
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHAA by the first {Mn(III)OH} complex with all O-donor ligands†
Shawn M.
Moore
a,
Chen
Sun
a,
Jennifer L.
Steele
a,
Ellen M.
Laaker
a,
Arnold L.
Rheingold
b and
Linda H.
Doerrer
 *a
*a
aBoston University, Chemistry Department, 590 Commonwealth Avenue, Boston, Massachusetts 02215, USA. E-mail: doerrer@bu.edu
bUniversity of California, San Diego Department of Chemistry and Biochemistry, 9500 Gilman Drive, La Jolla, California 92093, USA
First published on 12th July 2023
Abstract
There is considerable interest in MnOHx moieties, particularly in the stepwise changes in those O–H bonds in tandem with Mn oxidation state changes. The reactivity of aquo-derived ligands, {MOHx}, is also heavily influenced by the electronic character of the other ligands. Despite the prevalence of oxygen coordination in biological systems, preparation of mononuclear Mn complexes of this type with all O-donors is rare. Herein, we report several Mn complexes with perfluoropinacolate (pinF)2− including the first example of a crystallographically characterized mononuclear {Mn(III)OH} with all O-donors, K2[Mn(OH)(pinF)2], 3. Complex 3 is prepared via deprotonation of K[Mn(OH2)(pinF)2], 1, the pKa of which is estimated to be 18.3 ± 0.3. Cyclic voltammetry reveals quasi-reversible redox behavior for both 1 and 3 with an unusually large ΔEp, assigned to the Mn(III/II) couple. Using the Bordwell method, the bond dissociation free energy (BDFE) of the O–H bond in {Mn(II)–OH2} is estimated to be 67–70 kcal mol−1. Complex 3 abstracts H-atoms from 1,2-diphenylhydrazine, 2,4,6-TTBP, and TEMPOH, the latter of which supports a PCET mechanism. Under basic conditions in air, the synthesis of 1 results in K2[Mn(OAc)(pinF)2], 2, proposed to result from the oxidation of Et2O to EtOAc by a reactive Mn species, followed by ester hydrolysis. Complex 3 alone does not react with Et2O, but addition of O2 at low temperature effects the formation of a new chromophore proposed to be a Mn(IV) species. The related complexes K(18C6)[Mn(III)(pinF)2], 4, and (Me4N)2[Mn(II)(pinF)2], 5, have also been prepared and their properties discussed in relation to complexes 1–3.
Introduction
Manganese-dependent enzymes effect both oxidation and reduction reactions involving O2. From the dioxygenation of cis, cis-1,4-pentadiene-containing fatty acids to alkyl hydro-peroxides by manganese lipoxygenases (MnLOX),1–3 to the reduction of water in the oxygen evolving cluster (OEC) of photosystem II (PSII),4,5 manganese is essential for both breaking and forming the O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond. Mn-oxo, -hydroxo, and -aquo species, collectively {MnOHx} (x = 0, 1, 2), have been proposed to play critical roles in the function of several of these enzymes (Chart 1). For example, a mononuclear Mn(III)–OH moiety has been implicated in the catalytic cycle of Mn-superoxide dismutase (MnSOD),6 which defends aerobic organisms from reactive oxygen species by catalyzing the disproportionation of superoxide into H2O2 and O2. Likewise, {MnOHx} species have also been proposed in the active sites of both Mn-lipoxygenase and the OEC.7,8
O bond. Mn-oxo, -hydroxo, and -aquo species, collectively {MnOHx} (x = 0, 1, 2), have been proposed to play critical roles in the function of several of these enzymes (Chart 1). For example, a mononuclear Mn(III)–OH moiety has been implicated in the catalytic cycle of Mn-superoxide dismutase (MnSOD),6 which defends aerobic organisms from reactive oxygen species by catalyzing the disproportionation of superoxide into H2O2 and O2. Likewise, {MnOHx} species have also been proposed in the active sites of both Mn-lipoxygenase and the OEC.7,8
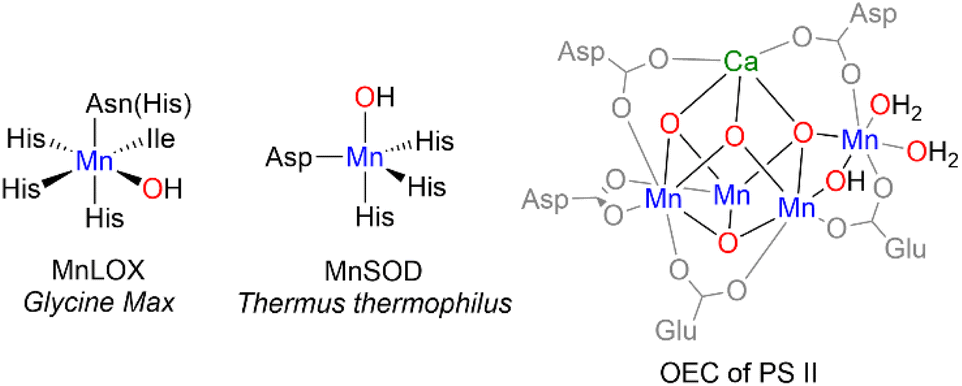 | ||
| Chart 1 Primary coordination spheres of three Mn containing enzymes. | ||
Importantly, each of these enzymes uses proton-coupled electron-transfer (PCET) reactions. The Mn center shuttles among the +2, +3, and +4 oxidation states while oxo, hydroxo, or aquo moieties are (de)protonated. This separable e−/H+ transfer reactivity is distinct from concerted e−/H+ transfer, also called a hydrogen atom transfer (HAT). In some cases, PCET mechanisms at {MOHx} sites cleave thermodynamically strong (>100 kcal mol−1) and kinetically inert C–H bonds.9 For example, Fe and Cu dependent methane monooxygenases catalyze the oxidation of CH4 (C–H BDFE = 105 kcal mol−1)10 to CH3OH via a PCET mechanism that invokes metal-oxo, -hydroxo, and -aquo species.11
As our understanding of the role of {MnOHx} species in biology has grown, so too has interest in the synthesis of metal complexes that mimic the structure and particularly the function of these enzymes. Histidine N-ligation is extremely widespread in Nature, including the MnLOX and MnSOD enzymes as shown in Chart 1. Therefore, several {MnOHx} complexes with N-donor ligands as models for histidine that can oxidize C–H and/or O–H bonds have been prepared, a selection of which are shown in Chart 2. The Borovik,12,13 Goldberg,14 Jackson,15 Kovacs,16 Nam,17 and Stack18 groups have each prepared mononuclear {MnOHx} complexes supported by amino, amido, and/or pyridyl ligands from Mn(II) starting materials using oxidants such as O2, H2O2, or PhIO. The reactivity of these complexes is dependent on the N-donor ligands. Donation from nitrogen lone pairs into the MnO π* orbital can weaken the MnO bond rendering it more reactive. High reactivity in model complexes, however, often leads to intra- or intermolecular ligand oxidation and makes isolation and characterization of reactive species a challenge.
