.-C. Zhang, L.-M. Su, X. Huang, D.-N. Zhang, G.-L. Yang and Y.-X. Liu, Bioorg. Med. Chem., 2013, 21, 7624–7627 CrossRef CAS PubMed.
- (a) A. Hoffmann-Röder and N. Krause, Angew. Chem., Int. Ed., 2004, 43, 1196–1216 CrossRef PubMed; (b) Y.-J. Jian and Y. Wu, Org. Biomol. Chem., 2010, 8, 1905–1909 RSC; (c) D. Xu, M. A. Drahl and L. J. Williams, Beilstein J. Org. Chem., 2011, 7, 937–943 CrossRef CAS PubMed; (d) P. Rivera-Fuentes and F. Diederich, Angew. Chem., Int. Ed., 2012, 51, 2818–2828 CrossRef CAS PubMed.
- (a) S. Ma, Acc. Chem. Res., 2003, 36, 701–712 CrossRef CAS PubMed; (b) S. Ma, Chem. Rev., 2005, 105, 2829–2872 CrossRef PubMed; (c) M. Jeganmohan and C.-H. Cheng, Chem. Commun., 2008, 3101–3117 RSC; (d) S. Ma, Acc. Chem. Res., 2009, 42, 1679–1688 CrossRef CAS PubMed; (e) N. Krause and C. Winter, Chem. Rev., 2011, 111, 1994–2009 CrossRef CAS PubMed; (f) S. Yu and S. Ma, Angew. Chem., Int. Ed., 2012, 51, 3074–3112 CrossRef CAS PubMed; (g) J. Ye and S. Ma, Acc. Chem. Res., 2014, 47, 989–1000 CrossRef CAS PubMed; (h) S. Kitagaki, F. Inagaki and C. Mukai, Chem. Soc. Rev., 2014, 43, 2956–2978 RSC; (i) M. Yang and A. S. K. Hashmi, Chem. Soc. Rev., 2014, 43, 2941–2955 RSC; (j) J. M. Alonso, M. T. Quirós and M. P. Muñoz, Org. Chem. Front., 2016, 3, 1186–1204 RSC; (k) B. Yang, Y. Qiu and J.-E. Bäckvall, Acc. Chem. Res., 2018, 51, 1520–1531 CrossRef CAS PubMed; (l) J. L. Mascareñas, I. Varela and F. López, Acc. Chem. Res., 2019, 52, 465–479 CrossRef PubMed.
- (a) M. Ogasawara, H. Ikeda, T. Nagano and T. Hayashi, J. Am. Chem. Soc., 2001, 123, 2089–2090 CrossRef CAS PubMed; (b) Y. Imada, K. Ueno, K. Kutsuwa and S.-I. Murahashi, Chem. Lett., 2002, 31, 140–141 CrossRef; (c) B. M. Trost, D. R. Fandrick and D. C. Dinh, J. Am. Chem. Soc., 2005, 127, 14186–14187 CrossRef CAS PubMed; (d) Y. Imada, M. Nishida, K. Kutsuwa, S.-I. Murahashi and T. Naota, Org. Lett., 2005, 7, 5837–5839 CrossRef CAS PubMed; (e) T. Nemoto, M. Kanematsu, S. Tamura and Y. Hamada, Adv. Synth. Catal., 2009, 351, 1773–1778 CrossRef CAS; (f) Q. Li, C. Fu and S. Ma, Angew. Chem., Int. Ed., 2012, 51, 11783–11786 CrossRef CAS PubMed; (g) Q. Li, C. Fu and S. Ma, Angew. Chem., Int. Ed., 2014, 53, 6511–6514 CrossRef CAS PubMed; (h) J. Dai, X. Duan, J. Zhou, C. Fu and S. Ma, Chin. J. Chem., 2018, 36, 387–391 CrossRef CAS; (i) S. Song, J. Zhou, C. Fu and S. Ma, Nat. Commun., 2019, 10, 507 CrossRef PubMed; (j) H.-C. Liu, Y.-Z. Hu, Z.-F. Wang, H.-Y. Tao and C.-J. Wang, Chem. Eur. J., 2019, 25, 8681–8685 CAS; (k) S. Song and S. Ma, Chin. J. Chem., 2020, 38, 1233–1238 CrossRef CAS; (l) J. Zhang, X. Huo, J. Xiao, L. Zhao, S. Ma and W. Zhang, J. Am. Chem. Soc., 2021, 143, 12622–12632 CrossRef CAS PubMed.
- (a) E. R. Ashley, E. C. Sherer, B. Pio, R. K. Orr and R. T. Ruck, ACS Catal., 2017, 7, 1446–1451 CrossRef CAS; (b) L.-X. Liu, W.-J. Huang, Q.-X. Xie, B. Wu, C.-B. Yu and Y.-G. Zhou, ACS Catal., 2021, 11, 12859–12863 CrossRef CAS; (c) Q.-X. Xie, L.-X. Liu, Z.-H. Zhu, C.-B. Yu and Y.-G. Zhou, J. Org. Chem., 2022, 87, 7521–7530 CrossRef CAS PubMed.
- W. Yang, Y. Yang and D.-M. Du, Org. Lett., 2013, 15, 1190–1193 CrossRef CAS PubMed.
- For Pd-catalyzed asymmetric allylic substitutions using thiol as nucleophile, see: (a) M. Frank and H.-J. Gais, Tetrahedron: Asymmetry, 1998, 9, 3353–3357 CrossRef CAS; (b) H.-J. Gais, N. Spalthoff, T. Jagusch, M. Frank and G. Raabe, Tetrahedron Lett., 2000, 41, 3809–3812 CrossRef CAS; (c) K. Ulbrich, P. Kreitmeier, T. Vilaivan and O. Reiser, J. Org. Chem., 2013, 78, 4202–4206 CrossRef CAS PubMed; (d) J. Cai, J. Cai, P. Zheng, X. Wang and X. Zhao, RSC Adv., 2017, 7, 256–259 RSC.
- D. A. Petrone, M. Isomura, I. Franzoni, S. L. Rössler and E. M. Carreira, J. Am. Chem. Soc., 2018, 140, 4697–4704 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and spectroscopic data of compounds. CCDC 2214958 (-)-3mk. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3sc01060k |
| This journal is © The Royal Society of Chemistry 2023 |