
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA redox-active inorganic crown ether based on a polyoxometalate capsule†
Nanako
Tamai
a,
Naoki
Ogiwara
 a,
Eri
Hayashi
b,
Keigo
Kamata
b,
Toshiyuki
Misawa
c,
Takeru
Ito
c,
Tatsuhiro
Kojima
d,
Mireia
Segado
e,
Enric
Petrus
e,
Carles
Bo
ef and
Sayaka
Uchida
*a
a,
Eri
Hayashi
b,
Keigo
Kamata
b,
Toshiyuki
Misawa
c,
Takeru
Ito
c,
Tatsuhiro
Kojima
d,
Mireia
Segado
e,
Enric
Petrus
e,
Carles
Bo
ef and
Sayaka
Uchida
*a
aDepartment of Basic Science, School of Arts and Sciences, The University of Tokyo, 3-8-1 Komaba, Meguro-ku, Tokyo 153-8902, Japan. E-mail: csayaka@g.ecc.u-tokyo.ac.jp
bLaboratory for Materials and Structures, Institute of Innovative Research, Tokyo Institute of Technology, Nagatsuta-cho 4259, Midori-ku, Yokohama 226-8503, Japan
cDepartment of Chemistry, School of Science, Tokai University, 4-1-1 Kitakaname, Hiratsuka 259-1292, Japan
dDepartment of Chemistry, Graduate School of Science, Osaka Univerisity, 1-1 Machikaneyamacho, Toyonaka, Osaka 560-0043, Japan
eInstitute of Chemical Research of Catalonia (ICIQ), The Barcelona Institute of Science and Technology (BIST), Av. Països Catalans, 16, 43007 Tarragona, Spain
fDepartament de Química Física i Inorgànica, Universitat Rovira i Virgili, Marcel lí Domingo s/n, 43007 Tarragona, Spain
First published on 26th April 2023
Abstract
Cation-uptake has been long researched as an important topic in materials science. Herein we focus on a molecular crystal composed of a charge-neutral polyoxometalate (POM) capsule [MoVI72FeIII30O252(H2O)102(CH3CO2)15]3+ encapsulating a Keggin-type phosphododecamolybdate anion [α-PMoVI12O40]3−. Cation-coupled electron-transfer reaction occurs by treating the molecular crystal in an aqueous solution containing CsCl and ascorbic acid as a reducing reagent. Specifically, multiple Cs+ ions and electrons are captured in crown-ether-like pores {MoVI3FeIII3O6}, which exist on the surface of the POM capsule, and Mo atoms, respectively. The locations of Cs+ ions and electrons are revealed by single-crystal X-ray diffraction and density functional theory studies. Highly selective Cs+ ion uptake is observed from an aqueous solution containing various alkali metal ions. Cs+ ions can be released from the crown-ether-like pores by the addition of aqueous chlorine as an oxidizing reagent. These results show that the POM capsule functions as an unprecedented “redox-active inorganic crown ether”, clearly distinguished from the non-redox-active organic counterpart.
Introduction
Cation-uptake has been long researched as an important topic in materials science.1,2 For example, crown ethers, which are a family of synthetic cyclic multidentate organic ligands receiving the Nobel Prize in Chemistry 1987, have attracted great interest due to their structural topologies and applications especially in selective cation-uptake.3 Crown ethers can bind alkali metal ions size-selectively with the oxygen donors in gas, solution, or solid phase.4 The denticity of the polyether influences the affinity toward various ions: 15-crown-5-ether,4 18-crown-6-ether,4 and dibenzo-30-crown-10-ether5 show high selectivity toward Na+, K+, and Cs+, ions respectively. Removal of radioactive Cs+ ion from wastewater has received significant attention, and high Cs+ ion adsorption selectivity is observed with a dibenzo-30-crown-10-ether immobilized mesoporous silica.6 Cryptands possess three-dimensional cavities with the aid of nitrogen atoms and encapsulate various metal ions more strongly and selectively than crown ethers.7Polyoxometalates (POMs) are robust, discrete, and structurally well-defined metal-oxide cluster anions that are mainly composed of high-valence transition metals and have stimulated research in broad fields of sciences.8–15 For example, POMs have been long researched as cation-exchangers for the removal of toxic metal ions.16 Cation-uptake by POM-based compounds can be categorized into three groups: (i) POM-based ionic solids as cation-exchangers,16,17 (ii) POM molecules as inorganic crown ethers or cryptands,18–22 and (iii) reduction-induced cation-uptake in POM-based compounds.23 Cation-exchangers (the first category), which are ubiquitous in clays and zeolites, have been widely applied in water softening and metal ion separation.24 In fact, mobile counter cations (Na+) of Keggin-type polyoxoniobates [XNbV12O40]n− (X = SiIV, GeIV, PV) can be exchanged with radionuclides (Sr2+, NpO2+, Pu4+), showing high potential in nuclear waste removal.17 An early example of inorganic crown ethers and cryptands (the second category) is an arsenotungstate [AsIII4WVI40O140]28−, which binds alkali and alkaline earth metal ions into the eight-coordinated central cryptand site.19 A more simple arsenotungstate [AsIIIWVI9O33]9− with Eu3+ ions self-assembles into a supramolecular ring structure by the encapsulation of Cs+ ion.20 A silicodecatungstate dimer possesses a rigid cavity with ten oxygen atoms (SiIV–O–WVI × 4 and WVI–O–WVI × 6) and captures Sr2+ ion more strongly than organic cryptands.21 As described above, the cation uptake characteristics of POM-based inorganic crown ethers and cryptands can be modulated by tailoring the denticity or coordination ability of the constituent metal ions. However, the utilization of their redox property for cation uptake has not been cultivated yet, which would contribute to distinguishing these compounds from their non-redox-active organic counterparts.