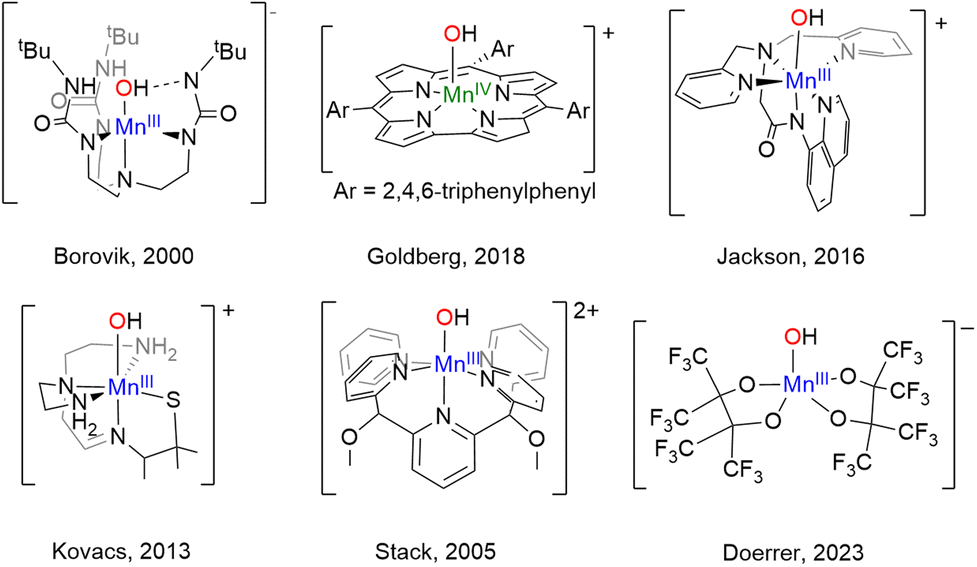 | ||
| Chart 2 Selected {Mn(OH)} complexes. | ||
In contrast to the N-donor rich coordination of MnLOX and MnSOD, the OEC shown on the right of Chart 1 features a CaMn4O5 cluster with Mn in an all O-coordination environment made up of -oxo, -hydroxo, and -aquo ligands in addition to aspartate and glutamate residues.4 Model complexes (structural, spectroscopic, or functional) with exclusively O-donor environments are much less common than their N-donor counterparts. To the best of our knowledge, the four-coordinate complex, [Mn(IV)(O)(ditox)3]−, from the Nocera19 group remains the only structurally characterized example of an O-coordinated mononuclear Mn complex with C–H bond activation abilities. In the trianionic, ∼C3v ligand field of the tris-ditox environment, the Mn(IV)O unit is sufficiently stable to permit isolation, but only reacts with C–H bonds having BDE <85 kcal mol−1. Computational studies have attributed the enhanced reactivity of this complex to low energy exited states that lower the barrier for reaction compared to the ground state.20 The dearth of oxidatively active O-ligated Mn complexes is due in large part to synthetic challenges posed by O-donor ligands. Neutral O-donors like H2O, ketones, and ethers form relatively weak Mn–O bonds leading to lability. Anionic alkoxides form stronger bonds, but π-donation from the oxygen lone pairs can lead to less reactive, coordinatively saturated alkoxide-bridged complexes. Using bulky alkoxides can prevent bridging, but excess bulk can inhibit substrate binding and prevent reactivity.21
Recently, the Kulik group carried out a computational screen of over 2500 mid-row 3d transition-metal (Cr, Mn, Fe, Co) complexes to uncover trends in reactivity.22 They found that stabilizing high-spin ground states enhances catalytic activity by enabling spin-allowed transitions to or from closed-shell substrates. Their analysis identified weak-field, O-coordinating ligands as promising candidates for future design of Fe(II) C–H activation catalysts, and for transition metal catalysts in general. This study underscores the need to develop ligand systems which leverage the promising effects of O-donor ligation on catalyst reactivity without the challenges posed by alkoxide ligation.
In the last decade, our group has demonstrated the ability of fluorinated alkoxides to support 3d metals in a variety of oxidation states and geometries.23–29 These ligands can stabilize the high oxidation states necessary for oxidation chemistry and the high-spin states that the Kulik group22 and others20,30 have shown to be beneficial to catalysis. We have prepared a highly-unusual square-planar S = 1 Co(III) complex,24 a high-spin S = 2 Fe(II) complex,27 and a bis-(μ2-OH) bridged V(IV) dimer that results from HAA (hydrogen atom abstraction) from alcohols.31 This latter process is part of catalytic oxidative alcohol dehydrogenation.31 Ligand fluorination decreases alkoxide bridging of metal centers by reducing π-bonding and Lewis basicity via the inductive effect. Fluorinated alkoxides are oxidatively robust when compared to their hydrogenated analogues due to the greater stability of C–F bonds versus C–H bonds. Ligand fluorination also increases water tolerance via the lower alcohol pKa values of up to 14 orders of magnitude.32 In some cases, transition metal complexes with fluorinated ligands have been able to oxidize C–H31,33 and O–H bonds.31
In this work we present the synthesis and characterization of several Mn complexes supported by the perfluoropinacolate (pinF) ligand. These complexes have all O-donor coordination leading to anionic complexes with supporting cations that form non-covalent K+⋯O and K+⋯F interactions with ligand and solvent molecules. These non-covalent interactions allowed, for the first time, isolation and crystallographic characterization of a {Mn(III)OH} complex with all O-donors, 3. This compound is a precursor to a potent oxidant of Et2O.
