
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTaming the parent oxoborane†
Gargi
Kundu
ab,
P. R.
Amrutha
a,
K. Vipin
Raj
 bc,
Srinu
Tothadi
d,
Kumar
Vanka
bc and
Sakya S.
Sen
*ab
bc,
Srinu
Tothadi
d,
Kumar
Vanka
bc and
Sakya S.
Sen
*ab
aInorganic Chemistry and Catalysis Division, CSIR-National Chemical Laboratory, Dr Homi Bhabha Road, Pashan, Pune 411008, India. E-mail: ss.sen@ncl.res.in
bAcademy of Scientific and Innovative Research (AcSIR), Ghaziabad 201002, India
cPhysical and Material Chemistry Division, CSIR-National Chemical Laboratory, Dr Homi Bhabha Road, Pashan, Pune 411008, India
dAnalytical and Environmental Sciences Division and Centralized Instrumentation Facility, CSIR-Central Salt and Marine Chemicals Research Institute, Gijubhai Badheka Marg, Bhavnagar-364002, India
First published on 26th April 2023
Abstract
Despite recent advancements in the chemistry of multiply bound boron compounds, the laboratory isolation of the parent oxoborane moiety, HBO has long remained an unsolved and well-recognized challenge. The reaction of 6-SIDipp·BH3 [6-SIDipp = 1,3-di(2,6-diisopropylphenyl)tetrahydropyrimidine-2-ylidene] with GaCl3 afforded an unusual boron–gallium 3c–2e compound (1). The addition of water to 1 resulted in the release of H2 and the formation of a rare acid stabilized neutral parent oxoborane, LB(H)![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O (2). Crystallographic and density functional theory (DFT) analyses support the presence of a terminal BO double bond. Subsequent addition of another equivalent of water molecule led to hydrolysis of the B–H bond to the B–OH bond, but the ‘BO’ moiety remained intact, resulting in the formation of the hydroxy oxoborane compound (3), which can be classified as a monomeric form of metaboric acid.
O (2). Crystallographic and density functional theory (DFT) analyses support the presence of a terminal BO double bond. Subsequent addition of another equivalent of water molecule led to hydrolysis of the B–H bond to the B–OH bond, but the ‘BO’ moiety remained intact, resulting in the formation of the hydroxy oxoborane compound (3), which can be classified as a monomeric form of metaboric acid.
Introduction
The parent oxoborane, HBO is a compound that features boron with both a –BO and a B–H functional group. This ephemeral compound has only been detected in the gas phase or by low-temperature matrix isolation studies.1 Significant effort has recently been focused on generating oxoborane derivatives RBO. The first report of a BO double bond (A) came from Cowley's group in 2005.2 Subsequently, the concept of donor/acceptor stabilization has been successfully employed by the groups of Cui, Kinjo, Rivard, and others (Scheme 1) to generate compounds with BO double bonds (B–D).3–5 The groups of Aldridge and Xie were able to isolate an acid-free, anionic and neutral BO doubly bound species, respectively (E & F).6,7 Braunschweig and coworkers adopted a different strategy for the isolation of multiply bound boron compounds by trapping them within the coordination sphere of a transition metal,8–10 and they were able to isolate the compounds with a B![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) O triple bond (G & H).11 Last year, Braunschweig and coworkers isolated a base-stabilized derivative of the parent borylene carbonyl (HBCO) and provided substantial evidence of the intermediacy of base-stabilized (cAACB(H)O; cAAC = cyclic alkyl amino carbene) and base-free oxoboranes (RBO) and these reported intermediates are also “parent” oxoboranes.12 Inspired by these reports, we wondered whether a parent oxoborane [LB(H)O] could be isolated (L 6-SIDipp; 6-SIDipp = 1,3-di(2,6-diisopropylphenyl)tetrahydropyrimidine-2-ylidene) and how the dominant cyclization reaction could be circumvented.
O triple bond (G & H).11 Last year, Braunschweig and coworkers isolated a base-stabilized derivative of the parent borylene carbonyl (HBCO) and provided substantial evidence of the intermediacy of base-stabilized (cAACB(H)O; cAAC = cyclic alkyl amino carbene) and base-free oxoboranes (RBO) and these reported intermediates are also “parent” oxoboranes.12 Inspired by these reports, we wondered whether a parent oxoborane [LB(H)O] could be isolated (L 6-SIDipp; 6-SIDipp = 1,3-di(2,6-diisopropylphenyl)tetrahydropyrimidine-2-ylidene) and how the dominant cyclization reaction could be circumvented.
