
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEnantioselective catalytic remote perfluoroalkylation of α-branched enals driven by light†‡
Matteo
Balletti
a,
Tommy
Wachsmuth
 a,
Antonio
Di Sabato
a,
Will C.
Hartley
a and
Paolo
Melchiorre
*b
a,
Antonio
Di Sabato
a,
Will C.
Hartley
a and
Paolo
Melchiorre
*b
aICIQ – Institute of Chemical Research of Catalonia, 43007 Tarragona, Spain
bDepartment of Industrial Chemistry ‘Toso Montanari’, University of Bologna, 40136 Bologna, Italy. E-mail: p.melchiorre@unibo.it
First published on 14th April 2023
Abstract
Herein, we report a photochemical organocatalytic method for the asymmetric introduction of perfluoroalkyl fragments (including the valuable trifluoromethyl moiety) at the remote γ-position of α-branched enals. The chemistry exploits the ability of extended enamines (dienamines) to form photoactive electron donor–acceptor (EDA) complexes with perfluoroalkyl iodides, which under blue light irradiation generate radicals through an electron transfer mechanism. The use of a chiral organocatalyst, derived from cis-4-hydroxy-L-proline, secures a consistently high stereocontrol while inferring complete site selectivity for the more distal γ position of the dienamines.
Introduction
The established benefit of fluorine-containing fragments in medicinal chemistry relies on their ability to alter the physicochemical and pharmacokinetic properties of organic compounds.1 In addition, it is acknowledged that increasing C(sp3) incorporation and the presence of stereogenic centres positively correlate with the clinical success of small molecule therapeutics.2 These aspects explain the importance of developing novel methods for the stereoselective incorporation of trifluoromethyl (CF3) and perfluoroalkyl (RF) units within organic molecules. However, only a few strategies are available for the catalytic production of perfluoroalkyl-containing stereogenicity.3 For example, the combination of enamine organocatalysis and photoredox catalysis served to develop the enantioselective α-perfluoroalkylation of aldehydes (Fig. 1a).4 In a different photochemical approach, the asymmetric α-perfluoroalkylation of β-ketoesters was reported under phase transfer catalysis (PTC, Fig. 1b).5 Both protocols used visible light to generate perfluoroalkyl radicals (RF˙, I) via single electron transfer (SET), which were then intercepted stereoselectively. The photochemical radical generation was mastered either by an external photocatalyst (Fig. 1a) or by excitation of a ground-state electron donor–acceptor (EDA) complex6 between perfluoroalkyl iodides and a chiral enolate (Fig. 1b). These methods led to the formation of C(sp3)–CF3 and C(sp3)–RF stereocenters at the α position of carbonyl compounds. In contrast, strategies that account for the stereoselective installation of perfluoroalkyl units at distal positions of the substrates are not available. Herein, we close this gap in synthetic methodology by disclosing a photochemical method for the asymmetric introduction of CF3 and RF groups at the remote γ position of α-branched enals 1 (Fig. 1c).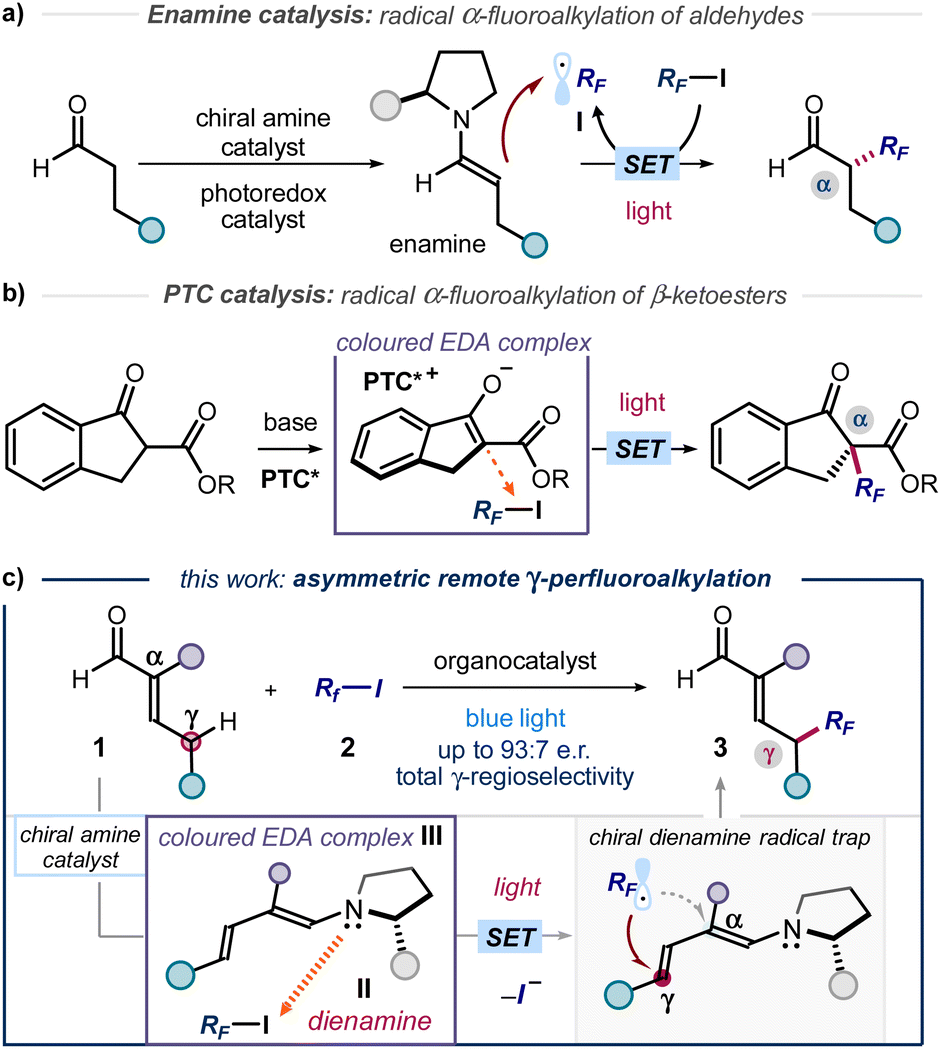 | ||