Meanwhile, reduction-induced cation-uptake (the third category) is unique to redox-active POMs and POM-based compounds. For instance, Keggin-type phosphododecamolybdate [α-PMoVI12O40]3− accommodates 24 electrons due to the reduction of twelve molybdenum atoms (MoVI/IV) coupled with Li+ uptake, as a component of a molecular cluster battery.25 We have previously reported a redox-active porous ionic crystal (PIC) An+1[CrIII3O(OOCH)6(4-methylpyridine)3]2[α-PMoVI12−nMoVnO40]·mH2O (A = alkali metal ion), which shows one-electron redox (MoVI/V) coupled with uptake/release of various alkali metal ions.26 This reaction is commonly known as cation-coupled electron-transfer (CCET) in relation with proton-coupled electron-transfer (PCET),27,28 which is ubiquitous in energy conversion and storage reactions concerning proteins and enzymes. The cation-uptake rate depended on the type of alkali metal ions (K+ < Rb+ < Cs+), which is in line with the hydration enthalpy of cations (K+ −84.1 kcal mol−1 < Rb+ −78.6 kcal mol−1 < Cs+ −73.1 kcal mol−1);29 it is energetically favorable to remove water molecules from the hydration sphere of Cs+ than K+, and Cs+ can more easily enter and diffuse through the crystal lattice.26 However, the cation-uptake process in the PIC consisted of simple cation-exchange (the first category) as well as CCET (the third category), and extracting the contribution of CCET from the whole cation-uptake process was impossible. Therefore, the states, dynamics, and locations of the cations and electrons participating in the CCET remain unexplored.
According to these observations, we have chosen a charge-neutral POM capsule firstly reported by Müller and co-workers [α-PMoVI12O40]3−⊂[MoVI72FeIII30O252(H2O)102(CH3CO2)15]3+·60H2O [I] (Fig. 1),30,31 which consists of 20 crown-ether-like pores {MoVI3FeIII3O6} with alternately-arranged corner-sharing [MoVIO6] and [FeIIIO6] units and an encapsulated [α-PMoVI12O40]3−. Müller and co-workers have reported the molecular structures of a large variety of POM capsules, and some capsules can incorporate cations and organic molecules through the crown-ether-like pores, while its redox property is unexplored.32–34 Due to the redox-active metal ions of I, we expect that reduction-induced cation-uptake based solely on CCET would occur, which is an integration of the functions represented by the second and third categories shown above. We note that I is suitable for use as a solid material for Cs+ ion adsorption owing to its insolubility in water, which may be attributed to the connections between adjacent POM capsules via Fe–O–Fe bonds.
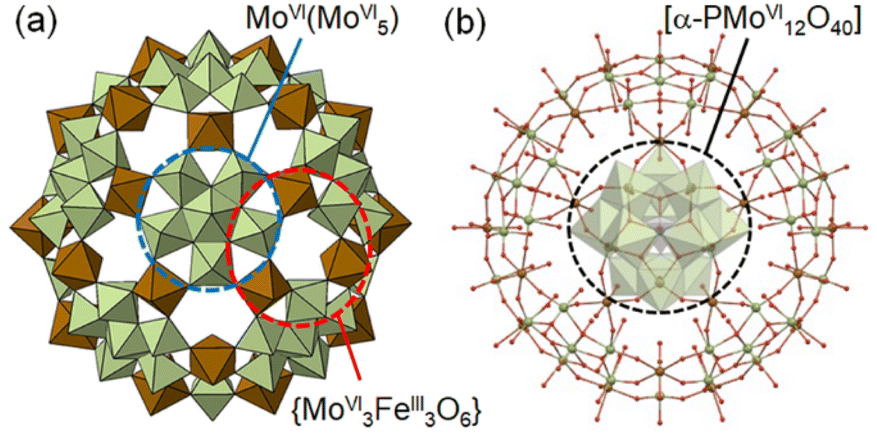 | ||
| Fig. 1 (a) Polyhedral and (b) ball-and-stick representation of the molecular structure of I. In (a), green and brown polyhedra show the [MoVIO6] and [FeIIIO6] units, respectively, and the encapsulated [α-PMoVI12O40]3− and acetates coordinating to the POM capsule are omitted for the clarity. In (b), green, brown, and red spheres show the Mo, Fe, and O atoms, respectively, and green and purple polyhedra show the [MoVIO6] and [PVO4] units of the encapsulated [α-PMoVI12O40]3−. | ||
Herein, we report that treating I with an aqueous solution containing CsCl and ascorbic acid as a reducing reagent leads to an uptake of ten Cs+ ions and electrons per I (i.e., 10 mol mol−1). Single crystal X-ray diffraction (SXRD) analysis shows that ten Cs+ ions are captured in the crown-ether-like pores, while four out of the ten Cs+ ions also serve to connect adjacent POM capsules. X-ray photoelectron spectroscopy (XPS) and density functional theory (DFT) calculations show that the electrons are stored in the Mo atoms. Selective uptake of Cs+ ion is observed from an aqueous solution containing various alkali metal ions. Upon oxidation with aqueous chlorine as an oxidation reagent, six Cs+ ions are released and the four Cs+ ions, which remain, are those connecting adjacent POM capsules. All these results show that the POM capsule can be regarded as a redox-active inorganic crown ether responding to external stimuli, which can be clearly distinguished from the non-redox-active organic counterpart.
