
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEngineering of antimicrobial peptide fibrils with feedback degradation of bacterial-secreted enzymes†
Fenghua
Wang‡
ab,
Wencheng
Xia‡
c,
Mingming
Zhang
c,
Rongrong
Wu
a,
Xiaolu
Song
a,
Yun
Hao
a,
Yonghai
Feng
a,
Liwei
Zhang
a,
Dan
Li
de,
Wenyan
Kang
fg,
Cong
Liu
 *cgh and
Lei
Liu
*a
*cgh and
Lei
Liu
*a
aInstitute for Advanced Materials, School of Materials Science and Engineering, Jiangsu University, Zhenjiang, Jiangsu 212013, China. E-mail: liul@ujs.edu.cn
bCollege of Aeronautical Engineering, Jiangsu Aviation Vocational and Technical College, Zhenjiang, Jiangsu 212134, China
cInterdisciplinary Research Center on Biology and Chemistry, Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, Shanghai 201210, China. E-mail: liulab@sioc.ac.cn
dBio-X Institutes, Key Laboratory for the Genetics of Developmental and Neuropsychiatric Disorders (Ministry of Education), Shanghai Jiao Tong University, Shanghai 200030, China
eZhangjiang Institute for Advanced Study, Shanghai Jiao Tong University, Shanghai 200240, China
fDepartment of Neurology and Institute of Neurology, Ruijin Hospital, Shanghai Jiao Tong University School of Medicine, Shanghai 200025, China
gDepartment of Neurology, Ruijin Hainan Hospital, Shanghai Jiao Tong University School of Medicine (Boao Research Hospital), Hainan, 571434, China
hState Key Laboratory of Bio-Organic and Natural Products Chemistry, Shanghai Institute of Organic Chemistry, University of Chinese Academy of Sciences, Shanghai, 200032, China
First published on 7th September 2023
Abstract
Proteins and peptides can assemble into functional amyloid fibrils with distinct architectures. These amyloid fibrils can fulfil various biological functions in living organisms, and then be degraded. By incorporating an amyloidogenic segment and enzyme-cleavage segment together, we designed a peptide (enzyme-cleavage amyloid peptides (EAP))-based functional fibril which could be degraded specifically by gelatinase. To gain molecular insights into the assembly and degradation of EAP fibrils, we determined the atomic structure of the EAP fibril using cryo-electron microscopy. The amyloidogenic segment of EAP adopted a β-strand conformation and mediated EAP-fibril formation mainly via steric zipper-like interactions. The enzyme-cleavage segment was partially involved in self-assembly, but also exhibited high flexibility in the fibril structure, with accessibility to gelatinase binding and degradation. Moreover, we applied the EAP fibril as a tunable scaffold for developing degradable self-assembled antimicrobial fibrils (SANs) by integrating melittin and EAP together. SANs exhibited superior activity for killing bacteria, and significantly improved the stability and biocompatibility of melittin. SANs were eliminated automatically by the gelatinase secreted from targeted bacteria. Our work provides a new strategy for rational design of functional fibrils with a feedback regulatory loop for optimizing the biocompatibility and biosafety of designed fibrils. Our work may aid further developments of “smart” peptide-based biomaterials for biomedical applications.
Introduction
The rational design and construction of peptide-based materials with a precise build-up manner and controllable degradation after use is attractive, but is highly challenging. Previously, peptide-based controllable “smart” bionanomaterials were mainly designed to stimulate peptide precursor molecules to self-assemble in situ and in vivo in response to different endogenous trigger factors (e.g., enzymes,1–3 pH,4–7 reactive oxygen species,8 others.9–12). These peptide-based smart materials could be applied to drug delivery for therapy against tumors and infections, and exhibited an assembly-induced retention (AIR) effect,13,14 targeted enhancement effect,15–17 multivalent bond effect,18,19 and membrane perturbation.20 In addition, the in situ self-sorting assembly system of smart responsive peptides could be constructed to target different organelles and achieve cooperative regulation of different biological processes, such as effective killing of tumor cells through the coordinated regulation of organelle dysfunction.21In contrast to intensive studies on developing controllable self-assembly of peptides as functional nanomaterials, the design of peptide-based nanomaterial in a controlled degradation manner has not been explored well and widely. Automatic degradation of bio-nanomaterials after use is important due to their potential cytotoxicity, including the metabolic risk of nano drugs in vivo,22,23 and the toxicity of nano aggregates.24–28 Remarkably, cells employ a complicated and precise “quality control” system to remove excessive and misfolded proteins/protein assemblies to maintain protein homeostasis after completion of the tasks of such proteins.29–32 This phenomenon inspired us to propose a strategy for developing degradable peptide-based materials in response to different factors (e.g., enzymes) in a controllable manner. To achieve this aim, two major points must be considered: self-assembly of functional peptides and selective degradation by enzymes. Accordingly, the sequences of peptide self-assemblies and enzymatic cleavage are required for peptide design. Previous efforts in the design of degradable peptides focused on hydrogel formation.33–35 However, a smarter and rational design showing a clear mechanism of self-assembly and degradation in a controllable manner would be preferable.
In this work, we sought to achieve a rational design and build-up peptide-based fibrils with controllable degradation using enzymes. By examining different peptide sequences, we identified enzyme-cleavage amyloid peptide-2 (EAP2). EAP2 could self-assemble into a well-ordered, twisted fibrillar structure and could be degraded effectively in a controllable manner by gelatinase. Cryo-emission microscopy (cryo-EM) was used to determine the atomic structure of these well-ordered twisted fibrils, revealing that the enzymatic-cleavage sequence was accessible to the enzyme for cleavage in the fibril structure. Based on this constructed smart system, we built a smart antimicrobial nanomaterial by integrating the antibacterial peptide melittin at the C-terminus of EAP2. The designed peptide-based fibril could kill bacteria efficiently. More importantly, the designed fibril could be degraded automatically by the gelatinase secreted from bacteria to abolish antimicrobial activity when bacteria had been killed completely, which could further reduce the potential toxicity of nanomaterials. Engineering peptide fibrils with feedback on degradation of a secreted enzyme is extremely important for: (i) mimicking an existing biosystem; (ii) the development of functional peptide-based smart materials with various applications (e.g., drug delivery and novel biosensors for specific enzymes).
Results and discussion
Peptide design and characterization of peptide self-assembly
By analyzing the amyloidogenic peptides reported previously to exhibit a strong capability to form fibrils, we identified an amyloidogenic segment (GLMVGGVVIA) (termed “AP”) which is highly prone to assemble into nanostructures with a β-sheet-rich conformation but also contains a homology sequence of the gelatinase-cleavage segment (PLGVRG).36–39 Based on the AP sequence, we designed three additional enzymatic-cleaved amyloid peptides (EAPs): EAP1 (GPLGMRGGVVIA), EAP2 (GPLGMLGGVVIA) and EAP3 (GPLGVLGGVVIA). This was achieved by considering the homology between the enzymatic-cleavage sequence (cleavage site indicated by a red line) and the amyloidogenic segment (GVVIA) (Fig. 1A and S1†).We synthesized and examined the self-assembly of EAP1, EAP2, EAP3 and AP. AFM and TEM revealed that only AP and EAP2 could form fibrils spontaneously after 24 h of incubation. EAP2 fibrils had a height of 12.3 ± 1.1 nm and width of 11.5 ± 6.9 nm. AP fibrils had a height of 3.9 ± 1.7 nm and diameter of 20.5 ± 4.0 nm (Fig. 1B and Table S1,† AP-Tris). EAP1 and EAP3 could not assemble into fibrils under test conditions (Fig. S2†). The thioflavin T (ThT) assay and Fourier transform infrared (FT-IR) spectroscopy showed that AP and EAP2 adopted a typical β-strand secondary structure (Fig. 1C and D) in fibril samples, represented by a classical emission peak at 495 nm and typical stretching vibration peaks at 1620–1640 cm−1. EAP1 and EAP3 featured a random-coil secondary structure with low emission at 495 nm and vibration peaks at 1640–1650 cm−1. Molecular dynamics (MD) simulations were used to investigate the conformation of the four designed peptides (AP, EAP1–EAP3) in solution. Interestingly, EAP2 and AP were prone to assemble into homo-dimers in water during the 100 ns simulation, with a mean interaction energy of −168.75 kJ mol−1 and −105.63 kJ mol−1, respectively. The interaction energy of EAP1 and EAP3 was much lower (Fig. 1G and S3†). Furthermore, we analyzed the binding free energy computed from the potential of mean force (PMF) using umbrella sampling methods. AP and EAP2 could form stable dimers and the binding energy (ΔG) between the stable dimers was −14.76 kcal mol−1 and −7.67 kcal mol−1, respectively. The ΔG of EAP1 and EAP3 was −0.41 kcal mol−1 and −4.69 kcal mol−1, respectively (Fig. 1H and S3†). Taken together, our results showed that EAP2 and AP tended to self-assemble into fibrils in solution.
