
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLaves phase Ir2Sm intermetallic nanoparticles as a highly active electrocatalyst for acidic oxygen evolution reaction†
Shuai
Zhang
a,
Leilei
Yin
a,
Qingqing
Li
a,
Siyuan
Wang
a,
Weihua
Wang
 b and
Yaping
Du
*a
b and
Yaping
Du
*a
aTianjin Key Lab for Rare Earth Materials and Applications, Center for Rare Earth and Inorganic Functional Materials, Haihe Laboratory of Sustainable Chemical Transformations, Smart Sensing Interdisciplinary Science Center, School of Materials Science and Engineering, National Institute for Advanced Materials, Nankai University, Tianjin 300350, China
bCollege of Electronic Information and Optical Engineering, Nankai University, Tianjin 300350, China
First published on 2nd May 2023
Abstract
Rare earth (RE) intermetallic nanoparticles (NPs) are significant for fundamental explorations and promising for practical applications in electrocatalysis. However, they are difficult to synthesize because of the unusually low reduction potential and extremely high oxygen affinity of RE metal–oxygen bonds. Herein, intermetallic Ir2Sm NPs were firstly synthesized on graphene as a superior acidic oxygen evolution reaction (OER) catalyst. It was verified that intermetallic Ir2Sm is a new phase belonging to the C15 cubic MgCu2 type in the Laves phase family. Meanwhile, intermetallic Ir2Sm NPs achieved a mass activity of 1.24 A mgIr−1 at 1.53 V and stability of 120 h at 10 mA cm−2 in 0.5 M H2SO4 electrolyte, which corresponds to a 5.6-fold and 12-fold enhancement relative to Ir NPs. Experimental results together with density functional theory (DFT) calculations show that in the structurally ordered intermetallic Ir2Sm NPs, the alloying of Sm with Ir atoms modulates the electronic nature of Ir, thereby reducing the binding energy of the oxygen-based intermediate, resulting in faster kinetics and enhanced OER activity. This study provides a new perspective for the rational design and practical application of high-performance RE alloy catalysts.
Introduction
In catalytic reactions, the electronic structure of the active sites plays a key role in the overall reactivity and selectivity of the catalysts. Alloying provides an effective strategy for modulating the electronic structure to improve the catalytic performance.1–3 RE alloys can integrate the unique electronic structure of RE into materials, resulting in significant changes in the electronic structure of catalytic materials.4–7 The unusually negative alloying energy and alloy formation enthalpy provide RE alloys with excellent stability under harsh conditions (strong acid or high-temperature environments).8,9 In addition to the stable structure of RE alloys, lanthanide contraction makes it possible to precisely tailor the electronic structure of the active site.10–12 The robust and tuneable catalytic properties of RE alloys make them one of the most promising advanced catalytic materials.13 However, the synthesis of RE alloy NPs for practical applications is still hindered by many factors. For example, the RE elements have an extremely low standard reduction potential (−2.30 V for Sm3+/Sm) and demand a strong reducing agent to be reduced to a metallic state; thus the traditional wet chemical synthesis route is not applicable.14 In addition, RE elements show extremely high oxygen affinity, and the alloying process needs to be carried out in an inert atmosphere.15 For the above reasons, the theoretical research and practical application of RE nano-alloys are severely limited.10,16,17 Therefore, it is necessary to deepen the understanding of the intermetallic RE systems and explore their application directions.Iridium-based materials are considered to have strong potential for use as OER catalysts in acidic media, but the problems of low mass activity and abundance hinder their wide application.18,19 Alloying with transition metals addresses these critical issues in two ways. Firstly, alloying greatly changes the electronic structure of Ir, thereby optimizing the adsorption and desorption of the surface adsorbates. Secondly, the introduction of foreign atoms can also reduce the use of Ir metal and thus reduce the cost of the catalyst. However, due to the disordered arrangement of solid solution alloys, corrosion and dissolution would inevitably occur, which compromises the benefits of alloying. Therefore, the development of an improved Ir alloy catalyst to exhibit higher activity and durability under acidic operating conditions is urgently needed.20–24 Theoretically, constructing an intermetallic with an ordered atomic arrangement can enhance the electrocatalytic activity and durability, and reduce the use of noble metals.25,26 Alloys modify the electronic state of catalytically active elements through electronic effects, and the degree of modification depends largely on the nature and composition of the second metal.27 RE elements have a unique electronic structure of [Xe]4fn–15d0–16s2 (n = 1–15), which can provide an additional route for adjusting the electronic structure of catalytically active elements.28,29 The electronic perturbation caused by RE doping in transition metals could optimize the valence band of transition metal-based materials.30–33 Due to the difference in electronegativity, electron transfer occurs between Sm and adjacent Ir atoms, which can optimize the electronic structure of Ir. The outer electron shell arrangement of Sm atoms is 4f66s2, which makes it possible to induce the 4f electron configuration to participate in the optimization and regulation of the Ir d-band center (εd) through alloying.
Herein, we synthesized a new phase of intermetallic Ir2Sm NPs on graphene through a facile solid-state synthesis strategy. The structure of Ir2Sm has been confirmed to be a cubic system belonging to the Fd![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m space group.34,35 The synthesized catalyst exhibited enhanced acidic OER activity. To further improve the performance of the catalyst, we optimized the particle size of the intermetallic Ir2Sm NPs. The Ir2Sm NPs achieved a much lower OER overpotential of 275 mV at 10 mA cm−2, better than those of the commercialized Ir/C benchmark (378 mV) and most documented Ir-based alloy catalytic materials. Meanwhile, intermetallic Ir2Sm NPs with 1–2 atomic layers of IrOx have robust stability, showing long-term stability with a current density of 10 mA cm−2 for more than 120 h. The alloying-induced electronic interaction between Sm and Ir tunes the εd of the Ir in intermetallic Ir2Sm, thereby accelerating the rate-determining step (RDS) of the OER and enhancing the OER activity.
m space group.34,35 The synthesized catalyst exhibited enhanced acidic OER activity. To further improve the performance of the catalyst, we optimized the particle size of the intermetallic Ir2Sm NPs. The Ir2Sm NPs achieved a much lower OER overpotential of 275 mV at 10 mA cm−2, better than those of the commercialized Ir/C benchmark (378 mV) and most documented Ir-based alloy catalytic materials. Meanwhile, intermetallic Ir2Sm NPs with 1–2 atomic layers of IrOx have robust stability, showing long-term stability with a current density of 10 mA cm−2 for more than 120 h. The alloying-induced electronic interaction between Sm and Ir tunes the εd of the Ir in intermetallic Ir2Sm, thereby accelerating the rate-determining step (RDS) of the OER and enhancing the OER activity.