Results and discussion
Thermodynamics and kinetics of reduction-induced uptake of Cs+ ions by I. Upon treating I with an aqueous solution containing CsCl and ascorbic acid as a reducing reagent, the color of I turned from yellow to dark blue suggesting that some of the Mo(VI) in the POM capsule are reduced. Note that I cannot be reduced without the addition of Cs+ ion (CsCl) to compensate the negative charge. Simple ion-exchange with Cs+ nor PCET takes place, owing to the absence of any counter cations in I and the fact that the hydration radius of H+ is too large to penetrate the crystal lattice of I. Fig. 2 shows the time course of reduction-induced uptake of Cs+ ions by I. The amounts of Cs+ ion uptake were quantitatively analyzed by atomic absorption spectrometry (AAS). The reduced compound will be denoted as I-red-Cs+ hereafter. AAS shows that adsorption equilibrium and saturation are reached in about 180 min with 10 mol mol−1 of Cs+ ions and no further increase with longer measurement time is observed. The time course of Cs+ ion uptake could be fitted with the Fick's diffusion equation in radial direction35 with a diffusion coefficient of D = 2.4 × 10−9 cm2 s−1 (Fig. 2, broken line). This value is larger by approximately 1–3 orders of magnitude than our previous work on Cs+ uptake with redox-active PICs based on a Keggin-type POM ([α-SiMo12O40]4−: 8.2 × 10−12 cm2 s−1 at 343 K)36 or Dawson-type POMs [α-P2MoVI18O62]6− and [α-P2WVI18O62]6−: 1.0–1.3 × 10−10 cm2 s−1 at 298 K).37 See Table S1† for the diffusion coefficients of Cs+ ion adsorption by various compounds. Infrared (IR) spectra (Fig. S1†) of I and I-red-Cs+ were basically identical, showing that the molecular structure is essentially maintained in the Cs+ ion uptake. In addition, the powder X-ray diffraction (PXRD) pattern (Fig. S2†) of I-red-Cs+ well agreed with that of I, which excludes biphase formation and supports uniform diffusion of Cs+ ions in the solid bulk. Fig. 3a shows the Cs+ ion adsorption isotherm, which could be well reproduced with the Langmuir model38 with a maximum adsorption capacity qm of 83 mg g−1 and an equilibrium constant KL of 2.4 × 10−3 L mg−1 (broken line). See Table S2† for the Langmuir parameters of Cs+ ion adsorption by various compounds. Fig. 3b shows the Langmuir linear regression fit with R2 = 0.9997, suggesting the homogeneity of Cs+ ion adsorption sites, where only one Cs+ ion each may be adsorbed.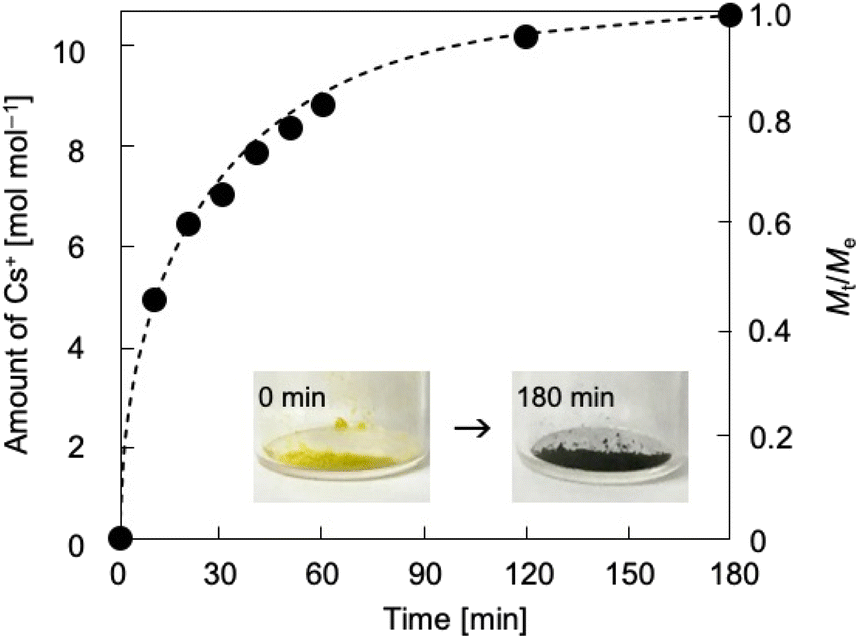 | ||
| Fig. 2 Time course of reduction-induced uptake of Cs+ ions in I at 298 K. Solid circles and broken line show the experimental data and the calculation according to the Fick's diffusion model, respectively. Mt and M∞ are the amounts of uptake at time t and equilibrium, respectively. Inset is the photo images of I before and after the treatment. | ||
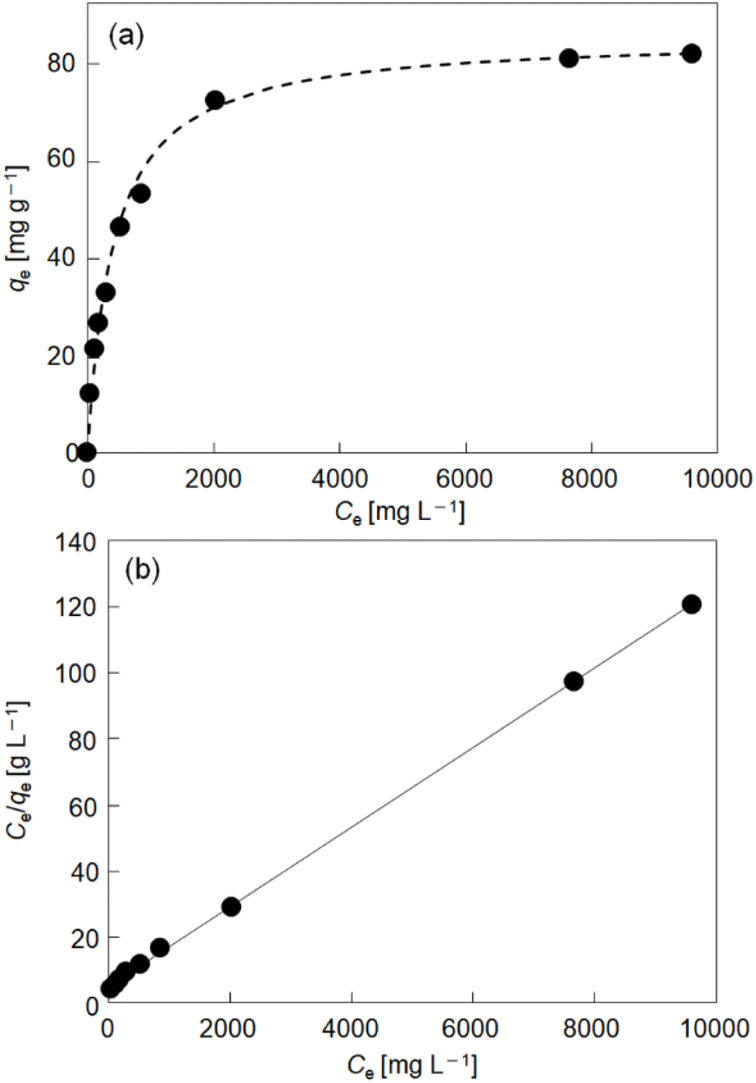 | ||
| Fig. 3 (a) Cs+ ion adsorption isotherm of I at 298 K. Solid circles and broken line show the experimental data and the calculation according to the Langmuir model, respectively. (b) Langmuir linear regression fit (R2 = 0.9997). | ||
Our previous results on redox-active PICs showed that the rate of reduction-induced alkali metal ion uptake was the fastest for Cs+ and depended on the dehydration enthalpy of cations.29 According to these observations, competitive adsorption of equimolar concentrations of alkali metal ions (Na+, K+, Rb+, Cs+) in water was carried out with ascorbic acid as a reducing reagent. As shown in Fig. S3,† the amounts of alkali metal ion uptakes were Na+ 0, K+ 0, Rb+ 1.1, and Cs+ 9.6 mol mol−1, and highly selective towards Cs+.