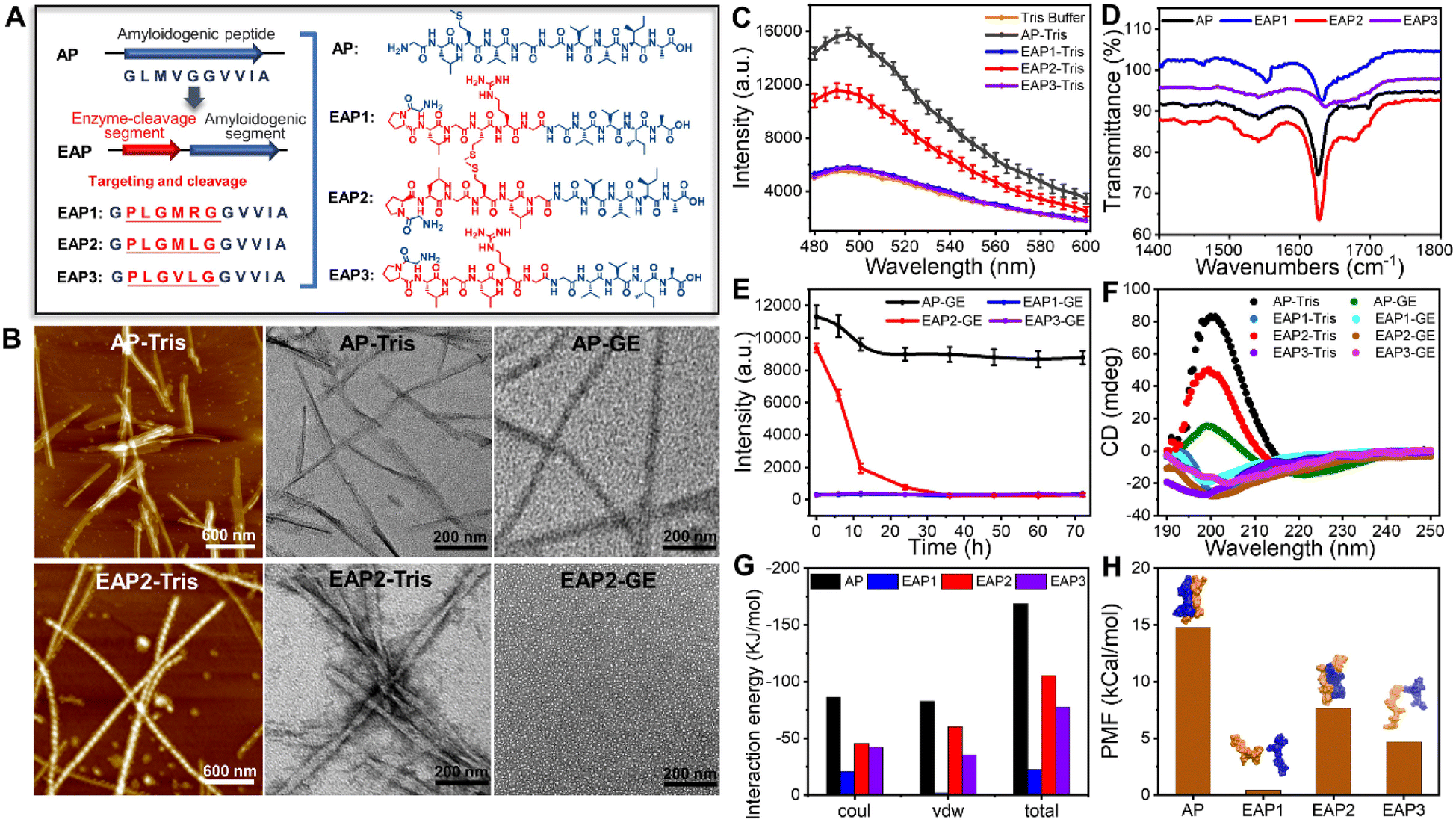 | ||
| Fig. 1 Design of a gelatinase-responsive peptide sequence (EAP1–EAP3) using amyloidogenic peptide (AP) as a reference peptide (schematic) (A). (B) AFM and TEM images of AP and EAP2 self-assembled fibrils in Tris buffer and degradation of fibrils in gelatinase buffer for 24 h. (C and D) ThT fluorescence analysis and characterization by FT-IR spectroscopy of the secondary structures of AP and EAP1–EAP3 in test samples. (E and F) ThT fluorescence assay and CD spectra of the secondary structure of AP and EAP1–EAP3 in Tris buffer and gelatinase solution, respectively. (G and H) Molecular dynamics simulation of the interaction energy and potential of mean force (PMF) of dimers of AP and EAP1–EAP3, with typical conformations of the four dimers. | ||
Gelatinase-mediated degradation of EAP2 fibrils
Next, we sought to investigate the enzyme-mediated degradation behavior of the fibrils formed by EAP2 and AP. Strikingly, we found that the self-assembled fibrils of EAP2 can be degraded effectively upon adding gelatinase (Fig. 1B, EAP2-GE) according to TEM and ThT measurement. AP fibrils were rarely degraded by gelatinase under the same condition. EAP2 fibrils could be degraded completely in 24 h with minimal ThT fluorescence intensity which was at the same level as EAP1 or EAP3 (Fig. 1E). The gelatinase cleavage site on EAP2 samples were measured further using LC-MS (Fig. S4†). EAP2 (10 mg mL−1) was degraded into shorter peptide fragments when cleaved from -L3-G4-, -G4-M5-, and -L6-G7- to -G7-G8- using gelatinase (2 U) for 12 h (details are shown in the ESI†).Circular dichroism (CD) spectroscopy further validated the secondary-structure changes of peptides in self-assembled and degraded states. Samples of AP and EAP2 fibrils displayed a typical β-strand secondary structure. After 24 h treatment in gelatinase solution, the EAP2 mixture featured a random-coil structure (positive peak at 192 nm, negative peak at 202 nm). AP retained a β-strand conformation under the same condition. As controls, EAP1 and EAP3 featured a random-coil conformation in solution (Fig. 1F). To further examine whether the enzyme-mediated degradation of EAP2 was specific to gelatinase as designed, we used other two proteases (trypsin and α-chymotrypsin) to treat EAP2 fibrils. Notably, EAP2 fibrils remained intact after treatment by these two proteases for 24 h (Fig. S5†). In general, our results demonstrated that EAP2 could self-assemble into fibrils that could be further degraded specifically by gelatinase.
Moreover, liquid-phase AFM was used to visualize, in situ, the gelatinase-mediated degradation of EAP2 fibrils in real time. We selected several EAP2 fibrils on the surface and monitored the degradation by three parallel tests (Fig. 2A). Interestingly, the number of EAP2 fibrils decreased markedly accompanied by an obviously shortened length of fibrils within 0.5 h treatment in all three tests. The coverage of fibrils in the observation window decreased from 19.88% to 0.56% within 2.5 h, and fibrils disappeared completely within 3 h (Fig. 2B). The decrease in fibril length and incubation time had a linear relationship (Fig. 2C). For EAP2, degradation was from the two open ends of the fibril (blue dotted box marks in Fig. 2A). In some cases, the disintegration and cleavage sites were observed in the middle of the fibril. These results confirm that the self-assembled fibrils of EAP2 could be degraded by gelatinase in a controllable manner (Fig. 2D).
