
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRuthenium-catalysed decarboxylative unsymmetric dual ortho-/meta-C–H bond functionalization of arenecarboxylic acids†
Xiankai
Li
,
Xiaofei
Wang
and
Jing
Zhang
 *
*
The Institute for Advanced Studies, Wuhan University, Wuhan, Hubei Province 430072, China. E-mail: jzhangwhu@whu.edu.cn
First published on 24th April 2023
Abstract
Here, we describe a ruthenium-catalysed decarboxylative unsymmetric ortho-C–H azaarylation/meta-C–H alkylation via a traceless directing group relay strategy. The installation of a 2-pyridyl functionality via carboxyl directed ortho-C–H activation is critical to promote decarboxylation and enable meta-C–H bond alkylation to streamline the synthesis of 4-azaaryl-benzo-fused five-membered heterocycles. This protocol is characterized by high regio- and chemoselectivity, broad substrate scopes, and good functional group tolerance under redox-neutral conditions.
Introduction
Polysubstituted arenes are frequently encountered in pharmaceuticals, agrochemicals, and functional materials,1–3 and thus their synthesis is of paramount interest in the community of synthetic chemistry.4–7 In recent decades, transition metal-catalysed C–H bond activation has become a powerful tool to selectively install various functionalities (Scheme 1A).2,8–13 Using this tactic, a number of dual C–H bond functionalization reactions have been achieved by repeated reaction with the same compounds in one step.2,13–17 In contrast, a method for selectively replacing multiple aryl C–H bonds with different functionalities remains challenging and highly desirable to streamline the synthesis of diversely functionalized aromatic compounds.9–11,18–23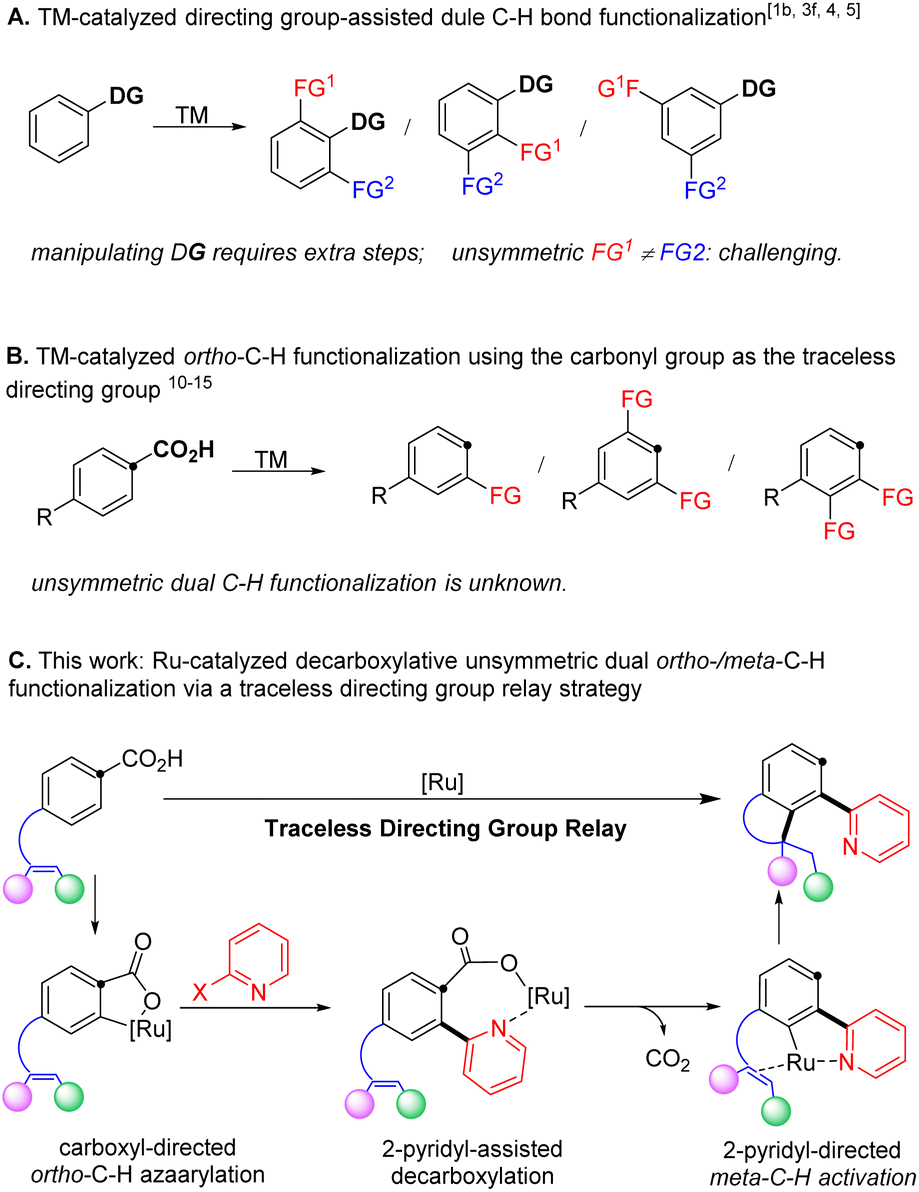 | ||
| Scheme 1 Synthesis of polysubstituted arenes via C–H bond functionalization. | ||
The employment of coordinating directing groups in transition metal-catalysed C–H bond functionalization is the predominant strategy to achieve regioselectivity.24–38 However, utilizing directing groups often adds additional synthetic steps for their installation and/or removal, thus reducing the overall synthetic economy. In this context, the carboxyl functionality represents a desirable directing group, because it is readily accessible39 and widely utilized to direct ortho-C–H bond functionalization,40–43 and can be completely removed to allow complementary regioselectivities in products (Scheme 1B).43–48 Indeed, utilizing the carboxyl functionality as a deciduous directing group has enabled a number of C–H bond functionalization reactions of arenecarboxylic acids with in situ elimination of CO2.49–60 Specifically, the recent advance in ruthenium catalysis, pioneered by Zhao and Hartwig,61 Gooβen62 and Ackermann,63 has enabled mild decarboxylative ortho-C–H bond alkenylation under redox neutral conditions, which eliminates the requirement of substrate activation and additives to facilitate decarboxylation in previous examples. However, so far, ruthenium catalysis is limited to C–H addition to unsaturated bonds such as alkynes,61–65 alkenes,66–70 and isocyanates.71 Besides, dual C–H bond functionalization is only applicable to repeated incorporation of the same functionality.72,73 Therefore, it is desirable to design new strategies to exploit the traceless carboxyl group that might selectively functionalize multiple C–H bonds with various functionalities to access polysubstituted arenes.