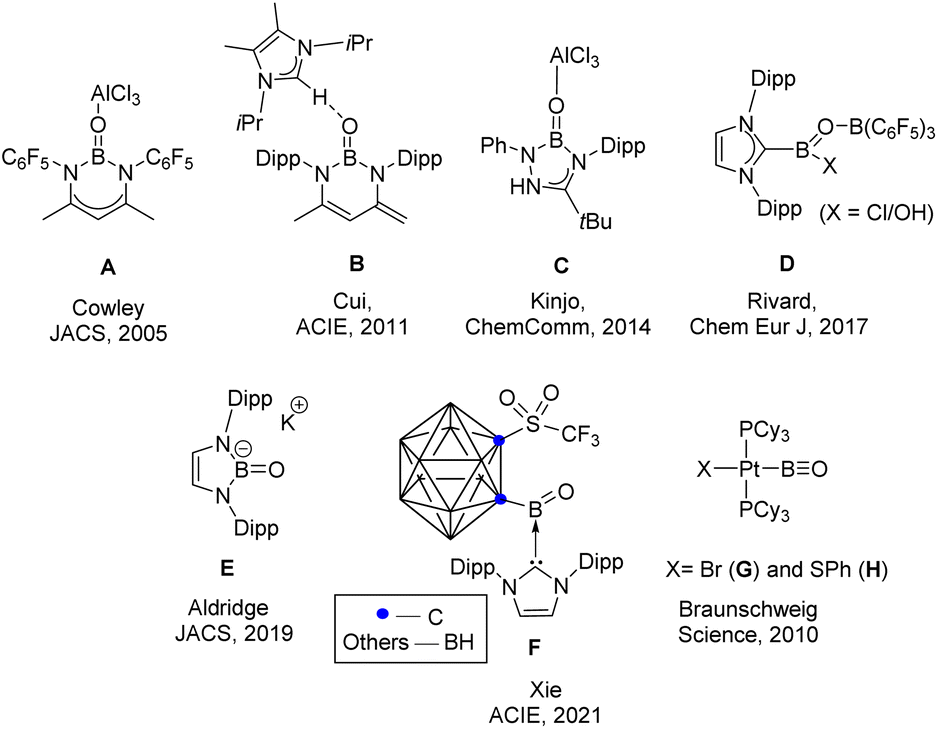 | ||
| Scheme 1 Examples of previously reported oxoborane and a compound with a B–O triple bond. The parent oxoborane is still elusive. | ||
Recently, a wide range of N-heterocyclic carbenes (NHCs) and cyclic alkyl amino carbenes (cAACs) have been utilized to form stable adducts with BH3, thanks to the initial works of Robinson13 and later from the groups of Fensterbank, Lacôte, Malacria, Curran, Braunschweig, and others.14,15 This has resulted in applications in the borylation of aromatic or aliphatic C–H bonds, the hydroboration of alkenes, and many other organic transformation.16 Beginning of the last year, we became interested in employing 6-SIDipp as a ligand in borane chemistry. We demonstrated substitution reactions of 6-SIDipp·BH3, ring expansion with 6-SIDipp·9BBN, and activation of the B–H bond of HBpin with 6-SIDipp.17–19 These studies have indicated that 6-SNHC has a notably higher donating capacity than 5-NHC. Our hypothesis was that by incorporating the 6-SNHC framework, along with bulky Dipp groups, and using a Lewis acid, we could potentially achieve the isolation of a single, parent oxoborane molecule.