| Fig. 1 Organocatalytic photochemical strategies for the stereoselective radical α-perfluoroalkylation of (a) aldehydes and (b) β-ketoesters. (c) Design plan for the stereocontrolled radical perfluoroalkylation of enals at the remote γ-carbon via excitation of an EDA complex III between a catalytic dienamine II and perfluoroalkyl iodides. PTC: phase transfer catalyst; EDA: electron donor–acceptor; RF: perfluoroalkyl chain. | ||
Design plan
Our plan for the remote perfluoroalkylation relied on the reactivity of chiral dienamines II, generated upon activation of α-branched enals 1 with a chiral amine catalyst (Fig. 1c). Intermediates II are characterized by vinylogous nucleophilicity,7 and have been used for the site-selective functionalization of enals 1 at their remote γ-carbon via traditional two-electron ionic pathways.8 Recently, we demonstrated that the dienamine activation platform could be translated successfully also to radical chemistry.9 The use of an external catalyst secured formation of electrophilic radicals, which were then stereoselectively intercepted by the chiral dienamine at remote position.9a In the context of C(sp3)–RF bond formation, we surmised that the electron-rich nature of II could be leveraged to form photoactive EDA complexes upon aggregation with perfluoroalkyl iodides.5,10 Visible light excitation of the EDA complex III would then drive the formation of perfluoroalkyl radical (RF˙, I), thus circumventing the need of an exogenous photocatalyst. Regio- and stereoselective trap would then offer a catalytic strategy for the remote γ-perfluoroalkylation of α-branched enals 1.Results and discussion
The feasibility of our plan was evaluated by reacting 2-phenyl pentenal 1a with nonafluoroiodobutane 2a in Et2O using the commercially available TMS-protected diphenyl prolinol catalyst A (20 mol%, TMS: trimethylsilyl) and 2,6-lutidine as the additive (Fig. 2a). We noticed that the reaction mixture turned immediately yellow right after addition of all the reaction components, which individually were colorless (see below for further details). Irradiation by a blue light LED afforded the desired product (R)-3a with complete γ-regioselectivity and good enantiomeric ratio, albeit in low yield (37% yield, 82![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :18 e. r.). Other solvents, including THF and dichloromethane, were suitable, but diethyl ether proved optimal (see Section E of the ESI‡ for full optimization studies).
:18 e. r.). Other solvents, including THF and dichloromethane, were suitable, but diethyl ether proved optimal (see Section E of the ESI‡ for full optimization studies).