We hypothesized that an unsymmetric dual ortho-/meta-C–H bond functionalization would be achieved via a directing group relay strategy by taking advantage of the traceless carboxyl group. Specifically, catalytic decarboxylative ortho-C–H bond azaarylation would install a 2-pyridyl functionality, which will serve as the directing group to assist the catalyst to functionalize the meta-C–H bond via an intramolecular alkene hydroarylation, furnishing chemo- and regio-selective unsymmetric dual C–H bond functionalization (Scheme 1C). We chose the 2-pyridyl group as the relay directing group because (1) a cyclic ruthenium carboxylate could be formed via coordinating to the nitrogen atom, which is expected to promote decarboxylation and avoid di-ortho-C–H azaarylation;12,16,61,80 (2) the 2-pyridyl group has been unambiguously demonstrated as a strong directing group in various ortho-C–H functionalization reactions, which will provide broad opportunities for the next C–H bond activation to introduce a different functionality.18 Following this design, we herein disclose a ruthenium-catalysed decarboxylative unsymmetric dual ortho-/meta-C–H bond functionalization of alkene-tethered arenecarboxylic acids via a directing group relay strategy. This protocol, for the first time, enables decarboxylative ortho-C–H azaarylation offering a new method for the construction of aryl-azaaryl structures, and streamlines the synthesis of 4-azaaryl-benzo-fused five-membered saturated heterocycles, which are privileged structural motifs in various natural products and pharmaceuticals.1,74
Results and discussion
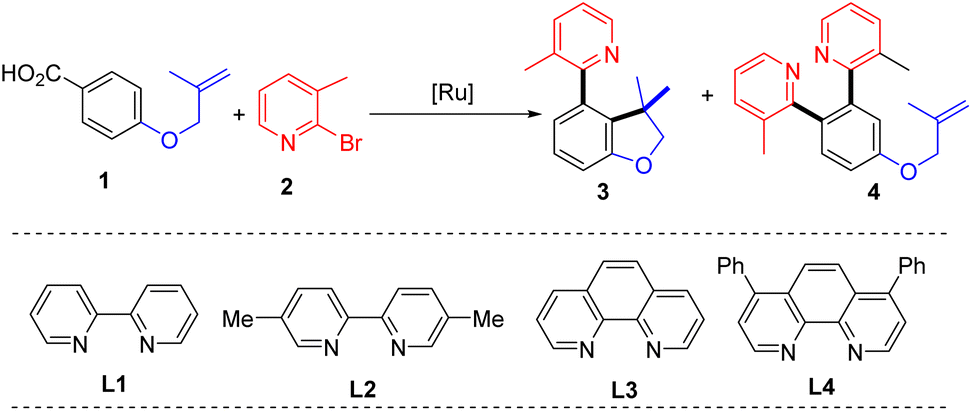
To verify our hypothesis, 4-((2-methylallyl)oxy)benzoic acid (1) and 2-bromo-3-methylpyridine (2) were selected as the model substrates to evaluate the reaction parameters. After extensive exploration (Tables S1–S6†), 73% of the desired 4-(2-pyridyl)-2,3-dihydrobenzofurane with a C3 quaternary carbon center (3) was obtained in the presence of 4 mol% of [Ru(p-cymene)Cl2]2, 8 mol% of bathophenanthroline (L4), and 5 mol% of ZnI2 and KOAc in 1,4-dioxane after 20 hours of reaction at 120 °C (entry 1). In the test of ligands, 2,2′-bipyridine (L1), 5,5′-dimethyl-2,2′-bipyridine (L2) and 1,10-phenanthropline (L3) led to inferior yields of 3 (entries 2–4). The effects of solvent show that NMP is also suitable and provides 3 in 57% yield (entry 5 and Table S1†). The reaction performed at 100 °C gave 63% of 3 (entry 6). A series of control experiments were conducted to elucidate the necessity of each component in the optimal system (Table S6†). The catalyst and base were proved indispensable for the reaction (entries 7 and 8), while the ligand played an essential role to get a high yield (Table 1, entry 9). In addition, ZnI2 is found to be capable of suppressing bis-2-pyridination product 4 (Table S5†).|