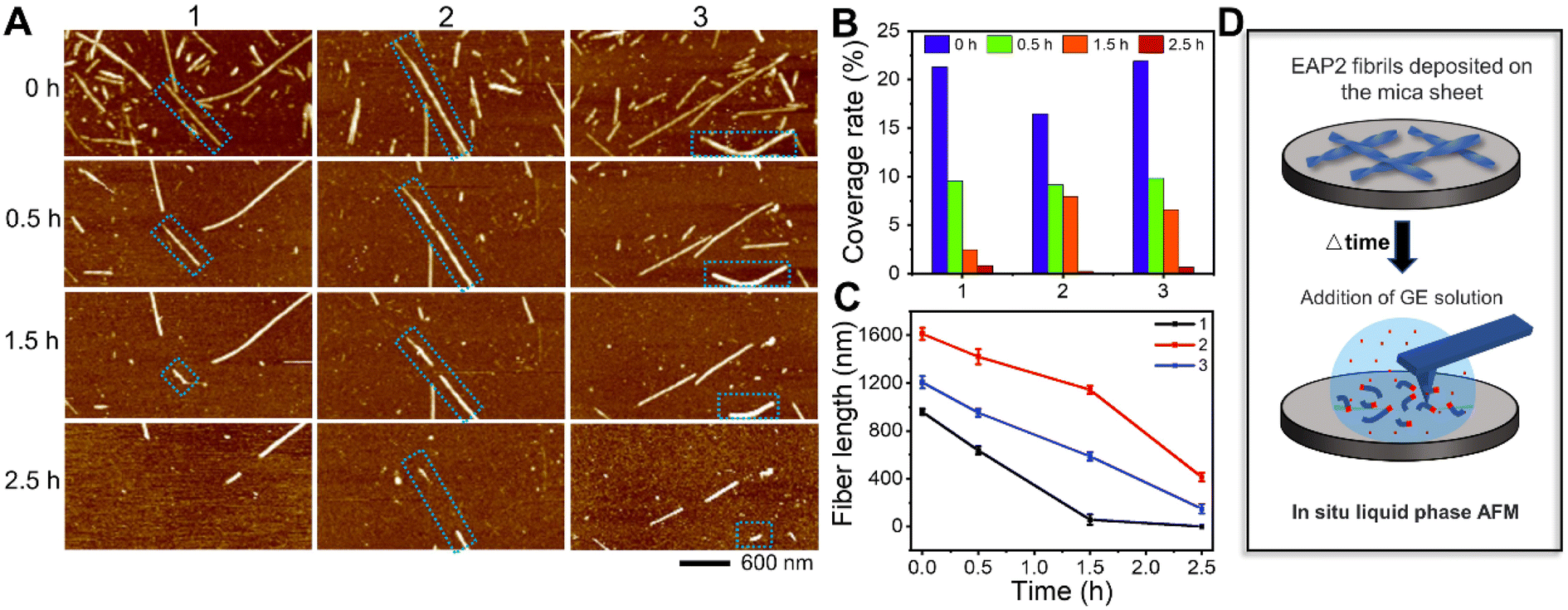 | ||
| Fig. 2 Characterization of the degradation of EAP2 fibrils in gelatinase solution by liquid-phase AFM (A) and changes in the coverage rate and length of fibrils with time (B and C). (D) In situ liquid-phase AFM for monitoring the degradation of EAP2 fibrils in gelatinase solution. | ||
Cryo-EM structures of the EAP2 fibril
To further explore the structural basis of the self-assembly of EAP2 into degradable fibrils, we carried out cryo-EM to determine the atomic structure of EAP2 fibrils. A sample of a EAP2 fibril was fixed on a carbon grid and frozen in liquid ethane. Then, cryo-EM data were collected using a direct detector (BioContinuum K3) on a cryo-transmission electron microscope (Titan Krios G4). We selected 58![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 888 fibrils derived from 4117 micrographs for the two-dimensional (2D) and 3D reconstruction of the EAP2 fibril (Fig. S3A and B†). Intriguingly, 2D classification revealed two major polymorphs of fibrils with a 980-pixel box size, which we termed EAP2 polymorph 1 (EAP2-P1) and EAP2 polymorph 2 (EAP2-P2). The proportion of EAP2-P1 and EAP2-P2 was ∼80.6%, and ∼10.2%, respectively.
888 fibrils derived from 4117 micrographs for the two-dimensional (2D) and 3D reconstruction of the EAP2 fibril (Fig. S3A and B†). Intriguingly, 2D classification revealed two major polymorphs of fibrils with a 980-pixel box size, which we termed EAP2 polymorph 1 (EAP2-P1) and EAP2 polymorph 2 (EAP2-P2). The proportion of EAP2-P1 and EAP2-P2 was ∼80.6%, and ∼10.2%, respectively.
Next, we reconstructed 3D density maps for the two polymorphs of the EAP2 fibril. The overall resolution of the density map for EAP2-P1 and EAP2-P2 was 2.4 Å and 2.6 Å, respectively (Fig. S6†). The EAP2-P1 featured a half pitch (one pitch represents a length of a 360° helical turn of the entire fibril) of 94 nm, a helical rise of 2.41 Å, and a helical twist of 179.54° (Fig. 3C). Similar to EAP2-P1, EAP2-P2 displayed a half pitch of 96 nm, a helical rise of 2.40 Å, and a twist of 179.55° (Fig. 3D). Moreover, both polymorphs shared the same left-handed twist and a pseudo-21 helical-screw symmetry (Fig. S3C and D†). The copy member, conformation, and spatial arrangement of EAP2 subunits in the two polymorphs were very different from each other. For each layer (cross-section) perpendicular to the fibril axis, EAP2-P1 consisted of 26 subunits of peptides, whereas EAP2-P2 contained 34 subunits of peptides (Fig. S3E, F and S7†).
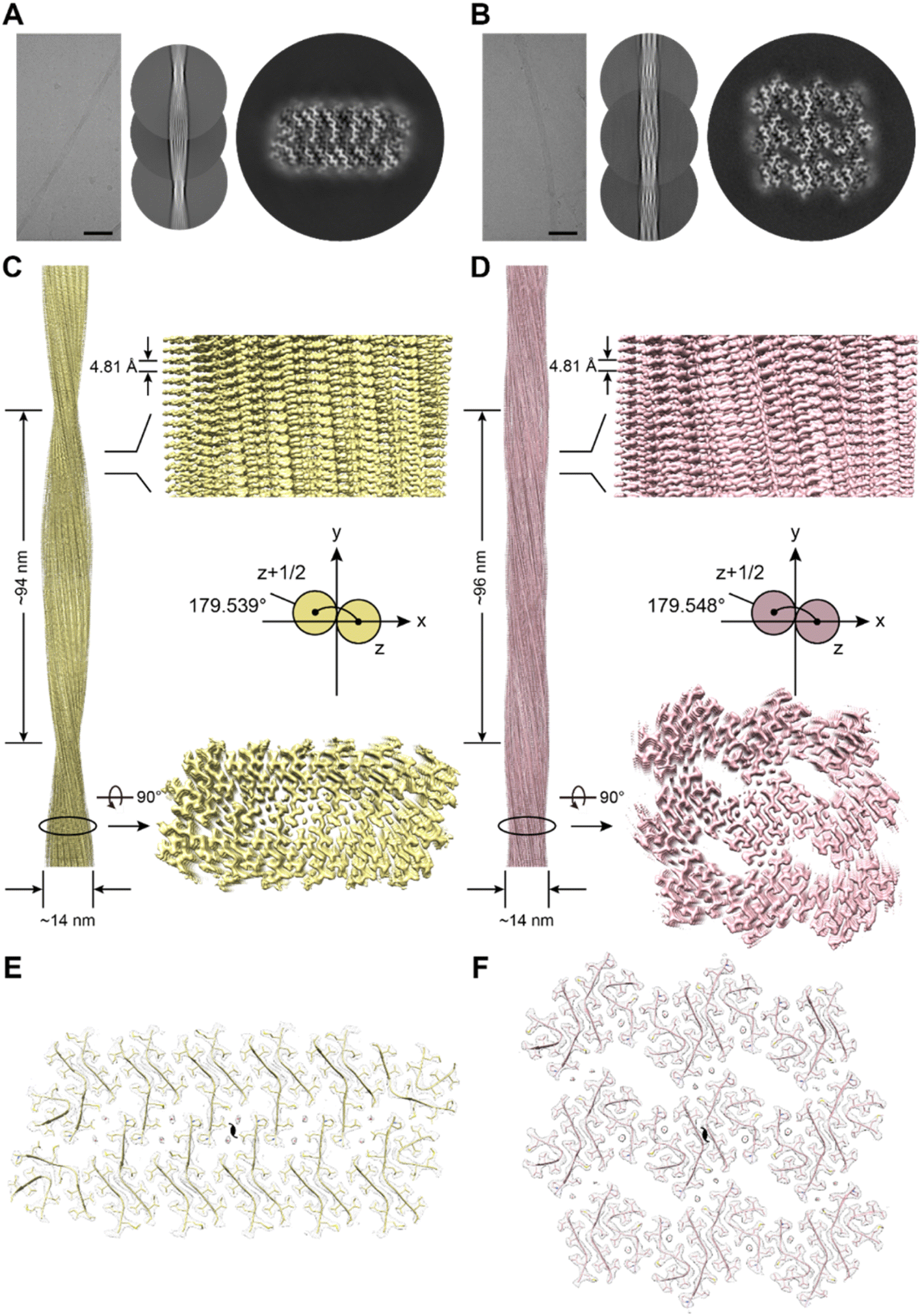 | ||
| Fig. 3 Structures of EAP2 fibril according to cryo-EM. (A and B) 2D and 3D structural reconstructions of two types of EAP2 fibrils by cryo-EM. (C and D) Density maps of the two EAP2 fibril polymorphs. EAP2-P1 is on the left, and EAP2-P2 is on the right. (E and F) Cross-sectional view of the two structural models fitted in the density maps of the two EAP2 fibril polymorphs. | ||