 | ||
| Fig. 2 (a–d) Catalyst optimization. Reactions performed for 20 h in 0.3 mL of Et2O using 0.2 mmol of 2a, 0.6 mmol of 1a, and 0.04 mmol of catalyst (20 mol%) under irradiation by a single high-power LED (λmax = 460 nm, irradiance = 100 mW cm−2). Yield of 3a determined by 1H NMR analysis of the crude mixture using trimethyl orthoformate as the internal standard. Enantiomeric ratio (e. r.) of 3a measured by chiral UPC2 analysis after derivatization to the corresponding 2,4-dinitrophenyl hydrazone. Note that catalysts A–H gave product 3a with an (R) absolute configuration; the figure shows the (S) major enantiomer of 3a afforded by catalysts I–M. TMS: trimethylsilyl; TBS: tert-butyldimethylsilyl; TIPS: triisopropylsilyl; TDS: thexyldimethylsilyl. | ||
To improve the stereoselectivity of the remote perfluoroalkylation, we modified the catalyst structure. Introduction of fluorinated aryl motifs (3,5-difluorophenyl and 3,5-bis(trifluoromethyl) substituents) in catalysts B and C improved the yield significantly, but the enantioselectivity remained unsatisfactory. Structural variation of the best-performing catalyst C focused on the effect of different silyl ether substituents (catalysts C–G). These studies, detailed in Fig. 2b, revealed that the smallest TMS group in catalyst C outperformed larger silyl ethers in terms of yield and enantioselectivity (93%, 84:16 e. r.). We next investigated the effect of a second stereogenic element on the catalyst's pyrrolidine core by synthesizing variants derived from 4-hydroxyproline (Fig. 2c).11 The tert-butyldimethylsilyl (TBS)-protected catalyst H, derived from trans-4-hydroxyproline, offered an increased stereocontrol (87:13 e. r.). The diastereomeric catalyst I, bearing the two pyrrolidine ring substituents in a cis relationship,12 led to a slightly increased stereocontrol (the opposite enantiomer of the product (S)-3a was formed in 88:12 e. r.). This result suggested us to further optimize the structure of catalysts derived from cis-4-hydroxyproline (Fig. 2d). Assessment of different silyl ether protecting groups at the 4-hydroxy moiety identified the bulky thexyldimethylsilyl (TDS)-protected catalyst M as the best-performing, since product 3a was obtained in 90:10 e. r. Lowering the reaction temperature to −10 °C afforded 3a in 93:7 e. r. and in excellent yield. Control experiments established that both the amine catalyst and light were essential for reactivity.