Results and discussion
Syntheses and crystallographic structures
The increased acidity afforded by perfluorination allows some pinF complexes to be prepared in water on the benchtop. Combining an aqueous solution of MnSO4 with an aqueous solution containing two equivalents of H2pinF and four of KOH in air affords a pink solution that darkens to a deep maroon over several hours, indicative of oxidation from Mn(II) to Mn(III) (Scheme 1, top). Purification results in brick red needles of K[Mn(OH2)(pinF)2], 1, that has a square pyramidal Mn(III) center with τ5 = 0.01 in an asymmetric unit containing three molecules of 1 and seven H2O (Fig. S1†). One of the Mn-based anions is shown in Fig. 1.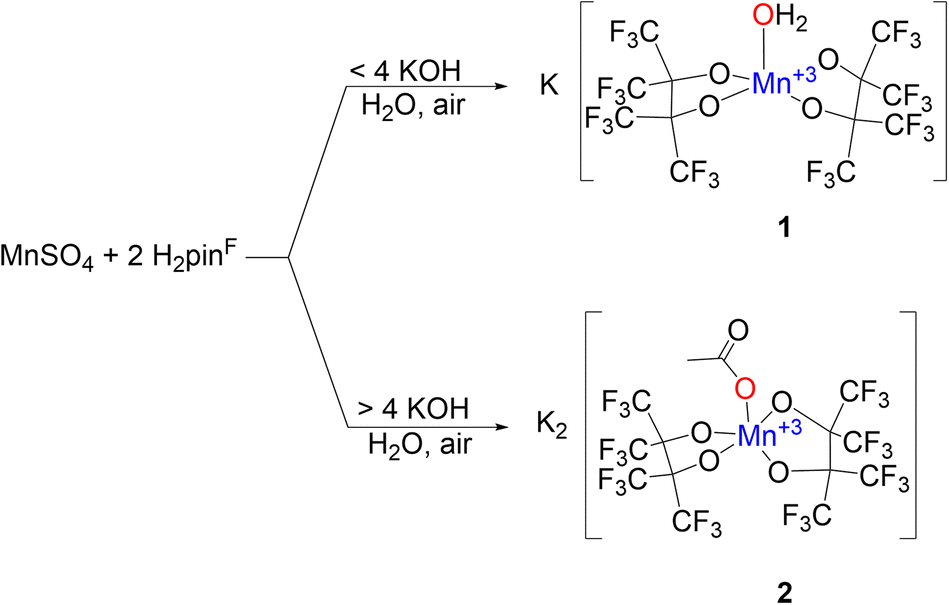 | ||
| Scheme 1 Synthesis of 1 (top) and 2 (bottom). | ||
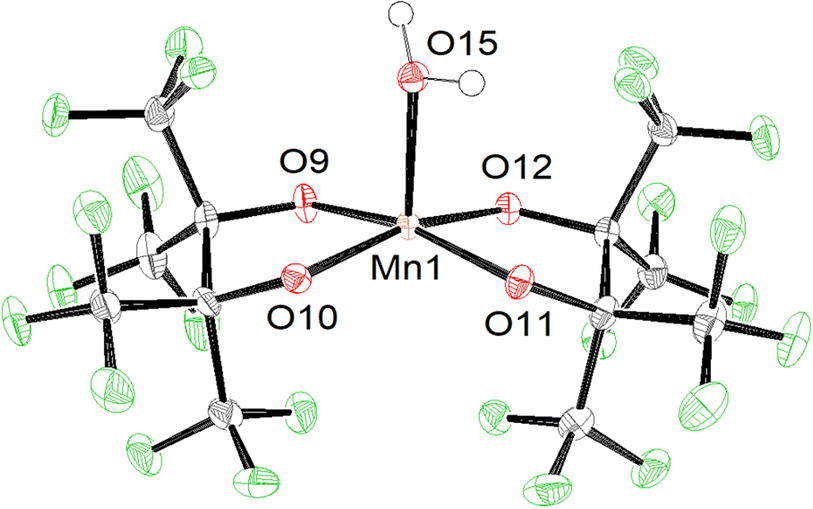 | ||
| Fig. 1 ORTEP of the 1 anion with cations and solvent molecules omitted for clarity. Ellipsoids are shown at the 50% probability level. | ||
One consequence of the use of alkoxides as ligands is the resulting complexes are all anions allowing for cations to play a role in structure and reactivity. Our group has previously described how K⋯O and K⋯F interactions influence the solubility, nuclearity, and geometry of these complexes.23 The chemistry community is increasingly interested in how non-covalent interactions with Lewis acidic, redox-inactive cations tune the reactivity of metal-oxo, -hydroxo, and -aquo complexes.34,35 Fluorinated alkoxide complexes provide an excellent model system to utilize these effects. For example, in 1 the three Mn–OH2 bonds average 2.18 Å, and the Mn–OpinF bonds are between 1.883(3) Å and 1.890(3) Å (Table S1†). The Mn(3)–O(5) bond is roughly 0.05 Å longer, however, than the other two Mn–OH2 bonds due to a 2.744(3) Å interaction between O(5) and K(3). Samples of 1 can be dried under vacuum and gentle heat to remove the lattice H2O molecules, but elemental analysis confirms that one molecule of H2O per Mn remains intact.
Using more than four equivalents of KOH per Mn when deprotonating H2pinF initially results in the formation of an orange precipitate, but after stirring overnight the solution darkens to a deep maroon (Scheme 1, bottom). Purification gives purple X-ray quality needles overnight which SCXRD reveal to be an unexpected acetate complex, K2[Mn(OAc)(pinF)2], 2 shown in Fig. 2. The asymmetric unit contains two molecules of 2 and two Et2O (Fig. S2†). The Mn(III) ion is square pyramidal with a τ5 value of 0.01. The Mn–OAc bonds average 2.09 Å, 0.1 Å shorter than the Mn–OH2 bonds in 1. The Mn–OpinF bonds are between 1.879(3) Å and 1.926(3) Å (Table S1†). Once again we see the structural role played by non-covalent interactions with cations as the two anions are bridged by K⋯OpinF and K⋯OAc interactions with K(3) and K(4) that make both K+ ions seven coordinate (Fig. S2†).