For EAP2-P1, peptides adopted three structural conformations, termed as subtype 1, 2, and 3 (Fig. 4A). Notably, the amyloidogenic segment –GVVIA and gelatinase-cleavage segment –LGML were directly involved in formation of the three subtypes in fibril spines. However, GVVIA displayed a β-strand conformation in all three subtypes, whereas LGML adopted a typical β-strand conformation only in subtype 1. Moreover, the N-terminal residues Gly, Pro, and Leu of EAP2 were highly flexible and were absent in the cryo-EM structure of subtype 2 and subtype 3. This finding strongly suggested that the two segments had distinct roles in the fibril formation of EAP2-P1. The three subtypes of the EAP2 monomer further assembled into three types of multimers: dimer 1 formed by subtype 1 and 2 of EAP2 monomers; dimer 2 formed by subtype 1 and 3 of EAP2 monomers; trimer 1 formed by three subtypes of EAP2 monomers (Fig. 4C).
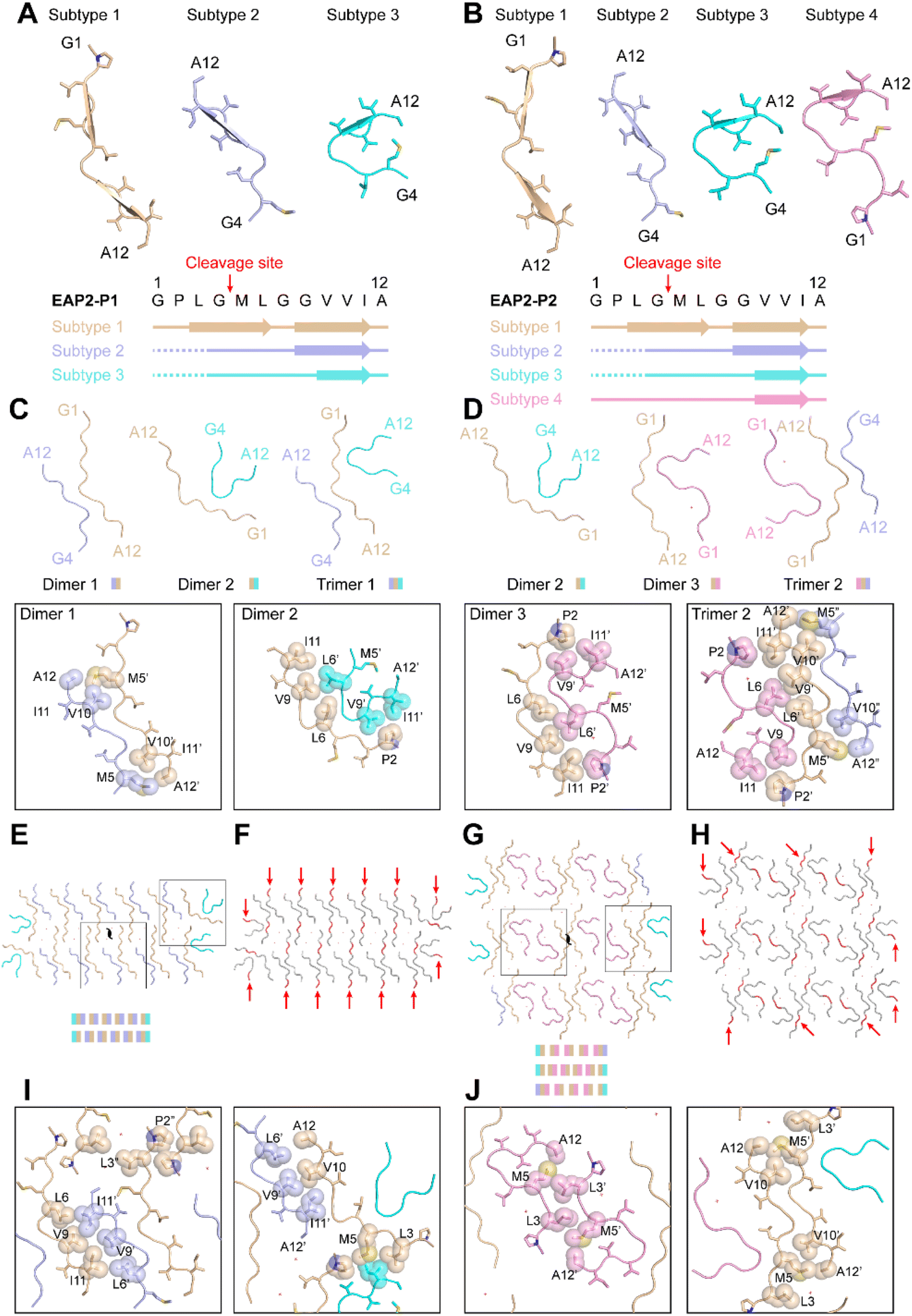 | ||
| Fig. 4 Structural analyses of EAP2 peptides in the fibril structures of EAP2-P1 and EAP2-P2. (A and B) Different structural conformations of the EAP2 subunit in EAP2-P1 and EAP2-P2 fibrils. Assignment of the secondary structure on different subtypes of EAP2 is shown on the bottom, with the gelatinase-cleavage site indicated by red arrows. (C and D) Dimeric and trimeric structures of EAP2 in two different fibrils. The side-chain interactions within different multimers are highlighted on the bottom. (E–H) Arrangement of different multimers within one cross-section of a fibril. Multiple gelatinase-cleavage sites are indicated by red arrows. Water molecules are marked by a red cross. The side-chain interactions between different multimers are highlighted in (I) and (J). | ||
The interfaces of these three multimers were mainly formed by hydrophobic interaction by residues Pro2, Met5, Leu6, Val9, and Val10 (Fig. 4C). As shown in Fig. 4E, eight copies of dimer 1, two copies of dimer 2, and two copies of trimer 1 jointly packed into the mature fibril spine with a two-fold symmetry in the center of the fibril structure. In addition, 10 water molecules were present within the interfaces of the adjacent dimer 1 (Fig. 4E). The interface between dimer 1 and dimer 1, dimer 1 and dimer 2, and dimer 1 and trimer 1 in the fibril spine structure was mainly formed by hydrophobic interaction (Fig. 4I). Notably, the major hydrophobic interface of the fibril spine was formed by the homotopical steric zipper of GVVIA segments of two neighboring molecules. The steric zipper structure recapitulated that in the full-length Aβ42 fibril-core structure (Fig. S8†). Importantly, the enzymatic cleavage sites of 14 copies out of 26 copies of EAP2 subunits were localized on the outer surface of the fibril spine with a highly flexible N-terminus flanking outside of the fibril, which was highly accessible for enzyme binding and cleavage on the EAP2 fibril (Fig. 4F).