To evaluate the generality of the method, we applied the optimized conditions and the cis-catalyst M to react enal 1a with iodides of different perfluoroalkyl chain lengths (Fig. 3a). Pleasingly, products 3a–g were obtained in good to high yields and enantiomeric ratios. Only reaction at the remote position occurred, exclusively leading to γ-functionalized products.
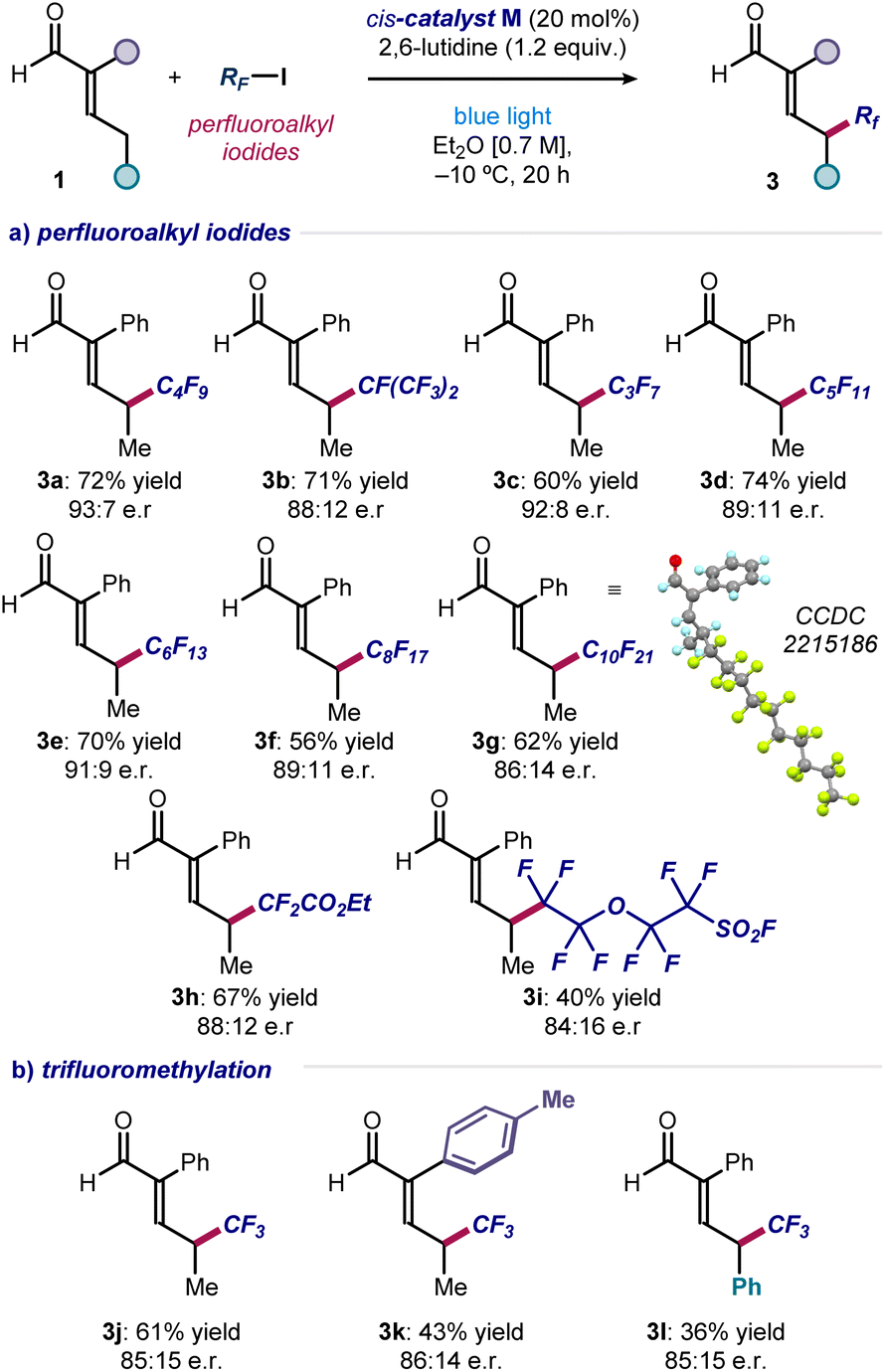 | ||
| Fig. 3 (a and b) Substrate scope for the asymmetric introduction of perfluoroalkyl and trifluoromethyl fragments at the remote γ position of α-branched enals. Reactions performed using 2 (0.2 mmol), enals 1 (0.6 mmol), 2,6-lutidine (0.24 mmol) in 0.3 mL of Et2O under illumination by a single high-power LED (λmax = 460 nm, irradiance = 100 mW cm−2). Yields of the isolated products 3 and enantiomeric ratios are reported below each entry. Enantiomeric ratios measured by UPC2 analysis on chiral stationary phase after derivatization of 3 to the corresponding 2,4-dinitrophenyl hydrazine derivatives. | ||
Crystals of compound 3g were suitable for X-ray crystallographic analysis, which allowed the (S) configuration of the newly formed stereocenter to be assigned unambiguously. Ethyl difluoroiodoacetate could be used as the radical precursor, enabling the synthesis of α-difluoro ester 3h in good yield and enantioselectivity. A sensitive sulfonyl fluoride was tolerated well, affording the heteroatom-dense product 3i. Importantly, the asymmetric formation of a C(sp3)–CF3 stereogenic center at the remote position of different enals was accomplished using CF3–I (products 3j–l). Other radical precursors, including perfluoroalkyl bromides and simple alkyl iodides, were unsuccessful, highlighting the need of both a weak carbon–iodine bond and adjacent fluorine atoms (see Section C of the ESI‡ for a list of unsuccessful substrates).
Next, a range of substituted and functionalized enals 1 was tested (Fig. 4). Extended alkyl chain products were obtained in good yield and enantioselectivity (3m & 3n), with terminal halide (adduct 3o), ester (3p), benzyl (3q) and phenyl (3r) groups were well tolerated. Various substituents on the α-phenyl group of enals were accepted, with products bearing alkyl (3s & 3t), 4-halogen (3u & 3v), and 4-methoxy (3w) groups all synthesized with consistently good results. In contrast, electron-withdrawing substituents (e.g., p-NO2 and p-CF3) completely inhibited the reactivity. With α-unsubstituted enals, achieving stereocontrol proved difficult, and the corresponding products 3x–z were formed essentially in racemic form but with complete remote regioselectivity. A