Results and discussion
In this study, sodium vapor was used to obtain intermetallic Ir2Sm NPs supported on graphene. As shown in Scheme 1, a mixture of graphene, H2IrCl6·6H2O, SmCl3, and cyanamide was used as the precursor. After annealing treatment, the graphene-supported intermetallic Ir2Sm NPs could be obtained by pickling and drying. Intermetallic Ir2Sm NPs with high crystallinity (denoted as Ir2Sm-L/G) were obtained using an annealing time of 6 h, which facilitated the determination of the crystal structure by XRD Rietveld refinement (Fig. 1a). The lattice parameters are summarized in Table S1.†Fig. 1b shows the unit cell structure, which indicates that Ir2Sm belongs to the C15 cubic MgCu2-type phase in the Laves phase family.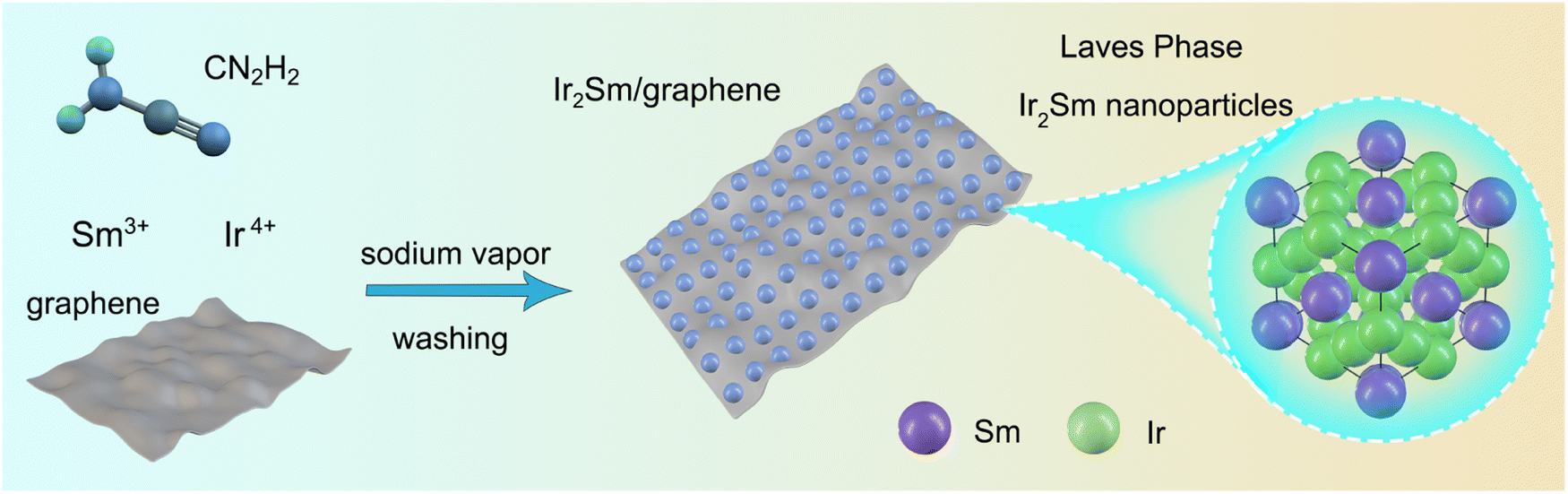 | ||
| Scheme 1 Schematic illustration of the synthesis method for intermetallic Ir2Sm NPs on graphene. | ||
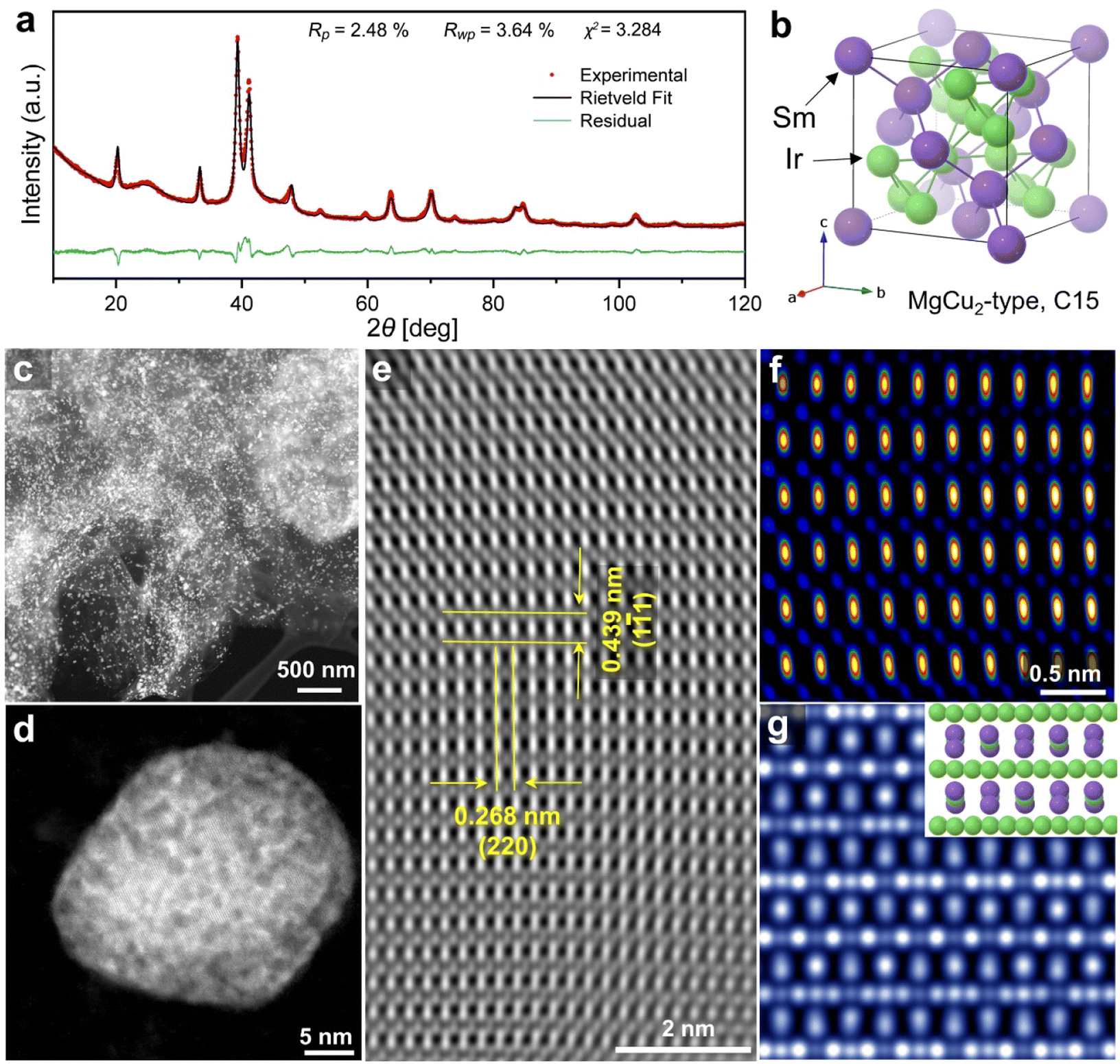 | ||
| Fig. 1 Structural characterization of Ir2Sm-L/G. (a) XRD Rietveld refinement result, (b) the corresponding crystal structure, (c–e) aberration-corrected HAADF-STEM images at different magnifications, (f) HAADF image, and (g) simulated HAADF image. Inset is the atomic model of Ir2Sm, where green and blue spheres are Ir and Sm atoms, respectively. | ||
HAADF-STEM images show that the Ir2Sm-L/G are dispersed on the graphene and exhibit a roughly spherical shape (Fig. 1c and d). Statistical analysis of 300 nanocrystals showed that Ir2Sm-L/G have an average concentration size distribution of 26.4 nm (ESI, Fig. S1†). As shown in Fig. 1e, the spatial distances between the two atoms measured along two orthogonal directions are 0.439 and 0.268 nm, corresponding to the interplanar spacing of the (1![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) 1) and (220) planes, respectively. The HAADF image was analyzed along the [12] zone axis of the intermetallic Ir2Sm NPs. To confirm that the experimentally resolved atomic configuration is consistent with that of the cubic crystal system (space group: Fdm), a HAADF image of [12] was simulated (Fig. 1g),36 and the results show the same brightness pattern as Fig. 1f. Furthermore, elemental mapping tested by energy-dispersive X-ray spectroscopy (EDS) revealed that Ir and Sm elements are homogeneously distributed in individual intermetallic NPs (ESI, Fig. S2 and S3†). In previous work on intermetallic NPs, under acidic conditions, the non-noble metal elements on the surface were etched to form a noble metal surface layer with a thickness of 1–2 atomic layers. Benefiting from higher thermodynamic stability, the inner alloy will stabilize the noble metal elements on the surface layer, thereby increasing the stability of the catalyst.37 There is a small amount of O atoms on the surface of the intermetallic Ir2Sm NPs, which may be caused by the oxidation of Ir element on the NPs surface.