|
|||
|---|---|---|---|
| Entry | Deviations | 3 (%) | 4 (%) |
| a Reaction conditions: 1a (0.2 mmol), 2a (0.2 mmol), KOAc (0.36 mmol), [Ru(p-cymene)Cl2]2 (4 mol%), bathophenanthroline (8 mol%), ZnI2 (5 mol%), 1,4-dioxane (2 mL), under argon, 120 °C, 20 hours. GC yields. Isolated yield in the parentheses. Nd: not detected. | |||
| 1 | None | 71 (73) | Trace |
| 2 | L1 instead of L4 | 30 | Nd |
| 3 | L2 instead of L4 | 43 | 5 |
| 4 | L3 instead of L4 | 64 | 5 |
| 5 | NMP as solvent | 57 | Nd |
| 6 | At 100 °C | 63 | Trace |
| 7 | Without [Ru(p-cymene)Cl2]2 | 0 | Nd |
| 8 | Without KOAc | 0 | Nd |
| 9 | Without L4 | 8 | Nd |
| 10 | Without ZnI2 | 67 | 13 |
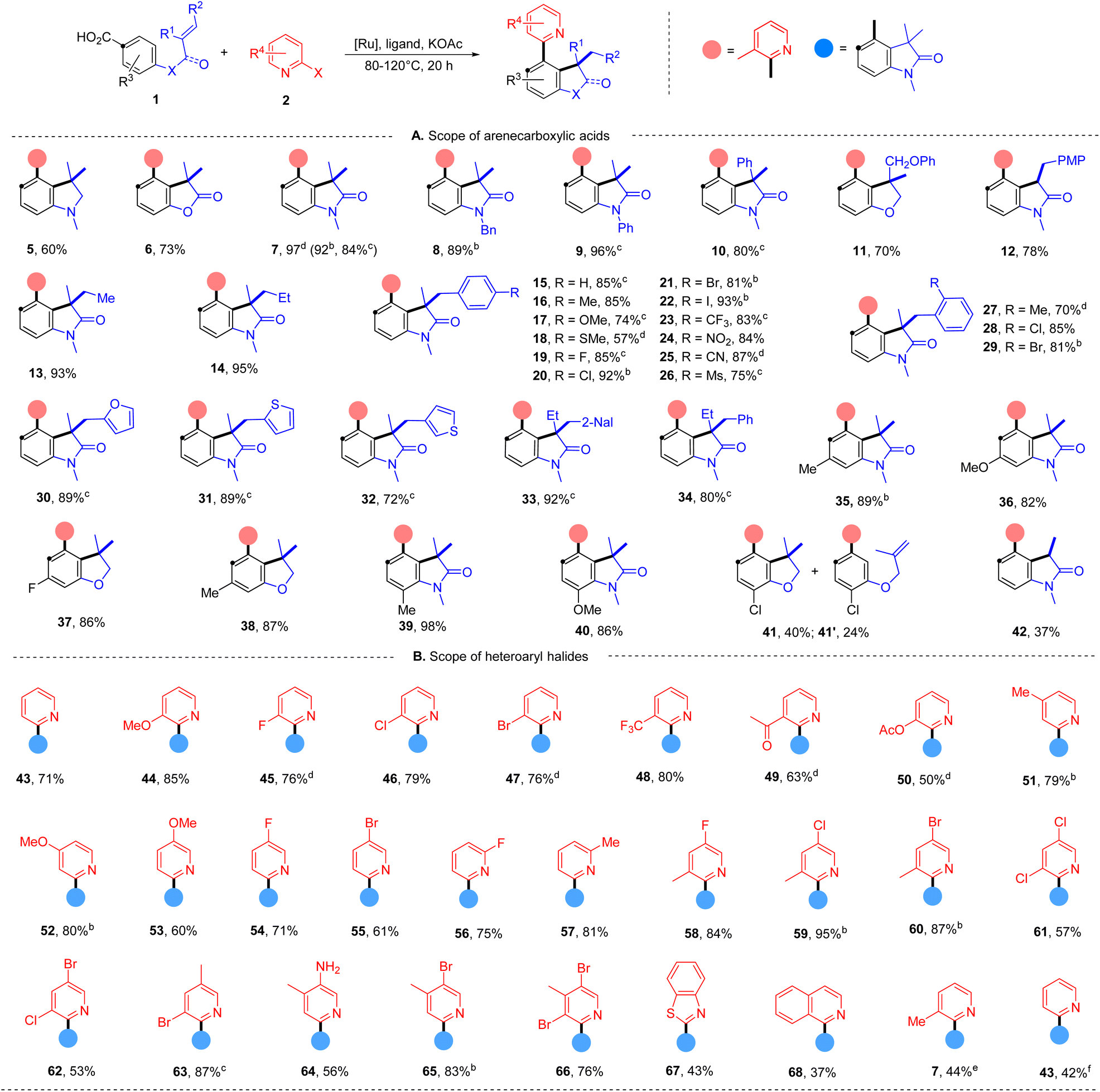
With the evaluated reaction conditions, we next explored the scope of alkene-tethered arenecarboxylic acids with 2-bromo-3-methylpyridine (2) (Table 2A). The linkage to the alkene unit could also be amine, ester and amide, affording the corresponding indoline (5), furanone (6), and oxindole (7) in good to high yields. Remarkably, substrates with amide linkers exhibit high reactivity and could produce oxindoles in high yields under much milder conditions (Table S7†). For example, 84% of 7 was obtained at 80 °C with a lower catalyst loading and simple 2,2′-bipydine ligand (L1) in the absence of ZnI2. Besides N-methyl amide, benzyl and phenyl-substituted amides were also suitable linkers producing 8 and 9 in high yields. Substrates with phenyl (10), phenoxymethyl (11), and ethyl (33 and 34) substituted alkene fragments also afforded high yields of products. In addition to substrates with 1,1-disubstituted alkenes, those with 1,2-disubstituted alkene are also amenable to the reaction giving 78% of 12. While those with 1,1,2-trisubstituted alkenes can efficiently deliver products in good to high yields (13–34). A wide range of functional groups with diverse electronic characters in different substitution patterns on the phenyl group of alkene were tolerated, including electron-donating methyl (16), methoxyl (17), methylthio (18) groups and electron-withdrawing trifluoromethyl (23), nitro (24), cyano (25), and mesyl (26) groups, furnishing products in good to high yields (57–93%). Aryl halides, including fluoride (19), chloride (20 and 28), bromide (21 and 29) and even iodide (22) remained intact during the reaction and delivered products in high yields (85–93%), highlighting the excellent chemoselectivity of this protocol and providing valuable handles to further functionalization.