selective γ-perfluoroalkylation took place also with an enal derived from the naturally-occurring diterpene phytol (product 3z). With respect to the enals amenable to this transformation, an aryl group is required at the α-position to gain high enantiocontrol, which ensures consistent configurational control over the dienamine intermediate. Enals bearing α-alkyl substituents are unsuitable substrates, which undergo reaction to produce a complex mixture of side-products.
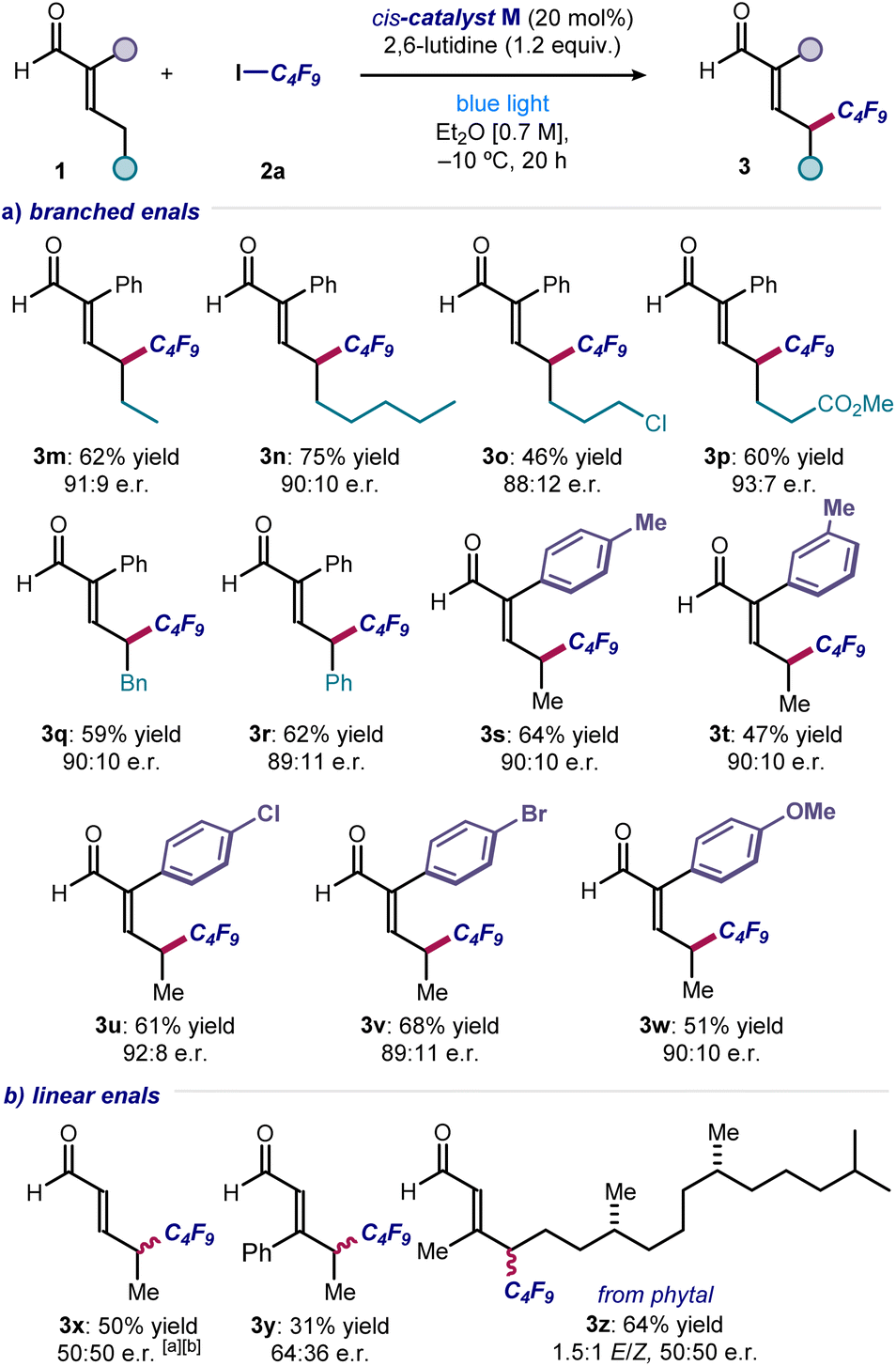 | ||
| Fig. 4 (a and b) Different enals that can participate in the light-driven remote perfluoroalkylation. Reactions performed using 2 (0.2 mmol), enals 1 (0.6 mmol), 2,6-lutidine (0.24 mmol) in 0.3 mL of Et2O under illumination by a single high-power LED (λmax = 460 nm, irradiance = 100 mW cm−2). Yields refer to the isolated products 3. Enantiomeric ratios measured by UPC2 analysis on chiral stationary phase after derivatization of 3 to the corresponding 2,4-dinitrophenyl hydrazine derivatives. a Carried out at room temperature. b Isolated as the corresponding 2,4-dinitrophenyl hydrazone. | ||
We then performed investigations to glean insight on the reaction mechanism (Fig. 5). UV-vis spectroscopic analysis confirmed that the individual components of the process were colorless, including the dienamine intermediate generated upon in situ from enal 1a and the amine catalyst (green line in Fig. 5a). Right upon mixing perfluoroalkyl iodide 2a, aminocatalyst, and enal 1a, a new absorption band was observed in the visible region, which was further corroborated by the appearance of a distinct yellow color of the reaction mixture. These observations are consonant with the formation of a visible-light absorbing EDA complex between the dienamine and RF-I.13 Mechanistically, we propose that light irradiation of the EDA complex III triggers an intracomplex SET, leading to the formation of perfluoroalkyl radical I after mesolysis of the C–I bond (Fig. 5b). The electrophilic RF˙ I is then intercepted by the chiral dienamine II, in a regio- and stereoselective fashion.14 The resulting α-amino radical IV would then either deliver an electron to 2via SET,15 or abstract an iodine atom from 2via ATRA (atom-transfer radical addition),16 to regenerate the perfluoroalkyl radical I thus propagating the radical chain. Hydrolysis of the iminium ion intermediate V releases product 3 and regenerates the organocatalyst. We measured an overall quantum yield for the reaction between enal 1a and nonafluoroiodobutane 2a of Φ = 1.14, which is congruent with a radical chain process being operational.17