Results and discussion
The groups of Vedejs and Curran attempted to synthesize the dihydrido borenium cation, [5-IDipp→BH2]+ starting from 5-Dipp·BH3 [IDipp = 1,3-bis(2,6-diisopropylphenyl)-imidazole-2-ylidene] and AlCl3, but they observed the formation of a cationic hydride-bridged dimer as an intermediate (Scheme 2).20 The latter was not isolated and [5-IDipp·BCl2]+ was obtained as the final product. However, such hydride-bridged dimer was reported by the Vedejs group by the treatment of R3N–BH3 with [Ph3C][B(C6F5)4]21 and by Alcarazo and coworkers upon reaction with 5-IDipp·BH3 with B(C6F5)3.22 The reaction of 6-SIDipp·BH3 with 1.2 equivalent of gallium trichloride in toluene at room temperature resulted in complete disappearance of the 11B{1H} NMR quartet of 6-SIDipp·BH3 at −31.3 ppm (JB–H = 84.1 Hz)17 and the appearance of a new multiplet at −27.4 ppm (JB–H = 72.1 Hz), which is in good agreement with several NHC supported boronium cations.22,23 Single crystal X-ray studies confirm the constitution of 1 (Fig. 1). The B–H–Ga binding mode in 1 can be best described as a three-center two-electron (3c–2e) bond. The location of the two bridging hydrogens and a single terminal hydrogen in each could be inferred from the difference Fourier map. The C1–B1 distance in 1 is 1.596(6) Å, which is good agreement with that in 6-SIDipp·BH3 [1.602(3) Å].17 The terminal B–H bond distance [1.10(4) Å] is slightly longer than those of bridging B–H bonds [1.08(3) Å]. The B–Ga distance for 1 is 2.280(5) Å, which is longer than the reported B–Ga coordinate bond in HC[MeC(2,6-iPr2C6H3)N]2Ga→B(C6F5)3 [2.156(1) and 2.142(3) Å],24 indicating the absence of any interaction between boron and the gallium center. The Ga–H bond distance is 1.83(3) Å, which is slightly shorter than in the Ga–H–Ga bridges.25 The geometry of the boron atom is distorted tetrahedral, while the gallium atom adopts a trigonal bipyramidal geometry.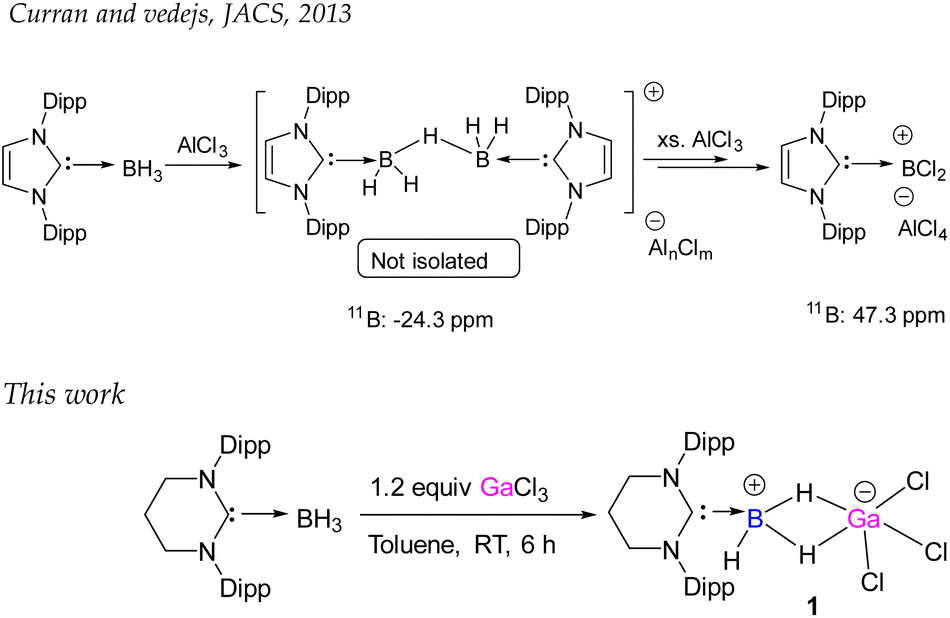 | ||
| Scheme 2 Diverse reactivity of NHC·BH3. The upper scheme describes the generation of 5-IDipp·BCl2 cation. The lower scheme shows the synthesis of boron–gallium 3c–2e complex (1). | ||
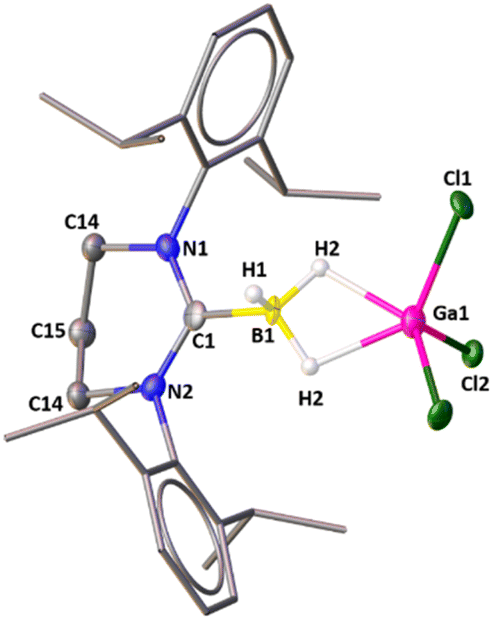 | ||
| Fig. 1 The molecular structure of 1. Hydrogen atoms (except the hydrogens attached with boron atom) are omitted for clarity. Selected bond distances (Å), bond angles (deg), and torsion angle (deg): C1–N1 1.337(3), C1–N2 1.337(3), B1–C1 1.596(6), B1–H1 1.10(4), B1–H2 1.08(3), Ga1⋯B1 2.280(5), Ga1–H2 1.83(3), Ga–Cl1 2.1755(8); N1–C1–N2 119.3(3), N2–C1–B1 120.29(16), N1–C1–B1 120.29(16), C1–B1–H2 112.3(17), C1–B1–H1 112(2). | ||
In order to investigate the electronic structure of compound 1, density functional theory (DFT) calculations were performed. The B3LYP functional and the def2-TZVP basis set have been employed in the calculations (please see the ESI† for further details). The NBO analysis shows that there are two significant electron donations from the BD orbital (which denotes a bonding orbital) of the B1–H2 bond to the vacant s and p orbitals of gallium in 1, with stabilization energies of 24.3 kcal mol−1 and 27.6 kcal mol−1 (see Fig. S17†). Similarly, the BD orbital of the second B–H bond (which is identically denoted as B1–H2 in Fig. 2) also shows two significant electron donations to the vacant s and p orbitals of gallium, with identical (to that of the first B–H bond) stabilization energies of 24.3 kcal mol−1 and 27.6 kcal mol−1 (see Fig. S17†). It should be noted that the NBO analysis in this case resulted in a misinterpretation of the nature of the Ga–Cl bonds as ionic instead of covalent, due to the limitations in NBO in accurately locating polar bonds. Conceptually, the B–H bonding orbitals can donate electron density to either an initially vacant p orbital located on Ga or to a Ga–Cl σ* antibonding orbital, in which the lobe is predominantly located on Ga. This corroborates the experimental observation of two identical B–H bonds and also suggests a 3c–2e bond character in B–H–Ga interactions.