| a Reaction conditions: 1 (0.20 mmol), 2a (0.20 mmol), [Ru(p-cymene)Cl2]2 (4 mol%), L4 (8 mol%), KOAc (0.36 mmol, 1.8 equiv.), ZnI2 (5 mol%), 1,4-dioxane (2 mL) under argon, 120 °C, 20 hours, isolated yields. b [Ru(p-cymene)Cl2]2 (3 mol%), L1 (6 mol%), KOAc (1.5 equiv.), dioxane (1 mL), 100 °C. c [Ru(p-cymene)Cl2]2 (3 mol%), L1 (6 mol%), KOAc (0.3 mmol), dioxane (1 mL), 80 °C. d [Ru(p-cymene)Cl2]2 (3 mol%), L1 (6 mol%), KOAc (0.3 mmol), dioxane (1 mL), 120 °C. e With 2-chloro-3-methylpyridine. f With 2-iodo-3-methylpyridine. |
|---|
|
Additionally, substrates with heteroaryl alkene fragments, such as 2-furyl (30), 2-thiophenyl (31), and 3-thiophenyl (32), were tolerated and underwent reaction with high efficiency. Oxindoles with a more crowed quaternary carbon center are also accessed in high yields (33 and 34). ortho-Substitutions to the carboxyl group were not detrimental to the reaction furnishing high yields of polysubstituted arenes (35–38). The effects of ortho-substitution to the tethered-alkene were found substrate-dependent. Those with the amide-linker gave high yields of oxindoles (39 and 40), and those with the ether-linker delivered 40% of benzodihydrofuran 41 with 24% of 2-arylpyridine 41′ indicating a hindered alkene hydroarylation.
Next, the scope of azaaryl halides was explored with 4-(N-methylmethacrylamido)benzoic acid (Table 2B). The reaction proved to be general providing a series of diversely functionalized 4-(2-pyridyl)oxindole structures in good to high yields (42–60). A broad array of functional groups in various substitution patterns is compatible, including methoxyl (44, 52 and 53), fluoride (45, 54 and 56), chloride (46, 59, 61 and 62), bromide (47, 55, 62, 63, 65 and 66), acyloxyl (50), trifluoromethyl (48), acetyl (49), acyloxy (50), and unprotected amino groups (64). Notably, only halide at the ortho-position of the nitrogen atom participates in the reaction, and both chloride and bromide substituents at other positions remained intact (45–47, 54–56, 58–62, 65 and 66), providing opportunities for orthogonal manipulations. Multi-substituted 2-bromopyridines also worked well (57–66, 50–95%). Other heterocyclic substrates, 2-bromo-benzothiazole and 1-bromo-isoquinoline, could also undergo the reaction, albeit with lower efficiency (67 and 68). Apart from azaaryl bromides, the corresponding chloride and iodide are also applicable in this transformation though leading to lower yields of products (7 and 43).
To demonstrate the practicability, a scaleup reaction with 1 and 2-bormopyridne was conducted and produced 43 in 74% yield. In addition, the synthetic applicability of this reaction was demonstrated by derivatization of 43 by a series of 2-pyridyl-directed ortho-C–H bond functionalization reactions (Scheme 2). Through a Rh-catalysed hydroarylation of terminal alkynes, 69 was produced from reaction with phenylacetylene in 91%.7569 could also be obtained from rhodium-catalysed ortho-vinylation with styrene.76ortho-Arylation of 43 was achieved by palladium catalysis with diphenyliodonium salt giving 70 in 78%.77 Additionally, via a ruthenium-catalysed C–H amidation with aryl isocyanate, 71 was obtained in 78% yield.78 The palladium-catalysed decarboxylative C–H benzoylation with benzoylformic acid introduced an ortho-benzoyl group forming 72 in 56%.79 At last, ortho-C–H cyanation was accomplished by a rhodium catalysis with N-cyano-N-phenyl-p-toluenesulfonamide (NCTS) affording 73 in 84%.