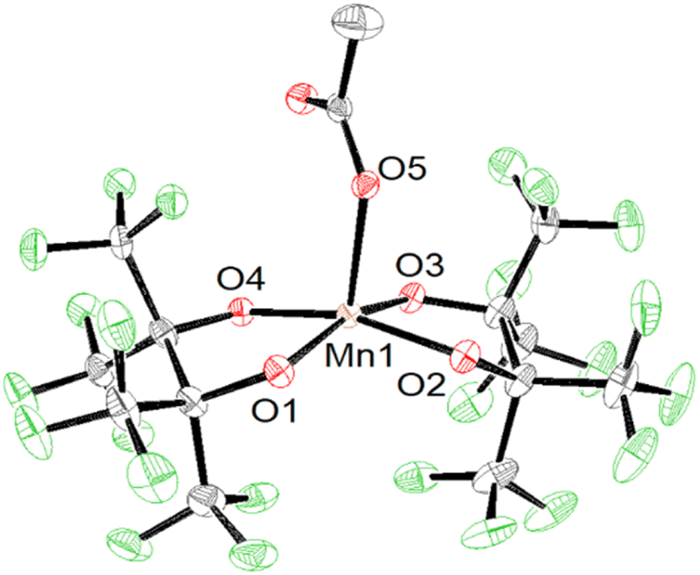 | ||
| Fig. 2 ORTEP of the anion of 2 with cations and solvent molecules omitted for clarity. Ellipsoids are shown at the 50% probability level. | ||
The presence of OAc− as a ligand was unexpected as there is no obvious source of a C2 fragment and this result is reproducible with yields as high as 58% ruling out contamination as the source. We hypothesize that OAc− is the result of oxidation of the crystallization solvent Et2O to EtOAc followed by hydrolysis to yield EtOH and OAc− (Scheme 2). There are numerous examples of Mn(III) complexes mediating oxidation reactions, most notably in the case of {Mn(OAc)3} which has been heavily studied for the promotion of oxidative free-radical reactions between carbonyl compounds and unsaturated molecules to form cyclic products.36 Busch and co-workers isolated the Mn(IV) complex [Mn(Me2EBC)(OH)(OAc)]+ with an unexpected acetate ligand proposed to be derived from oxidation of the reaction solvent, EtOH, by [Mn((Me2EBC))(OH)2]+.37
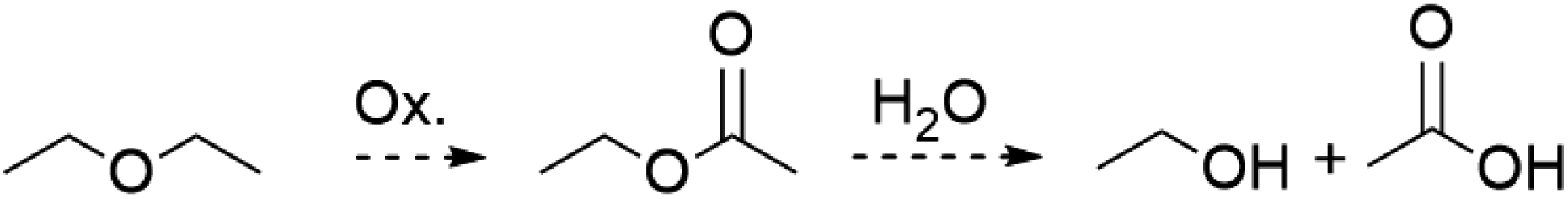 | ||
| Scheme 2 General reaction by which Et2O may be oxidized to ethyl acetate then hydrolysed to ethanol and acetic acid that is proposed to be the source of the OAc− anion in 2. | ||
Formation of 2 in the presence of excess base led us to investigate deprotonation of 1 under more controlled conditions. Under a N2 atmosphere reacting 1 with an equivalent of KN(SiMe3)2 in methyl tert-butyl ether (MTBE) (Scheme 3) immediately causes a color change from red to blue. Purification resulted in the formation of blue single crystalline blocks shown by SCXRD to be a rare example of a mononuclear Mn(III) hydroxide complex K2[Mn(OH)(pinF)2]·3 MTBE, 3·3 MTBE (Fig. 3).
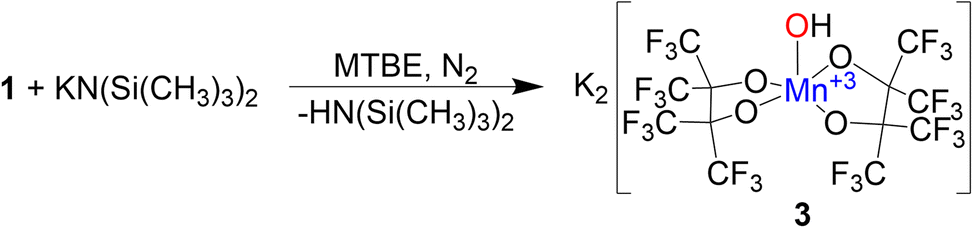 | ||
| Scheme 3 Synthesis of 3. | ||
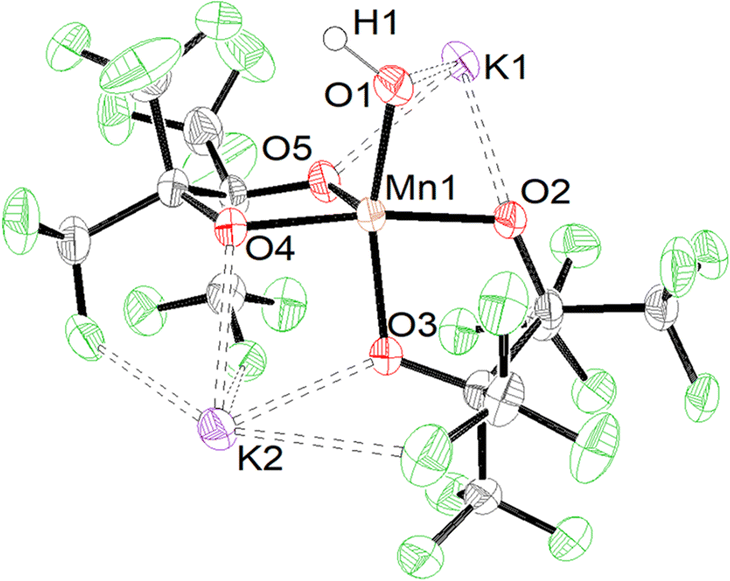 | ||
| Fig. 3 ORTEP of 3 with solvent molecules omitted for clarity. Ellipsoids are shown at the 50% probability level. | ||
Like 1 and 2 the Mn atom in 3 is square pyramidal, although more distorted, with a τ5 value of 0.35. The Mn–OH bond in 3 is 1.857(3) Å (Table S1†), ∼0.3 Å shorter than the Mn–OH2 bond in 1 and is consistent with that found in other Mn(III) hydroxide complexes.12,15–18,38 Complex 3 is air and moisture sensitive but remains stable in the solid state over months in a N2 filled dry box. Polar coordinating solvents such as Et2O, THF, or MTBE are required to solubilize 3 which does not dissolve in CH2Cl2 or toluene. In MeCN, 3 irreversibly changes color from blue to red/brown which we hypothesize is the result of nucleophilic attack on the nitrile group. For this reason, all remaining characterization and reactivity with 3 was performed in THF. Complex 3 is the first example of a {Mn(III)(OH)} complex with all O-donors. No other {Mn(III)OH} is reported to act as a nucleophile, a property that may be attributable to the anionic charge of the complex enabled by the use of alkoxide ligands. The synthesis of 3 is unique among {Mn(III)OH} complexes in that it is achieved by deprotonation of the corresponding {Mn(III)OH2} complex. Existing complexes of this type are prepared by the oxidation of a Mn(II) complex with O2 or PhIO.