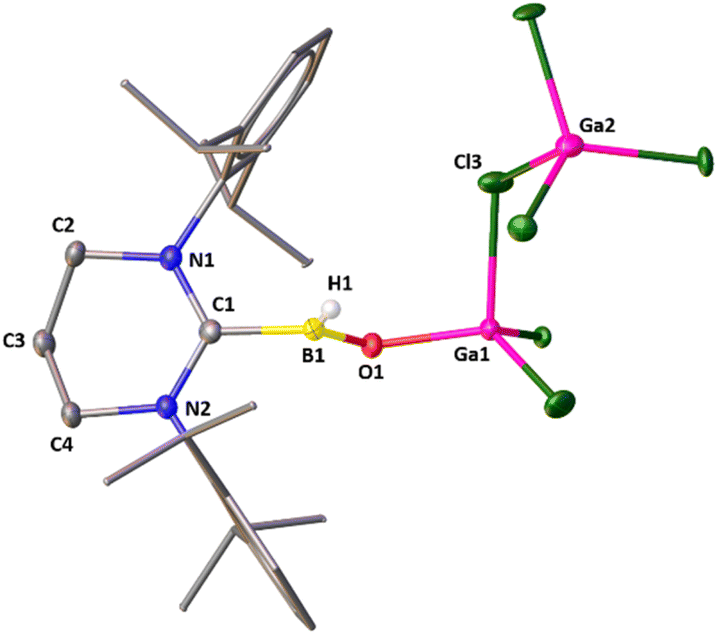 | ||
| Fig. 2 The molecular structure of 2·GaCl3. Hydrogen atoms (except the hydrogen attached with boron atom) are omitted for clarity. Selected bond distances (Å), bond angles (deg), and torsion angle (deg): C1–N1 1.328(3), C1–N2 1.328(3), B1–C1 1.621(4), B1–O1 1.307(3), B1–H1 1.09(3), Ga1–O1 1.807(2), Ga1–Cl3 2.3015(10); N1–C1–N2 122.1(2), N1–C1–B1 118.5(2), C1–B1–O1 118.3(2), C1–B1–H1 117.6(17), B1–O1–Ga1 127.97(18), Ga1–Cl3–Ga2 117.49(4). | ||
Boranes have been identified as promising candidates for facilitating hydrogen release from water, as documented in several early microwave spectroscopic studies, and theoretical calculations.26 It is known that BH3 reacts with H2O to give B(OH)3 with concomitant dihydrogen liberation. Recently, Kraka and co-workers investigated the mechanism of H2 release from BH3 in water and demonstrated that one H2O molecule interacting with BH3 led to an activation energy of 24.9 kcal mol−1.27
We have reacted 1 with 1.1 equivalent of H2O, which resulted in the formation of 2 with simultaneous elimination of dihydrogen (Scheme 3). 2 is a heterodinuclear compound that can be considered as the acid-stabilized parent oxoborane containing an unusual H(BO) linkage. The elimination of dihydrogen was detected using NMR spectroscopy and GC-MS. The amount of simultaneous hydrogen production is quite large (2.1 mmol h−1 g−1). Most importantly, the NHC moiety remained intact during the reaction, which was not the case for 5-IDipp·BH2I and 5-IDipp·BH2OTf as their reactions with water (or methanol) resulted in fast hydrolysis (or methanolysis) to the corresponding imidazolium salts.28 The 11B{1H} NMR spectrum of 2 displays a broad signal at 21.6 ppm for the BO moiety, which falls in the range of 19.7–32.3 ppm, typically found in oxoboranes. The characteristic terminal ν(B–O) stretch frequency appeared at 1665 cm−1 for 2, which is in good agreement with the previously reported oxoboranes (1558–1646 cm−1).5,6
 | ||
| Scheme 3 Stepwise synthesis of -hydrido and -hydroxy oxoboranes, 2 and 3 from the reaction of 1 with water. | ||
Attempts to crystallize 2 resulted in two types of colorless single crystals: one is block shaped, and the other one is needle shaped. While the needle shaped crystals belong to 2, the block shaped crystals account for 2·GaCl3. Though the constitution and the connectivity of 2 can be unambiguously established from the single crystal X-ray data (Fig. S16, ESI†), we refrain from a discussion