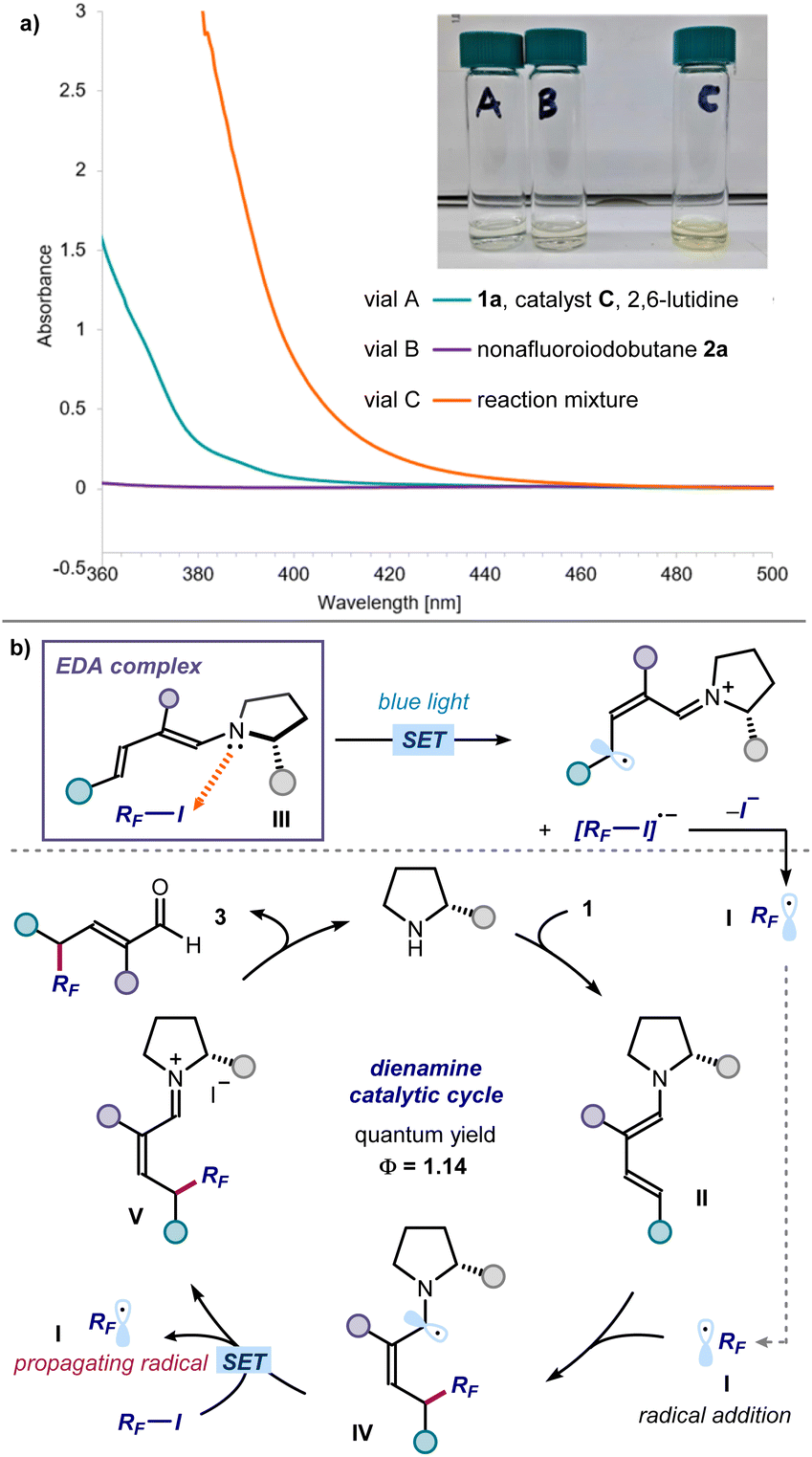 | ||
| Fig. 5 Mechanistic studies. (a) UV-vis measurements acquired on a Shimadzu UV-2401PC spectrophotometer with a quartz cuvette with path length of 1 cm. All samples in Et2O. Vial A (green line): [catalyst C] = 0.6 M, [1a] = 0.6 M and [2,6-lutidine] = 0.6 M; vial B (purple line): [2a] = 0.6 M; vial C: [catalyst C] = 0.6 M, [1a] = 0.6 M, [2a] = 0.6 M and [2,6-lutidine] = 0.6 M. While the individual reaction components are all colourless, the vial C mixture containing all reaction components displays a yellow colour and an absorption band from approximately 450 nm. (b) Proposed radical propagation mechanism triggered by the excitation of the dienamine-based EDA complex III, which serves as the initiation event. | ||
Conclusions
In summary, we have developed the first enantioselective methodology that accounts for the remote installation of perfluoroalkyl-containing stereogenicity. The chemistry exploits the ability of chiral catalytic dienamines to form photoactive electron donor–acceptor (EDA) complexes with perfluoroalkyl iodides, which under blue irradiation generate radicals through a SET mechanism. Key to achieving high levels of enantiocontrol and complete site selectivity for the more distal γ position of enals was the use of a sterically encumbered cis-4-hydroxyprolinol-derived catalyst.Data availability
Crystallographic data for compound 3g has been deposited as CCDC 2215186. Additional experimental details and data are provided in the ESI,‡ including experimental procedures, full characterization data, and copies of NMR spectra (PDF).Author contributions
M. B., T. W., A. S and W. C. H. performed the experiments. M. B. and P. M. conceptualized the project. P. M. directed the project. P. M. and W. C. H. wrote the manuscript with assistance from all authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support was provided by Agencia Estatal de Investigación (PID2019-106278GB-I00 and CTQ2016-75520-P). W. C. Hartley thanks the EU for a Horizon 2020 Marie Skłodowska-Curie Fellowship (H2020-MSCA-IF-2020, 101031533).References
- (a) K. Müller, C. Faeh and F. Diederich, Science, 2007, 317, 1881–1886 CrossRef PubMed; (b) W. K. Hagmann, J. Med. Chem., 2008, 51, 4359–4369 CrossRef CAS PubMed; (c) S. Purser, P. R. Moore, S. Swallow and V. Gouverneur, Chem. Soc. Rev., 2008, 37, 320–330 RSC; (d) N. A. Meanwell, J. Med. Chem., 2018, 61, 5822–5880 CrossRef CAS PubMed.