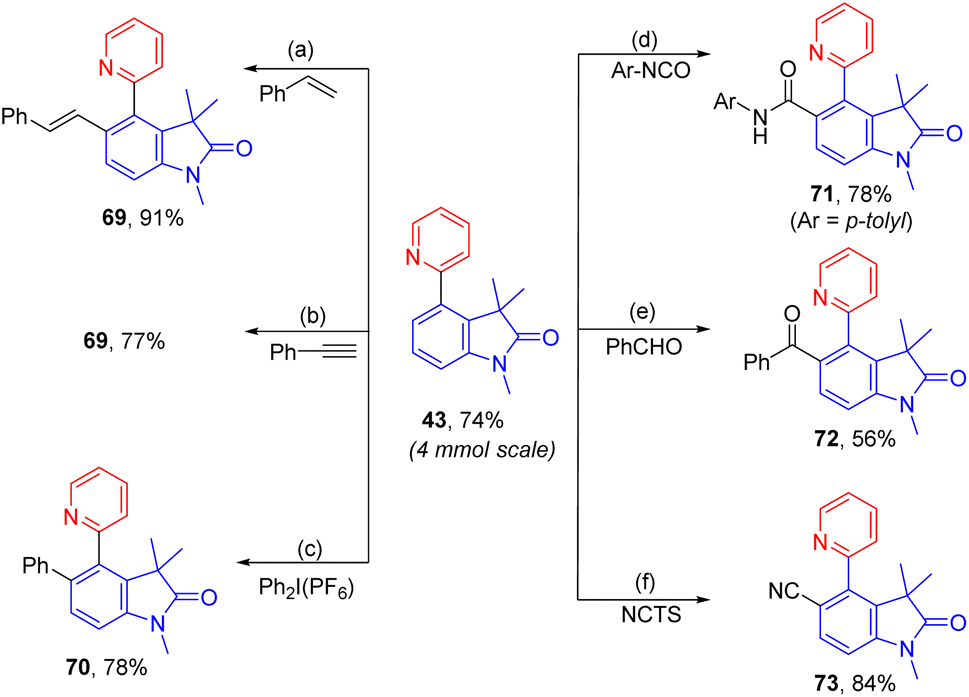 | ||
| Scheme 2 Scaleup reaction and derivatization for synthesizing polysubstituted arenes (a styrene (1.5 equiv.), [Cp*RhCl2]2 (2.5 mol%), Cu(OAc)2·H2O (1 equiv.), DMF, 100 °C, 24 h; b ethynylbenzene (1.5 equiv.), [Cp*RhCl2]2 (2.5 mol%), AgSbF6 (15 mol%), AcOH (0.1 M), 100 °C, 24 h; c (c) Ph2IPF6 (1.5 equiv.), Pd(OAc)2 (5 mol%), AcOH, 100 °C, 24 h; d 4-Me-PhNCO (1.8 equiv.), [Ru(p-cymene)Cl2]2 (5 mol%), AgSbF6 (20 mol%), 2-NO2-PhCO2H (30 mol%), DCE, 50 °C, 24 h; e PhCHO (1.5 equiv.), t-BuOOH (3.0 equiv.), Pd(OAc)2 (10 mol%), MeCN, r.t., 24 h; f NCTS (2.0 equiv.), [Cp*RhCl2]2 (1 mol%), AgSbF6 (10 mol%), PhMe, 120 °C, 24 h.). | ||
While the mechanistic hypothesis guided our thinking in the design of this system, further mechanistic investigations will be necessary to evaluate its validity (Scheme 3). To identify the intermediate, the reaction with 1 and 2 under optimal conditions was halted at the early stage. Aside from 3 (15%), 3′ was detected in 7% yield without hydroarylation of the tethered alkene (Scheme 3A). A significant amount of this type of byproduct 41′ was also observed when cyclization was hindered (Table 2). Then 3′ was subjected to the standard conditions, giving 3 quantitatively (Scheme 3B). These results suggest that 3′ is likely to be the intermediate connecting successive C–H bond functionalization, while, under identical conditions, 1 failed intramolecular hydroarylation, but gave an alkene isomerization product 1′ (56%, Scheme 3C). Moreover, the standard reaction with 1′′ and 2 produced a mixture of isomers of mono- and di-azaarylation products (Scheme 3D). These results suggest the unsymmetric dual C–H bond functionalization is a tandem process via a directing group relay. When the reaction of 1 and 2 was carried out in the presence of D2O, the meta- and ortho-C–H bonds to the 2-pyridyl substituent in 3-D were found to be highly deuterated (Scheme 3E). When 3′ was subjected to the same reaction conditions, 3-D′ was obtained with 73% of deuterium decoration of the ortho-C–H bond These results illustrate reversible C–H bond activation directed by the carboxyl and 2-pyridyl groups. Moreover, the absence of 2-pyridyl arenecarboxylic acids in all reactions implies a swift decarboxylation, as expected from the design of the formation of the 2-pyridyl coordinated cyclic ruthenium carboxylate intermediate.
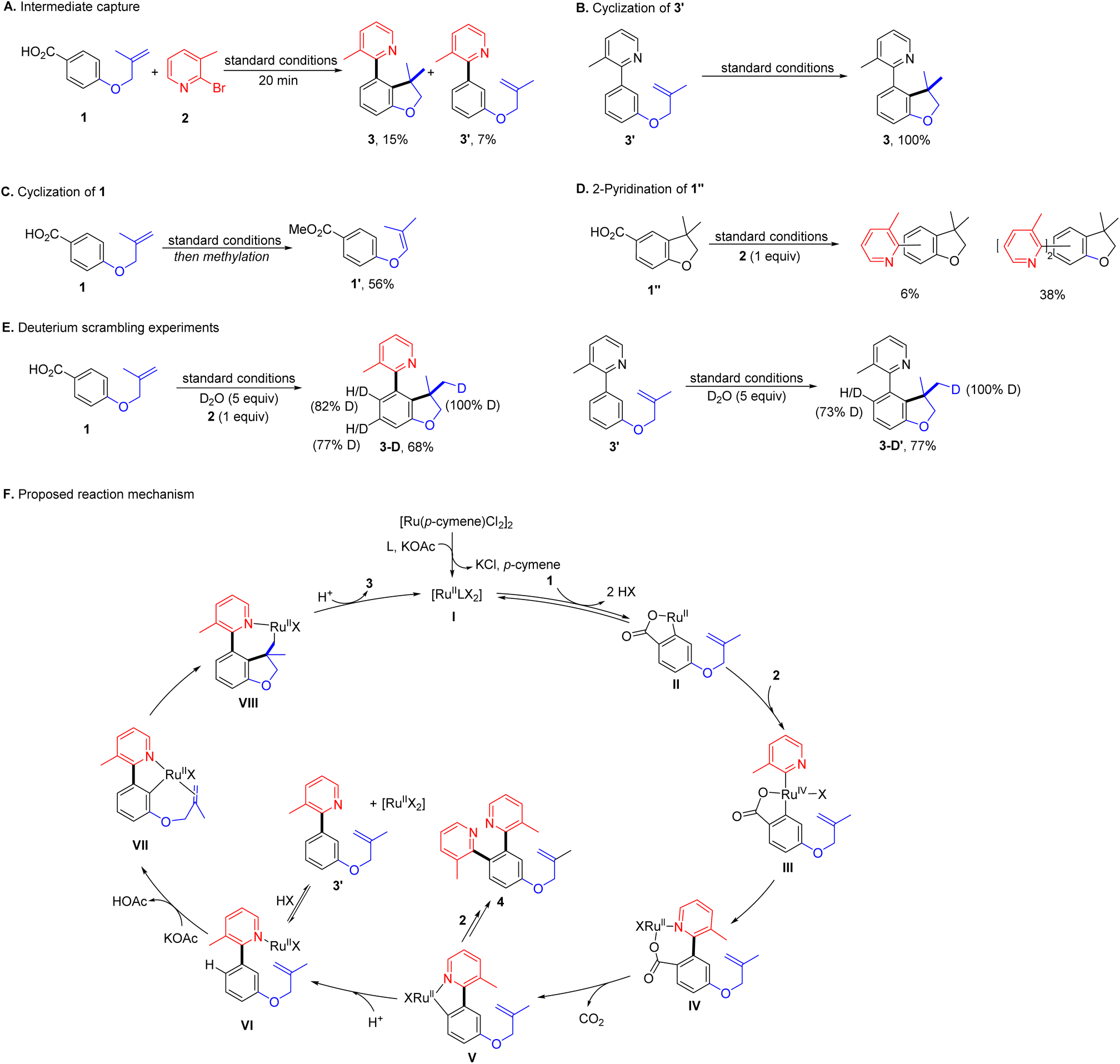 | ||
| Scheme 3 Mechanistic investigations and proposed reaction mechanism. | ||
Based on the results from mechanistic studies, we proposed a plausible reaction mechanism for this ruthenium-catalysed decarboxylative unsymmetric dual ortho-/meta-C–H bond functionalization as shown in Scheme 3F. At the beginning, the ruthenium catalyst precursor, the ligand and the base would generate an active catalytic complex, [RullLX2]. In the catalytic cycle, the dinitrogen ligand would be omitted and the anion would be labelled as X to get a clear view of reaction. The arenecarboxylic acid 1 would exchange with X to coordinate to the ruthenium, followed by a reversible ortho-C–H bond activation generating a five-membered ruthenium carboxylate II. Then II undergoes oxidative addition with 2 