
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMasked cerulenin enables a dual-site selective protein crosslink†
Ziran
Jiang
 a,
Aochiu
Chen
a,
Jeffrey
Chen
a,
Arman
Sekhon
a,
Gordon V.
Louie
b,
Joseph P.
Noel
b,
James J.
La Clair
a and
Michael D.
Burkart
*a
a,
Aochiu
Chen
a,
Jeffrey
Chen
a,
Arman
Sekhon
a,
Gordon V.
Louie
b,
Joseph P.
Noel
b,
James J.
La Clair
a and
Michael D.
Burkart
*a
aDepartment of Chemistry and Biochemistry, University of California San Diego, La Jolla, CA 92093-0358, USA. E-mail: mburkart@ucsd.edu
bThe Salk Institute for Biological Studies, Jack H. Skirball Center for Chemical Biology and Proteomics, La Jolla, CA 92037, USA
First published on 8th September 2023
Abstract
Protein-reactive natural products such as the fungal metabolite cerulenin are recognized for their value as therapeutic candidates, due to their ability to selectively react with catalytic residues within a protein active site or a complex of protein domains. Here, we explore the development of fatty-acid and polyketide-synthase probes by synthetically modulating cerulenin's functional moieties. Using a mechanism-based approach, we reveal unique reactivity within cerulenin and adapt it for fluorescent labeling and crosslinking of fatty-acid and iterative type-I polyketide synthases. We also describe two new classes of silylcyanohydrin and silylhemiaminal masked crosslinking probes that serve as new tools for activity and structure studies of these biosynthetic pathways.
Introduction
First discovered in 1960 from cultures of Cephalosporium caerulens,1 (2R,3S)-2,3,epoxy-4-oxo-7,10-dodecadienoyl amide, or cerulenin (Fig. 1a), was one of the first antibiotics identified to target fatty acid biosynthesis.2 Its activity arises through covalent modification of ketosynthase (KS) domains in fatty acid synthases (FASs) and polyketide synthases (PKSs).3 Initial identification along with synthetic4–7 and medicinal chemistry8–10 revealed a set of structure–activity relationships (SARs)11 resulting in covalent modification of the active site cysteine residues in FAS and PKS KSs.12,13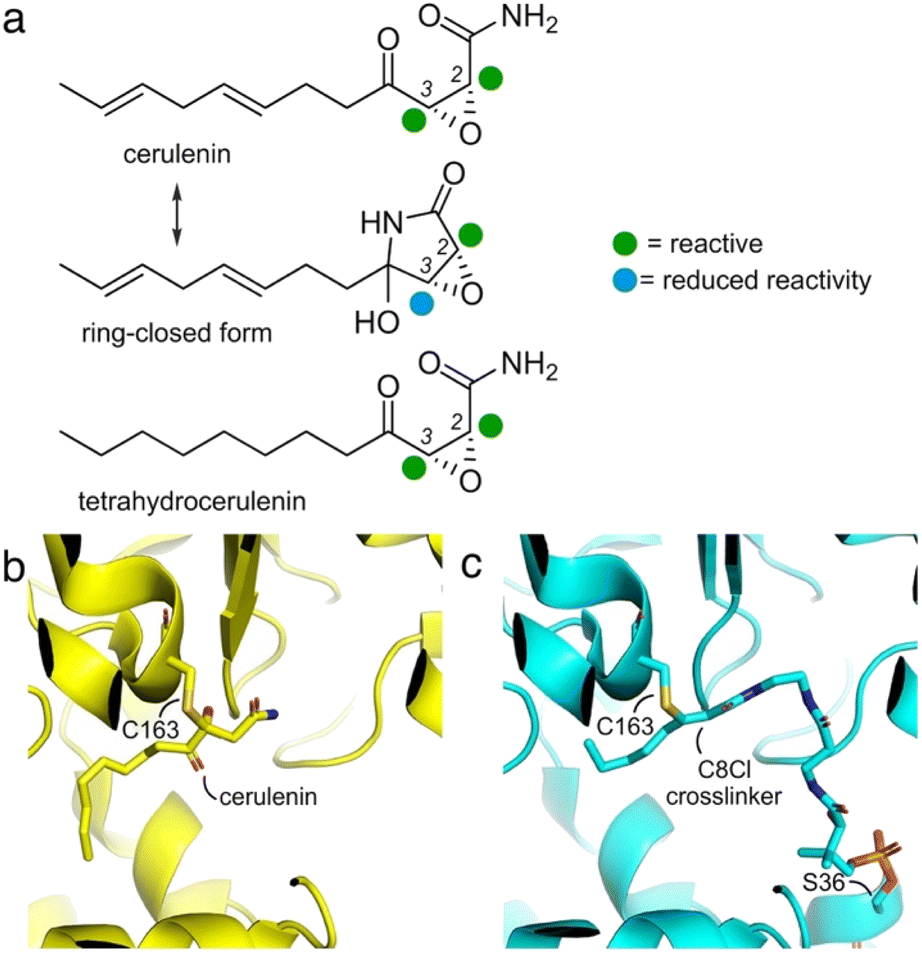 | ||
| Fig. 1 Cerulenin and ketosynthase inhibition. (a) Cerulenin exists as a mixture of ring-open and ring-closed tautomers through the formation of a hemiaminal. Colored spheres indicate suggested reactivity at each of the two-epoxide centers. Recent studies by Hansen and Rovira et al. have provided methods to predict the electrophilicity of three-membered heterocycles.24 While accurate for many epoxides, the additional α-keto and α-amido substituents further complicate the ability to provide effective calculations of the relative electrophilicity of the C2 and C3 positions in cerulenin, and these spheres represent suggestions. An electrostatic map of the cerulenin tautomers have been provided in ESI Fig. S1.† (b) Structures of cerulenin-bound FabF at C163 (PDB: 4LS8) and (c) crosslinked complex of FabF (via C163) and the E. coli acyl carrier protein AcpP (via S36) (PDB: 7L4L), demonstrating how cerulenin mimics native substrate binding.18–21 The structure in (c) was obtained by a C8-chloroacrylate crosslinker (C8Cl). Expansions of (b) and (c) are available in ESI Fig. S2.† | ||
As illustrated in Fig. 1a, two electrophilic positions reside in each of the linear and ring-closed tautomers of cerulenin.14 Three of these positions (green, Fig. 1a) are predicted to be more reactive due to their positioning adjacent to a carbonyl moiety; however, selective tuning of reactivity, except with small thiols,15–17 has yet to be demonstrated. With the fatty-acid biosynthetic enzyme FabF (β-ketoacyl-acyl carrier protein synthase) from E. coli, structural comparisons of cerulenin-bound FabF (PDB: 4LS8) with a recently elucidated crosslinked complex FabF = AcpP (PDB: 7L4L) (Fig. 1b and c) demonstrate how cerulenin mimics positioning of the native AcpP-bound substrate.18–21
Fundamental studies by Ōmura and Kawaguchi et al. to understand cerulenin reactivity used NMR analyses of adducts with free cysteine in aqueous buffers, identifying regioselective thiol attack at C2.22 Similar NMR methods were also used to demonstrate that the linear tautomer was favored in aprotic solvents, while the ring closed tautomer was favored in protic media.23
Later, structural studies of cerulenin reactivity reported in multiple deposited crystal structures of cerulenin-bound KS complexes further exemplified the mechanism of cerulenin structure–activity relationships (SARs). Although these reported high-resolution protein–cerulenin complex structures include protein covalent complexes, selectivity does not appear to be uniform. For instance, Bacillus subtilis FabF (PDB ID: 4LS8, 2.1 Å)21 captured in covalent complex with cerulenin (Fig. 1b) showed a covalent linkage with the active site Cys at C3, while E. coli FabB (PDB ID: 1FJ8, 2.3 Å)15 shows bond formation at C2.
Although the reacted linear tautomer of cerulenin (Fig. 1a) and unreacted linear tautomer in crystal soaking study were observed in these KS structures, the reacted or unreacted ring-closed tautomer known to predominate in aqueous media was not observed within KS pockets.14,15,21 These structures have identified a complexity within the regioselection at C2 or C3. Current hypotheses suggest nucleophilic addition of Cys thiol side chains within KS pockets to C2, due to the masking of the C3 position in the aqueous-favored, ring-closed tautomer. However, solution-based reactivity may not recapitulate the environment of an enzyme pocket, where local effects may result in enhanced tautomerization to the linear form thus enabling both C2 and C3 reactivity. Here, we describe how enzyme active site reactivity can be exploited to tune KS reactive probes and crosslinking agents.