With respect to the second polymorphic structure of the EAP2 fibril, EAP2-P2 contained four subtypes of the EAP2 monomer: subtypes 1–3 (which were highly similar to that of EAP2-P1) and subtype 4 (Fig. 4B). In the structure of subtype 4, the GPLGMLGGVVIA segment adopted a similar conformation to that of subtype 3, whereas the N-terminal residues Leu3, Gly4, Met5, and Leu6 formed an additional β-turn structure. The four subtypes assemble into three types of multimers: dimer 2 and dimer 3 (which were observed in EAP2-P1) and trimer 2. The latter was formed by subtypes 1, 2, and 4 via hydrophobic interaction (Fig. 4D). Moreover, three multimers (four copies of dimer 2, 10 copies of dimer 3, and two copies of trimer 2) packed into the fibril spin of EAP2-P2 (Fig. 4G). The detailed hydrophobic interactions of the interfaces of different multimers are shown in Fig. 4J. Twenty-four water molecules were found within the fibril spine (Fig. 4G). Moreover, the gelatinase cleavage sites of the 10 copies of the EAP2 monomer were distributed evenly on the surface of the EAP2-P2 fibril spine and accessible for enzyme binding (Fig. 4H).
Taken together, the cryo-EM study revealed that the EAP2 subunit could adopt several conformations which further assembled into two architectures of a fibril spine. Amyloidogenic and gelatinase cleavage-segments were involved in formation of the fibril core. Importantly, the amyloidogenic segment GVVIA mainly adopted a β-conformation in different subtypes and played a key part in mediating inter-subunit assembly of EAP2 via hydrophobic interaction. Gelatinase-cleavage segments were more flexible with distinct conformations in different subtypes, and were partially distributed on the outer surface of fibril spines. This structural arrangement made it highly accessible for gelatinase binding and degradation. This was especially true for the two open ends of the EAP2 fibril, where gelatinase could approach the enzyme cleavage-segment without steric hindrance from the neighboring segments packed in the fibril. Thus, the cryo-EM structure provided a molecular basis for EAP2 packing within fibrils as well as its binding and degradation by gelatinase. Furthermore, cryo-EM updated information on the multistage process of peptide self-assembly.40 Structural characteristics at high resolution were revealed. In addition, the roles of the amyloidogenic segment and enzyme-cleavage segment in the degradable EAP2 fibril were clarified.
Design of degradable self-assembled antimicrobial fibrils (SANs)
Next, we sought to apply the gelatinase-degraded fibril to construct smart antibacterial materials through physicochemical alterations caused by enzymes.41 The rationale was to design an antimicrobial fibril with an EAP2 fibril spine as a scaffold. The latter could eliminate bacteria and then be degraded automatically by the gelatinase secreted from bacteria to abolish its antimicrobial activity to avoid potential toxicity. This feedback loop could ensure spontaneous self-cleaning of antimicrobial fibrils once targeted bacteria had been eliminated.We fused an antimicrobial peptide (melittin (Mel)) to our designed EAP2 at the C-terminus with three glycine residues as a spacer in between them, which we named EAP2-Mel (Fig. S9†). An antimicrobial moeity could be a short peptide (i.e., Mel42,43) with an α-helical secondary structure, but not be able to self-assemble.44 EAP2-Mel was anticipated to self-assemble into fibrils with Mel flanking outside the fibril core. Like other short fibrillizing peptides45–47 and peptide-amphiphiles,48,49 the self-assembled peptides acted as adjustable scaffolds to display functional sequences or chemical groups onto the surface of their self-assembled β-sheet-rich nanofibers. As designed, EAP2-Mel could assemble spontaneously into fibrils, as characterized by AFM and TEM. More importantly, these fibrils could be degraded efficiently by gelatinase (Fig. S10A†). CD spectroscopy confirmed the secondary-structure conversion of peptides from β-sheets to random coils in SANs after degradation by gelatinase (Fig. S10B†). This finding strongly suggested that incorporation of Mel had a negligible effect on the fibrillation and degradation behavior of EAP2.
Next, we examined the antimicrobial activity and degradability of SANs in the presence of bacteria. To evaluate the antimicrobial activity, we measured the minimum inhibitory concentration (MIC) of Mel monomer, EAP2 fibrils, EAP2-Mel monomer, and SANs (Table 1). As shown in Fig. S11,† the EAP2-Mel monomer and SANs (Mel) exhibited superior antimicrobial activity against Gram-negative (Escherichia coli) and Gram-positive (Staphylococcus aureus) bacteria. Fusion of Mel to EAP reduced the antimicrobial activity of Mel according to the MIC. Nevertheless, they showed a comparable ability to kill bacteria according to SEM and staining experiments to identify live/dead bacteria. Significant destructive effects on bacterial cell membranes were detected, and most bacteria were killed within 2 h. These data indicated that Mel, EAP2-Mel, and SANs exhibited potent activity for killing bacteria, rather than EAP2 fibrils.
| Peptide | Gram-negative | Gram-positive | ||||
|---|---|---|---|---|---|---|
| E. coli | P. aeruginosa | S. typhi | M. luteus | B. subtilis | S. aureus | |
| EAP2 fibrils | >160 | >160 | >160 | >160 | >160 | >160 |
| Mel monomer | 10 | 5 | 40 | 5 | 2.5 | 2.5 |
| EAP2-Mel monomer | 40 | 20 | 80 | 20 | 5 | 5 |
| SANs | 40 | 40 | 20 | 10 | 5 | 5 |
Furthermore, a N-phenyl-1-naphthylamine (NPN) probe was used to evaluate the penetration level and extent of disruption of the outer membrane of bacteria. We monitored the enhanced fluorescence of NPN upon binding to the hydrophobic region of the disrupted outer membrane (see the Experimental section of the ESI†). Mel monomer, EAP2-Mel monomer, and SANs showed similar destructive effects on bacterial outer membranes. This destruction reached the same level gradually by increased intensity from low to high as time increased (Fig. 5A). The internal cytosolic β-galactosidase of bacteria metabolized ONPG to the chromophore ortho-nitrophenol (ONP) in the cytoplasm. This action resulted in obvious UV-vis absorption at 420 nm, which could be used to indicate ONPG intake when the inner membrane of bacteria was disrupted (see the Experimental section of the ESI†). In descending order, Mel monomer, EAP2-Mel monomer, and SANs could disrupt bacterial inner membranes, but the difference between them was not large (Fig. 5B). This order was very similar for their ability to destroy bacterial outer membranes. However, EAP2 fibrils had no destructive effects on bacterial outer or inner membranes. In general, SANs retained excellent antimicrobial activity and had a similar antibacterial mechanism to that of Mel peptides.