Complex 3 crystalizes in pairs (Fig. S3†) of crystallographically identical molecules bridged by K⋯OpinF, K⋯OH, and K⋯F interactions. While we have described the hydroxide as terminal, it is formally a μ3-OH in the solid state with two K⋯OH interactions measuring 2.655(3) Å and 3.110(4) Å, but these bonds are unlikely to persist when 3 is dissolved in polar organic solvents. The distances in 3 are consistent with the few crystallographically characterized K(μ2-OH)TM structures, e.g., K(DME)[tpaMesFe(OH)].40 Attempts to encapsulate the K+ ions with DME or 18C6 invariably lead to degradation of 3 demonstrating the importance of the K⋯O and K⋯F interactions in allowing the isolation of 3. The hydroxyl proton was located in the difference map and was refined independently to a length of 1.02(15) Å.
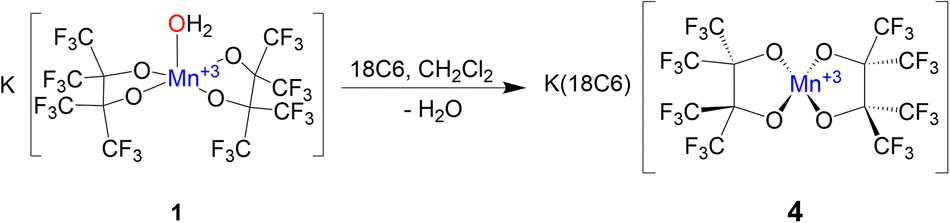
The solid-state structures of 1–3 demonstrate how non-covalent interactions with "naked" K+ can play integral roles in bonding and even allow for the isolation of reactive species by stabilizing metal hydroxide complexes. Interested in how reducing these interactions would alter the structure of the resulting complex, we chose to encapsulate the K+ in 1 with a crown ether. Complex 1 is insoluble in CH2Cl2, but addition of one equivalent of 18C6 to a suspension of 1 in CH2Cl2 allows for complete solubility. After filtering and drying the red solution an orange powder is produced which was subsequently crystalized from dry CH2Cl2 resulting in orange crystals of K(18C6)[Mn(pinF)2], 4, in greater than 80% yield (Scheme 4).
 | ||
| Scheme 4 Synthesis of complex 4. | ||
SCXRD of 4 shows a square-planar Mn(III) center (Fig. 4) with two pairs of equivalent Mn–OpinF bonds measuring 1.8588(15) Å and 1.8574(14) Å respectively (Table S2†). None of the K⋯O and K⋯F interactions seen in the structures of 1–3 persist in 4 (Fig. S4†). To the best of our knowledge, 4 is the only crystallographically characterized {Mn(III)O4} complex with square planar geometry (τ4 = 0.00) and this geometry is perhaps influenced by a Jahn–Teller distortion in this high-spin (S = 2) d4 ion. Complex 1, with its naked K+, is resistant to dehydration even under vacuum and mild heat, while transient {K(18C6)}[Mn(OH2)(pinF)2] is easily dehydrated under a gentle air stream at RT. We attribute this contrast in ligand lability to K⋯OpinF interactions in 1 resulting in a more electrophilic metal center. Without the K⋯OpinF interactions in 4, lone pairs from the alkoxide ligands donate more strongly into the Mn center and reduce its Lewis acidity.
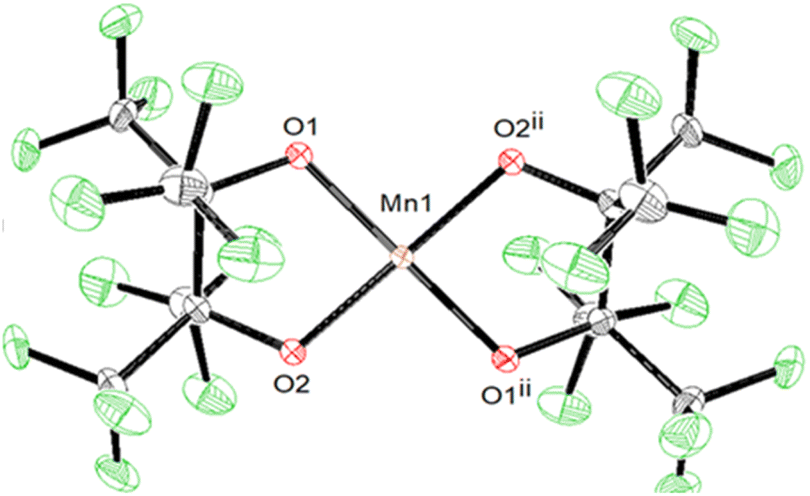 | ||
| Fig. 4 ORTEP of the anion of 4. Ellipsoids are shown at the 50% probability level. | ||
In addition to the four Mn(III) complexes discussed above, the Mn(II) complex (Me4N)2[Mn(pinF)2], 5, was prepared according to Scheme 5. This colorless compound consists of a pseudo-tetrahedral anion (τ4 = 0.43), shown in Fig. 5, with Mn–OpinF bonds that average 2.042 Å which is 0.18 Å longer than in 4 (Table S2†). The anions of 4 and 5 differ by just a single electron but show a dramatic difference in geometry. There are no interactions between the Me4N+ and the anion (Fig. S5†).