1) and (220) planes, respectively. The HAADF image was analyzed along the [12] zone axis of the intermetallic Ir2Sm NPs. To confirm that the experimentally resolved atomic configuration is consistent with that of the cubic crystal system (space group: Fdm), a HAADF image of [12] was simulated (Fig. 1g),36 and the results show the same brightness pattern as Fig. 1f. Furthermore, elemental mapping tested by energy-dispersive X-ray spectroscopy (EDS) revealed that Ir and Sm elements are homogeneously distributed in individual intermetallic NPs (ESI, Fig. S2 and S3†). In previous work on intermetallic NPs, under acidic conditions, the non-noble metal elements on the surface were etched to form a noble metal surface layer with a thickness of 1–2 atomic layers. Benefiting from higher thermodynamic stability, the inner alloy will stabilize the noble metal elements on the surface layer, thereby increasing the stability of the catalyst.37 There is a small amount of O atoms on the surface of the intermetallic Ir2Sm NPs, which may be caused by the oxidation of Ir element on the NPs surface.
The atomic resolution HAADF-STEM (Fig. 2a) image of the Ir2Sm-L/G shows that the Ir2Sm NPs have an orderly atomic arrangement, and the nanopore structure existing on the NPs still shows the intermetallic structure of Ir2Sm NPs, which indicates that the intermetallic NPs after pickling still maintained the intermetallic compound structure. The surface of the intermetallic NPs has only about 0.23–0.42 nm of amorphous structure (Fig. 2b), and the phase structure of the Ir2Sm NPs is still well maintained. ICP measurement showed that the atomic ratio of Ir and Sm is about 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (ESI, Table S2†). These results indicate that the alloy can exist stably under acidic conditions. Considering the presence of few oxygen atoms on the catalyst surface (ESI, Fig. S2c†), the amorphous surface layer of the NPs consists of Ir and O atoms. To explore the reason for the formation of porous structures in the intermetallic NPs, the EDS elemental mapping of the sample Ir2Sm-L/G precursor (after sodium vapor reduction, without acid etching) was investigated (ESI, Fig. S4 and S5†), where the sample was prepared for TEM testing with ultradry tetrahydrofuran in an argon atmosphere. STEM-EDS line scan showed that Na element is distributed in the intermetallic NPs, which indicates that the nanoporous structure on the alloy surface is caused by the dissolution of the Na remnant during the pickling process.
:1 (ESI, Table S2†). These results indicate that the alloy can exist stably under acidic conditions. Considering the presence of few oxygen atoms on the catalyst surface (ESI, Fig. S2c†), the amorphous surface layer of the NPs consists of Ir and O atoms. To explore the reason for the formation of porous structures in the intermetallic NPs, the EDS elemental mapping of the sample Ir2Sm-L/G precursor (after sodium vapor reduction, without acid etching) was investigated (ESI, Fig. S4 and S5†), where the sample was prepared for TEM testing with ultradry tetrahydrofuran in an argon atmosphere. STEM-EDS line scan showed that Na element is distributed in the intermetallic NPs, which indicates that the nanoporous structure on the alloy surface is caused by the dissolution of the Na remnant during the pickling process.