Results and discussion
Probe design and synthesis
We first compared the structure of cerulenin-bound KS domains (cerulenin·KS) and the structures of E. coli KS domains crosslinked to their associated acyl carrier protein, AcpP (AcpP = KS). As shown in Fig. 1b and c and ESI Fig. S2,† the crosslinking arm in AcpP = KS overlaps with the structure of KS bound cerulenin (cerulenin·KS), suggesting that cerulenin may serve as a reactive probe and crosslinker suitable for further development. We next designed a series of motifs 1–10 (Fig. 2), where the reactivity of 2–10 would likely arise from a single position within a single tautomer (C2 in Fig. 1a) and began with motif 1.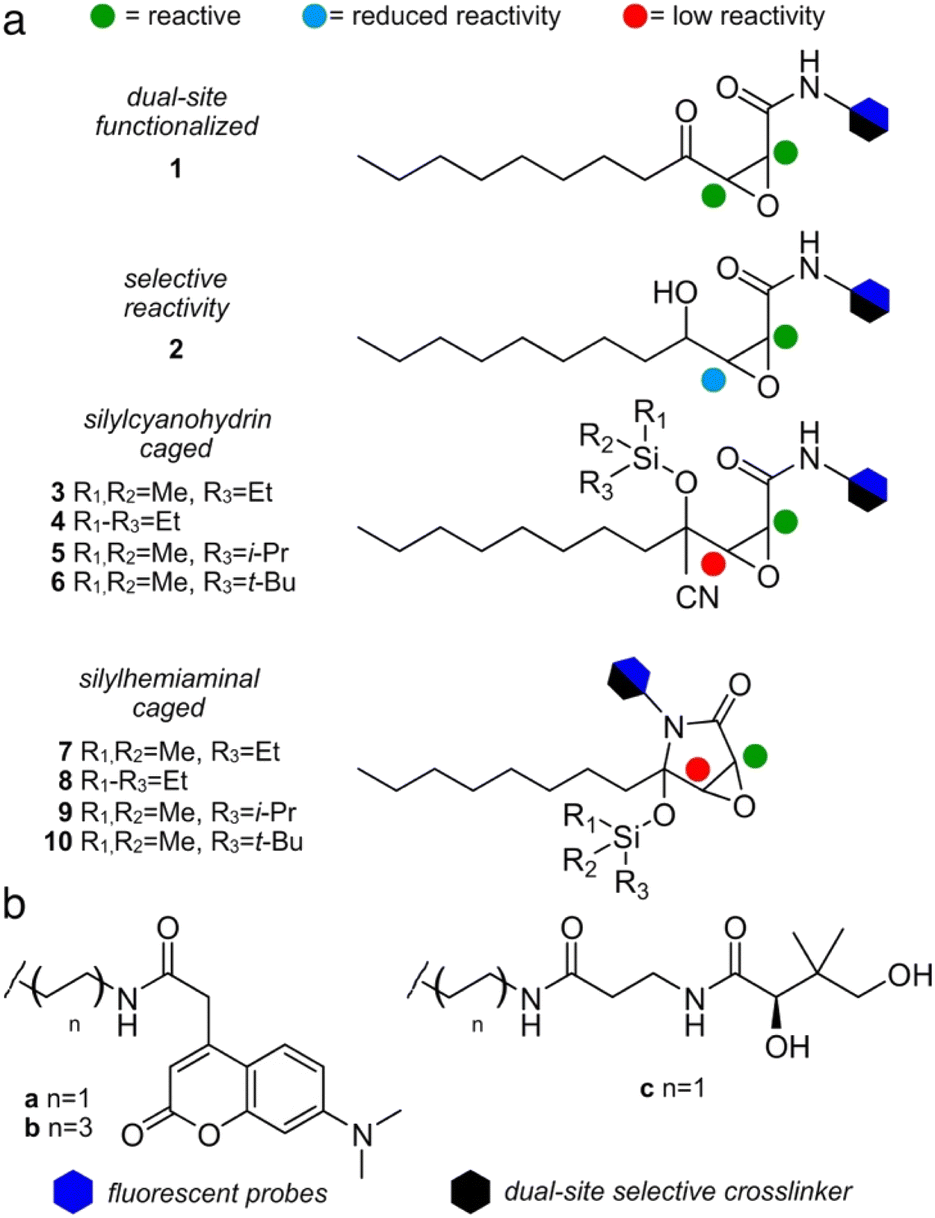 | ||
| Fig. 2 Cerulenin probe design. (a) To evaluate the role of complex tautomeric functionality, we designed a series of reactive (1 or 2) and silicon-masked (3–10) probes. Colored spheres denote the suggested reactivity at each center (a). Hexagons denote the position where fluorescent (blue) or crosslinking (black) functional groups are appended. (b) Fluorescent tags a or b were attached by 2- or 6-carbon chains, respectively. A single 2-carbon chain length was used in crosslinker c. Compounds are referred to herein by their reactive group (number, 1–10) and tag (letter, a–c). For instance, 1a denotes a compound with C2-fluorescent tag a on reactive group 1. | ||
As depicted in Fig. 2, motif 1 mimics the cerulenin γ-ketoepoxyamide, and therefore, should display comparable reactivity at C2 and C3. We evaluated two additional strategies to mask reactivity at C3. As shown in 3–6 (Fig. 2a), conversion of the γ-ketone to a silylcyanohydrin25 reduces the reactivity at C3. Silylcyanohydrins 3–6 are functional groups trapped as linear tautomers, thus predictably tuning and reducing their reactivity. Moreover, inclusion of a reactive functionality within the alkyl positions of the silyl group can be used to tune the stability and deprotection ability of dimethylethylsilyl (DMES), triethylsilyl (TES), isopropyldimethylsilyl (IPDMS), and tert-butyldimethylsilyl (TBS).25 A second set of probes natively traps the cyclic hemiaminal as a silyl ether. As shown in Fig. 2a, silylhemiaminal masked 7–10 also direct reactivity to C2. These designed silyl ethers can thus resolve the tautomerization of 1 to linear trapped silylcyanohydrins 3–6 and ring closed silylhemiaminals 7–10.
We next turned to SAR data11 focused on the acyl chain of cerulenin indicating that the fully reduced tetrahydrocerulenin (Fig. 1a) retains activity. We also noted a lack of elucidation of chirality effects within the epoxide, and, therefore, developed methods to investigate with both the natural (2R,3S)- and synthetic (2S,3R)-epoxides of the fully saturated alkyl analogues of cerulenin. We selectively prepared intermediate 14 and its pure enantiomers (2R,3S,4R)-14 and (2S,3R,4S)-14. As shown in Scheme 1, 14 is prepared in 44% overall yield in 4-steps from nonanal beginning with addition of vinyl magnesium bromide. The resulting allyl alcohol 11 was esterified by acryloyl chloride to 12. Ring closing metathesis with ester 12 was achieved with Hoveyda–Grubbs II to furnish butenolide 13. Sequentially, sodium hypochlorite was used to epoxidize 13 into 14.