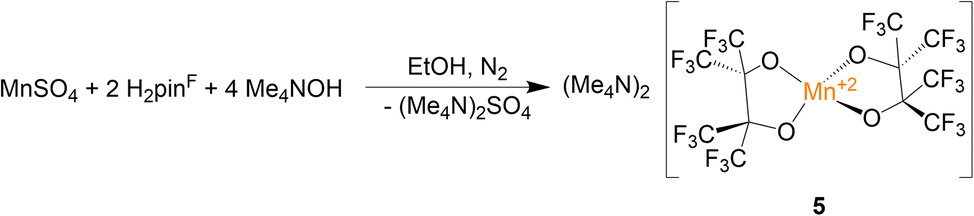 | ||
| Scheme 5 Synthesis of complex 5. | ||
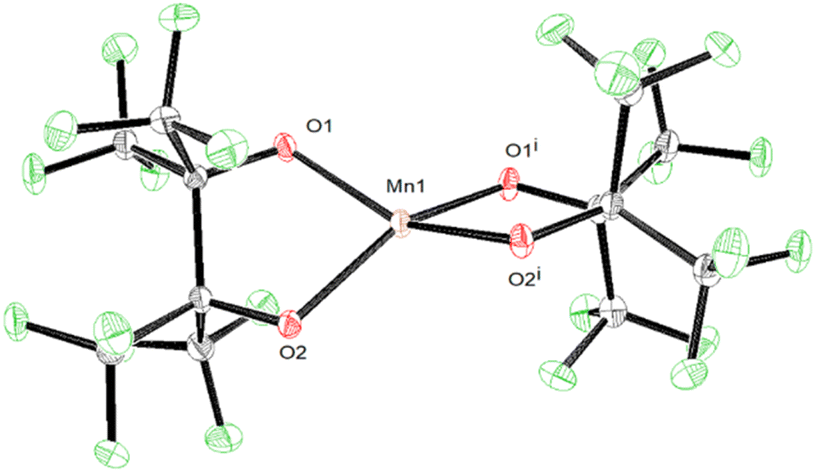 | ||
| Fig. 5 ORTEP of the anion of 5 with cations and solvent molecule excluded for clarity. Ellipsoids are shown at the 50% probability level. | ||
Proton, electron, and H-atom transfer
Compound 1 can be deprotonated by titrating a THF solution of 1 (Fig. 6, red) with KOtBu which affords 3 (Fig. 6, blue) and displays beautiful isosbestic behavior. More than one equivalent of KOtBu is required to fully react with 1 suggesting that an equilibrium is established between the Mn–OH2 species and KOtBu. For this to be true, the pKa of the Mn bound H2O must be similar in value to that of HOtBu. Efforts to directly determine the pKa of the coordinated H2O in 1 by spectrophotometric titration were thwarted by the reactivity of 3 with MeCN and DMSO which have well established pKa scales. We instead bracketed the pKa by reactions with stoichiometric amounts of bases with known pKa values. One equivalent of KN(SiMe3)2 (pKa = 26) is sufficient to completely deprotonate 1, while one equivalent of KOtBu (pKa = 17) only results in partial deprotonation (Fig. 6). Similarly, reaction with up to 30 equivalents of 7-methyl-1,5,7-triazabicyclo[4.4.0]dec-5-ene (MTBD, pKa = 18.0) results in only partial conversion to 3 (Fig. S6†). Based on these observations, we estimate the pKa of the O–H bond in 1 to be 18.3 ± 0.3 in THF.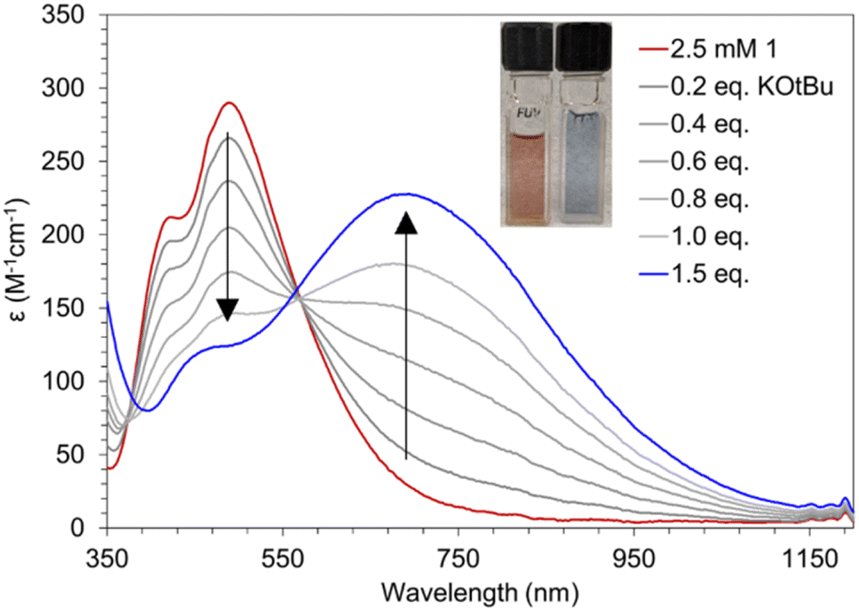 | ||
| Fig. 6 UV-vis spectra of titration of 1 (2 mM) to form 3 with portions of KOtBu in THF. | ||
Determining the pKa of a metal bound H2O is extremely challenging. Complex 1 is the only {Mn(III)OH2} complex for which characterization has been performed in THF. Nam and co-workers have measured the pKa of the H2O in [Mn(III)(OH2)(dpaqH)]+ to be 6.78 in MeCN.39 The Kovacs group found that pKa = 5.3 in H2O for [Mn(II)(SMe2N4(tren))(H2O)]+ which results from PCET by [Mn(III)(SMe2N4(tren))(OH)]+.16 There have also been pKa measurements reported for metal bound hydroxide Mn–O(H) complexes. Borovik and co-workers have reported the pKa of [MnIIIH3buea(OH)]− to be 28 in DMSO.41 The Borovik group also reported that 22 < pKa < 24.4 of [Mn(III)H3bpuea-R(OH)]2− (R = OMe, H, F, Cl, CF3, 5F) in DMSO with the more electron withdrawing ligands resulting in a lower pKa.42 The pKa of a metal bound water or hydroxide is anticorrelated with an increase in reduction potential. Having a large pKa can allow a complex to abstract H atoms despite unfavorable thermodynamics for electron transfer. For example, the basicity of the oxido ligand in [MnIIIH3buea(O)]2− allows it to abstract H-atoms from 9–10 dihydroanthracene (DHA, BDFEC–H = 76 kcal mol−1) despite its unfavorable reduction potential of −2.0 V vs. Fc+/0.41 One would expect that the pKa of Mn–OH > Mn–OH2 and, thus, that the Mn–O would be a better oxidant.
The redox behavior of 1 and 3 was investigated with cyclic voltammetry, as shown in Fig. 7. The voltammogram for 1 in THF (Fig. 7, red) shows a quasi-reversible feature with Ep,c = −0.98 V and Ep,a = 1.43 V at a scan rate of 1 V s−1. Potentials have been referenced to Fc+/0 using an internal standard and the reversibility was consistent over a range of scan rates (Fig. S7–S9†). The voltammogram displays an unusually large peak separation of 2.41 V attributed to a large inner sphere reorganization (loss of coordinated H2O) that occurs upon reduction from Mn(III) to Mn(II). This proposal is supported by the difference in geometry (square planar vs. tetrahedral) for the [Mn(pinF)2]n− anion in Mn(III) 4 and Mn(II) 5 respectively. In a less coordinating solvent such as the atypical 60![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :40 CH2Cl2:Et2O mixture (Fig. S10†) a single broad reduction event is observed with Ep,c = −1.70 V corresponding to the reduction of five-coordinate Mn(III). In the oxidative direction, however, two events can be seen at Ep,a = −0.23 V and −0.033 V corresponding to the oxidation of both a proposed transient solvated Mn(II) and the known four-coordinate species, indicative of weak ligand binding in the axial position upon reduction to Mn(II) (Scheme S11†).