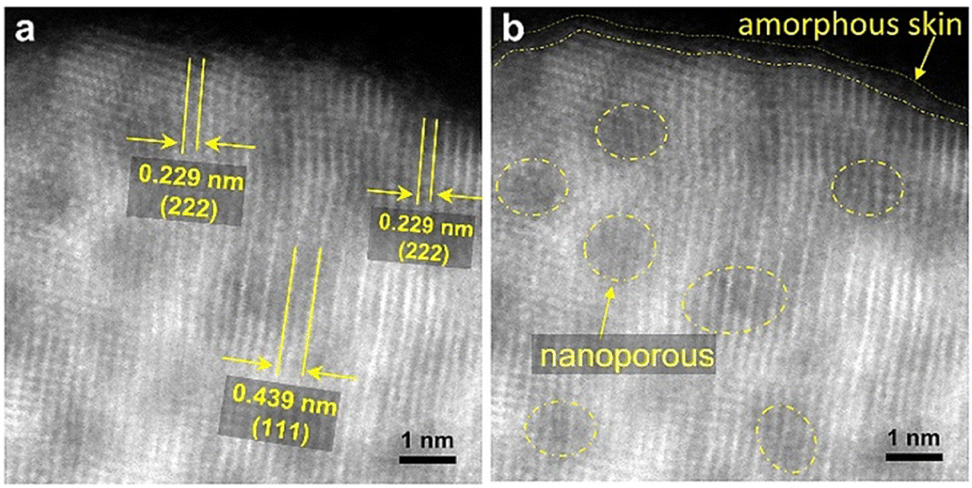 | ||
| Fig. 2 HAADF-STEM images of Ir2Sm-L/G. (a) Crystal face information and (b) nanoporous and amorphous skin distribution. | ||
After confirming the structure of the intermetallic Ir2Sm, the synthesis process was optimized to obtain smaller intermetallic NPs and further increase the number of active sites of the catalyst (ESI, Fig. S6†). As shown in Fig. S7 (ESI†), the average concentrated size distribution of the 300 nanoparticles statistically analyzed was 5.9 nm (denoted as Ir2Sm-S/G). STEM images show that the intermetallic Ir2Sm NPs are uniformly dispersed on the graphene. The lattice stripes shown in Fig. S6c (ESI†) easily point to the (311) and (111) planes of the intermetallic Ir2Sm, and the EDS elemental mapping shows that the Ir and Sm elements are uniformly distributed in a single intermetallic particle.
Furthermore, pure Ir NPs were also prepared in the absence of Sm precursor and characterized (denoted as Ir/G). X-ray photoelectron spectroscopy (XPS) analysis showed partial spontaneous oxidation on the intermetallic NPs surface (Fig. 3). Compared with the Ir4+ valence state signal of Ir/G, the Ir 4f signal of the intermetallic Ir2Sm is shifted to lower binding energies, which indicates the existence of electron transfer from Sm to Ir in the intermetallic.38–40 In the Sm 3d spectrum, the main peak ΔE of 27.5 eV indicates that Sm is mainly in the metallic valence state.30
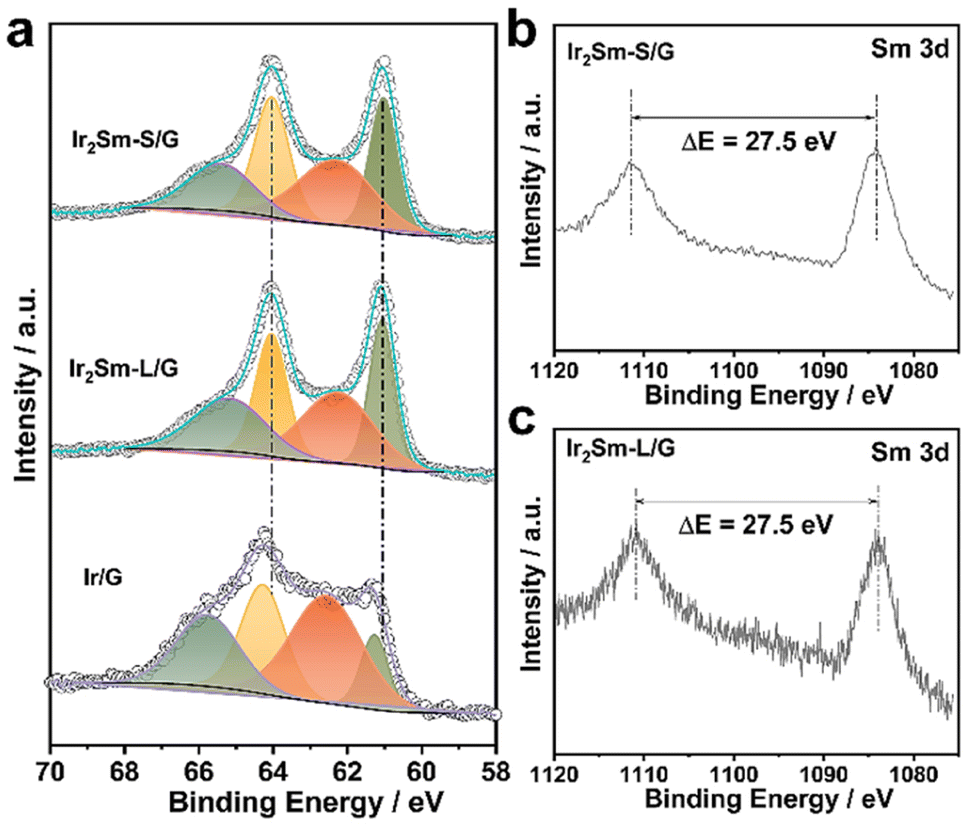 | ||
| Fig. 3 High-resolution XPS spectra and local structure analysis. (a) Ir 4f core levels of Ir2Sm-S/G, Ir2Sm-L/G and Ir/G, (b) Sm 3d core levels of Ir2Sm-S/G, (c) Sm 3d core levels of Ir2Sm-L/G. | ||
Next, the OER performance of Ir2Sm-L/G, Ir2Sm-S/G, Ir/G and commercial Ir/C catalysts was investigated using a typical three-electrode setup in 0.5 M H2SO4 (see ESI† for more details). Fig. 4a shows the linear sweep voltammetry (LSV) curves at 5 mV s−1 for various catalysts. It can be observed that the overpotentials at a current density of 10 mA cm−2 are 275 mV (Ir2Sm-S/G) and 310 mV (Ir2Sm-L/G), which are much lower than those for Ir/G (389 mV) and Ir/C (379 mV), respectively, indicating that the alloying of Sm and Ir effectively enhances the OER activity in acidic medium. Compared with other Ir-based alloy materials, including Ir-Cu, Ir-Ni, and Ir-Rh, Ir2Sm-S/G could work at much lower overpotentials (Table S3†).23,41–45 As shown in Fig. 4b, the intermetallic Ir2Sm NPs exhibit lower Tafel slopes (72.9 mV dec−1 for Ir2Sm-S/G and 81.3 mV dec−1 for Ir2Sm-L/G), indicating that the Sm element effectively modulates the reaction kinetics of Ir in the acidic OER.