of bonding parameters because of the low quality of the data. The molecular structure of 2·GaCl3 is displayed in Fig. 2. One of the GaCl3 molecule cocrystallizes with 2. The main feature of 2·GaCl3 is that the central boron atom has bonded to one terminal hydrogen atom and oxygen atom which is connected to the gallium center. The boron atom exhibits a highly distorted trigonal planar coordination site with the carbon atom of NHC, the hydride ligand, and the (μ-O) atom. The gallium atom has a distorted tetrahedral coordination sphere comprised of the (μ-O) atom and the three chloride ligands. The B–H [1.09 (3) Å] and B–C [1.621(4) Å] bond lengths compare well with their typical bond lengths. It is noted that the BO distance in 2·GaCl3 [1.307(3) Å] is in good agreement with the reported acid stabilized oxoboranes,3–6 slightly longer compared to Aldridge's acid free anionic oxoboranes [1.256(3) Å, 1.273(8) Å and 1.287 (2) Å],7 and substantially longer than Braunschweig's BO triply bound complex.11 The Ga–O bond length in 2·GaCl3 [1.807(2) Å] is in good agreement with that in Roesky's Ga(Me)–O–Bi linkage [1.815(13) Å].29
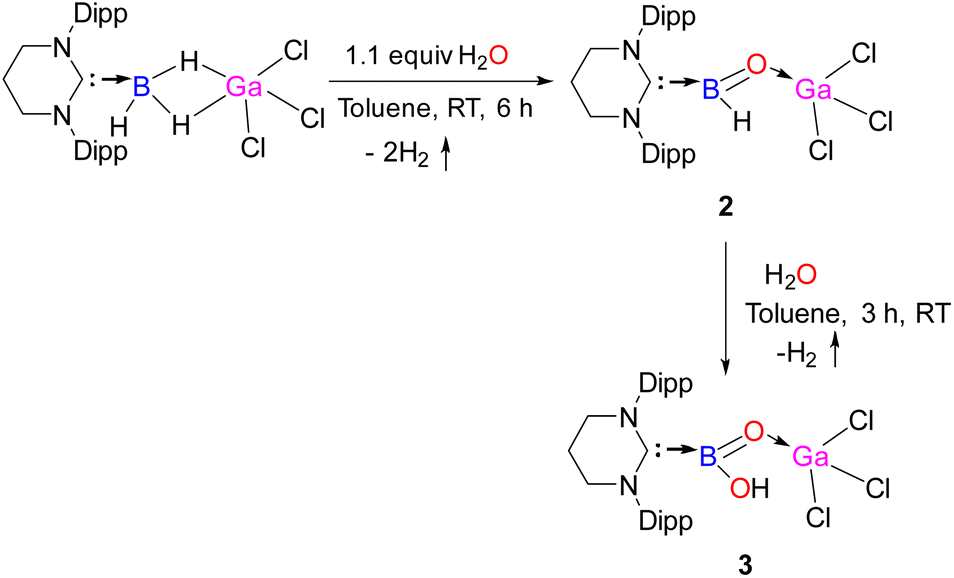
Having found that water undergoes facile dehydrocoupling with the bridging hydride of 1, it seemed plausible that further addition of H2O may lead to the release of another molecule of H2 and convert the terminal B–H bond to a B–OH bond to generate a (hydroxy)oxoborane. Gratifyingly, the reaction of 2 with another molecule of H2O led to the formation of 3 containing a B(OH)O–GaCl3 motif with concomitant elimination of H2 (Scheme 3). The NHC moiety and the (BO)–GaCl3 remained intact, while the B–H bond underwent hydrolysis with H2O to generate the terminal B–OH bond. 3 possesses a rare unit of [B(OH)O], and it is the second example of a monomeric boracarboxylic acid complex that has been isolated, followed by Rivard's discovery of D.5 The [B(OH)O] unit is transient and can only be observed in the gas phase during the thermal conversion of orthoboric acid (B(OH)3) to metaboric acid [(HOBO)3].30 The boron atom resonates at 23.6 ppm. The solid-state structure of 3 is displayed in Fig. 3. The terminal B–O bond length [1.334(10) Å] in 3 is very similar with the B–O double bond [1.320(9)] Å as they overlap within three times of estimated standard deviation. For a note, the B–O double bond in 3 is also comparable to that in 2 [1.307(3) Å]. The characteristic terminal ν(BO) stretching frequency of 3 appeared at 1595 cm−1.