- (a) F. Lovering, J. Bikker and J. Humblet, J. Med. Chem., 2009, 52, 6752–6756 CrossRef CAS PubMed; (b) F. Lovering, MedChemComm, 2013, 4, 515–519 RSC.
- (a) A. E. Allen and D. W. C. MacMillan, J. Am. Chem. Soc., 2010, 132, 4986–4987 CrossRef CAS PubMed; (b) Q.-H. Deng, H. Wadepohl and L. H. Gade, J. Am. Chem. Soc., 2012, 134, 10769–10772 CrossRef CAS PubMed; (c) H. Huo, X. Huang, X. Shen, K. Harms and E. Meggers, Synlett, 2015, 27, 749–753 CrossRef; (d) J. Liu, W. Ding, Q.-Q. Zhou, D. Liu, L.-Q. Lu and W.-J. Xiao, Org. Lett., 2018, 20, 461–464 CrossRef CAS PubMed; (e) C. Jiang, L. Wang, H. Zhang, P. Chen, Y.-L. Guo and G. Liu, Chem, 2020, 6, 2407–2419 CrossRef CAS; (f) P. Xu, W. Fan, P. Chen and G. Liu, J. Am. Chem. Soc., 2022, 144, 13468–13474 CrossRef CAS PubMed.
- D. A. Nagib, M. E. Scott and D. W. C. MacMillan, J. Am. Chem. Soc., 2009, 131, 10875–10877 CrossRef CAS PubMed.
- Ł. Woźniak, J. J. Murphy and P. Melchiorre, J. Am. Chem. Soc., 2015, 137, 5678–5681 CrossRef PubMed.
- G. E. M. Crisenza, D. Mazzarella and P. Melchiorre, J. Am. Chem. Soc., 2020, 142, 5461–5476 CrossRef CAS PubMed.
- For reviews on aminocatalytic remote functionalisation strategies, see: (a) J. J. Hao, L. Albrecht and K. A. Jørgensen, Chem. Sci., 2013, 4, 2287–2300 RSC; (b) I. Jurberg, I. Chatterjee and P. Melchiorre, Chem. Commun., 2013, 49, 4869–4883 RSC . For a review on dienamine catalysis:; (c) V. Marcos and J. Aleman, Chem. Soc. Rev., 2016, 45, 6812–6832 RSC . For pioneering studies on dienamine catalysis, see:; (d) A. G. Nigmatov and E. P. Serebryakov, Russ. Chem. Bull., 1993, 42, 213 CrossRef; (e) S. Bertelsen, M. Marigo, S. Brandes, P. Diner and K. A. Jørgensen, J. Am. Chem. Soc., 2006, 128, 12973–12980 CrossRef CAS PubMed.
- For selected examples: (a) K. Liu, A. Chougnet and W. Woggon, Angew. Chem., Int. Ed., 2008, 47, 5827–5829 CrossRef CAS PubMed; (b) G. Bergonzini, S. Vera and P. Melchiorre, Angew. Chem., Int. Ed., 2010, 49, 9685–9688 CrossRef CAS PubMed; (c) J. Stiller, E. Marqués-López, R. P. Herrera, R. Fröhlich, C. Strohmann and M. Christmann, Org. Lett., 2011, 13, 70–73 CrossRef CAS PubMed; (d) M. Silvi, C. Cassani, A. Moran and P. Melchiorre, Helv. Chim. Acta, 2012, 95, 1985–2006 CrossRef CAS; (e) G. Talavera, E. Reyes, J. L. Vicario and L. Carrillo, Angew. Chem., Int. Ed., 2012, 51, 4104–4107 CrossRef CAS PubMed; (f) B. S. Donslund, K. S. Halskov, L. A. Leth, B. M. Paz and K. A. Jørgensen, Chem. Commun., 2014, 50, 13676–13679 RSC; (g) C. Martín-Santos, C. Jarava-Barrera, S. del Pozo, A. Parra, S. Díaz-Tendero, R. Mas-Ballesté, S. Cabrera and J. Alemán, Angew. Chem., Int. Ed., 2014, 53, 8184–8189 CrossRef PubMed; (h) L. Naesborg, K. S. Halskov, F. Tur, S. M. N. Mønsted and K. A. Jørgensen, Angew. Chem., Int. Ed., 2015, 54, 10193–10197 CrossRef CAS PubMed.