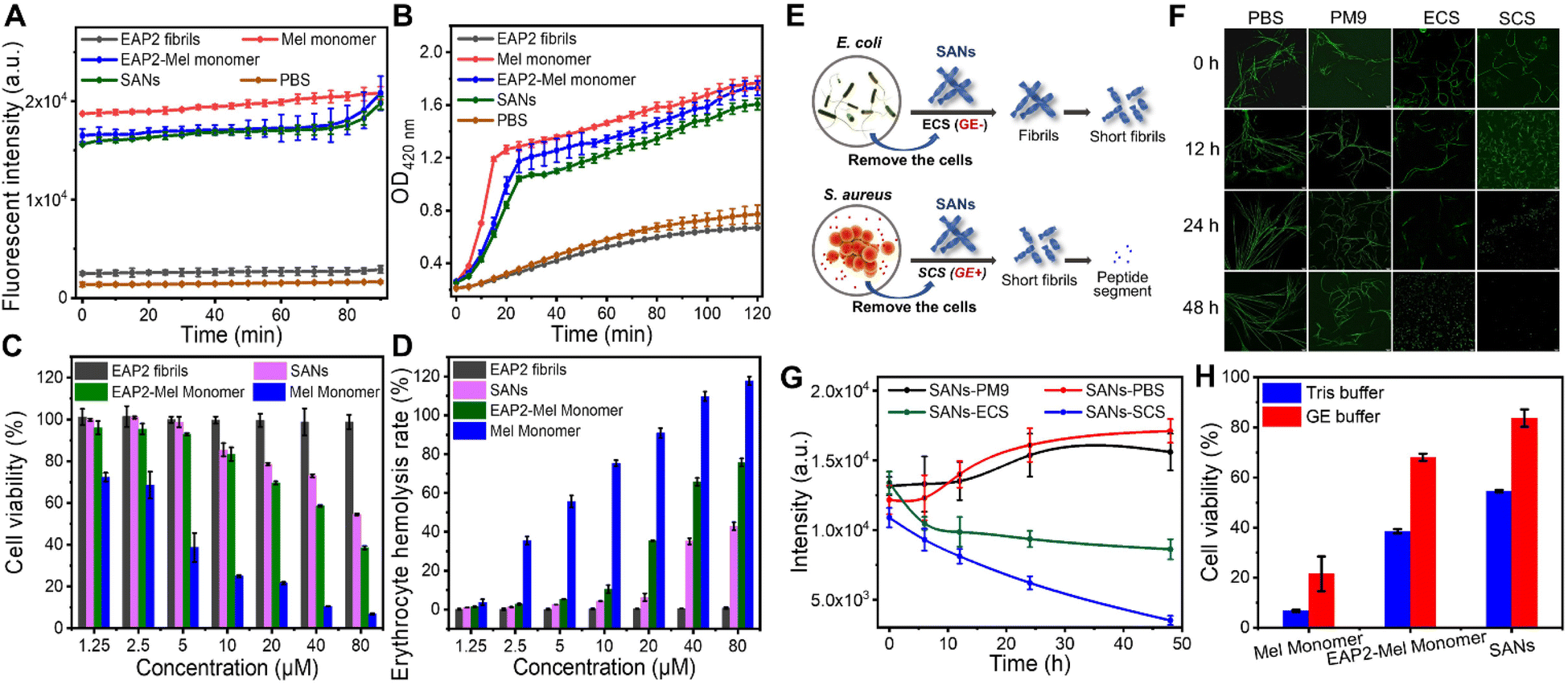 | ||
| Fig. 5 Verification of antimicrobial mechanism and biocompatibility of self-assembled antimicrobial fibrils formed by EAP2-Mel. (A and B) Effect of EAP2 fibrils, Mel monomer, EAP2-Mel monomer, SANs, and PBS on the E. coli outer-membrane (NPN fluorescence-labelled) and inner-membrane (ONPG intake) permeability. (C and D) Cytotoxicity and hemolytic properties of EAP2 fibrils, Mel monomer, EAP2-Mel monomer, and SANs. The degradation of SANs treated by the medium secreted by bacteria as well as the cytotoxicity and antibacterial properties of EAP2 fibrils, Mel monomer, EAP2-Mel monomer, and SANs after treatment by the medium secreted by E. coli and S. aureus. (E) Degradation of SANs fibrils in cell-free culture solutions of E. coli (ECS, GE−) and S. aureus (SCS, GE+). (F and G) Fluorescence images and intensity of SANs stained by ThT in ECS (GE−), SCS (GE+), Tris buffer, or PM9 solution. (H) Cytotoxicity comparison of Mel monomer, EAP2-Mel monomer, and SANs, all at 80 μM, treated by gelatinase and Tris buffer for 24 h. | ||
Next, to decipher the antibacterial mechanism of action of our designed SANs, we examined Ca2+ loss in bacteria.50 The intracellular Ca2+ concentration of bacteria treated by Mel monomer, EAP2-Mel monomer, and SANs was measured. The residual Ca2+ concentration of bacteria decreased markedly and was significantly lower than that of control bacteria (p < 0.0001) (Fig. S12†), indicating that Ca2+ efflux was induced by antimicrobial peptides or SANs. The developed SANs showed a comparable effect of Mel on the Ca2+ homeostasis of bacteria, suggesting that they had a common antibacterial mechanism. In general, the designed SANs displayed an antimicrobial activity mainly due to the Mel moiety. This mechanism is different to that of 2D nanomaterials, which act by physical penetrations, or producing oxidative-stress reactions through physicochemical interactions with microbes. Also, peptide nanotubes can upregulate expression of genes to break bacterial cell membranes.51,52
With regard to potential clinical applications, cytotoxicity is a major concern for most natural and synthetic AMPs and antimicrobial materials.53 Based on our results from membrane-permeation assays, we hypothesized that the structural constraints and partial shielding of SANs on Mel might alleviate toxicity towards mammalian cells. We undertook in vitro cytotoxicity assays for Mel monomer, EAP2 fibrils, EAP2-Mel, and SANs towards mouse fibroblast (L929) cells. As shown in Fig. 5C, all Mel-grafted SANs demonstrated greatly reduced cytotoxicity compared with that of free Mel, and the difference was much more obvious at higher Mel doses. Hemocompatibility was assessed by incubating mouse red blood cells with different concentrations of Mel, EAP2-Mel, and SANs for 1 h, and the hemoglobin released was measured by UV spectroscopy (Fig. 5D). The percentage of hemolyzed cells due to the EAP2-Mel monomer and SANs was much lower than that of Mel monomer, and met the requirement of <5% for percent hemolysis. In comparison, EAP2 fibrils as scaffolds also exhibited low cytotoxicity and percentage of hemolyzed cells.
Degradation of SANs in the bacterial environment
Finally, we investigated SANS degradation in a bacterial environment in which gelatinase could be secreted efficiently from bacteria as an extracellular enzyme. Remarkably, SANs could be degraded into short fibrils in a S. aureus culture solution (SCS) with gelatinase (GE+) within 12 h, and degraded completely within 48 h. In comparison, the SANs fibril was not degraded readily in E. coli culture solution (ECS) without gelatinase (GE−) in 24 h. We examined the fluorescence images of SANs stained by ThT and measured the mean fluorescence intensity (MFI) of SANs via the ThT assay (Fig. S5E–G†). The MFI decreased dramatically and linearly with the time for SANs in SCS (GE+), but not for SANs in ECS (GE−). As a control, we examined the degradation performance of SANs in phosphate-buffered saline and PM9 medium solution used for culturing S. aureus and E. coli: they could not be degraded within 48 h. These results showed that the designed SANs could be degraded efficiently in the presence of gelatinase-secreting bacteria.The antimicrobial activity and toxicity of degraded SANs were examined further. Importantly, SANs treated by gelatinase decreased the cytotoxicity significantly compared with those that did not receive enzyme treatment (Fig. 5H). In contrast, Mel monomer continued to display the lower cell viability. MIC calculations for various Gram-positive and Gram-negative bacteria showed that degraded SANs almost lost their antibacterial activities and stopped working, and the performance of the EAP2-Mel monomer also decreased significantly, but the activity of Mel was maintained (Table 2). Therefore, the degradation effect of the enzyme on the antimicrobial performance and cytotoxicity of as-constructed materials was verified. The time needed to kill bacteria by SANs was much shorter than that of the degradation of SANs. However, this strategy worked well because most gelatinase had been secreted from bacteria present in the microenvironment, and could act on SANs even if bacteria had been killed. Killing bacteria by SANs in a short time could also make disinfection efficient before SANs are degraded over a relatively long time.
| Peptide | Gram-negative | Gram-positive | ||||
|---|---|---|---|---|---|---|
| E. coli | P. aeruginosa | S. typhi | M. luteus | B. subtilis | S. aureus | |
| Mel monomer | 20 | 20 | 40 | 20 | 10 | 10 |
| EAP2-Mel monomer | >160 | >160 | >160 | >160 | 160 | 160 |
| SANs | >160 | >160 | >160 | >160 | >160 | >160 |
In summary, we constructed an enzyme-controlled degradable peptide-based fibril system. Then, we applied it to develop degradable antimicrobial nanomaterials that could kill bacteria. We demonstrated that the designed fibrils could kill bacteria in a short time and were degraded automatically by the already secreted gelatinase of bacteria, thereby maximizing the biosafety of these nanomaterials (Fig. 6). This feedback-loop strategy could be used to develop biocompatible, self-cleaning, smart nanomaterials with different biological activities. We demonstrated one application of this enzyme-controlled degradable peptide-based fibril system. More bio-inspired smart peptide-based nanosystems will be designed and developed in our future work. Our data pave the way for developing smart peptide-based nanomaterials for different biomedical and clinical applications, such as smart biosensing and drug delivery.