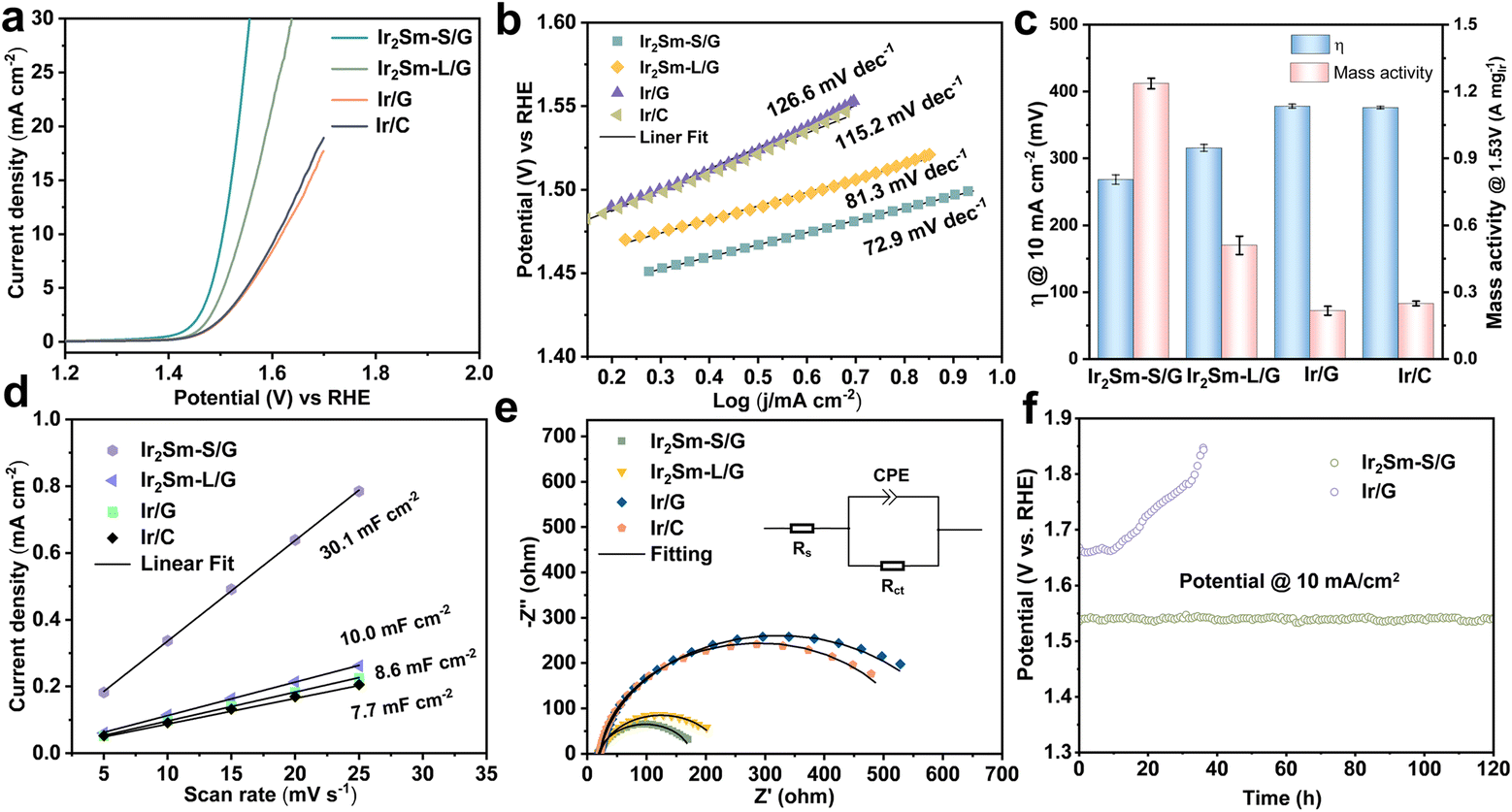 | ||
| Fig. 4 The electrochemical performance of Ir2Sm-L/G, Ir2Sm-S/G, Ir/G and Ir/C in 0.5 M H2SO4 electrolyte. (a) IR-corrected OER polarization curves and (b) Tafel plots. (c) Overpotentials at a current density of 10 mA cm−2 and current density at 1.53 V vs. RHE. (d) Cdl curves by plotting current density versus scan rate. (e) The Nyquist plots. Inset is an equivalent circuit. (f) Chronopotentiometry curves for Ir2Sm-S/G and Ir/G. | ||
The double-layer capacitance (Cdl) of the catalysts derived from the cyclic voltammetry (CV) curves (ESI, Fig. S8†) was used to calculate the electrochemically active surface area (ECSA).46–49 As shown in Fig. 4c, Ir2Sm-S/G has the largest ECSA, which indicates that the synthesis of small-sized intermetallic NPs effectively increases the ECSA (Fig. 4d). To quantitatively evaluate the OER activity of Ir in the catalyst, the current density and the mass activity of Ir at 1.53 V (vs. RHE) were compared. Ir2Sm-L/G yielded a current density of 8.47 mA cm−2 and a mass activity of 0.59 A mgIr−1 at 1.53 V (vs. RHE). Ir2Sm-S/G has a current density of 18.27 mA cm−2 and a mass activity of 1.24 A mgIr−1, which are 5.2 and 5.6 times higher than that of Ir/G (3.52 mA cm−2, 0.22 A mgIr−1), respectively. This confirms the significant enhancement of the electrocatalytic activity after the increase of the number of active sites in the intermetallic Ir2Sm NPs. In addition, the EIS results of Ir2Sm-S/G, Ir2Sm-L/G, Ir/G and Ir/C in Fig. 4e exhibit semicircular curves. The semicircle radius of the intermetallic Ir2Sm NPs is smaller than that of Ir/G and Ir/C, which indicates that the alloying of Ir with Sm atoms effectively accelerates the electron transfer in OER electrolysis. There was little difference in the solution resistances (Rs) of all of the samples. Ir2Sm-S/G has the smallest charge transfer resistance (Rct) of 157.5 Ω, compared with Ir/G (603.0 Ω), Ir/C (532.9 Ω) and Ir2Sm-L/G (206.8 Ω), indicating the excellent charge transfer rate and electron conductivity (Table S4†).
The chronopotentiometry curve was used to test the stability of the catalyst. Ir2Sm-S/G showed long-term stability with a current density of 10 mA cm−2 for more than 120 h (Fig. 4f), while Ir/G started to deactivate after 10 h. After 120 h of OER testing, the stability of Ir2Sm-S/G was determined from the crystal structure and chemical state of the samples. As shown in Fig. S9a and b,† the morphology and phase of the Ir2Sm NPs catalyst remained stable, similar to the pristine sample. XPS analysis (ESI, Fig. S9c and d†) showed that the Ir 4f and Sm 3d spectra of Ir2Sm-S/G were similar before and after electrocatalysis, except for a slight increase of Ir4+ species. These results further demonstrated the stability of Ir2Sm-S/G. The above results indicate that the alloying of Ir with Sm atoms improves both the catalytic activity and the stability.