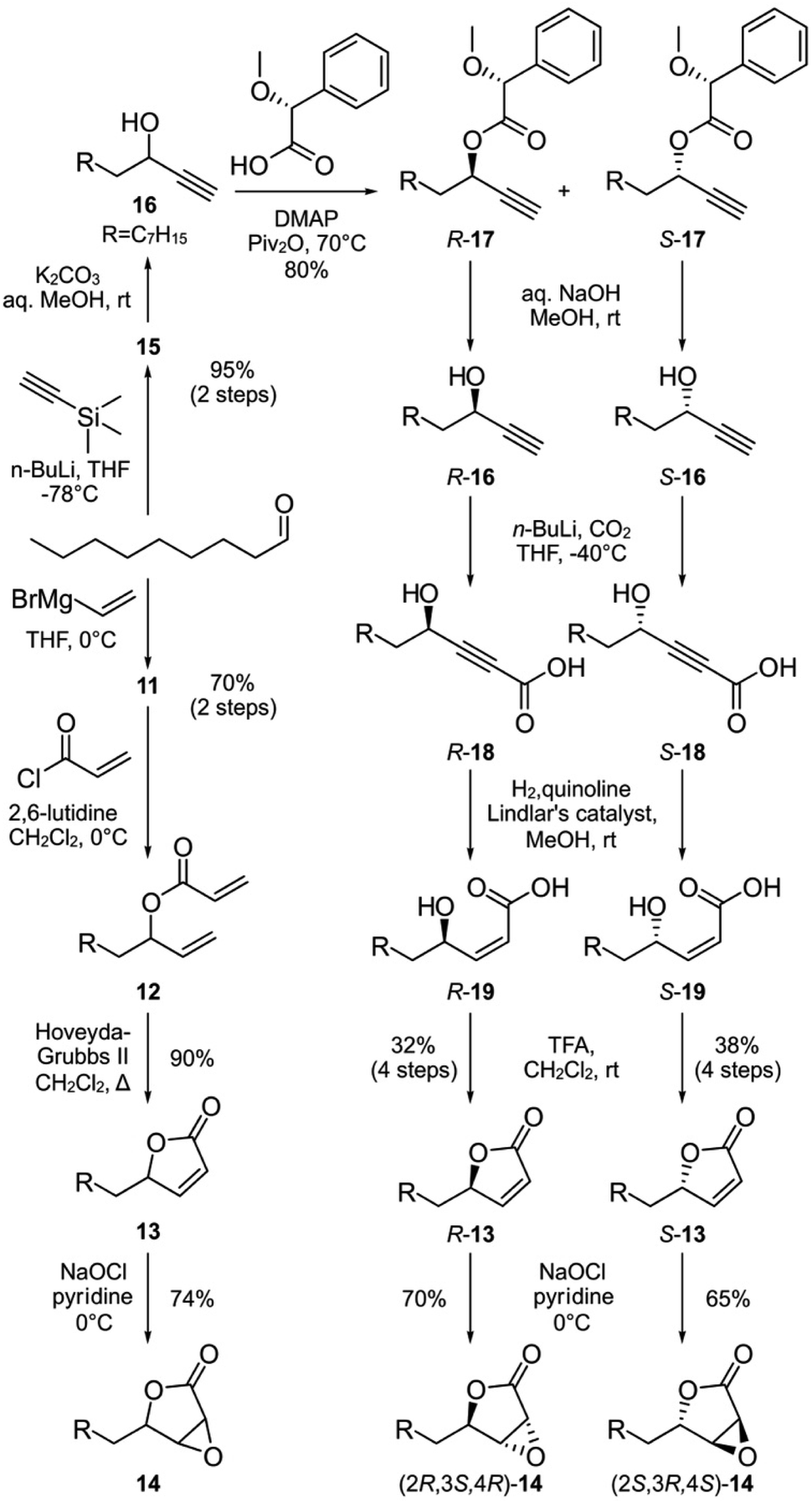 | ||
| Scheme 1 Parallel syntheses of racemic epoxylactone 14 and enantiopure (2R,3S,4R)-14 and (2S,3R,4S)-14 from nonanal. R denotes a n-heptyl group, which represents the side chain of tetrahydrocerulenin (Fig. 1a). | ||
We next prepared enantiopure (2R,3S,4R)-14 and (2S,3R,4S)-14. This route began with addition of trimethylsilylacetylene to nonanal followed by deprotection of 15 to 16 under mildly basic conditions (Scheme 1). Racemic 16 was resolved into chromatographically separable (4R)-17 and (4S)-17 using (R)-α-methoxyphenylacetic acid.26 The esters were hydrolyzed by NaOH to give (R)-undec-1-yn-3-ol ((4R)-16) and (S)-undec-1-yn-3-ol ((4S)-16), respectively. The absolute stereochemistry of the propargyl alcohols were determined through Mosher analysis (see ESI†).27 With the enantiomeric propargyl alcohols in hand, lithiation followed by carboxylation with CO2 (dry ice) yielded propargylic acids (4R)-18 or (4S)-18 (Scheme 1). Careful hydrogenation in the presence of Lindlar's catalyst afforded cis-α,β-alkenyl carboxylic acids (4R)-19 or (4S)-19, which were lactonized under acidic conditions to afford (4R)-13 or (4S)-13 over 4-steps from (4R)-17 and (4S)-17, respectively in 32–38% enantioselective yield. For stereospecific epoxidation, we adopted the protocol by Tishler4 using sodium hypochlorite to give (2R,3S,4R)-14 and (2S,3R,4S)-14.
With racemic and enantiopure 14, we then converted these materials into a set of probes (Fig. 2). Leveraging prior experience, we prepared fluorescently labeled probes (blue hexagon, Fig. 2b) for imaging specific ketosynthase enzymes, and crosslinking probes (black hexagon, Fig. 2b) for evaluation of the interaction between the ACPs and their partner ketosynthases.28–31 The fluorescent probes evaluated the domain reactivity and the crosslinkers probed protein·protein interactions. To further test the domain reactivity, we chose both a short C2 chain (a, n = 1, Fig. 2b) and a longer C6 chain (b, n = 3, Fig. 2b) for the fluorescent probes (ESI Fig. S1†). To mimic the structure of the pantetheine linker, the crosslinking probes were prepared with only one length (c, n = 1, Fig. 2b). With this design, we turned our attention to prepare different cerulenin motifs (1–10, Fig. 2a).
We began by developing a route to convert 14 to 1a and 1b. As shown in Scheme 2, a two-step process was developed that began by incorporating the fluorescent tag a or b by amidation of the lactone in 14 to 2a and 2b, followed by subsequent oxidation to the respective probes 1a and 1b. Using this approach, we also prepared (2R,3S)-1b, and (2S,3R)-1b from (2R,3S,4R)-14 and (2S,3R,4S)-14, respectively. NMR analyses on these probes showed equilibrium between the ring closed and linear tautomers (see ESI†). Taking advantage of the self-cyclization equilibrium (Fig. 1a), a single step silylation successfully trapped 1b in the ring-closed tautomer affording masked 7b and 8b (Scheme 2b). Overall, the synthetic pathway generated ten probes 1a, 1b, (2R,3S)-1b, (2S,3R)-1b, 2a, 2b, (2R,3S,4R)-2b, (2S,3R,4S)-2b, 7b, and 8b in 5–10 steps with overall yields of 6–11% (Scheme 2a and b).
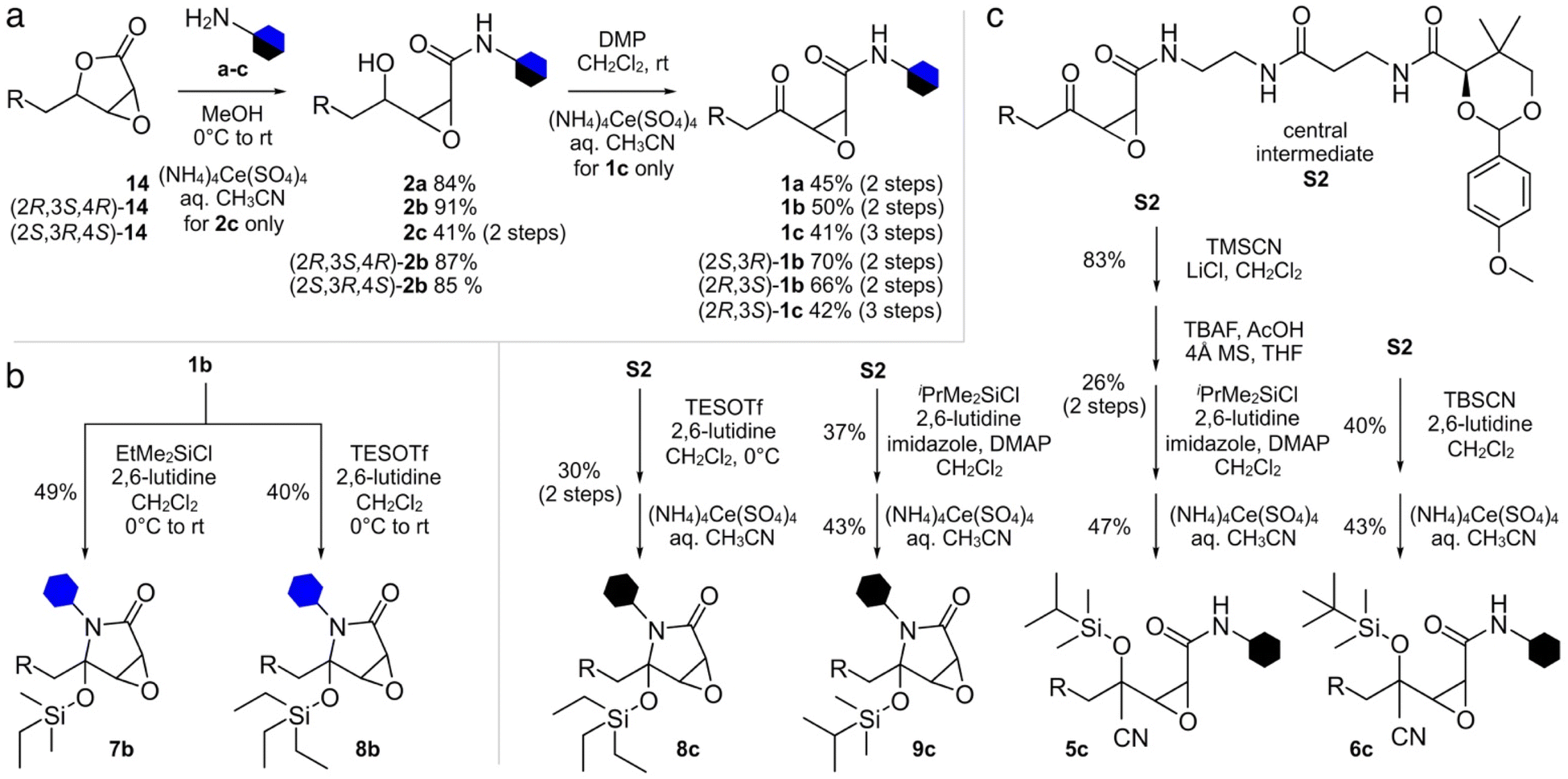 | ||
| Scheme 2 Synthesis of cerulenin-based probes and crosslinkers. (a) Synthesis of ketoamide and γ-alcohol probes and crosslinkers. Four ketoamide probes 1a, 1b, (2R,3S)-1b, and (2S,3R)-1b were prepared from 14, (2R,3S,4R)-14 or (2S,3R,4S)-14 in 2 steps. Two ketoamide crosslinkers 1c and (2R,3S)-1c were prepared in 3 steps from 14 or (2R,3S,4R)-14, respectively. Four γ-alcohol probes 2a, 2b, (2R,3S,4R)-2b and (2S,3R,4S)-2b were prepared in one step from 14, (2R,3S,4R)-14 or (2S,3R,4S)-14. (b) Silylhemiaminal probes 7b and 8b were prepared in 1 step from 1b. (c) Silylhemiaminal crosslinkers 8c and 9c were prepared in 2 steps from central intermediate S2 (see ESI†). Silylcyanohydrin crosslinkers 5c and 6c were also prepared from S2 through 2 or 4 steps. | ||
The synthesis of crosslinkers 1c and 2c began with epoxylactone 14 (Scheme 2a). A lactone ring opening of 14 with pantetheine amine c (Fig. 2b) and a chemoselective deprotection of the p-anisaldehyde acetal using (NH4)4Ce(SO4)4 in aq. CH3CN yielded 2c. For crosslinker 1c, N-alkylation of 14 with pantetheine amine c, oxidation of the resulting γ-alcohol, and deprotection of the p-anisaldehyde acetal provided 1c. Crosslinker (2R,3S)-1c was accomplished by similar methods from (2R,3S,4R)-14. The synthesis of masked crosslinkers 8c and 9c (Scheme 2c) arose from S2 through a single step silylation followed by acetal deprotection. The synthesis of linear crosslinker 5c began with cyanosilylation of S2 and sequential desilylation to give cyanohydrin intermediate S6 (see ESI†). Re-silylation with i-PrMe2SiCl and deprotection of the p-anisaldehyde acetal delivered 5c. Linear crosslinker 6c was prepared by silyl-cyanohydrin generation with TBSCN and acetal deprotection from intermediate S2. Overall, the methods in Scheme 2 enabled the preparation of three types of crosslinking agent, two with tunable masks, and afforded a total of 8 crosslinks between ACP domains and their partner proteins.