 | ||
| Fig. 6 Design of an antimicrobial peptide fibril under feedback-loop regulation (schematic). The rational design of EAP fibrils and EAP-based antimicrobial fibrils, and their application in killing bacteria and auto-degradation by an enzyme with a feedback loop, are shown. | ||
Materials and methods
The materials and methods section is available in the ESI.†Data availability
Cryo-EM 3D density maps of EAP2 fibrils have been deposited in the Electron Microscopy Data Bank with entry codes of EMD-34392 for EAP2 polymorph 1 and EMD-34393 for EAP2 polymorph 2. The corresponding structural models have been deposited in the Worldwide Protein Data Bank with entry codes of 8GZ8 for EAP2 polymorph 1 and 8GZ9 for EAP2 polymorph 2. Additional data that support the findings of this study are available from the corresponding authors upon reasonable request.Author contributions
The manuscript was written through the contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
We thank the Cryo-EM Microscopy Center at Interdisciplinary Research Center on Biology and Chemistry, Shanghai Institute of Organic Chemistry, for help with data collection. This work was supported financially by the National Natural Science Foundation of China (22072060, 21573097, 82188101, 32170683, 31872716 and 32171236), Science and Technology Commission of Shanghai Municipality (20XD1425000, 2019SHZDZX02 and 22JC1410400), and Shanghai Pilot Program for Basic Research-Chinese Academy of Science, Shanghai Branch (CYJ-SHFY-2022-005).References
- H. He, J. Guo, X. Lin and B. Xu, Enzyme-Instructed Assemblies Enable Mitochondria Localization of Histone H2B in Cancer Cells, Angew Chem. Int. Ed. Engl., 2020, 59, 9330–9334 Search PubMed.
- C. Liang, Z. Wang, T. Xu, Y. Chen, D. Zheng, L. Zhang, W. Zhang, Z. Yang, Y. Shi and J. Gao, Preorganization Increases the Self-Assembling Ability and Antitumor Efficacy of Peptide Nanomedicine, ACS Appl. Mater. Interfaces, 2020, 12, 22492–22498 Search PubMed.
- T. Xu, Y. Cai, X. Zhong, L. Zhang, D. Zheng, Z. Gao, X. Pan, F. Wang, M. Chen and Z. Yang, β-Galactosidase instructed supramolecular hydrogelation for selective identification and removal of senescent cells, Chem. Commun., 2019, 55, 7175–7178 Search PubMed.
- Z. Y. Qiao, C. Y. Hou, D. Zhang, Y. Liu, Y. X. Lin, H. W. An, X. J. Li and H. Wang, Self-assembly of cytotoxic peptide conjugated poly(β-amino ester)s for synergistic cancer chemotherapy, J. Mater. Chem. B, 2015, 3, 2943–2953 Search PubMed.
- Z. Y. Qiao, W. J. Zhao, Y. J. Gao, Y. Cong, L. Zhao, Z. Hu and H. Wang, Reconfigurable Peptide Nanotherapeutics at Tumor Microenvironmental pH, ACS Appl. Mater. Interfaces, 2017, 9, 30426–30436 Search PubMed.
- K. Zhang, P. P. Yang, P. P. He, S. F. Wen, X. R. Zou, Y. Fan, Z. M. Chen, H. Cao, Z. Yang, K. Yue, X. Zhang, H. Zhang, L. Wang and H. Wang, Peptide-Based Nanoparticles Mimic Fibrillogenesis of Laminin in Tumor Vessels for Precise Embolization, ACS Nano, 2020, 14, 7170–7180 Search PubMed.
- Y. Cong, L. Ji, Y. J. Gao, F. H. Liu, D. B. Cheng, Z. Hu, Z. Y. Qiao and H. Wang, Microenvironment-Induced In Situ Self-Assembly of Polymer-Peptide Conjugates That Attack Solid Tumors Deeply, Angew Chem. Int. Ed. Engl., 2019, 58, 4632–4637 Search PubMed.
- D. B. Cheng, X. H. Zhang, Y. J. Gao, L. Ji, D. Hou, Z. Wang, W. Xu, Z. Y. Qiao and H. Wang, Endogenous Reactive Oxygen Species-Triggered Morphology Transformation for Enhanced Cooperative Interaction with Mitochondria, J. Am. Chem. Soc., 2019, 141, 7235–7239 Search PubMed.
- Y. Fan, X. D. Li, P. P. He, X. X. Hu, K. Zhang, J. Q. Fan, P. P. Yang, H. Y. Zheng, W. Tian, Z. M. Chen, L. Ji, H. Wang and L. Wang, A biomimetic peptide recognizes and traps bacteria in vivo as human defensin-6, Sci. Adv., 2020, 6, eaaz4767 Search PubMed.
- X. X. Hu, P. P. He, G. B. Qi, Y. J. Gao, Y. X. Lin, C. Yang, P. P. Yang, H. Hao, L. Wang and H. Wang, Transformable Nanomaterials as an Artificial Extracellular Matrix for Inhibiting Tumor Invasion and Metastasis, ACS Nano, 2017, 11, 4086–4096 Search PubMed.
- B. N. Li, P. P. He, P. P. Yang, J. P. Zhang, L. Wang and H. Wang, In situ construction of nanonetworks from transformable nanoparticles for anti-angiogenic therapy, J. Mater. Chem. B, 2018, 6, 5282–5289 Search PubMed.
- H. Wang, Z. Feng, W. Tan and B. Xu, Assemblies of d-Peptides for Targeting Cell Nucleolus, Bioconjugate Chem., 2019, 30, 2528–2532 Search PubMed.
- Q. Cai, Y. Fei, L. Hu, Z. Huang, L. L. Li and H. Wang, Chemotaxis-Instructed Intracellular Staphylococcus aureus Infection Detection by a Targeting and Self-Assembly Signal-Enhanced Photoacoustic Probe, Nano Lett., 2018, 18, 6229–6236 Search PubMed.
- X. X. Zhao, L. L. Li, Y. Zhao, H. W. An, Q. Cai, J. Y. Lang, X. X. Han, B. Peng, Y. Fei, H. Liu, H. Qin, G. Nie and H. Wang, In Situ Self-Assembled Nanofibers Precisely Target Cancer-Associated Fibroblasts for Improved Tumor Imaging, Angew Chem. Int. Ed. Engl., 2019, 58, 15287–15294 Search PubMed.
- E. B. Peters, N. D. Tsihlis, M. R. Karver, S. M. Chin, B. Musetti, B. T. Ledford, E. M. Bahnson, S. I. Stupp and M. R. Kibbe, Atheroma Niche-Responsive Nanocarriers for Immunotherapeutic Delivery, Adv. Healthcare Mater., 2019, 8, e1801545 Search PubMed.
- D. B. Cheng, X. H. Zhang, Y. J. Gao, D. Wang, L. Wang, H. Chen, Z. Y. Qiao and H. Wang, Site-Specific Construction of Long-Term Drug Depot for Suppression of Tumor Recurrence, Small, 2019, 15, e1901813 Search PubMed.
- D. B. Cheng, D. Wang, Y. J. Gao, L. Wang, Z. Y. Qiao and H. Wang, Autocatalytic Morphology Transformation Platform for Targeted Drug Accumulation, J. Am. Chem. Soc., 2019, 141, 4406–4411 Search PubMed.
- G. B. Qi, D. Zhang, F. H. Liu, Z. Y. Qiao and H. Wang, An On-Site Transformation Strategy for Treatment of Bacterial Infection, Adv. Mater., 2017, 29, 1703461 Search PubMed.
- A. P. Xu, P. P. Yang, C. Yang, Y. J. Gao, X. X. Zhao, Q. Luo, X. D. Li, L. Z. Li, L. Wang and H. Wang, Bio-inspired metal ions regulate the structure evolution of self-assembled peptide-based nanoparticles, Nanoscale, 2016, 8, 14078–14083 Search PubMed.
- Z. Wang, H. W. An, D. Hou, M. Wang, X. Zeng, R. Zheng, L. Wang, K. Wang, H. Wang and W. Xu, Addressable Peptide Self-Assembly on the Cancer Cell Membrane for Sensitizing Chemotherapy of Renal Cell Carcinoma, Adv. Mater., 2019, 31, e1807175 Search PubMed.