To further understand the properties of intermetallic Ir2Sm NPs towards the acidic OER, DFT calculations were employed to reveal the active sites and the reaction mechanisms. Based on the surface features obtained from experiments, the Ir (111) and Ir2Sm (111) were used to simulate the surfaces. Considering the slight oxidation of the intermetallic NPs surface and the corrosion of the Sm atoms in the surface layer, Ir atoms are enriched on the surface of Ir2Sm (111). The Ir atoms on the intermetallic NPs surface are slightly oxidized, but mainly keep a metallic state before and after the OER test, thus allowing partial oxidation of the surface of the model (ESI, Fig. S10†). First, by comparing the PDOS of the Ir-5d orbital, the εd was identified, revealing the binding strength and catalytic activity of the Ir sites (Fig. 5a). The f–d orbital coupling further shows that Sm modified the d-band center of Ir through the ligand and strain effect. Compared with pure Ir metal (εd, −2.72 eV), the εd of the Ir2Sm intermetallic is shifted up to −2.14 eV, and the Ir-5d orbitals become less dispersed, which is attributed to the ligand and strain effect in Ir2Sm.
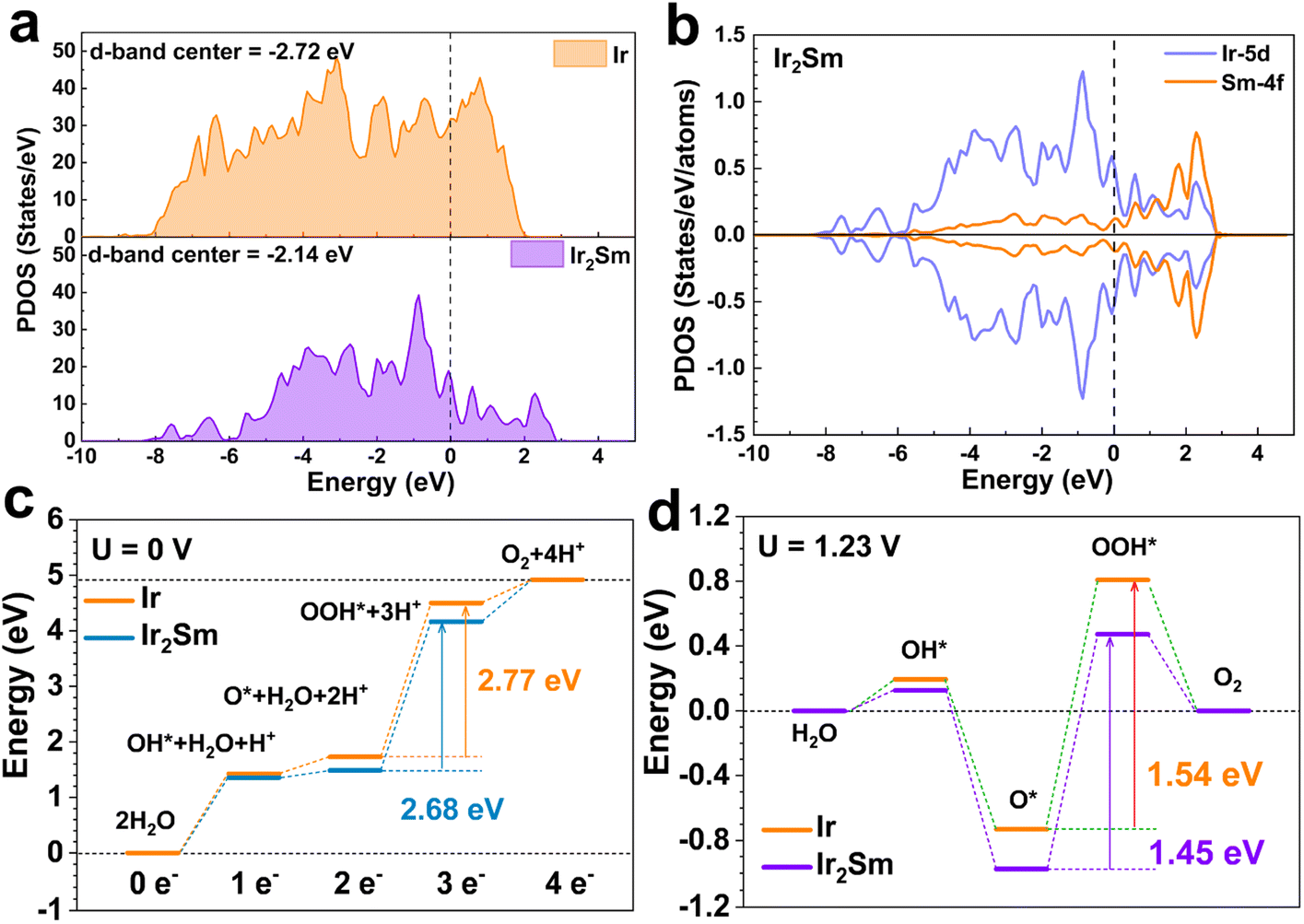 | ||
| Fig. 5 Theoretical calculations. (a) Density of states of d-orbitals of the Ir sites on Ir (111) and Ir2Sm (111) surfaces. (b) The PDOS of Ir-5d and Sm-4f orbitals in Ir2Sm (111). (c) Pathways of the acidic OER at U = 0 V and (d) U = 1.23 V (vs. RHE). | ||
In intermetallic Ir2Sm, the Sm-4f orbitals show splitting near the Fermi level (EF), which indicates the existence of electronic interaction between Ir and Sm atoms, and the overlap of the Ir-5d and Sm-4f orbitals supports fast electron transfer and protects Ir sites under acidic media (Fig. 5b). The εd is enhanced by alloying and the electrocatalytic activity of Ir is activated. Furthermore, the Ir-5d orbitals are up-shifted and relatively localized, supporting enhanced electrical activity to guarantee an efficient OER process. For OER, free energy maps of the Ir (111) and Ir2Sm (111) surfaces at U = 0 V vs. RHE and 1.23 V vs. RHE are shown in Fig. 5c and d, respectively. The first step is the adsorption of water molecules on the Ir2Sm (111) surface to form OH*, and the second step is the deprotonation of OH* species to form O*. Then, the adsorption of another water molecule occurs to form OOH*. Finally, molecular oxygen is generated. The free energy diagrams (Fig. 5c and d) show that the step of forming the *OOH group from the *O group is strongly suppressed, which is considered to be the RDS. The initial adsorption and dissociation of H2O is more favored on the Ir2Sm than the initial oxidation step on Ir, indicating a stronger reaction tendency. The ΔG value of the RDS corresponding to Ir (111) is 1.54 eV, and the ΔG value of the RDS corresponding to Ir2Sm (111) is 1.45 eV, indicating that the OER of Ir2Sm is more favorable. The alloying-induced electronic interaction between Sm and Ir changes the position of the Ir εd in the Ir2Sm alloy, which affects the adsorption strength of the adsorbate on the catalyst surface. The DFT calculations are in good agreement with the experimental results.