 | ||
| Fig. 3 The molecular structure of 3. Hydrogen atoms (except the hydrogens attached with oxygen atom) are omitted for clarity. Selected bond distances (Å), bond angles (deg), and torsion angle (deg): C20–N1 1.334(5), B1–C20 1.631(9), B1–O2 1.320(9), B1–O1 1.334(10), O1–H1 0.97(2), O2–Ga1 1.822(5), Ga1–Cl1 2.1803(14), Ga1–Cl2 2.159(2); N1–C20–N2 120.4(6), N1–C20–B1 119.7(3), O1–B1–O2 130.2(7), B1–O2–Ga1 131.1(5), Cl2–Ga1–Cl2 111.74(5). | ||
To investigate the electronic structure of 2 and 3, density functional theory (DFT) calculations were performed. The B3LYP functional and the def2-TZVP basis set have been employed in the calculations (see the ESI†for further details). The Wiberg Bond Index (WBI) of the B–O bond in 2 was determined to be 1.19. Moreover, the natural population analysis (NPA) charges on B and O were found to be +0.77 and −1.04, respectively, indicating a multiple bond character of the B–O bond, which is polarized towards oxygen. Similarly, the Wiberg Bond Index (WBI) of the B–O bond in compound 3 is 1.09, which is slightly lower than that of compound 2, indicating a weaker B–O bond in compound 3. The natural population analysis (NPA) charges on B and O were determined to be +1.08 and −1.06, respectively. The higher positive charge on the boron atom in compound 3 is likely due to its bonding with two oxygen atoms, in contrast to one oxygen and one hydrogen in compound 2.
Furthermore, the NBO analysis shows a significant electron donation from the LP orbital (which denotes a lone pair orbital) of oxygen to the vacant p orbital of boron in 2, with a stabilization energy of 85.4 kcal mol−1 (see Fig. 4). This indicates significant B–O π bonding. In the case of 3, the corresponding interaction was weaker, with a stabilization energy of 66.1 kcal mol−1 (see Fig. S18†), indicating a weaker π bonding interaction between boron and oxygen in 3 compared to 2.
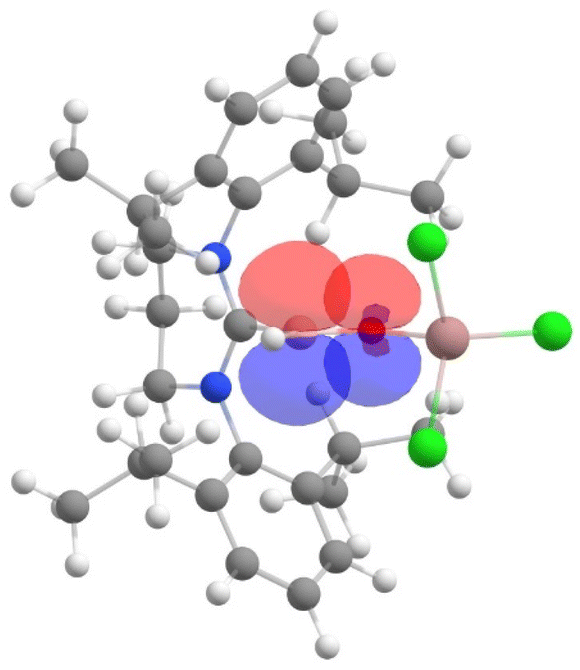 | ||
| Fig. 4 The NBO plot of the desired intramolecular donor–acceptor interactions (LP orbital of oxygen to vacant p orbital of boron) in 2. | ||
Conclusions
In summary, we synthesized the first parent oxoborane derivative (2) by adopting the textbook reaction of BH3 with water. We accessed 2 by reacting an unusual 3c–2e boron–gallium compound (1) with water as an oxygen source, with concomitant elimination of H2. The strong σ-donation from 6-SIDipp and coordination of the BO unit to gallium can be attributed for the stabilization of 2. Further reaction of 2 with another molecule of water resulted in a bora-carboxylic acid derivative (3), which is a monomer of metaboric acid. Investigation of the reactivity 2 and synthesis of other multiply bound boron compounds is underway in our laboratory, and will be published in due course.