- (a) M. Balletti, E. Marcantonio and P. Melchiorre, Chem. Commun., 2022, 58, 6072–6075 RSC . For the direct excitation of dienamines, which was limited to the use of tertiary bromomalonates as the only suitable radical precursors, see:; (b) M. Silvi, E. Arceo, I. D. Jurberg, C. Cassani and P. Melchiorre, J. Am. Chem. Soc., 2015, 137, 6120–6123 CrossRef CAS PubMed; (c) During the preparation of this manuscript, a photoredox/enamine dual catalysis protocol was reported detailing two entries for the remote perfluoroalkylation of α-branched enals with poor reactivity but good stereocontrol, see: M. Briand, L. D. Thai, F. Bourdreux, N. Vanthuyne, X. Moreau, E. Magnier, E. Anselmi and G. Dagousset, Org. Lett., 2022, 24, 9375–9380 CrossRef CAS PubMed.
- (a) E. Arceo, I. D. Jurberg, A. Álvarez-Fernández and P. Melchiorre, Nat. Chem., 2013, 5, 750–756 CrossRef CAS PubMed; (b) G. A. Russell and K. Wang, J. Org. Chem., 1991, 56, 3475–3479 CrossRef CAS; (c) D. Cantacuzène, C. Wakselman and R. Dorme, J. Chem. Soc., Perkin Trans. 1, 1977, 1365–1371 RSC; (d) A. Postigo, Eur. J. Org. Chem., 2018, 46, 6391–6404 CrossRef.
- (a) T. Ishino and T. Oriyama, Chem. Lett., 2007, 36, 550–551 CrossRef CAS; (b) H. Sato, F. Nagashima and T. Oriyama, Chem. Lett., 2010, 39, 379–381 CrossRef CAS; (c) C. Ma, Z.-J. Jia, J.-X. Liu, Q.-Q. Zhou, L. Dong and Y.-C. Chen, Angew. Chem., Int. Ed., 2013, 52, 948–951 CrossRef CAS PubMed.
- (a) I. Arenas, A. Ferrali, C. Rodríguez-Escrich, F. Bravo and M. A. Pericàs, Adv. Synth. Catal., 2017, 359, 2414–2424 CrossRef CAS; (b) T. Inoshita, K. Goshi, Y. Morinaga, Y. Umeda and H. Ishikawa, Org. Lett., 2019, 21, 2903–2907 CrossRef CAS PubMed.
- The EDA complex interaction could be elicited by halogen bonding between the nitrogen lone pair of the dienamine and the σ-hole on the iodine atom of RF-I. Our efforts to obtain suitable crystals of the EDA aggregate, which would have provided clear insights on the nature of this interaction, had unfortunately met with failure. For a review on the importance of halogen bonding in EDA complexes involving perfluoroalkyl iodides, see: A. Postigo, Eur. J. Org. Chem., 2018, 6391–6404 CrossRef CAS.
- The sense of asymmetric induction, as deduced by the absolute configuration of product 3g based on X-ray analysis, is consonant with the dienamine reacting with an s-cis geometry, as depicted for intermediate II in Fig. 5. This conformational behaviour of dienamines formed from α-phenyl enals is a consequence of 1,3-allylic strain; its effect on the stereochemical outcome of dienamine-promoted asymmetric processes has been already observed: C. Cassani and P. Melchiorre, Org. Lett., 2012, 14, 5590–5593 CrossRef CAS PubMed.
- (a) D. D. M. Wayner, J. J. Dannenberg and D. Griller, Chem. Phys. Lett., 1986, 131, 189–191 CrossRef CAS; (b) C. P. Andrieux, L. Gelis, M. Medebielle, J. Pinson and J. M. Saveant, J. Am. Chem. Soc., 1990, 112, 3509–3520 CrossRef CAS.
- (a) A. Bahamonde and P. Melchiorre, J. Am. Chem. Soc., 2016, 138, 8019–8030 CrossRef CAS PubMed; (b) F. Juliá, T. Constantin and D. Leonori, Chem. Rev., 2022, 122, 2292–2352 CrossRef PubMed.
- (a) L. Buzzetti, G. E. M. Crisenza and P. Melchiorre, Angew. Chem., Int. Ed., 2019, 58, 3730–3747 CrossRef CAS PubMed; (b) M. A. Cismesia and T. P. Yoon, Chem. Sci., 2015, 6, 5426–5434 RSC.
Footnotes |
| † Dedicated to the 70th birthday of Professor Dennis Curran. |
| ‡ Electronic supplementary information (ESI) available: details of experimental procedures, full characterization data, and copies of NMR spectra (PDF). Crystallographic data for compound 3g has been deposited with the Cambridge Crystallographic Data Centre. CCDC 2215186. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3sc01347b |
| This journal is © The Royal Society of Chemistry 2023 |