Conclusions
In summary, a new Laves phase of intermetallic Ir2Sm NPs was successfully synthesized, which belongs to the Fdm space group. Through optimizing the NPs size, the Ir2Sm-S/G exhibits enhanced OER activity in 0.5 M H2SO4 electrolyte. Specifically, the Ir2Sm-S/G delivers a mass activity of 1.24 A mgIr−1 at 1.53 V and excellent stability for 120 h at 10 mA cm−2. Experimental results together with theoretical studies show that the alloying of Ir and Sm atoms tailors the electronic properties of Ir, resulting in favorable adsorption energies for OER intermediates and further lowering the reaction barrier for the RDS. The synthesis of intermetallic RE NPs will inspire a new strategy for the rational design of efficient electrocatalysts.
Data availability
The data that support the finding of this study are available in the main text and the ESI.†Author contributions
Y. D. proposed the whole research direction and guided the project. S. Z., L. Y., S. W., Q. L. and Y. D. designed and carried out all the synthesis, characterizations and electrochemical measurements. W. W. helped to analyze the theoretical part. All the authors contributed to the formal analysis and writing – original draft.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge the support from the Natural Science Foundation of China (21971117), Functional Research Funds for the Central Universities, Nankai University (63186005), Tianjin Key Lab for Rare Earth Materials and Applications (ZB19500202), 111 Project (No. B18030) from China, the Outstanding Youth Project of Tianjin Natural Science Foundation (20JCJQJC00130), the Key Project of Tianjin Natural Science Foundation (20JCZDJC00650), the National Postdoctoral Program for Innovative Talents (BX20220157), Open Foundation of State Key Laboratory of Featured Metal Materials and Life-cycle Safety for Composite Structures (Grant No. 2022GXYSOF07), Tianjin “131” Innovative Talent Team Construction Project, and Haihe Laboratory of Sustainable Chemical Transformations. This work was also supported by the PhD Candidate Research Innovation Fund of NKU School of Materials Science and Engineering.References
- T. N. Ye, S. W. Park, Y. Lu, J. Li, M. Sasase, M. Kitano, T. Tada and H. Hosono, Nature, 2020, 583, 391–395 CrossRef CAS.
- J. Guo, Y. Zheng, Z. Hu, C. Zheng, J. Mao, K. Du, M. Jaroniec, S. Z. Qiao and T. Ling, Nat. Energy, 2023, 8, 264–272 CAS.
- M. Zhou, C. Li and J. Fang, Chem. Rev., 2021, 121, 736–795 CrossRef CAS PubMed.
- B. Zheng, J. Fan, B. Chen, X. Qin, J. Wang, F. Wang, R. Deng and X. Liu, Chem. Rev., 2022, 122, 5519–5603 CrossRef CAS PubMed.
- S. Zhang, S. E. Saji, Z. Yin, H. Zhang, Y. P. Du and C. H. Yan, Adv. Mater., 2021, 33, 2005988 CrossRef CAS PubMed.
- Y. T. Gong, J. Z. Wu, M. Kitano, J. J. Wang, T. N. Ye, J. Li, Y. Kobayashi, K. Kishida, H. Abe, Y. Niwa, H. S. Yang, T. Tada and H. Hosono, Nat. Catal., 2018, 1, 178–185 CrossRef CAS.
- Y. Yan, J. S. Du, K. D. Gilroy, D. Yang, Y. Xia and H. Zhang, Adv. Mater., 2017, 29, 1605997 CrossRef PubMed.
- S. G. Peera, T. G. Lee and A. K. Sahu, Sustain. Energy Fuels, 2019, 3, 1866–1891 RSC.
- C. Kim, F. Dionigi, V. Beermann, X. Wang, T. Moller and P. Strasser, Adv. Mater., 2019, 31, 1805617 CrossRef.
- S. Zhang, Z. Zeng, Q. Li, B. Huang, X. Zhang, Y. P. Du and C. H. Yan, Energy Environ. Sci., 2021, 14, 5911–5918 RSC.
- J. Shan, C. Ye, C. Zhu, J. Dong, W. Xu, L. Chen, Y. Jiao, Y. Jiang, L. Song, Y. Zhang, M. Jaroniec, Y. Zhu, Y. Zheng and S. Z. Qiao, J. Am. Chem. Soc., 2022, 144, 23214–23222 CrossRef CAS PubMed.
- H. Mizoguchi, S. W. Park, K. Kishida, M. Kitano, J. Kim, M. Sasase, T. Honda, K. Ikeda, T. Otomo and H. Hosono, Zeolitic Intermetallics: LnNiSi (Ln = La-Nd), J. Am. Chem. Soc., 2019, 141, 3376–3379 CrossRef CAS PubMed.
- M. E. Escribano, P. Malacrida, M. H. Hansen, U. G. V. Hansen, A. V. Palenzuela, V. Tripkovic, J. Schiotz, J. Rossmeisl, I. E. L. Stephens and I. Chorkendorff, Science, 2016, 352, 73–76 CrossRef PubMed.
- Y. Hu, J. O. Jensen, L. N. Cleemann, B. A. Brandes and Q. Li, J. Am. Chem. Soc., 2020, 142, 953–961 CrossRef CAS PubMed.
- J. S. Kanady, P. Leidinger, A. Haas, S. Titlbach, S. Schunk, K. S. Arndt, E. J. Crumlin, C. H. Wu and A. P. Alivisatos, J. Am. Chem. Soc., 2017, 139, 5672–5675 CrossRef CAS PubMed.
- Y. Hu, J. O. Jensen, P. Norby, L. N. Cleemann, F. Yang and Q. Li, Chem. Mater., 2021, 33, 535–546 CrossRef CAS.
- R. Ryoo, J. Kim, C. Jo, S. Han, W. J. C. Kim, H. Park, J. Han, H. S. Shin and J. W. Shin, Nature, 2020, 585, 221–224 CrossRef CAS PubMed.
- J. Wang, L. Han, B. Huang, Q. Shao, H. L. Xin and X. Huang, Nat. Commun., 2019, 10, 5692 CrossRef CAS PubMed.