Locations of electrons upon reduction-induced uptake of Cs+ ions by I. XPS was utilized to characterize the composition and electronic state of I-red-Cs+. Wide scan XPS (Fig. S4†) confirmed the existence of Cs+ in I-red-Cs+, and no signal due to Cl (2p3/2: 198.7 ± 0.6 eV) was observed. The narrow-scan XPS of I-red-Cs+ in the Fe 2p region exhibited a signal attributable to Fe(III) (2p3/2: 710–712 eV) and not Fe(II) (2p3/2: <710 eV) (Fig. S5†). These results suggest that Fe atoms remain as Fe(III) and Cl− from CsCl is not incorporated by the reduction-induced uptake of Cs+ ion. Fig. S6† shows the narrow-scan XPS of I and I-red-Cs+ in the Mo 3d region together with peak deconvolution. The Mo 3d5/2 and 3d3/2 signals of I were well reproduced with a single peak due to Mo(VI). These signals broadened toward the lower energies upon forming I-red-Cs+, showing that some of the Mo(VI) have been reduced,36 which is in line with the color change (Fig. 2, inset). Deconvolution of the Mo 3d5/2 and 3d3/2 signals shows that the Mo(V) fraction in I-red-Cs+ was 12.6%, suggesting ten electron reduction of the Mo(VI) in I by the reduction-induced Cs+ uptake. According to these results, the reduction-induced Cs+ ion uptake can be formulated as follows,
| PMoVI12O40]3− ⊂ [MoVI72FeIII30O252(H2O)102(CH3CO2)15]3+ + 10Cs+ + 10e− → [α-PMoV2MoVI10O40]5− ⊂ Cs10[MoV8MoVI64FeIII30O252(H2O)102(CH3CO2)15]5+, | (1) |
| {PMoVI12}3− ⊂ {MoVI72FeIII30}3+ + 10Cs+ + 10e− → {PMoV2MoVI10}5− ⊂ Cs10{MoV8MoVI64FeIII30}5+. | (2) |
As indicated in the chemical formula, I possess two types of Mo species; those constructing the crown-ether-like pores {MoVI3FeIII3O6} and/or pentagonal MoVI(MoVI5) units (external Mo atoms: 72 out of 84, I-capsule), or those of the encapsulated [α-PMoVI12O40]3− (internal Mo atoms: 12 out of 84, I-Keggin) (see Fig. 1). Since XPS or other experimental methods cannot differentiate which kind of Mo species is reduced, we relied on electronic structure studies at the DFT level. We note that it is intrinsically impossible experimentally to decide which Mo in the Keggin anion or the POM capsule is reduced because the electrons are delocalized among the Mo atoms, as indicated by the blue color of I-red-Cs+ due to inter-valence charge-transfers.26,39 The fully oxidized state with no additional electrons and +15 charge PMoVI12O40 MoVI72FeIII30O252(H2O)60]15+ model denoted as I-all as well as the reduced states (1, 6, 12, 18, and 24-electron reduction) were considered (see ESI†). Because of the large size of the system, density of states (DOS) plots, which are more typical of bands in solids, were utilized to visualize the electronic structure.40 In the total density of states (TDOS) (Fig. S7†), the deepest bands around −25 eV comprise 2s oxygen electrons and some contribution of p metal orbitals, and the wide band from −15 eV till below the HOMO area known as the oxo-band corresponds to 2p oxygen electrons, which mix with empty d metal orbitals. Then, the partial density of states (PDOS) was computed to investigate the locations of electrons upon reduction of I-all. Fig. 4a shows the PDOS at different reduced states, where only contributions of metal atoms are plotted. Note that the shape of these bands hardly changes upon reduction. The vertical red lines in Fig. 4a, which mark the position of the HOMO for each reduced state, show that the whole electronic system causes a systematic upward-shift in energy upon reduction and that the first-electron reduction causes the largest effect in energy.
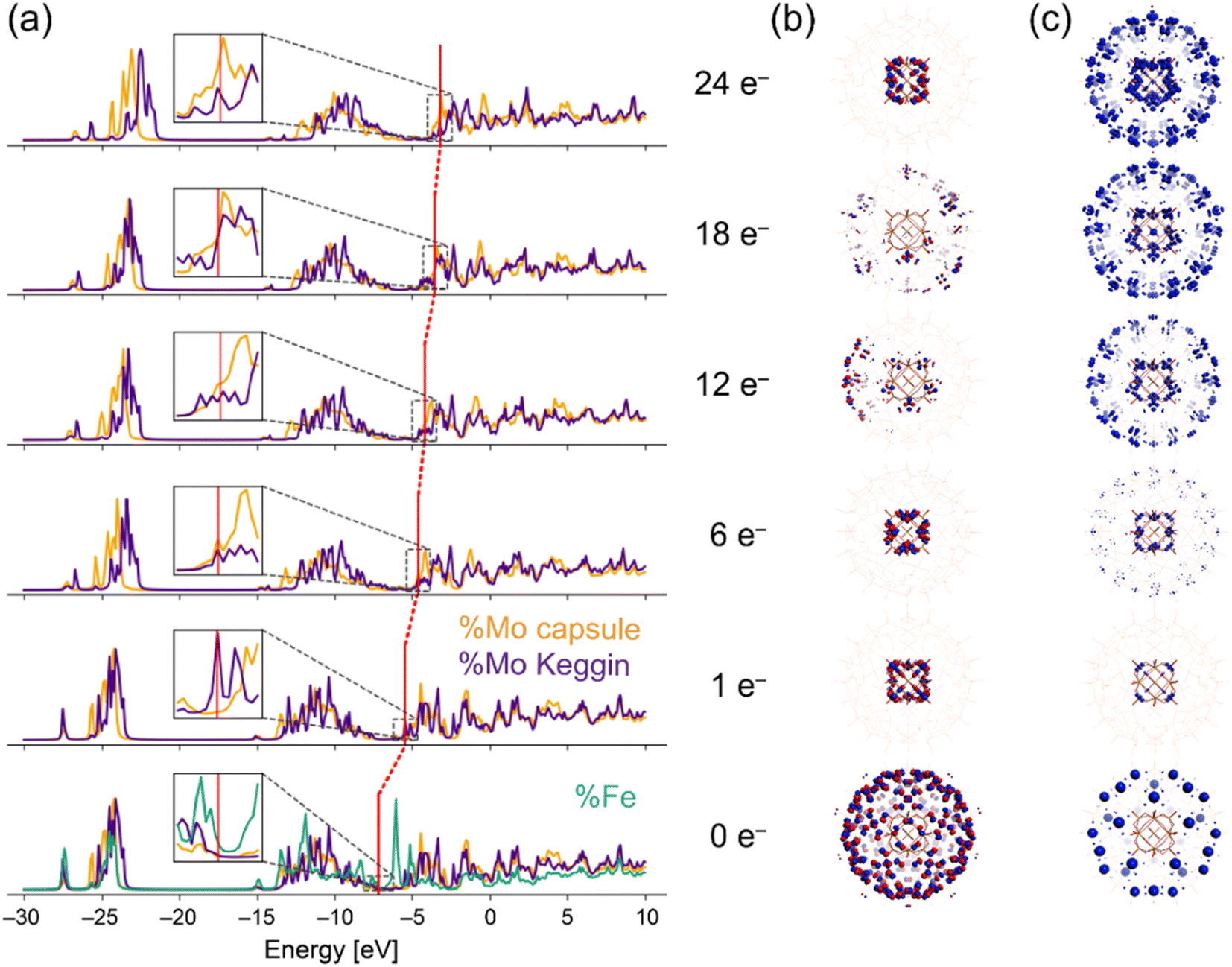 | ||