presumably facilitated by the coordinating nitrogen atom, and affords the high valent ruthenium complex III. Subsequent reductive elimination produces a seven-membered ruthenium carboxylate IV, which possesses the favourable structure for decarboxylation.61,80 In the next step, a five-membered ruthenacycle V is generated after elimination of CO2. There are two possible reaction paths of V, one would lead to the bis-2-pyridination product by reacting with 2 producing 4. The other would deliver intermediate 3′via protonation completing the decarboxylative ortho-2-azaarylation. Next, the 2-pyridyl group serves as the directing group and assists the catalyst to activate the meta-C–H bond reversibly. The resulting ruthenacycle-VI undergoes migratory insertion with the tethered alkene furnishing a seven-membered alkyl ruthenium complex Vll. Finally, protonation of Vll yields the desired product 3 and completes the catalytic cycle.
Conclusions
In summary, we have developed a ruthenium-catalysed decarboxylative unsymmetric dual ortho-/meta-C–H functionalization of arenecarboxylic acids via a traceless directing group relay strategy. This transformation includes the first example of transition metal-catalysed decarboxylative ortho-C–H bond azaarylation, which is ascribed to the formation of a seven-membered ruthenium carboxylate assisted by the 2-pyridyl group. This protocol provides a streamlined path to a series of 4-azaaryl-benzo-fused five-membered heterocycles with a C3 quaternary center and features high regio- and chemo-selectivity, broad substrate scope and excellent functional group tolerance. We expect this protocol would inspire more efforts to exploit the deciduous carboxyl group to develop methods for the synthesis of polysubstituted arenes.Data availability
All experimental procedures and characterization for this study, can be found in the ESI.†Author contributions
Xiankai Li and Xiaofei Wang conducted and analyzed the synthetic experiments. Xiankai Li and Jing Zhang planned the project. Jing Zhang designed and directed the project and wrote the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful to the National Natural Science Foundation of China (22071185; 22271224), the Fundamental Research Funds for the Central Universities (2042019kf0008), and Wuhan University Startup funding for financial support.Notes and references
- R. D. Taylor, M. MacCoss and A. D. G. Lawson, J. Med. Chem., 2014, 57, 5845 CrossRef CAS PubMed.
- Y. Segawa, T. Maekawa and K. Itami, Angew. Chem., Int. Ed., 2015, 54, 66 CrossRef CAS PubMed.
- A. Nilova, L.-C. Campeau, E. C. Sherer and D. R. Stuart, J. Med. Chem., 2020, 63, 13389 CrossRef CAS PubMed.
- P. Bamfield and P. F. Gordon, Chem. Soc. Rev., 1984, 13, 441 RSC.
- V. Snieckus, Chem. Rev., 1990, 90, 879 CrossRef CAS.
- S. Saito and Y. Yamamoto, Chem. Rev., 2000, 100, 2901 CrossRef CAS PubMed.
- T. J. Donohoe, A. J. Orr and M. Bingham, Angew. Chem., Int. Ed., 2006, 45, 2664 CrossRef CAS PubMed.
- S. H. Cho, J. Y. Kim, J. Kwak and S. Chang, Chem. Soc. Rev., 2011, 40, 5068 RSC.
- A. Mandal, S. Dana, D. Chowdhury and M. Baidya, Chem.–Asian J., 2019, 14, 4074 CrossRef CAS PubMed.
- M. Murai and K. Takai, Synthesis, 2019, 51, 40 CrossRef CAS.