Data availability
Synthetic details and analytical data, including depictions of all spectra and detailed accounts on the methods applied are documented in the ESI.† Crystallographic data are made available via the CCDC. Coordinate data of all computationally optimised species are provided as a separate xyz-file in the ESI.†Author contributions
G. K. and S. S. S. conceived the idea. G. K. and P. R. A. have carried out all the experiments. G. K. and S. T. performed the single-crystal XRD measurements. K. V. R. and K. V. have performed the theoretical calculations. All the authors have contributed in writing the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
S. S. S. is thankful for SJF Grant SB/SJF/2021-22/06, GOI for providing financial assistance. G. K. and K. V. R. thank CSIR, India for their research fellowships. K. V. and P. R. A. are grateful to the CSIR-FIRST scheme for providing financial assistance. S. T. is grateful to AESD&CIF, CSIR-CSMCRI for instrumentation facilities and infrastructure. The support and the resources provided by ‘PARAM Brahma Facility’ under the National Supercomputing Mission, Government of India at the Indian Institute of Science Education and Research (IISER) Pune are gratefully acknowledged.Notes and references
- (a) B. Pachaly and R. West, J. Am. Chem. Soc., 1985, 107, 2987–2988 CrossRef CAS; (b) P. Paetzold, S. Neyses and L. Geret, Z. Anorg. Allg. Chem., 1995, 621, 732–736 CrossRef CAS; (c) M. Ito, N. Tokitoh and R. Okazaki, Tetrahedron Lett., 1997, 38, 4451–4454 CrossRef CAS; (d) S. A. Wescott, Angew. Chem., Int. Ed., 2010, 49, 9045–9046 CrossRef PubMed.
- D. Vidovic, J. A. Moore, J. N. Jones and A. H. Cowley, J. Am. Chem. Soc., 2005, 127, 4566–4567 CrossRef CAS PubMed.
- Y. Wang, H. Hu, J. Zhang and C. Cui, Angew. Chem., Int. Ed., 2011, 50, 2816–2819 CrossRef CAS PubMed.
- Y. K. Loh, C. C. Chong, R. Ganguly, Y. Li, D. Vidovic and R. Kinjo, Chem. Commun., 2014, 50, 8561–8564 RSC.
- A. K. Swarnakar, C. Hering-Junghans, M. J. Ferguson, R. McDonald and E. Rivard, Chem.–Eur. J., 2017, 23, 8628–8631 CrossRef CAS PubMed.
- H. Wang, J. Zhang, J. Yang and Z. Xie, Angew. Chem., Int. Ed., 2021, 60, 19008–19012 CrossRef CAS PubMed.
- Y. K. Loh, K. Porteous, M. Á. Fuentes, D. C. H. Do, J. Hicks and S. Aldridge, J. Am. Chem. Soc., 2019, 141, 8073–8077 CrossRef CAS PubMed.
- H. Braunschweig, K. Radacki, D. Rais and K. Uttinger, Angew. Chem., Int. Ed., 2006, 45, 162–165 CrossRef CAS PubMed.
- J. Brand, H. Braunschweig, F. Hupp, A. K. Phukan, K. Radacki and S. S. Sen, Angew. Chem., Int. Ed., 2014, 53, 2240–2244 CrossRef CAS PubMed.
- T. E. Stennett, J. D. Mattock, I. Vollert, A. Vargas and H. Braunschweig, Angew. Chem., Int. Ed., 2018, 57, 4098–4102 CrossRef CAS PubMed.
- H. Braunschweig, K. Radacki and A. Schneider, Science, 2010, 328, 345–347 CrossRef CAS PubMed.
- A. Stoy, M. Härterich, R. D. Dewhurst, J. O. C. Jiménez-Halla, P. Endres, M. Eyßelein, T. Kupfer, A. Deissenberger, T. Thiess and H. Braunschweig, J. Am. Chem. Soc., 2022, 144, 3376–3380 CrossRef CAS PubMed.
- Y. Wang, B. Quillian, P. Wei, C. S. Wannere, Y. Xie, R. B. King, H. F. Schaefer III, P. v. R. Schleyer and G. H. Robinson, J. Am. Chem. Soc., 2007, 129, 12412–12413 CrossRef CAS PubMed.