| Fig. 4 (a) PDOS metal atoms contributions for I-all at the fully oxidized (0e−) and reduced (1, 6, 12, 18, and 24e−) states. Fe: green; internal Mo atoms (I-Keggin): purple; external Mo atoms (I-capsule): yellow. Vertical red lines mark HOMO level positions. Fe PDOS is shown for only the 0e− state. The inset shows the magnified views of the HOMO areas. (b) HOMO plots for each state. (c) Spin density iso-surfaces for the 0e− state, and difference spin densities of the reduced capsules with respect to the spin density of the 0e− state. | ||
In the fully oxidized state (0e−), the HOMO of I-all contains 150 unpaired electrons at the 30 octahedral Fe(III) centers (Fig. 4b), which agrees with the work reported by Kuepper et al.,41 and the LUMO corresponds to the empty d-levels of Fe and Mo. When the system is reduced, only the Mo levels are filled, and the first electron is located at the internal Mo atoms (I-Keggin). As shown in Fig. 4c, the difference spin density of the 1e− reduced state with respect to the 0e− state illustrates where the additional electron is located. Only the first two added electrons are located in I-Keggin, as can be deduced from the inset plots of PDOS in Fig. 4a and much easier from the spin density plots in Fig. 4c. As for the 6e− reduced state, the spin density appears in the d-orbitals of external Mo atoms (I-capsule), which accept 4 electrons. At higher reduction degrees, the filled orbitals show larger contribution of I-capsule, and this trend is maintained for the highest (24e−) reduced state, where integration of the spin density enabled assigning 20 out 24 electrons to I-capsule (Fig. S8†). The PDOS results suggest that in I-red-Cs+, 2 and 8 electrons are located in the I-Keggin and I-capsule, respectively. Therefore, the reduction-induced Cs+ ion uptake proceeds according to the following reaction,
| {PMoVI12}3− ⊂ {MoVI72FeIII30}3+ + 10Cs+ + 10e− → {PMoV2MoVI10}5− ⊂ Cs10{MoV8MoVI64FeIII30}5+. | (3) |
Crystal structure of I-red-Cs+: a POM capsule as a redox-active inorganic crown ether
Fig. 5a shows the molecular unit and local structures of I-red-Cs+ analyzed by SXRD analysis (Table S3†). The molecular unit of I-red-Cs+ possesses four crystallographically independent Cs sites, and multiple measurements have indicated that ten Cs per molecular unit could be reasonably located, which agrees with the amount of uptake at adsorption equilibrium. Cs3 and Cs1 have site occupancies of 1.0, and these Cs (six Cs per molecular unit) are captured in the crown-ether-like pores of {Mo3Fe3O6} with Cs–O distances of 3.171–3.273 Å (Cs3) and 3.167–3.271 Å (Cs1). The openings of these crown-ether-like pores, assuming from the distances between the two oxygens across the pore, are 5.5–5.8 Å. Therefore, the pore size is smaller than twice the Cs–O distance so that Cs do not fit in but are located slightly above the pore. The coordination environments of Cs3 and Cs1 can be compared to alkali metal ions captured in 18-crown-6; for example, K+ ion shows the best fit among the alkali metal ions into the cavity of 18-crown-6 with K–O distances of 2.825–2.968 Å in K(18-crown-6)(CH3COO).42 The Cs–O distances in Cs(18-crown-6)I are 3.032–3.244 Å,43 which fairly agree with those of I-red-Cs+ (3.167–3.273 Å). Therefore, the POM capsule can selectively capture Cs+ ions on the crown-ether-like pores of {Mo3Fe3O6} as a charge compensation to the electrons stored in the encapsulated phosphododecamolybdate via reduction. This observation can be recognized as the first report of a redox-active inorganic crown ether, which clearly distinguished from the non-redox-active organic counterpart.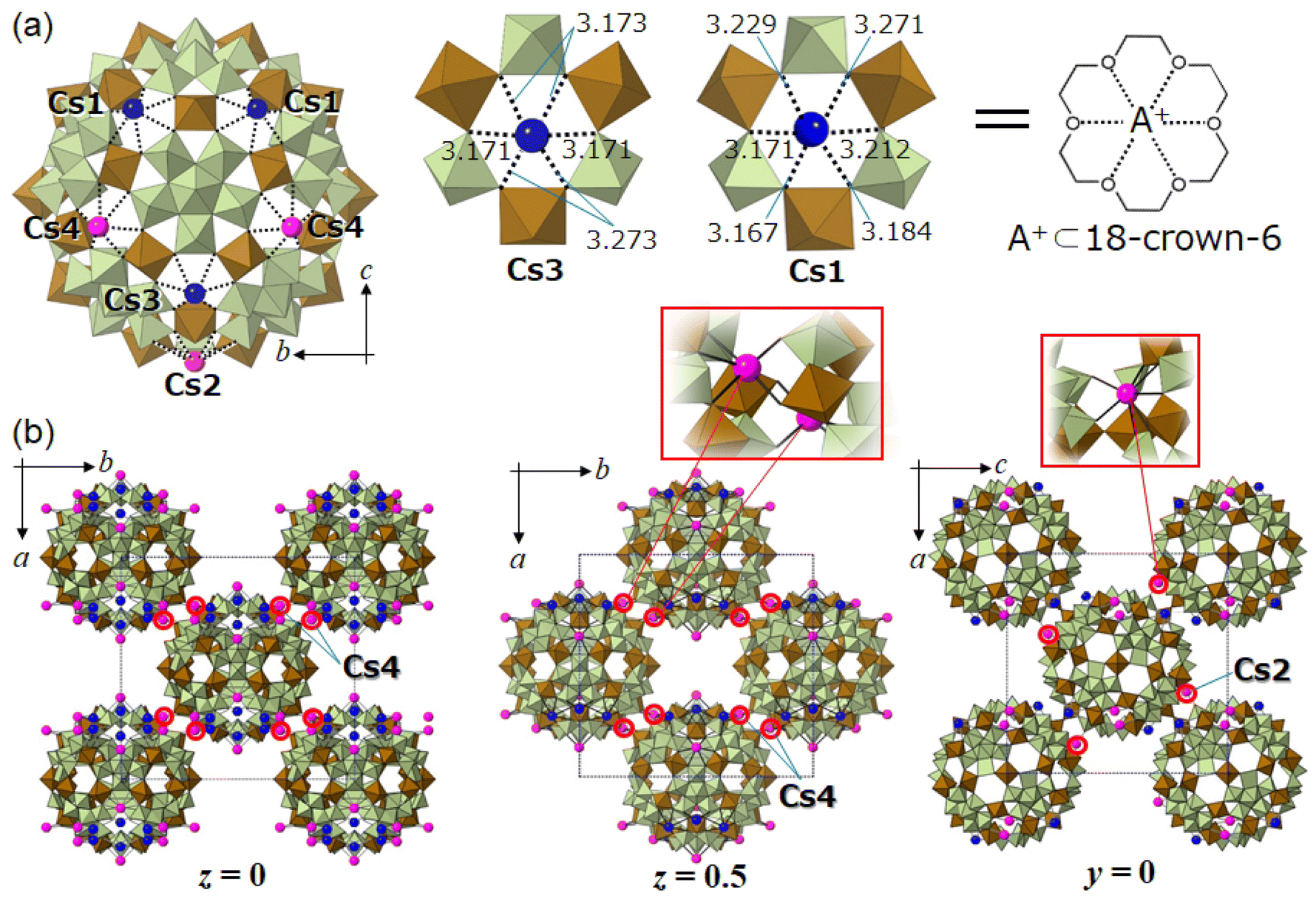 | ||