Additionally, attempts were made to prepare crosslinker 4c (Fig. 2a), however the trimethyl silyl group was not stable and deprotected during handling. Similar issues existed for the 3c as similarly protected materials such as intermediate S5 (see ESI†) were only useful as intermediates in the synthetic process. Similar issues were also observed in our attempts to prepare 7c, which was far less stable than 8c which underwent partial hydrolysis in buffer. Similar synthetic limitations were observed for materials with very large protecting groups such as 10c. While further screening may provide methods to obtain these materials, the fact that our data has shown that these materials would not be of use due to lability or lack of reactivity.
Fluorescent probes identify regioselective KS reactivity and protein binding selectivity
To validate the potential for C3 reactivity, fluorescent labeling experiments were performed on warhead 1 and 2 with type-II ketosynthases (monofunctional enzymes) FabB, FabF, and FabH, and type-I (multi-domain) mycocerosic acid synthase (MAS). As expected, γ-ketone probes 1a and 1b labeled FabB, FabF, FabH, and MAS, but not MASΔKS-AT (Fig. 3a–c). The absence of fluorescent labeling for MASΔKS-AT implies 1a and 1b do not react with ketoreductase (KR), dehydratase (DH), and enoylreductase (ER) domains within MAS (Fig. 3c). Compared with 1b, 1a with shorter carbon linker was more efficient at labeling FabB and FabH (Fig. 3a and b). We found that the linker length effect was less pronounced for FabF. It was surprising that FabH was labeled with 1a and 1b as cerulenin was not previously reported as an efficient inhibitor of FabH.15 γ-Alcohol probes 2a and 2b did not label FabB, FabF, FabH, MAS or MASΔKS-AT (Fig. 3c and ESI Fig. S4†), implying that the presence of a γ-ketone proximal to the epoxide is required for activity. As reported,15,21 covalent complexes of FabB and FabF with cerulenin indicate that the enzyme-bound form was the linear tautomer, however the resolution in these structures was not sufficient to definitively identify linkage to C2 or C3. Our probes 2a and 2b suggested that C3 was the reactive position (ESI Fig. S3†).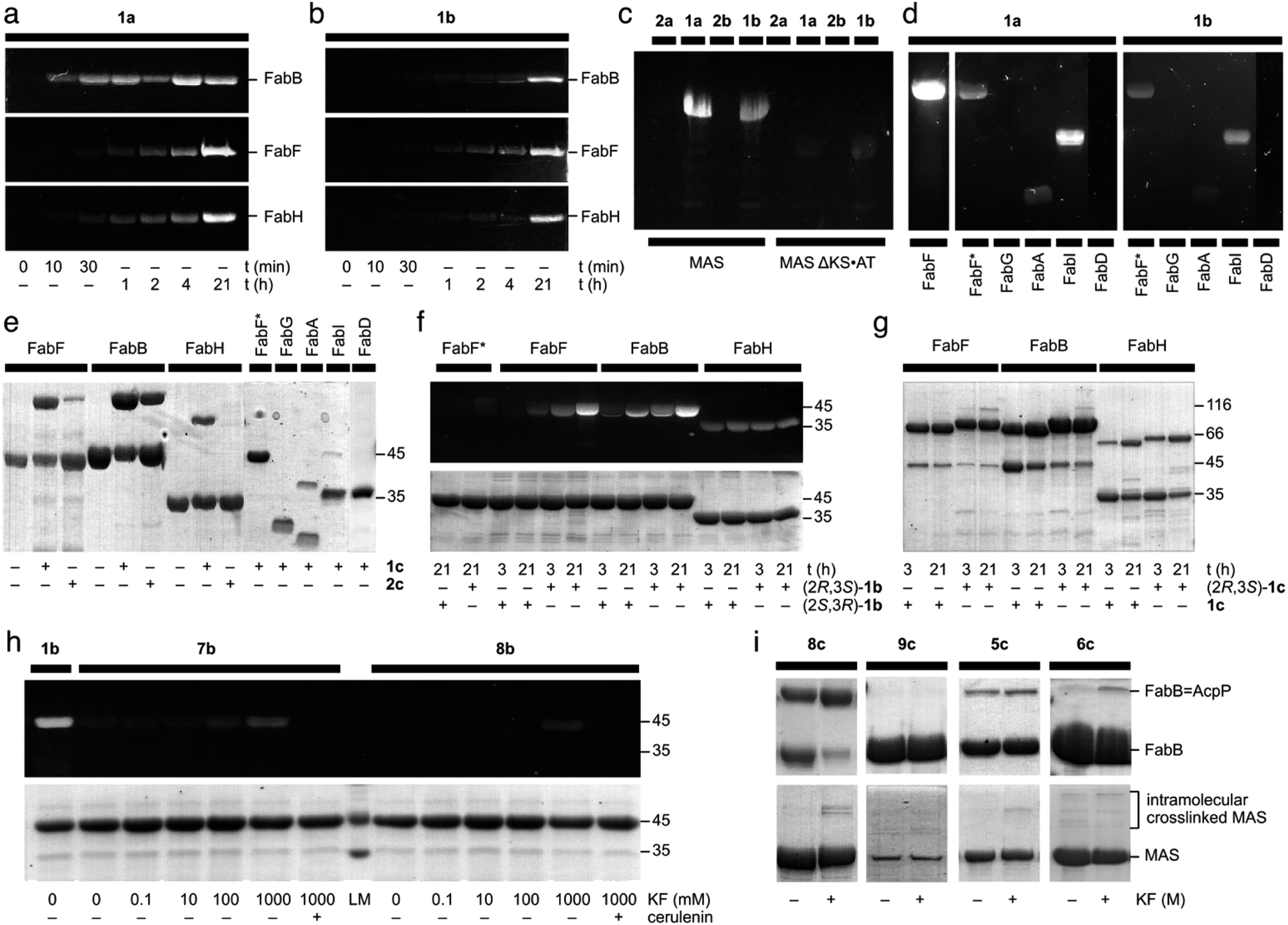 | ||
| Fig. 3 Evaluation of cerulenin-based fluorescent probes and crosslinkers. (a) and (b) 12% SDS-PAGE gels demonstrate the time course fluorescent labeling of 20 μM FabF, 20 μM FabB, or 20 μM FabH with 100 μM 1a or 100 μM 1b. Similar fluorescent labeling was performed with 100 μM 2a, or 100 μM 2b (ESI Fig. S3†). (c) An 8% SDS-PAGE depicts the incubation of 50 μM MAS or 50 μM of the modifying compartment MASΔKS·AT with 100 μM 1a, 100 μM 1b, 100 μM 2a, or 100 μM 2b. (d) 12% SDS-PAGE gels demonstrate the fluorescent labeling of 20 μM FabF, 20 μM FabF* (a C163A mutant of FabF), 20 μM FabG (KR), 20 μM FabA (DH), 20 μM FabI (ER), or 20 μM FabD (AT) with 100 μM 1a or 100 μM 1b. (e) 12% SDS-PAGE gels depict the crosslinking of 50 μM crypto-AcpP loaded with 1c or 2c with 20 μM FabF, 20 μM FabB, or 20 μM FabH. They also demonstrate crosslinking of 20 μM FabF*, 20 μM FabG (KR), 20 μM FabA (DH), 20 μM FabI (ER), or 20 μM FabD (AT) with 100 μM 1ccrypto-AcpP. (f) A 12% SDS-PAGE gel depicting the fluorescent labeling of 50 μM FabF*, 50 μM FabF, 50 μM FabB, and 50 μM FabH with 250 μM (2R,3S)-1b or 250 μM (2S,3R)-1b at 3 h or 21 h. (g) A 12% SDS-PAGE gel shows the crosslinking of 20 μM FabF, FabB, and FabH with 100 μM (2R,3S)-1c or 100 μM racemic 1ccrypto-AcpP at 3 h or 21 h. (h) A 12% SDS-PAGE gel shows the activation of fluorescent labeling of 20 μM FabF with 100 μM 7b or 100 μM 8b with KF in 100 mM Tris·HCl pH 7.5 at 25 °C for 21 h. The preincubation with 200 μM (+)-cerulenin was conducted 30 min prior to the labeling experiments as a means of control. (i) The 12% SDS-PAGE gels (top) demonstrate the crosslinking of 50 μM crypto-AcpP bearing 5c, 6c, 8c, or 9c with 10 μM FabB in 100 mM Tris·HCl pH 7.5 with or without KF (ESI†) at 25 °C for 21 h. AcpP = FabB denotes the crosslinked species of crypto-AcpP and FabB. The 8% SDS-PAGE gels (bottom) depict the incubation of 4.4 μM crypto-MAS bearing 5c, 6c, 8c, or 9c with 10 μM FabB in 100 mM Tris·HCl pH 7.5 with or without KF (ESI†) at 25 °C for 21 h. The level of fluorescent intensity was used as a means of calibrating the successful labeling of a protein. A successful crosslinking experiment is noted by the appearance of a higher molecular weight band. Unless otherwise noted, all experiments were conducted in 50 mM Tris·HCl 150 mM NaCl 10% glycerol pH 7.5 at 25 °C for 21 h. The uncropped gels for the corresponding gels shown here have been provided in ESI Fig. S3–S7.† The purity of each AcpP protein state including apo-AcpP, holo-AcpP, and each crypto-AcpP has been provided in ESI Fig. S8.† A summary of the experimental and structural data for each interaction has been tabulated in ESI Fig. S9.† | ||