:40 CH2Cl2:Et2O mixture (Fig. S10†) a single broad reduction event is observed with Ep,c = −1.70 V corresponding to the reduction of five-coordinate Mn(III). In the oxidative direction, however, two events can be seen at Ep,a = −0.23 V and −0.033 V corresponding to the oxidation of both a proposed transient solvated Mn(II) and the known four-coordinate species, indicative of weak ligand binding in the axial position upon reduction to Mn(II) (Scheme S11†).
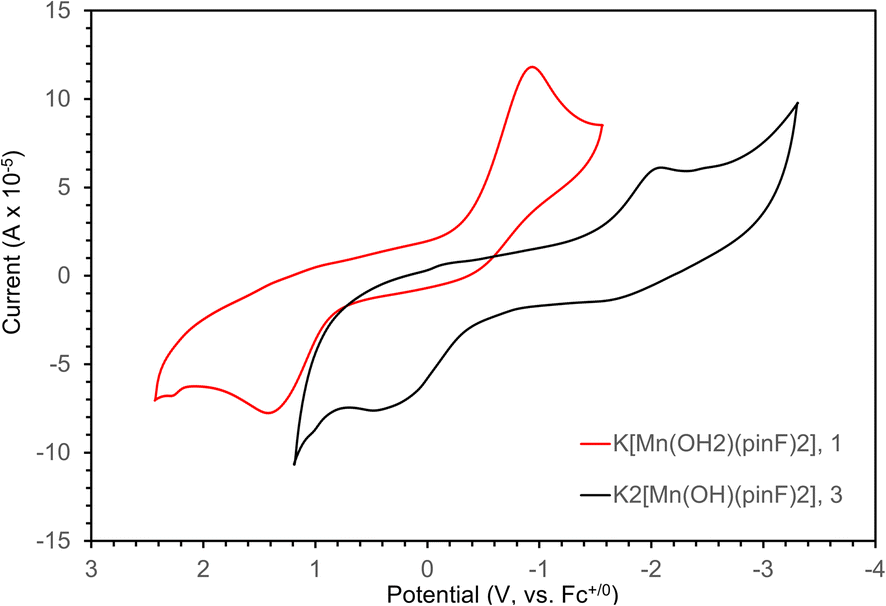 | ||
| Fig. 7 Cyclic voltammograms for 5 mM solutions of 1 (red) and 3 (black) in THF with 250 mM TBAPF6 as a supporting electrolyte. Scan rate is 1 V s−1 and potentials are referenced to Fc+/0. | ||
The voltammogram of 3 (Fig. 7, black) also displays a quasi-reversible feature with a reduction event at −2.05 V coupled to a broad oxidative feature at 0.39 V. The measured potentials were referenced to Fc+/0 using a 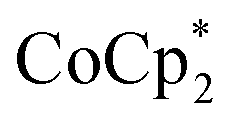 internal standard and the reversibility confirmed over scan rates of 0.1 to 2.0 V s−1 (Fig. S12–S14†). Once again, the peak separation of 2.44 V is unusually large and is likely due to a chemical transformation upon reduction to Mn(II). Complex 3 is more difficult to reduce than 1 consistent with the increase in energy necessary to reduce a complex with a greater negative charge.
internal standard and the reversibility confirmed over scan rates of 0.1 to 2.0 V s−1 (Fig. S12–S14†). Once again, the peak separation of 2.44 V is unusually large and is likely due to a chemical transformation upon reduction to Mn(II). Complex 3 is more difficult to reduce than 1 consistent with the increase in energy necessary to reduce a complex with a greater negative charge.
The hydrogen atom abstraction (HAA) ability of 3 was investigated with stoichiometric amounts of substrates with weak X–H bonds (X = C, N, O). Reactions with 1,2-diphenylhydrazine (DPH, BDFEN–H = 64.0 kcal mol−1) are complete after roughly 100 minutes at RT producing azobenzene and [Mn(II)(pinF)2]2− along with an equivalent of water (Fig. 8). Similarly, reactions with 1-hydroxy-2,2,6,6-tetramethyl-piperidine (TEMPOH, BDFEO–H = 66.5 kcal mol−1 in MeCN) are rapid at RT to produce the (2,2,6,6-tetramethylpiperidin-1-yl)oxyl (TEMPO) radical as evidenced by the characteristic feature at 440 nm (Fig. S15†). Oxidation of TEMPOH is strongly biased to occur via PCET due to its thermodynamically unfavorable pKa (41 in MeCN) and one e− reduction potential (−1.85 V vs. Fc+/0 in MeCN).43 Reactivity of 3 with TEMPOH supports a PCET mechanism for other reactions but does not rule out asynchronous H+/e− transfer. Complex 3 also reacts with 2,4,6-tri-tert-butylphenol (2,4,6-TTBP, O–H BDFE = 74.4 kcal mol−1).43 Monitoring the reaction of 3 with 10 eq. 2,4,6-TTBP by electronic absorption spectroscopy shows the decay of 3 and the formation of the 2,4,6-tri-t-butylphenoxyl radical (λmax = 630 nm, Fig. 9).