- (a) S.-H. Ueng, M. M. Brahmi, É. Derat, L. Fensterbank, E. Lacôte, M. Malacria and D. P. Curran, J. Am. Chem. Soc., 2008, 130, 10082–10083 CrossRef CAS PubMed; (b) M. M. Brahmi, J. Monot, M. D.-E. Murr, D. P. Curran, L. Fensterbank, E. Lacôte and M. Malacria, J. Org. Chem., 2010, 75, 6983–6985 CrossRef CAS PubMed; (c) A. Solovyev, Q. Chu, S. J. Geib, L. Fensterbank, M. Malacria, E. Lacôte and D. P. Curran, J. Am. Chem. Soc., 2010, 132, 15072–15080 CrossRef CAS PubMed; (d) W. Dai, S. J. Geib and D. P. Curran, J. Org. Chem., 2018, 83, 8775–8779 CrossRef CAS PubMed; (e) D. P. Curran, A. Solovyev, M. M. Brahmi, L. Fensterbank, M. Malacria and E. Lacôte, Angew. Chem., Int. Ed., 2011, 50, 10294–10317 CrossRef CAS PubMed.
- D. Auerhammer, M. Arrowsmith, H. Braunschweig, R. D. Dewhurst, J. O. C. Jiménez-Halla and T. Kupfer, Chem. Sci., 2017, 8, 7066–7071 RSC.
- (a) A. Prokofjevs, A. Boussonnière, L. Li, H. Bonin, E. Lacôte, D. P. Curran and E. Vedejs, J. Am. Chem. Soc., 2012, 134, 12281–12288 CrossRef CAS PubMed; (b) A. Prokofjevs and E. Vedejs, J. Am. Chem. Soc., 2011, 133, 20056–20059 CrossRef CAS PubMed.
- G. Kundu, V. S. Ajithkumar, K. V. Raj, K. Vanka, S. Tothadi and S. S. Sen, Chem. Commun., 2022, 58, 3783–3786 RSC.
- G. Kundu, K. Balayan, S. Tothadi and S. S. Sen, Inorg. Chem., 2022, 61, 12991–12997 CrossRef CAS PubMed.
- G. Kundu, R. Dixit, S. Tothadi, K. Vanka and S. S. Sen, Dalton Trans., 2022, 51, 14452–14457 RSC.
- A. Prokofjevs, J. W. Kampf, A. Solovyev, D. P. Curran and E. Vedejs, J. Am. Chem. Soc., 2013, 135, 15686–15689 CrossRef CAS PubMed.
- T. S. De Vries and E. Vedejs, Organometallics, 2007, 26, 3079–3081 CrossRef CAS PubMed.
- B. Inés, M. Patil, J. Carreras, R. Goddard, W. Thiel and M. Alcarazo, Angew. Chem., Int. Ed., 2011, 50, 8400–8403 CrossRef PubMed.
- C. Chen, J. Li, C. G. Daniliuc, C. Mück-Lichtenfeld, G. Kehr and G. Erker, Angew. Chem., Int. Ed., 2020, 59, 21460–21464 CrossRef CAS PubMed.
- (a) N. J. Hardman, P. P. Power, J. D. Gorden, C. L. B. Macdonald and A. H. Cowley, Chem. Commun., 2001, 1866–1867 RSC; (b) N. Dettenrieder, C. Schädle, C. Maichle-Mössmer, P. Sirsch and R. Anwander, J. Am. Chem. Soc., 2014, 136, 886–889 CrossRef CAS PubMed.
- (a) C. R. Pulham, A. J. Downs, M. J. Goode, D. W. H. Rankin and H. E. Robertson, J. Am. Chem. Soc., 1991, 113, 5149–5162 CrossRef CAS; (b) A. J. Downs, S. Parsons, C. R. Pulham and P. F. Souter, Angew. Chem., Int. Ed. Engl., 1997, 36, 890–891 CrossRef CAS.
- S. Swinnen, V. S. Nguyen, S. Sakai and M. T. Nguyen, Chem. Phys. Lett., 2009, 472, 175–180 CrossRef CAS.
- S. Nanayakkara, M. Freindorf, Y. Tao and E. Kraka, J. Phys. Chem. A, 2020, 124, 8978–8993 CrossRef CAS PubMed.
- https://d-scholarship.pitt.edu/10833/1/Andrey_Solovyev_PhD_Dissertation_2012.pdf .
- B. Nekoueishahraki, A. Jana, H. W. Roesky, L. Mishra, D. Stern and D. Stalke, Organometallics, 2009, 28, 5733–5738 CrossRef CAS.
- (a) L. Andrews and T. R. Burkholder, J. Chem. Phys., 1992, 97, 7203 CrossRef CAS; (b) D. White, D. E. Mann, P. N. Walsh and A. Sommer, J. Chem. Phys., 1960, 32, 488 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, spectroscopic data, theoretical calculations, and representative NMR spectra. CCDC 2234583 (1), 2234584 (2) and 2234585 (3). For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3sc01544k |
| This journal is © The Royal Society of Chemistry 2023 |