To further investigate the warhead selectivity, we tested 1a and 1b with a panel of E. coli fatty-acid biosynthetic enzymes (Fig. 3d). This study included a ketoreductase (FabG), dehydratase (FabA), enoyl reductase (FabI), and a malonyl CoA-ACP transacylase (FabD). Both probes 1a and 1b had comparable labeling with FabA, FabI, and the FabF active site cysteine mutant (FabF*, a C163A mutant), while no labeling was observed with FabG and FabD (Fig. 3d). We observed that probes 1a and 1b were able to non-selectively label C58, C375, or C395 in FabF* (wild type FabF contains 4 Cys including C163 and C395, within the pocket, and two others at C58 and C375, residing on the surface). Similar labeling and crosslinking experiments were performed with preincubation of cerulenin or iodoacetamide demonstrated the observation of fluorescent labeling on other cysteines besides the reactive cysteine (ESI Fig. S11†). It is worth noting that precipitation was formed immediately upon the addition of iodoacetamide into FabF, which might lead to the discrepancy in the FabF results. This suggested that the use of excess probe (>5 eq.) could lead to off target labeling non-active site cysteine residues.
Crosslinking selectivity and stereoselectivity confirm the substrate guidance by an ACP
To examine the translation of the reactive warhead from fluorescent probes to crosslinkers,32–35 we tested AcpP loaded with 1c and 2c against different partner proteins. AcpP loaded with 1c crosslinked with FabF, FabB, and FabH, demonstrating that the reactivity of the warhead was translatable from fluorescent to crosslinking experiments (Fig. 3e). AcpP loaded with 1c did not show significant activity with the other partner proteins. Only mild crosslinking activity was only observed with FabA and FabI. Through this result, AcpP-mediated reaction of crosslinker 1c was site-specific to KS domains. Whereas the reduction to a γ-alcohol in 2a and 2b (ESI Fig. S3†) completely eradicated KS labeling, crosslinker 2c tethered AcpP successfully crosslinked with FabB and FabF (Fig. 3e). This suggested that protein·protein interactions between AcpP and its cognate KS domain can encourage crosslinking for agents with reduced activity.In contrast to the off-target labeling, AcpP loaded with 1c did not show significant crosslinking with other partner proteins (Fig. 3e), indicating that non-selective fluorescent labeling would not translate to non-specific crosslinking. Only a low level of crosslinking activity was observed between AcpP bearing 1c and FabA or FabI, with yields of 15.5% and 5.9%, respectively (Fig. 3e). Similar results were observed using 10 eq. of 1c-crypto-AcpP (ESI Fig. S12†). Upon the preincubation of FabB/FabF/FabH with cerulenin or iodoacetamide, no crosslinking was observed (ESI Fig. S13†). This finding indicated that the AcpP-mediated crosslinking reaction was site-specific to KS domains.
Next, we explored the effect of the epoxide stereochemistry on cerulenin reactivity with ketosynthases. The enantiopure fluorescent probes (2R,3S)-1b and (2S,3R)-1b and crosslinker (2R,3S)-1c were screened for their reactivity with FabB, FabF, and FabH. Fluorescent labeling with the (2R,3S)-configuration, as found in natural (+)-cerulenin, showed higher efficiency with both FabB and FabF compared to the unnatural antipode (Fig. 3f). Interestingly, FabH reacted identically with both fluorescent enantiomers, ((2R,3S)-1bversus (2S,3R)-1b in Fig. 3f) and lacked selectivity in its crosslinking ((2R,3S)-1cversus racemic 1c in Fig. 3g). In contrast, FabB and FabF (particularly FabB) showed a clear preference for the natural (2R,3S)-1b (Fig. 3f) and (2R,3S)-1c (Fig. 3g). The latter result suggests that the natural (2R,3S) configuration was more KS-reactive, particularly when delivered by a cognate ACP for crosslinking experiments. Time course experiments (at 1, 15, 30, and 60 minutes) were also conducted depicting that (2R,3S)-1c crosslinker is the preferred substrate for FabB and FabF while FabH did not show any preference (ESI Fig. S13†).
After revealing the crucial role that C3 of the epoxide plays in protein labeling, we proceeded to use silicon masking as a tool to harness the C3 reactivity. Fluorescent probes 7b (DMES) and 8b (TES) trapped in the ring-closed form were evaluated for their reaction with FabF. Probe 7b exhibited tunable reactivity with increasing KF concentrations, while 8b was switchable only at high KF concentrations (Fig. 3h). This result suggested that steric hindrance from the silyl group can tune the reactivity of C3. FabF was pre-incubated with (+)-cerulenin, followed by labeling with 7b or 8b to examine off-target labeling (comparable studies were conducted for 1a, 1b and 1c, ESI Fig. S11†). As shown in Fig. 3h, no labeling was observed even at the high concentrations of KF.