- K. Ghosh, R. K. Rit, M. Shankar, K. Mukherjee and A. K. Sahoo, Chem. Rec., 2020, 20, 1017 CrossRef CAS PubMed.
- O. Baudoin, Angew. Chem., Int. Ed., 2020, 59, 17798 CrossRef CAS PubMed.
- A. Saha, M. Shankar, S. Sau and A. K. Sahoo, Chem. Commun., 2022, 58, 4561 RSC.
- S. Motohiro, K. Fumitoshi, C. Naoto and M. Shinji, Bull. Chem. Soc. Jpn., 1997, 70, 3117 CrossRef.
- B. Li, C. B. Bheeter, C. Darcel and P. H. Dixneuf, ACS Catal., 2011, 1, 1221 CrossRef CAS.
- M. Shankar, A. Saha, S. Sau, A. Ghosh, V. Gandon and A. K. Sahoo, Chem. Sci., 2021, 12, 6393 RSC.
- D. Wang, M. Li, X. Chen, M. Wang, Y. Liang, Y. Zhao, K. N. Houk and Z. Shi, Angew. Chem., Int. Ed., 2021, 60, 7066 CrossRef CAS PubMed.
- K. M. Engle, D.-H. Wang and J.-Q. Yu, Angew. Chem., Int. Ed., 2010, 49, 6169 CrossRef CAS PubMed.
- N. Umeda, K. Hirano, T. Satoh and M. Miura, J. Org. Chem., 2009, 74, 7094 CrossRef CAS PubMed.
- D. Sarkar, F. S. Melkonyan, A. V. Gulevich and V. Gevorgyan, Angew. Chem., Int. Ed., 2013, 52, 10800 CrossRef CAS PubMed.
- J. Lin, L. Hu, C. Chen, H. Feng, Y. Yu, Y. Yang and B. Zhou, Org. Lett., 2021, 23, 1194 CrossRef CAS PubMed.
- L. Sun, Y. Zhao, B. Liu, J. Chang and X. Li, Chem. Sci., 2022, 13, 7347 RSC.
- Dattatri, M. Kumar Reddy Singam, S. Vavilapalli, J. B. Nanubolu and M. Sridhar Reddy, Angew. Chem., Int. Ed., 2023, e202215825 CAS.
- D. A. Colby, R. G. Bergman and J. A. Ellman, Chem. Rev., 2010, 110, 624 CrossRef CAS PubMed.
- T. W. Lyons and M. S. Sanford, Chem. Rev., 2010, 110, 1147 CrossRef CAS PubMed.
- F. Zhang and D. R. Spring, Chem. Soc. Rev., 2014, 43, 6906 RSC.
- Z. Chen, B. Wang, J. Zhang, W. Yu, Z. Liu and Y. Zhang, Org. Chem. Front., 2015, 2, 1107 RSC.
- C. Sambiagio, D. Schoenbauer, R. Blieck, T. Dao-Huy, G. Pototschnig, P. Schaaf, T. Wiesinger, M. F. Zia, J. Wencel-Delord, T. Besset, B. U. W. Maes and M. Schnuerch, Chem. Soc. Rev., 2018, 47, 6603 RSC.
- J. Yang, Org. Biomol. Chem., 2015, 13, 1930 RSC.
- X.-C. Wang, W. Gong, L.-Z. Fang, R.-Y. Zhu, S. Li, K. M. Engle and J.-Q. Yu, Nature, 2015, 519, 334 CrossRef CAS PubMed.
- A. Dey, S. Agasti and D. Maiti, Org. Biomol. Chem., 2016, 14, 5440 RSC.
- A. Dey, S. Maity and D. Maiti, Chem. Commun., 2016, 52, 12398 RSC.
- S. Sasmal, S. K. Sinha, G. K. Lahiri and D. Maiti, Chem. Commun., 2020, 56, 7100 RSC.
- N. Goswami, T. Bhattacharya and D. Maiti, Nat. Rev. Chem., 2021, 5, 646 CrossRef CAS PubMed.
- U. Dutta, S. Maiti, T. Bhattacharya and D. Maiti, Science, 2021, 372, 701 CrossRef PubMed.
- S. Sasmal, U. Dutta, G. K. Lahiri and D. Maiti, Chem. Lett., 2020, 49, 1406 CrossRef CAS.
- G. Rani, V. Luxami and K. Paul, Chem. Commun., 2020, 56, 12479 RSC.
- S. K. Sinha, S. Guin, S. Maiti, J. P. Biswas, S. Porey and D. Maiti, Chem. Rev., 2022, 122, 5682 CrossRef CAS PubMed.
- T. Kalliokoski, ACS Comb. Sci., 2015, 17, 600 CrossRef CAS PubMed.
- L. Ackermann, Chem. Rev., 2011, 111, 1315 CrossRef CAS PubMed.
- G. Shi and Y. Zhang, Carboxylate-Directed C–H Functionalization, Adv. Synth. Catal., 2014, 356, 1419 CrossRef CAS.
- M. P. Drapeau and L. J. Goossen, Chem. – Eur. J., 2016, 22, 18654 CrossRef PubMed.
- M. Font, J. M. Quibell, G. J. P. Perry and I. Larrosa, Chem. Commun., 2017, 53, 5584 RSC.
- S. Mochida, K. Hirano, T. Satoh and M. Miura, Org. Lett., 2010, 12, 5776 CrossRef CAS PubMed.
- S. Mochida, K. Hirano, T. Satoh and M. Miura, J. Org. Chem., 2011, 76, 3024 CrossRef CAS PubMed.
- L. Huang, D. Hackenberger and L. J. Gooßen, Angew. Chem., Int. Ed., 2015, 54, 12607 CrossRef CAS PubMed.