- S. Hao, H. Sheng, M. Liu, J. Huang, G. Zheng, F. Zhang, X. Liu, Z. Su, J. Hu, Y. Qian, L. Zhou, Y. He, B. Song, L. Lei, X. Zhang and S. Jin, Nat. Nanotechnol., 2021, 16, 1371–1377 CrossRef CAS PubMed.
- Y. Ge, X. Wang, B. Chen, Z. Huang, Z. Shi, B. Huang, J. Liu, G. Wang, Y. Chen, L. Li, S. Lu, Q. Luo, Q. Yun and H. Zhang, Adv. Mater., 2022, 34, 2107399 CrossRef CAS PubMed.
- L. Li, P. Wang, Q. Shao and X. Huang, Adv. Mater., 2021, 33, 2004243 CrossRef CAS PubMed.
- C. Tang, L. Chen, H. Li, L. Li, Y. Jiao, Y. Zheng, H. Xu, K. Davey and S. Z. Qiao, J. Am. Chem. Soc., 2021, 143, 7819–7827 CrossRef CAS PubMed.
- J. Feng, F. Lv, W. Zhang, P. Li, K. Wang, C. Yang, B. Wang, Y. Yang, J. Zhou, F. Lin, G. C. Wang and S. Guo, Adv. Mater., 2017, 29, 1703798 CrossRef PubMed.
- J. Kibsgaard and I. Chorkendorff, Nat. Energy, 2019, 4, 430–433 CrossRef.
- H. Zhang, P. Shi, X. Ma, C. Ma, S. Han, C. He, H. Wu, L. Zhu, B. Zhang, Y. Lu, W. Cao, H. Yin, X. Meng, J. Xia, J. Zhang, A. L. Wang and Q. Lu, Adv. Energy Mater., 2022, 13, 2202703 CrossRef.
- J. Li, S. Sharma, X. Liu, Y. T. Pan, J. S. Spendelow, M. Chi, Y. Jia, P. Zhang, D. A. Cullen, Z. Xi, H. Lin, Z. Yin, B. Shen, M. Muzzio, C. Yu, Y. S. Kim, A. A. Peterson, K. L. More, H. Zhu and S. Sun, Joule, 2019, 3, 124–135 CrossRef CAS.
- Y. Nakaya and S. Furukawa, Chem. Rev., 2023, 123, 5859–5947 CrossRef CAS PubMed.
- B. Zheng, J. Fan, B. Chen, X. Qin, J. Wang, F. Wang, R. Deng and X. Liu, Chem. Rev., 2022, 122, 5519–5603 CrossRef CAS PubMed.
- C. Fan, X. Wang, X. Wu, Y. Chen, Z. Wang, M. Li, D. Sun, Y. Tang and G. Fu, Adv. Energy Mater., 2022, 32, 2203244 Search PubMed.
- X. Sun, K. Yuan and Y. W. Zhang, J. Rare Earth, 2020, 38, 801–818 CrossRef CAS.
- X. Shi, B. Cao, J. Liu, J. Zhang and Y. P. Du, Small, 2021, 17, 2005371 CrossRef CAS PubMed.
- Q. Miao, S. Yang, Q. Xu, M. Liu, P. Wu, G. Liu, C. Yu, Z. Jiang, Y. Sun and G. F. Zeng, Small Struct., 2022, 3, 2100225 CrossRef CAS.
- Y. Peng, Y. Bai, C. L. Liu, S. Cao, Q. Q. Kong and H. Pang, Coord. Chem. Rev., 2022, 466, 214602 CrossRef CAS.
- A. Morozkin, Y. D. Seropegin, A. Gribanov and J. Barakatova, J. Alloys Compd., 1997, 256, 175–191 CrossRef CAS.
- Z. Blazina, R. C. Mohanty and A. Raman, Int. J. Mater. Res., 1989, 80, 192–196 CrossRef CAS.
- J. Barthel, Ultramicroscopy, 2018, 193, 1–11 CrossRef CAS PubMed.
- Q. Shi, C. Zhu, D. Du and Y. Lin, Chem. Soc. Rev., 2019, 48, 3181–3192 RSC.
- Y. Inami, H. Ogihara, S. Nagamatsu, K. Asakura and I. Yamanaka, ACS Catal., 2019, 9, 2448–2457 CrossRef CAS.
- T. Toliński, G. Chełkowska, B. Andrzejewski, A. Kowalczyk, M. Kováč and J. Timko, Phys. Status Solidi A, 2003, 196, 294–296 CrossRef.
- H. Jin, X. Liu, P. An, C. Tang, H. Yu, Q. Zhang, H.-J. Peng, L. Gu, Y. Zheng, T. Song, K. Davey, U. Paik, J. Dong and S. Z. Qiao, Nat. Commun., 2023, 14, 354 CrossRef CAS PubMed.
- M. Li, Z. Zhao, Z. Xia, M. Luo, Q. Zhang, Y. Qin, L. Tao, K. Yin, Y. Chao and L. J. Gu, Angew. Chem., Int. Ed., 2021, 133, 8324–8331 CrossRef.
- Y. Pi, J. Guo, Q. Shao and X. Huang, Chem. Mater., 2018, 30, 8571–8578 CrossRef CAS.
- J. Zhu, M. Xie, Z. Chen, Z. Lyu, M. Chi, W. Jin and Y. J. Xia, Adv. Energy Mater., 2020, 10, 1904114 CrossRef CAS.
- D. Cao, H. Xu and D. J. Cheng, Adv. Energy Mater., 2020, 10, 1903038 CrossRef CAS.
- H. Y. Guo, Z. W. Fang, H. Li, D. Fernandez, G. Henkelman, S. M. Humphrey and G. H. Yu, ACS Nano, 2019, 13, 13225–13234 CrossRef CAS PubMed.
- L. Li and Z. C. Zhang, Rare Met., 2022, 41, 3943–3945 CrossRef CAS.
- J. M. Luo, E. Matios, H. Wang, X. Y. Tao and W. Y. Li, InfoMat, 2020, 2, 1057–1076 CrossRef CAS.
- H. X. Huang, J. J. Zha, S. S. Li and C. L. Tan, Chin. Chem. Lett., 2022, 33, 163–176 CrossRef CAS.
- Y. Zhang, L. X. Xue, H. B. Liang, Y. Z. Chen, J. J. Liu, C. Shen, Q. Li, Y. Q. Cai, C. L. Tan and Z. M. Luo, Appl. Surf. Sci., 2021, 561, 150079 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sc01052j |
| This journal is © The Royal Society of Chemistry 2023 |