| Fig. 5 Molecular unit, crystal and local structures of I-red-Cs+. Green and brown polyhedra show the [MoO6] and [FeO6] units, respectively. Blue and pink spheres show the Cs captured in the crown-ether-like pores of {Mo3Fe3O6} and those connecting adjacent POM capsules, respectively. (a) Left: molecular unit. Middle to right: coordination environments (Cs–O) and bond lengths (Å) of Cs (Cs3 and Cs1) captured in the crown-ether-like pores, which can be compared to an alkali-metal ion captured in 18-crown-6. (b) Left and middle: crystal structures in the ab-plane at z = 0 and 0.5, respectively, assuming a pseudo-fcc structure. Right: crystal structure in the ac-plane at y = 0 assuming a pseudo-fcc structure. Local structures of Cs (Cs4 and Cs2) connecting adjacent POM capsules are shown in the red rectangles. | ||
Fig. 5b shows the crystal structure of I-red-Cs+. While the crystal system of I-red-Cs+ is orthorhombic, the lengths of a- (35.0800(7) Å), b- (36.9265(7) Å), and c- (34.8114(6) Å) axes are close to each other, and the POM capsules are located on the eight vertices and six face-centers of the unit cell. Therefore, the crystal structure of I-red-Cs+ can be explained as a pseudo-face centered cubic (fcc) structure. The left and middle images in Fig. 5b show the crystal structures in the ab-plane at z = 0 and 0.5, respectively, indicating that the POM capsules are located on the vertices and face-centers of the pseudo-fcc cell. Cs2 and Cs4 have site occupancies of 1.0 and 0.5, respectively, and these Cs (four Cs per molecular unit) occupy the crown-ether-like-pores with Cs–O distances of 3.140–3.438 Å. Additionally, Cs2 and Cs4 connect adjacent POM capsules within the ab- and ac-planes with Cs–O distances of 3.059 Å and 2.761–3.085 Å, respectively. These bonds are shorter than those coordinating to the crown-ether-like pores, suggesting that the POM capsules are strongly connected to each other by Cs2 and Cs4. Local structures of Cs2 and Cs4 are shown in the inset of Fig. 5b, and the bond distances are summarized in Fig. S10.†
Oxidation-induced release of Cs+ ions from I-red-Cs+. Finally, oxidation-induced release of Cs+ ions from I-red-Cs+ was investigated with aqueous chlorine as an oxidation reagent. The color of I-red-Cs+ turned from dark blue to a yellowish color suggesting the oxidation of Mo species. Fig. S11† shows the time course of the oxidation-induced release of Cs+ ions. AAS shows that Cs+ desorption equilibrium is reached in about 60 min; six Cs+ ions per molecular unit are released and four Cs+ ions remain in the solid after the oxidation. These numbers were not affected by the concentration and/or the stoichiometric amount of aqueous chlorine. The compound after the oxidation-induced release of Cs+ ions will be denoted as I-ox-Cs+ hereafter. Deconvolution of the narrow-scan XPS of I-ox-Cs+ (Fig. S6†) shows that the Mo(V) fraction is 5.4% suggesting four electron reduction of the Mo species, which is in line with the fact that four Cs+ ions remain in I-ox-Cs+. All these results show that the oxidation-induced Cs+ ion release proceeds according to the following reaction,
| {PMoV2MoVI10}5− ⊂ Cs10{MoV8MoVI64FeIII30}5+ − 6Cs+ − 6e− → {PMoV2MoVI10}5− ⊂ Cs4{MoV2MoVI70FeIII30}5+. | (4) |
Schematic illustration of eqn (4) together with the local structure and coordination environment of I-ox-Cs+ are shown in Fig. S12.† Four Cs per molecular unit, which connect adjacent POM capsules and are six-coordinated to the crown-ether-like pores, could be reasonably assigned by preliminary SXRD analysis. These Cs cannot be removed by increasing the concentration and/or stoichiometric amount of the oxidation reagent and are likely crucial to maintaining the crystal structure. Such site-specific coordination has been also observed for Sr2+ ions bridging anionic POM capsules comprising 132 Mo atoms.44 Upon treatment of I-ox-Cs+ with an aqueous solution containing CsCl and ascorbic acid, six Cs+ ions and electrons are re-introduced into the solid-state structure forming I-red-Cs+, showing that the CCET reaction occurs reversibly.