- A. J. S. Johnston, K. B. Ling, D. Sale, N. Lebrasseur and I. Larrosa, Org. Lett., 2016, 18, 6094 CrossRef CAS PubMed.
- D. Lee and S. Chang, Chem. – Eur. J., 2015, 21, 5364 CrossRef CAS PubMed.
- J. Cornella, M. Righi and I. Larrosa, Angew. Chem., Int. Ed., 2011, 50, 9429 CrossRef CAS PubMed.
- D. Nandi, Y.-M. Jhou, J.-Y. Lee, B.-C. Kuo, C.-Y. Liu, P.-W. Huang and H. M. Lee, J. Org. Chem., 2012, 77, 9384 CrossRef CAS PubMed.
- D. Nandi, Y.-M. Jhou, J.-Y. Lee, B.-C. Kuo, C.-Y. Liu, P.-W. Huang and H. M. Lee, J. Org. Chem., 2012, 77, 9384 CrossRef CAS PubMed.
- S. Bhadra, W. I. Dzik and L. J. Goossen, Angew. Chem., Int. Ed., 2013, 52, 2959 CrossRef CAS PubMed.
- S. Bhadra, W. I. Dzik and L. J. Goossen, Synthesis, 2013, 45, 2387 CrossRef CAS.
- J. Luo, S. Preciado and I. Larrosa, Chem. Commun., 2015, 51, 3127 RSC.
- X. Qin, D. Sun, Q. You, Y. Cheng, J. Lan and J. You, Org. Lett., 2015, 17, 1762 CrossRef CAS PubMed.
- Y. Zhang, H. Zhao, M. Zhang and W. Su, Angew. Chem., Int. Ed., 2015, 54, 3817 CrossRef CAS PubMed.
- J. Tang, D. Hackenberger and L. J. Goossen, Angew. Chem., Int. Ed., 2016, 55, 11296 CrossRef CAS PubMed.
- A. Biafora and L. J. Goossen, Synlett, 2017, 28, 1885 CrossRef CAS.
- M. Font, A. R. A. Spencer and I. Larrosa, Chem. Sci., 2018, 9, 7133 RSC.
- S. Hazra, K. Hirano and M. Miura, Org. Lett., 2021, 23, 1388 CrossRef CAS PubMed.
- J. Zhang, R. Shrestha, J. F. Hartwig and P. Zhao, Nat. Chem., 2016, 8, 1144 CrossRef CAS PubMed.
- L. Huang, A. Biafora, G. Zhang, V. Bragoni and L. J. Gooßen, Angew. Chem., Int. Ed., 2016, 55, 6933 CrossRef CAS PubMed.
- N. Y. P. Kumar, A. Bechtoldt, K. Raghuvanshi and L. Ackermann, Angew. Chem., Int. Ed., 2016, 55, 6929 CrossRef CAS PubMed.
- A. R. A. Spencer, R. Korde, M. Font and I. Larrosa, Chem. Sci., 2020, 11, 4204 RSC.
- A. Biafora, B. A. Khan, J. Bahri, J. M. Hewer and L. J. Goossen, Org. Lett., 2017, 19, 1232 CrossRef CAS PubMed.
- N. Y. P. Kumar, T. Rogge, S. R. Yetra, A. Bechtoldt, E. Clot and L. Ackermann, Chem. – Eur. J., 2017, 23, 17449 CrossRef CAS PubMed.
- A. Mandal, H. Sahoo, S. Dana and M. Baidya, Org. Lett., 2017, 19, 4138 CrossRef CAS PubMed.
- F. Pu, Z.-W. Liu, L.-Y. Zhang, J. Fan and X.-Y. Shi, ChemCatChem, 2019, 11, 4116 CrossRef CAS.
- K. R. Bettadapur, V. Lanke and K. R. Prabhu, Chem. Commun., 2017, 53, 6251 RSC.
- F. Li, X. Li, T. Gong and Y. Fu, ChemCatChem, 2019, 11, 5124 CrossRef CAS.
- X.-Y. Shi, X.-F. Dong, J. Fan, K.-Y. Liu, J.-F. Wei and C.-J. Li, Sci. China Chem., 2015, 58, 1286 CrossRef CAS.
- Y. Honjo, Y. Shibata, E. Kudo, T. Namba, K. Masutomi and K. Tanaka, Chem. – Eur. J., 2018, 24, 317 CrossRef CAS PubMed.
- K. Ueura, T. Satoh and M. Miura, J. Org. Chem., 2007, 72, 5362 CrossRef CAS PubMed.
- E. Vitaku, D. T. Smith and J. T. Njardarson, J. Med. Chem., 2014, 57, 10257 CrossRef CAS PubMed.
- C.-L. Duan, X.-Y. Liu, Y.-X. Tan, R. Ding, S. Yang, P. Tian and G.-Q. Lin, Synlett, 2019, 30, 932 CrossRef CAS.
- N. Umeda, K. Hirano, T. Satoh and M. Miura, J. Org. Chem., 2009, 74, 7094 CrossRef CAS PubMed.
- D. Kalyani, N. R. Deprez, L. V. Desai and M. S. Sanford, J. Am. Chem. Soc., 2005, 127, 7330 CrossRef CAS PubMed.
- A. I. McKay, W. A. O. Altalhi, L. E. McInnes, M. L. Czyz, A. J. Canty, P. S. Donnelly and R. A. J. O'Hair, J. Org. Chem., 2020, 85, 2680 CrossRef CAS PubMed.
- A. Hossian, M. K. Manna, K. Manna and R. Jana, Org. Biomol. Chem., 2017, 15, 6592 RSC.
- J.-L. Yu, S.-Q. Zhang and X. Hong, J. Am. Chem. Soc., 2017, 139, 7224 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sc01226c |
| This journal is © The Royal Society of Chemistry 2023 |