We then turned to crosslinking to evaluate the masking process (Fig. 4). We were interested in understanding how silicon-masked cyanohydrin or hemiaminal masking could be used to enable sequential site-selective crosslinking. The process began by loading a crosslinker onto the active site Ser residue on an ACP. As shown in Fig. 4, this was then followed by sequential unmasking and ACP = KS crosslinking.25 As shown in Scheme 2c, we were able to prepare four silicon-masked crosslinkers including TES hemiaminal 8c, IPDMS hemiaminal 9c, IPDMS cyanohydrin 5c, and TBS cyanohydrin 6c.
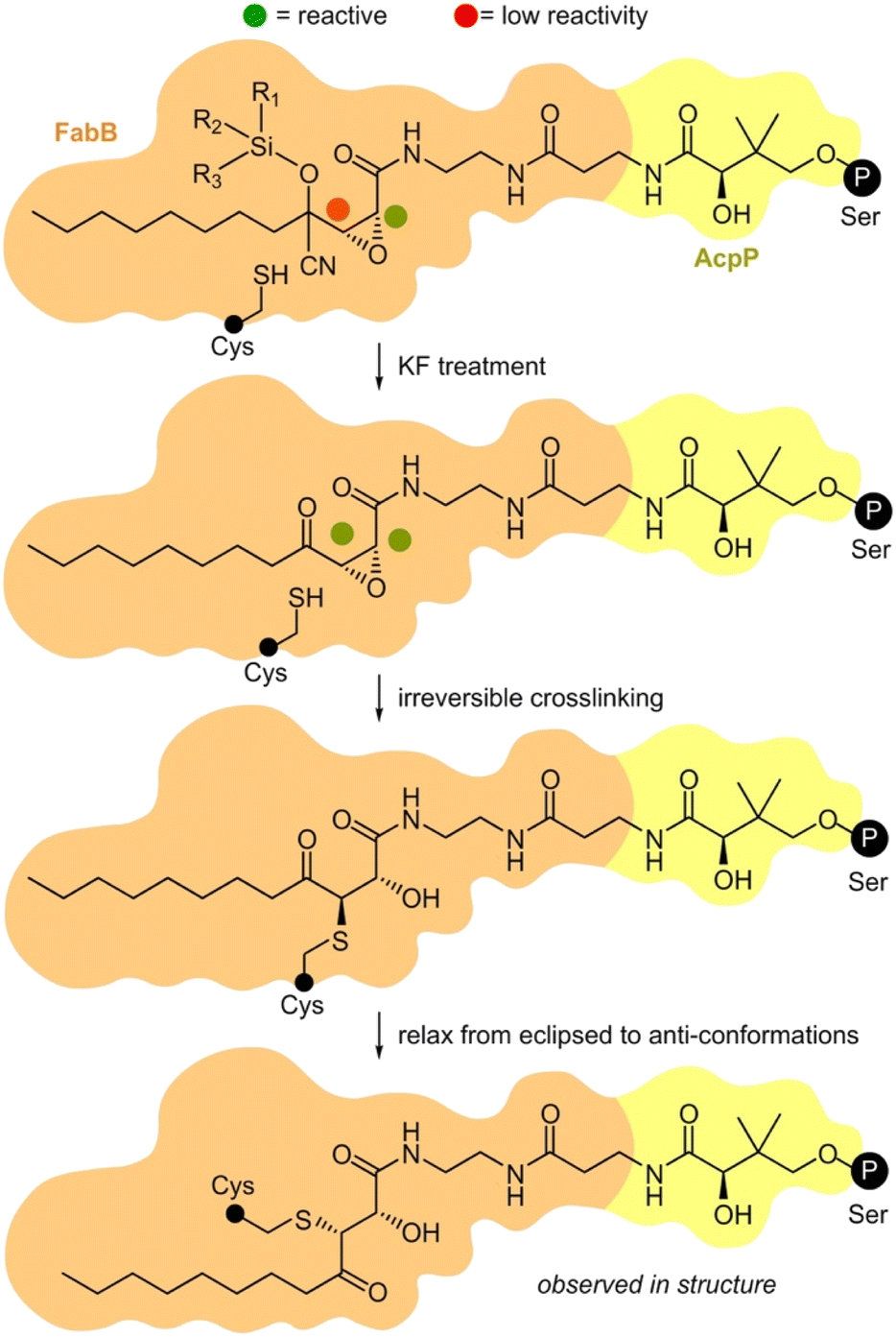 | ||
| Fig. 4 Proposed mechanism of masked AcpP (green) crosslinking with a KS domain (blue). An AcpP-tethered silyl cyanohydrin crosslinker was delivered into the binding pocket of a KS domain by interaction with AcpP. The C3 was protected by a silyl cyanohydrin preventing it from reacting with active site Cys residue within the KS. The addition of KF allows the desilylation and the elimination of HCN to restore activity at C3. Next, the activated C3 position can alkylate the active site residue cysteine of KS. Black circle P denotes the position of the 4′-phosphopantetheinyl-linkage to the active site Ser residue on an ACP domain. The small black circle denotes the KS active site Cys. Further details on the steps of this process have been provided in ESI Fig. S10.† | ||
Each of these crosslinkers was then screened for its ability to crosslink AcpP with FabB. With enhanced lability, TES 8c provided nearly complete crosslinking in the presence of KF (Fig. 3i). Although switchable, 8c was found to be partially labile, as evidenced by LC/MS analyses (see ESI†) showing hydrolysis of the TES group upon storage in labeling buffer (50 mM Tris·HCl 150 mM NaCl 10% glycerol pH 7.5) at 25 °C for 21 h. Based on this, it was not surprising that 8c crosslinked AcpP and FabB without KF. Bulkier IPDMS 9c did not show crosslinking under these conditions (Fig. 3i) and displayed improved buffer stability with 30% deprotection after at 25 °C over 21 h. Under comparable conditions (Fig. 3i), linear crosslinkers IPDMS 5c and TBS 6c underwent crosslinking with 6c showing KF induced selectivity. While this process would require further optimization for downstream structural applications, these studies demonstrate the unique ability to tune the reactivity of these probes by adjusting the substitution within their silyl ether.
While the lack of switching in these studies was not detrimental to the ultimate crosslinking result, as noted by the formation of AcpP = FabB (Fig. 3i), synthases where the ACP = KS crosslinking would occur within a single protein, such as type I FAS or PKS systems, pose challenges. Here, the two-step loading and crosslinking process cannot be conducted in trans, thus leading to shunt product formation from KS labeling before the ACP was loaded (ESI Fig. S10†). We explored these considerations with the type-I PKS protein mycocerosic acid synthase (MAS). As apparent in Fig. 3i, we observed switchable crosslinking with 5c. The fact that we did not observe high levels of crosslinking in this study could arise from the fact that the KS·ACP interaction in MAS may be short-lived or unable to meet the reaction times required for robust crosslinking (Fig. 3i).