Conclusion
We have demonstrated that a charge-neutral POM capsule encapsulating a Keggin-type phosphododecamolybdate anion [α-PMoVI12O40]3− ⊂ [MoVI72FeIII30O252(H2O)102(CH3CO2)15]3+·60H2O [I] selectively captured Cs+ ions into the crown-ether-like pores via cation-coupled electron-transfer (CCET) reaction, functioning as an unprecedented redox-active inorganic crown ether. The thermodynamic and kinetic aspects of Cs+ ion uptake were analyzed experimentally by the Fickian diffusion and Langmuir-type model, respectively. XPS revealed that electrons were incorporated cooperatively with the Cs+ ions. DFT studies showed that these electrons were located at the Mo atoms of both the capsule and the phosphododecamolybdate anion. The CCET reaction with Cs+ ions and electrons occurs reversibly and in a single-crystal-to-single-crystal manner, which was traced by SXRD analysis. SXRD analysis revealed that the Cs+ ions are tightly bound to the crown-ether-like pores, indicating that the POM capsule is geometrically ideal for Cs+ ion uptake. These results can be clearly distinguished from those of the widely-known non-redox-active organic crown ethers and cryptands, because not only the denticity or coordination ability but also the redox property of the constituent metal ions of POMs is utilized in the cation-uptake. Our next aim is to fine-tune the CCET reaction by appropriately choosing the elements and structures of POMs to realize the selective uptake and release of multivalent metal ions. Preparation of soluble redox-active inorganic crown ethers will be another branch of future work. We believe that redox-active inorganic crown ethers may open new opportunities to solve environmental issues as well as to discover post-synthetic methods for novel solid-state materials.Data availability
All data associated with this publication are provided in the ESI.†Author contributions
N. T. performed the syntheses and characterization of the compounds; N. T., T. M., T. I., T. K., and S. U. carried out the single-crystal X-ray diffraction measurements and analysis; E. H. and K. K. carried out the XPS measurements. N. O. analyzed the XPS spectra. M. S. and E. P. carried out, and C. B. supervised the DFT calculations. N. T., N. O., and S. U. wrote the manuscript. S. U. conceived and designed the project. All authors discussed the results and commented on the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by JSPS Grants-in-Aid for Scientific Research JP19K23648, JP19H04564, JP19H04686, JP20H02750, and core-to-core program on “International Network on Polyoxometalate Science for Advanced Functional Energy Materials” from MEXT of Japan. X-ray diffraction measurements with synchrotron radiation (2019-1st-2D-015, 2019-2nd-2D-025) were performed at the Pohang Accelerator Laboratory (2D Beamline) supported by Pohang University of Science and Technology (POSTECH), and were additionally performed at the BL02B1 of SPring-8 with the approval of the Japan Synchrotron Radiation Research Institute (JASRI) (Proposal No. 2021A1034). The authors thank the ICIQ Foundation, CERCA Program and AGAUR (grant 2017SGR00290) of the Generalitat de Catalunya, and the Spanish Ministerio de Ciencia e Innovacion through projects PID2020-112806RB-I00 and the Severo Ochoa Excellence Accreditation 2020–2023 CEX2019-000925-S.Notes and references
- B. A. Moyer, J. F. Birdwell Jr, P. V. Bonnesen, S. H. Bruffey, L. H. Delmau, N. C. Duncan, D. D. Ensor, T. G. Hill, D. L. Lee, A. Rajbanshi, B. D. Roach, P. L. Szczygiel, F. V. Sloop Jr, E. L. Stoner and N. J. Williams, Report Number: ORNL/TM-2014/22, https://www.osti.gov/servlets/purl/116700.
- A. Tripathi, D. G. Medvedev, J. Delgado and A. Clearfield, J. Solid State Chem., 2004, 177, 2903–2915 CrossRef CAS.
- C. J. Pedersen, J. Am. Chem. Soc., 1967, 89, 7017–7036 CrossRef CAS.
- G. W. Gokel, W. M. Leevy and M. E. Weber, Chem. Rev., 2004, 104, 2723–2750 CrossRef CAS PubMed.
- D. Live and S. I. Chan, J. Am. Chem. Soc., 1976, 98, 3769–3778 CrossRef CAS.
- M. R. Awual, Chem. Eng. J., 2016, 303, 539–546 CrossRef CAS.
- J. M. Lehn, Acc. Chem. Res., 1978, 11, 49–57 CrossRef CAS.
- M. T. Pope and A. Müller, Angew. Chem., Int. Ed., 1991, 30, 34–48 CrossRef.
- C. L. Hill, Chem. Rev., 1998, 98, 1–390 CrossRef CAS PubMed.
- Y. Song and R. Tsunashima, Chem. Soc. Rev., 2012, 41, 7384–7402 RSC.
- L. Vilà-Nadal and L. Cronin, Nat. Rev. Mater., 2017, 2, 17054 CrossRef.
- A. Misra, K. Kozma, C. Streb and M. Nyman, Angew. Chem., Int. Ed., 2020, 59, 596–612 CrossRef CAS PubMed.
- A. V. Anyushin, A. Kondinski and T. N. Parac-Vogt, Chem. Soc. Rev., 2020, 49, 382–432 RSC.