- X. Liu, M. Li, J. Liu, Y. Song, B. Hu, C. Wu, A. A. Liu, H. Zhou, J. Long, L. Shi and Z. Yu, In Situ Self-Sorting Peptide Assemblies in Living Cells for Simultaneous Organelle Targeting, J. Am. Chem. Soc., 2022, 144, 9312–9323 Search PubMed.
- F. Peng, M. I. Setyawati, J. K. Tee, X. Ding, J. Wang, M. E. Nga, H. K. Ho and D. T. Leong, Nanoparticles Promote in Vivo Breast Cancer Cell Intravasation and Extravasation by Inducing Endothelial Leakiness, Nat. Nanotechnol., 2019, 14, 279–286 Search PubMed.
- B. Wang, X. He, Z. Zhang, Y. Zhao and W. Feng, Metabolism of Nanomaterials in Vivo: Blood Circulation and Organ Clearance, Acc. Chem. Res., 2013, 46, 761–769 Search PubMed.
- F. Chiti and C. M. Dobson, Protein Misfolding, Functional Amyloid, and Human Disease, Annu. Rev. Biochem., 2006, 75, 333–366 Search PubMed.
- F. Chiti and C. M. Dobson, Protein Misfolding, Amyloid Formation, and Human Disease: A Summary of Progress Over the Last Decade, Annu. Rev. Biochem., 2017, 86, 27–68 Search PubMed.
- P. Ciryam, G. G. Tartaglia, R. I. Morimoto, C. M. Dobson and M. Vendruscolo, Widespread Aggregation and Neurodegenerative Diseases are Associated with Supersaturated Proteins, Cell Rep., 2013, 5, 781–790 Search PubMed.
- C. M. Dobson, Protein Folding and Misfolding, Nature, 2003, 426, 884–890 Search PubMed.
- D. A. White, A. K. Buell, T. P. Knowles, M. E. Welland and C. M. Dobson, Protein Aggregation in Crowded Environments, J. Am. Chem. Soc., 2010, 132, 5170–5175 Search PubMed.
- W. E. Balch, R. I. Morimoto, A. Dillin and J. W. Kelly, Adapting Proteostasis for Disease Intervention, Science, 2008, 319, 916–919 Search PubMed.
- N. Iwata, S. Tsubuki, Y. Takaki, K. Shirotani, B. Lu, N. P. Gerard, C. Gerard, E. Hama, H. J. Lee and T. C. Saido, Metabolic Regulation of Brain Abeta by Neprilysin, Science, 2001, 292, 1550–1552 Search PubMed.
- S. Li and Y. Yao, Proteolysis in Bacteria-A Review, Acta Microbiol. Sin., 2015, 55, 521–528 Search PubMed.
- S. A. Mahmoud and P. Chien, Regulated Proteolysis in Bacteria, Annu. Rev. Biochem., 2018, 87, 677–696 Search PubMed.
- M. C. Giano, D. J. Pochan and J. P. Schneider, Controlled biodegradation of self-assembling β-hairpin peptide hydrogels by proteolysis with matrix metalloproteinase-13, Biomaterials, 2011, 32, 6471–6477 Search PubMed.
- X. Li, J. Li, Y. Gao, Y. Kuang, J. Shi and B. Xu, Molecular nanofibers of olsalazine form supramolecular hydrogels for reductive release of an anti-inflammatory agent, J. Am. Chem. Soc., 2010, 132, 17707–17709 Search PubMed.
- M. J. Webber, C. J. Newcomb, R. Bitton and S. I. Stupp, Switching of Self-Assembly in a Peptide Nanostructure with a Specific Enzyme, Soft Matter, 2011, 7, 9665–9672 Search PubMed.
- A. S. Duarte, A. Correia and A. C. Esteves, Bacterial collagenases - A review, Crit. Rev. Microbiol., 2016, 42, 106–126 Search PubMed.
- L. L. Li, G. B. Qi, F. Yu, S. J. Liu and H. Wang, An adaptive biointerface from self-assembled functional peptides for tissue engineering, Adv. Mater., 2015, 27, 3181–3188 Search PubMed.
- L. Liu, Q. Li, S. Zhang, X. Wang, S. V. Hoffmann, J. Li, Z. Liu, F. Besenbacher and M. Dong, Identification of a Novel Parallel β-Strand Conformation within Molecular Monolayer of Amyloid Peptide, Adv. Sci., 2016, 3, 1500369 Search PubMed.
- X. Wang, J. K. Weber, L. Liu, M. Dong, R. Zhou and J. Li, A novel form of β-strand assembly observed in Aβ33–42 adsorbed onto graphene, Nanoscale, 2015, 7, 15341–15348 Search PubMed.
- Y. Song, B. Dai, Y. Wang, C. Liu, P. Gourdon, L. Liu, K. Wang and M. Dong, Identifying Heterozipper β-Sheet in Twisted Amyloid Aggregation, Nano Lett., 2022, 22(9), 3707–3712 Search PubMed.
- J. Y. Quek, E. Uroro, N. Goswami and K. Vasilev, Design principles for bacteria-responsive antimicrobial nanomaterials, Mater. Today Chem., 2022, 23, 100606 Search PubMed.
- J. Hong, X. Lu, Z. Deng, S. Xiao, B. Yuan and K. Yang, How Melittin Inserts into Cell Membrane: Conformational Changes, Inter-Peptide Cooperation, and Disturbance on the Membrane, Molecules, 2019, 24, 1775 Search PubMed.
- L. Ramirez, A. Shekhtman and J. Pande, Nuclear Magnetic Resonance-Based Structural Characterization and Backbone Dynamics of Recombinant Bee Venom Melittin, Biochemistry, 2018, 57, 2775–2785 Search PubMed.
- L. C. Dorman and L. D. Markley, Solid phase synthesis and antibacterial activity of N-terminal sequences of melittin, J. Med. Chem., 1971, 14, 5–9 Search PubMed.
- A. Horii, X. Wang, F. Gelain and S. Zhang, Biological designer self-assembling peptide nanofiber scaffolds significantly enhance osteoblast proliferation, differentiation and 3-D migration, PLoS One, 2007, 2, e190 Search PubMed.
- S. L. Gras, A. K. Tickler, A. M. Squires, G. L. Devlin, M. A. Horton, C. M. Dobson and C. E. MacPhee, Functionalised amyloid fibrils for roles in cell adhesion, Biomaterials, 2008, 29, 1553–1562 Search PubMed.
- J. S. Rudra, Y. F. Tian, J. P. Jung and J. H. Collier, A self-assembling peptide acting as an immune adjuvant, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 622–627 Search PubMed.
- V. M. Tysseling-Mattiace, V. Sahni, K. L. Niece, D. Birch, C. Czeisler, M. G. Fehlings, S. I. Stupp and J. A. Kessler, Self-assembling nanofibers inhibit glial scar formation and promote axon elongation after spinal cord injury, J. Neurosci., 2008, 28, 3814–3823 Search PubMed.
- J. D. Hartgerink, E. Beniash and S. I. Stupp, Self-assembly and mineralization of peptide-amphiphile nanofibers, Science, 2001, 294, 1684–1688 Search PubMed.
- X. Sha, P. Li, Y. Feng, D. Xia, X. Tian, Z. Wang, Y. Yang, X. Mao and L. Liu, Self-Assembled Peptide Nanofibrils Designed to Release Membrane-Lysing Antimicrobial Peptides, ACS Appl. Bio Mater., 2020, 3, 3648–3655 Search PubMed.
- B. Li, Y. Luo, Y. Zheng, X. Liu, L. Tan and S. Wu, Two-dimensional antibacterial materials, Prog. Mater. Sci., 2022, 130, 100976 Search PubMed.
- L. Schnaider, S. Brahmachari, N. W. Schmidt, B. Mensa, S. Shaham-Niv, D. Bychenko, L. Adler-Abramovich, L. J. W. Shimon, S. Kolusheva, W. F. DeGrado and E. Gazit, Self-assembling Dipeptide Antibacterial Nanostructures with Membrane Disrupting Activity, Nat. Commun., 2017, 8, 1365 Search PubMed.
- W. Aoki, K. Kuroda and M. Ueda, Next generation of antimicrobial peptides as molecular targeted medicines, J. Biosci. Bioeng., 2012, 114, 365–370 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sc01089a |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2023 |