Validation through X-ray crystallographic structure determination of crosslinked FabB = AcpP
To validate the mechanism shown in Fig. 4, we obtained a crystal structure of FabB crosslinked with (2R,3S)-1c-crypto-AcpP at a resolution of 1.93 Å (Fig. 5, ESI Table S1 and Fig. S14†). Exploration of the pocket bearing the 4′-phosphopantetheine arm revealed an unambiguous electron density for covalent linkages of the pantetheine arm between the post-translationally modified S36 in AcpP and active site C163 of FabB (Fig. 5b, ESI Fig. S15†). The density within this appendage (Fig. 5b) clearly showed reactivity of C163 of FabB with the C3 position of (2R,3S)-1c, as depicted in Fig. 4. This structure not only provided support for the activation step (step 1, Fig. 4), where the C3 position remains unreactive until released by fluoride (experimentally demonstrated in Fig. 3i) but also reported on the stereochemical and regiochemical selectivity of this reaction. As depicted in Fig. 4 and demonstrated in Fig. 5b, we were able to observe addition of C163 from the back face of the epoxide to generate a trans-adduct which relaxed from its eclipsed to anti-conformation (observed structurally, Fig. 5b). Here, the γ-ketone of (2R,3S)-1c falls into the oxyanion hole formed by the backbone amides of C163 and F392 of FabB (Fig. 5b) instead of the hydroxyl group formed by epoxide opening.15 This result provided definitive evidence for the masking, release, and capture process.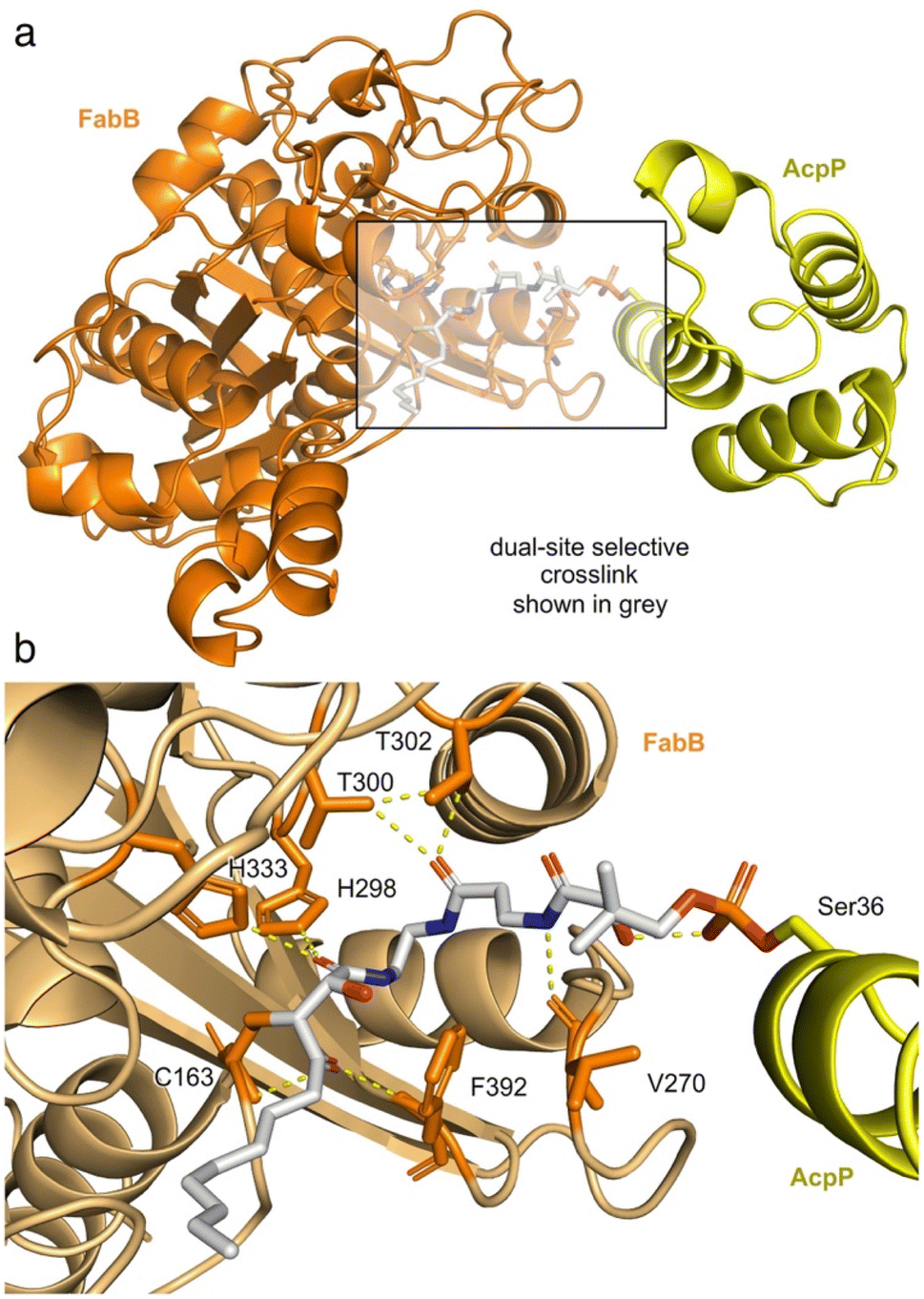 | ||
| Fig. 5 Structure of crosslinked FabB = AcpP. (a) Monomeric unit of the dimeric 1.93 Å resolution X-ray crystal structure of FabB (orange) crosslinked with (2R,3S)-1c-crypto-AcpP (yellow). The 4′-phosphopantetheine arm is shown in stick from (grey). (b) An expansion of the crosslinking region delineating the covalent linkages at the AcpP active residue S36 and FabB active residue C163 formed by crosslinker (2R,3S)-1c. Key residues interacting with 4-phosphopantetheine arm are noted. | ||
In addition to this validation, this structure revealed critical details as to how the pantetheine arm delivers its substrate from its ACP to its KS processing partner. As shown in Fig. 5b, the pantetheine arm was anchored at the entrance of the substrate binding tunnel of FabB. Here, a polar contact was also observed between the first pantetheine amide and the main chain carbonyl of V270 of FabB. The carbonyl of the second amide formed hydrogen bonding with two highly conserved threonine residues T300 and T302 of FabB. Such protein–substrate interactions lock the pantetheine arm in place at the spacious entrance of the binding tunnel.
The crosslinked FabB = AcpP structure further detail the positioning of the cerulenin moiety as it formed its reactive adduct with FabB. As shown in Fig. 5b, the C1 amide of the 4-phosphopantetheine-linked tetrahydrocerulenin mimics the thioester in the natural acyl substrate delivered by AcpP. This was verified by the coordination between the C1 amide carbonyl and H298 and H333 in the FabB His–His–Cys catalytic triad.20,36 To verify if crosslinker 1c mimics the cerulenin binding mode as it was designed, we overlayed the structures of cerulenin bound FabB (PDB: 1FJ8), FabB crosslinked with C12αBr-crypto-AcpP (PDB: 6OKC), and FabB crosslinked with (2R,3S)-1c-crypto-AcpP (PDB: 8MSM). Crosslinker (2R,3S)-1c trapped the crosslinked complex in its catalytic relevant state sharing similar substrate–protein interactions (C163, V270, H298, T300, T302, H333, and F392) with the previously reported structures (ESI Fig. S16†).
This state-trapped structure (Fig. 5a) also reveal details of the AcpP·FabB interaction as well as providing further support for its gated mechanism.15,36,37 As shown in ESI Fig. S17,† the FabB = AcpP crosslinked structure revealed the electrostatic interactions that FabB (positively charged) and AcpP (negatively charged) formed within R62, K63, R66, R124, K127, Y132 of FabB and Q14, D35, D38, E47 of AcpP. Here, prior studies have shown that near the binding tunnel, gating loops 1 G399–G402 (GFGG) and loop 2 D265–E275 (DGADMVAPSGE) facilitate the substrate delivery (‘gate-open’ form) and the catalysis of the transacylation and condensation half reactions (‘gate-closed’ form).15,38 Based on this evidence, our structure delineates a ‘gate closed’ conformation (ESI Fig. S18†). To govern the substrate chain length specificity, FabB contains a ‘double swinging-door enzyme gate’ which comprises two sets of Q113 and E200. These four residues can expand one of the binding pockets while restricting the size of the other binding pocket,38 ultimately providing a means to regulate the positioning of the fatty acid side chains based on their length. Here, our data show that Q113 of FabB (chain B) is more proximal to the dimer interface expanding the binding pocket while narrowing the other side (ESI Fig. S19†), therein providing direct evidence for the negative cooperativity, and ultimately supporting this structure as an active, trapped state.
Conclusions
These studies revealed new concepts and tool compounds to regio- and stereo-selectively guide KS protein conjugation with an epoxide. Specifically, we were interested in using the knowledge on the mechanisms of cerulenin reactivity learned herein to guide the selective targeting and crosslinking of ACP to KS domains. Using this selectivity, we defined a method to selectively conjugate active site residues, as demonstrated by their fluorescent labeling and crosslinking to their associated ACPs. We further demonstrated a role for silicon containing moieties as masking agents whose reactivity can be triggered with the addition of fluoride ion to initiate stepwise site-selective crosslinking. These studies further illustrate how silicon protection offers a general tool to guide the reactivity of covalently reactive natural products, an observation that can be accommodated by a variety of other functional modifications.39To further validate the developed crosslinker, we report a 1.93 Å FabB crosslinked structure with (2R,3S)-1c-crypto-AcpP. This atomic resolution data demonstrates that the tetrahydrocerulenin crosslinker (2R,3S)-1c is a site specific crosslinker for FabB and AcpP by mimicking substrate binding (nucleophilic attack on C3 of the epoxide) at the condensation transition state in fatty acid chain elongation. The crystal structure revealed the fundamental structural elements of FabB catalysis. Going forward, we are exploiting these tools to further elucidate the mechanisms that guide large biosynthetic proteins such as type I PKS.