- N. Ogiwara, T. Iwano, T. Ito and S. Uchida, Coord. Chem. Rev., 2022, 462, 214524 CrossRef CAS.
- K. Xia, K. Yamaguchi and K. Suzuki, Angew. Chem., Int. Ed., 2023, 62, e202214506 CAS.
- V. Pekárek and V. Veselý, Talanta, 1972, 19, 1245–1283 CrossRef PubMed.
- M. Nyman, C. R. Powers, F. Bonhomme, T. M. Alam, E. J. Maginn and D. T. Hobbs, Chem. Mater., 2008, 20, 2513–2521 CrossRef CAS.
- W. G. Klemperer and G. Westwood, Nat. Mater., 2003, 2, 780–781 CrossRef CAS PubMed.
- F. Robert, M. Leyrie, G. Hervé, A. Tézé and Y. Jeannin, Inorg. Chem., 1980, 19, 1746–1752 CrossRef CAS.
- K. Fukaya and T. Yamase, Angew. Chem., Int. Ed., 2003, 42, 654–658 CrossRef CAS PubMed.
- A. Yoshida, Y. Nakagawa, K. Uehara, S. Hikichi and N. Mizuno, Angew. Chem., Int. Ed., 2009, 48, 7055–7058 CrossRef CAS PubMed.
- M. Barsukova-Stuckart, N. V. Izarova, R. A. Barrett, Z. Wang, J. van Tol, H. W. Kroto, N. S. Dalal, P. Jiménez-Lozano, J. J. Carbó, J. M. Poblet, M. S. von Gernler, T. Drewello, P. de Oliveira, B. Keita and U. Kortz, Inorg. Chem., 2012, 51, 13214–13228 CrossRef CAS PubMed.
- S. Uchida, Chem. Sci., 2019, 10, 7670–7679 RSC.
- A. Clearfield, Chem. Rev., 1988, 88, 125–148 CrossRef CAS.
- H. Wang, S. Hamanaka, Y. Nishimoto, S. Irle, T. Yokoyama, H. Yoshikawa and K. Awaga, J. Am. Chem. Soc., 2012, 134, 4918–4924 CrossRef CAS PubMed.
- R. Kawahara, S. Uchida and N. Mizuno, Chem. Mater., 2015, 27, 2092–2099 CrossRef CAS.
- D. R. Weinberg, C. J. Gagliardi, J. F. Hull, C. F. Murphy, C. A. Kent, B. C. Westlake, A. Paul, D. H. Ess, D. G. McCafferty and T. J. Meyer, Chem. Rev., 2012, 112, 4016–4093 CrossRef CAS PubMed.
- P. R. D. Murray, J. H. Cox, N. D. Chiappini, C. B. Roos, E. A. MnLoughlin, B. G. Hejna, S. T. Nguyen, H. H. Ripberger, J. M. Ganley, E. Tsui, N. Y. Shin, B. Koronkiewicz, G. Qiu and R. R. Knowles, Chem. Rev., 2022, 122, 2017–2291 CrossRef CAS PubMed.
- E. R. Nightingale Jr, J. Phys. Chem., 1959, 63, 1381–1387 CrossRef.
- A. Müller, S. K. Das, H. Bögge, M. Schmidtmann, A. Botar and A. Patrut, Chem. Commun., 2001, 657–658 RSC.
- K. Tandekar, P. Naulakha and S. Supriya, Inorg. Chim. Acta, 2020, 511, 119729 CrossRef CAS.
- A. Müller, E. Krickemeyer, H. Bögge, M. Schmidtmann, S. Roy and A. Berkle, Angew. Chem., Int. Ed., 2002, 41, 3604–3609 CrossRef.
- A. Müller, S. K. Das, S. Talismanov, S. Roy, E. Beckmann, H. Bögge, M. Schmidtmann, A. Merca, A. Berkle, L. Allouche, Y. Zhou and L. Zhang, Angew. Chem., Int. Ed., 2003, 42, 5039–5044 CrossRef PubMed.
- A. Ziv, A. Grego, S. Kopilevich, L. Zeiri, P. Miro, C. Bo, A. Müller and I. A. Weinstock, J. Am. Chem. Soc., 2009, 131, 6380–6382 CrossRef CAS PubMed.
- J. Crank, The Mathematics of Diffusion, Oxford University Press, London, 1956, ch. 6 Search PubMed.
- S. Seino, R. Kawahara, Y. Ogasawara, N. Mizuno and S. Uchida, Angew. Chem., Int. Ed., 2016, 55, 3987–3991 CrossRef CAS PubMed.
- S. Hitose and S. Uchida, Inorg. Chem., 2018, 57, 4833–4836 CrossRef CAS PubMed.
- P. Atkins and J. de Paula, Physical Chemistry, Oxford University Press, London, 8th edn, 2006, ch. 25 Search PubMed.
- X. Kong, G. Wan, B. Li and L. Wu, J. Mater. Chem. B, 2020, 8, 8189–8206 RSC.
- D. Melgar, N. A. G. Bandeira and C. Bo, Chem.–Eur. J., 2017, 23, 5338–5344 CrossRef CAS PubMed.
- K. Kuepper, C. Derks, C. Taubitz, M. Prinz, L. Joly, J. P. Kappler, A. Postnikov, W. Yang, T. V. Kuznetsova, U. Wiedwald, P. Ziemann and M. Neumann, Dalton Trans., 2013, 42, 7924–7935 RSC.
- P. Liebing, A. Zaeni, F. Olbrich and F. T. Edelmanna, Acta Crystallogr., Sect. E: Crystallogr. Commun., 2016, 72, 1757–1761 CrossRef CAS PubMed.
- T. H. Kim, S. S. Lee, J. S. Kim and J. Kim, Anal. Sci., 2001, 17, 573–574 CrossRef CAS PubMed.
- M. Sato and T. Ozeki, Dalton Trans., 2012, 41, 9846–9848 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures; diffusion coefficients and Langmuir parameters of Cs+ ion adsorption by various compounds; crystallographic data; IR, PXRD, XPS, TG, theoretical calculation, competitive adsorption of alkali metal ions, and oxidation-induced release of Cs+ ion. CCDC 1981851. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3sc01077e |
| This journal is © The Royal Society of Chemistry 2023 |