Data availability
All experimental and characterization data, as well as NMR spectra are available in the ESI.†Author contributions
Z. J. synthesized the probes, Z. J. and J. J. L. collected and assigned the spectral data characterizing these probes and crosslinkers, Z. J., A. C., J. C., and A. S. prepared the proteins and conducted the protein labeling and crosslinking studies. Z. J., J. C., and G. V. L. collected and analyzed the protein crystallographic results. All authors participated in the design of the studies. Z. J. and J. J. L. drafted the manuscript. The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by grants from the NIH (R01-GM095970 and R21-AI156484). We thank Drs Xuemei Huang, Anthony Mrse, and Yongxuan Su for assistance with acquisition of NMR and MS data.Notes and references
- A. Matsumae, Y. Kamio and T. Hata, J. Antibiot., 1963, 16, 236–238 CAS.
- S. Nomura, T. Horiuchi, S. Ōmura and T. Hata, J. Biochem., 1972, 71, 783–796 CrossRef CAS PubMed.
- S. Ōmura, Methods Enzymol., 1981, 72, 520–532 CrossRef.
- A. A. Jakubowski, F. S. Guziec Jr and M. Tishler, Tetrahedron Lett., 1977, 28, 2399–2402 CrossRef.
- A. A. Jakubowski, F. S. Guziec Jr, M. Sugiura, C. C. Tam and M. Tishler, J. Org. Chem., 1982, 47, 1221–1228 CrossRef CAS.
- N. S. Mani and C. A. Townsend, J. Org. Chem., 1997, 62, 636–640 CrossRef CAS PubMed.
- T. E. Kedar, M. W. Miller and L. S. Hegedus, J. Org. Chem., 1996, 61, 6121–6126 CrossRef CAS PubMed.
- F. P. Kuhajda, E. S. Pizer, J. N. Li, N. S. Mani, G. L. Frehywot and C. A. Townsend, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 3450–3454 CrossRef CAS PubMed.
- B. Schmid, J. F. Rippmann, M. Tadayyon and B. S. Hamilton, Biochem. Biophys. Res. Commun., 2005, 328, 1073–1082 CrossRef CAS PubMed.
- L. E. Landree, A. L. Hanlon, D. W. Strong, G. Rumbaugh, I. M. Miller, J. N. Thupari, E. C. Connolly, R. L. Huganir, C. Richardson, L. A. Witters, F. P. Kuhajda and G. V. Ronnett, J. Biol. Chem., 2004, 279, 3817–3827 CrossRef CAS PubMed.
- D. S. Lawrence, J. T. Zilfou and C. D. Smith, J. Med. Chem., 1999, 42, 4932–4941 CrossRef CAS PubMed.
- P. Johansson, B. Wiltschi, P. Kumari, B. Kessler, C. Vonrhein, J. Vonck, D. Oesterhelt and M. Grininger, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 12803–12808 CrossRef CAS PubMed.
- M. Hiltunen and K. Söderhäll, Appl. Environ. Microbiol., 1992, 58, 1043–1045 CrossRef CAS PubMed.
- H. Funabashi, S. Iwasaki, S. Okuda and S. Ōmura, Tetrahedron Lett., 1983, 24, 2673–2676 CrossRef CAS.
- A. C. Price, K. H. Choi, R. J. Heath, Z. Li, S. W. White and C. O. Rock, J. Biol. Chem., 2001, 276, 6551–6559 CrossRef CAS PubMed.
- M. Moche, G. Schneider, P. Edwards, K. Dehesh and Y. Lindqvist, J. Biol. Chem., 1999, 274, 6031–6034 CrossRef CAS PubMed.
- A. Kawaguchi, H. Tomoda, S. Nozoe, S. Ōmura and S. Okuda, J. Biochem., 1982, 92, 7–12 CrossRef CAS PubMed.
- J. T. Mindrebo, A. Chen, W. E. Kim, R. N. Re, T. D. Davis, J. P. Noel and M. D. Burkart, ACS Catal., 2021, 11, 6787–6799 CrossRef CAS PubMed.
- J. T. Mindrebo, A. Patel, W. E. Kim, T. D. Davis, A. Chen, T. G. Bartholow, J. J. La Clair, J. A. McCammon, J. P. Noel and M. D. Burkart, Nat. Commun., 2020, 11, 1727 CrossRef CAS PubMed.
- A. Chen, Z. Jiang and M. D. Burkart, Chem. Sci., 2022, 13, 4225–4238 RSC.
- F. Trajtenberg, S. Altabe, N. Larrieux, F. Ficarra, D. de Mendoza, A. Buschiazzo and G. E. Schujman, FEBS J., 2014, 281, 2324–2338 CrossRef CAS PubMed.
- H. Funabashi, A. Kawaguchi, H. Tomoda, S. Ōmura, S. Okuda and S. Iwasaki, J. Biochem., 1989, 105, 751–755 CrossRef CAS PubMed.
- B. H. Ariso and S. Ōmura, J. Antibiot., 1974, 27, 28–30523 CrossRef PubMed.
- T. Hansen, A. Nin-Hill, J. D. C. Codée, T. A. Hamlin and C. Rovira, Chem.–Eur. J., 2022, 28, e202201649 CrossRef CAS PubMed.
- S. Konno, J. J. La Clair and M. D. Burkart, Angew Chem. Int. Ed. Engl., 2018, 57, 17009–17013 CrossRef CAS PubMed.
- W. C. Chan, J. J. La Clair, B. León, K. A. Trieger, M. Q. Slagt, M. T. Verhaar, D. U. Bachera, M. T. Rispens, R. M. Hofman, V. L. de Boer, R. der Hulst, R. Bus, P. Hiemstra, M. L. Neville, K. A. Mandla, J. S. Figueroa, C. Jamieson and M. D. Burkart, Cell Rep. Phys. Sci., 2020, 1, 100277 CrossRef CAS.
- T. R. Hoye, C. S. Jeffrey and F. Shao, Nat. Protoc., 2007, 2, 2451–2458 CrossRef CAS PubMed.
- K. Charov and M. D. Burkart, Methods Enzymol., 2020, 638, 321–340 CAS.
- K. Charov and M. D. Burkart, ACS Infect. Dis., 2019, 5, 1518–1523 CrossRef CAS PubMed.
- A. C. Mercer, J. L. Meier, J. W. Torpey and M. D. Burkart, ChemBioChem, 2009, 10, 1091–1100 CrossRef CAS PubMed.
- J. J. La Clair, T. L. Foley, T. R. Schegg, C. M. Regan and M. D. Burkart, Chem. Biol., 2004, 11, 195–201 CrossRef CAS PubMed.
- F. Ishikawa, R. H. Haushalter, D. J. Lee, K. Finzel and M. D. Burkart, J. Am. Chem. Soc., 2013, 135, 8846–8849 CrossRef CAS PubMed.
- S. Kapur, A. Worthington, Y. Tang, D. E. Cane, M. D. Burkart and C. Khosla, Bioorg. Med. Chem. Lett., 2008, 18, 3034–3038 CrossRef CAS PubMed.
- G. H. Hur, J. L. Meier, J. Baskin, J. A. Codelli, C. R. Bertozzi, M. A. Marahiel and M. D. Burkart, Chem. Biol., 2009, 16, 372–381 CrossRef CAS PubMed.
- W. E. Kim, A. Patel, G. H. Hur, P. Tufar, M. G. Wuo, J. A. McCammon and M. D. Burkart, ChemBioChem, 2019, 20, 147–152 CrossRef CAS PubMed.
- A. Chen, J. T. Mindrebo, T. D. Davis, W. E. Kim, Y. Katsuyama, Z. Jiang, Y. Ohnishi, J. P. Noel and M. D. Burkart, Acta Crystallogr., Sect. D: Struct. Biol., 2022, 78, 1171–1179 CrossRef CAS PubMed.
- P. von Wettstein-Knowles, J. G. Olsen, K. A. McGuire and A. Henriksen, FEBS J., 2006, 73, 695–710 CrossRef PubMed.
- J. T. Mindrebo, A. Chen, W. E. Kim, R. N. Re, T. D. Davis, J. P. Noel and M. D. Burkart, ACS Catal., 2021, 11, 6787–6799 CrossRef CAS PubMed.
- M. Gersch, J. Kreuzer and S. A. Sieber, Nat. Prod. Rep., 2021, 29, 659–682 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures for protein expression, protein purification, chemoenzymatic reaction for the loading of chemical probes onto ACP, chemical probe synthesis, NMR analysis for the synthesized probes, LC-MS analysis for the hydrolysis of two of the caged-crosslinking probes, protein labeling and crosslinking studies along with copies of select NMR spectra from the intermediates and products. See DOI: https://doi.org/10.1039/d3sc02864j |
| This journal is © The Royal Society of Chemistry 2023 |