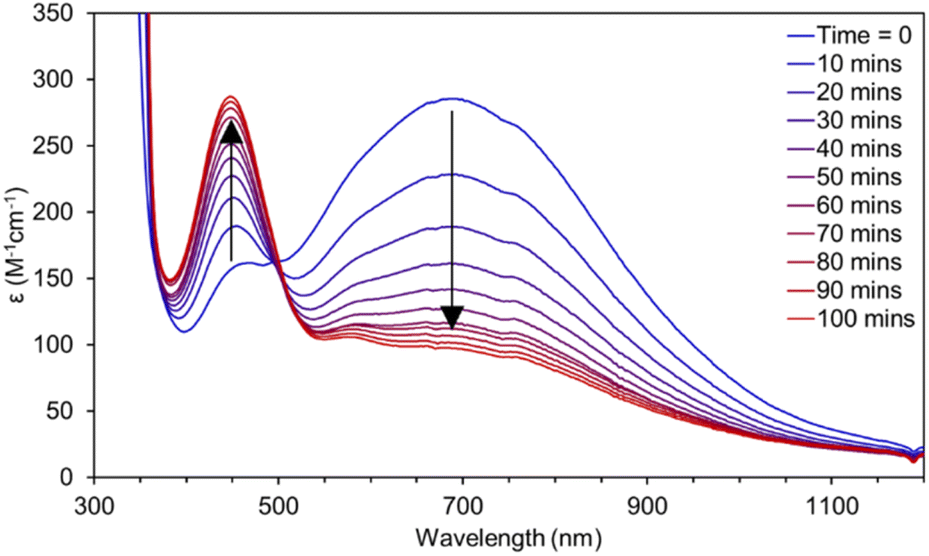 | ||
| Fig. 8 UV-vis spectra for the reaction of a 2 mM solution of 3 in THF with 1/2 equivalent of 1,2-diphenylhydrazine. | ||
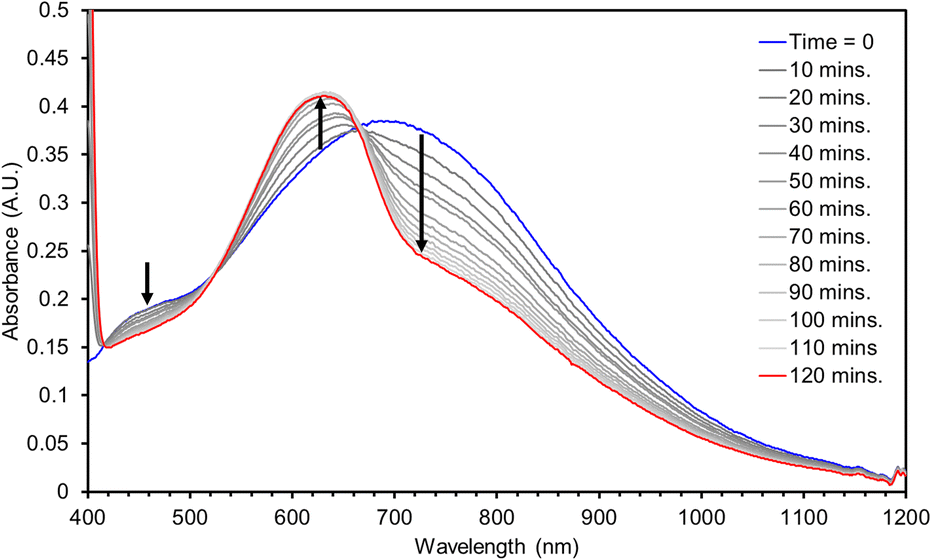 | ||
| Fig. 9 UV-vis spectra for the reaction of a 2 mM solution of 3 in MTBE with 10 eq. TTBP. | ||
No reaction has been observed at RT with either the C–H bonds of 9,10-dihydroanthracene (DHA, BDFEC–H = 72.9 kcal mol−1) or 1,4-cyclohexadiene (CHD, BDFEC–H = 67.8 kcal mol−1). Lack of reaction with DHA or CHD despite the ability to oxidize the stronger O–H bond in 2,4,6-TTBP suggests that substrate BDFE may not be the perfect descriptor for understanding the thermodynamics of HAA by 3.
| BDFE = 23.06E° + 1.37(pKa) + C(g,sol) | (1) |
The O–H bond BDFE in the hypothesized, transient {Mn(II)OH2} moiety can be calculated using the Bordwell equation (eqn (1)).43,44 The C(g,sol) term is a composite, solvent dependent, free-energy constant that represents the energy of solvation for an H-atom in the solvent of interest (59.9 in THF).43 For systems which undergo ideal one e− redox processes, E° can be approximated as E1/2 from CV. However, the large ΔE in the CV of 1 casts doubt on the reliability of E1/2 as an approximation of E°. If instead we choose E at Ip,c from the voltammogram recorded at 100 mV s−1 (−0.725 V) we calculate a BDFE of 67–70 kcal mol−1. This range does not overlap exactly with the demonstrated reactivity with 2,4,6-TTBP (BDFE = 74.4 kcal mol−1). The value in our Bordwell equation with the greatest uncertainty is the reduction potential of {Mn(III)(OH)2}, 1, to {Mn(II)}, 5, which is accompanied by a change in geometry and coordination number. With this uncertainty, the calculated BDFE range is reasonable.
The Bordwell thermochemical cycle for the H-atom transfer reaction is shown in Scheme 6. Complex 3 is the only {Mn(III)(OH)} complex for which thermodynamic data have been reported in THF. While pKa and E1/2 values are strongly solvent dependent, it has recently been shown that BDFEs are nearly independent of solvent.43
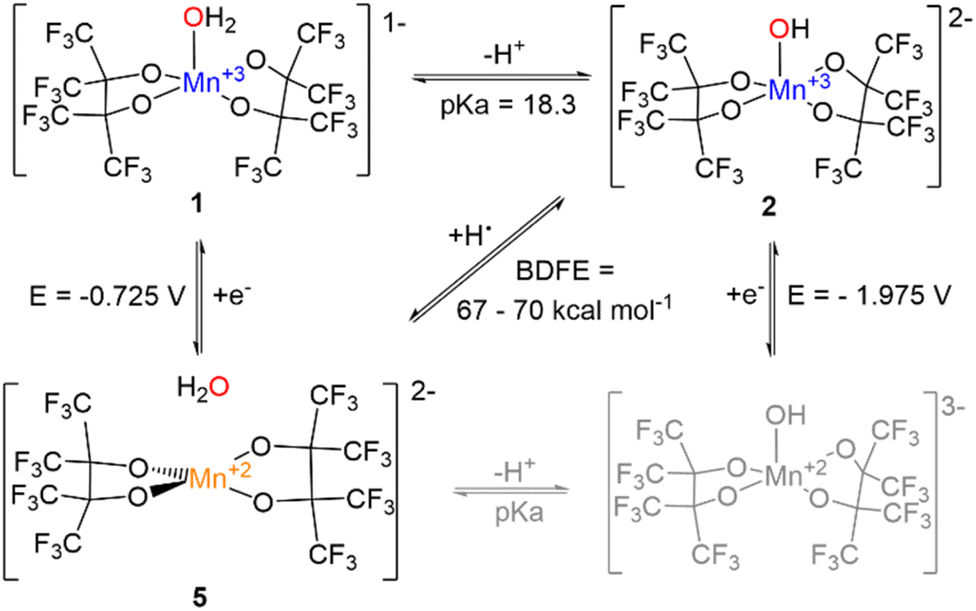 | ||
| Scheme 6 Bordwell scheme depicting the proton, electron, and hydrogen atom transfer events and their respective thermodynamic parameter for the [Mn(OHx)(pinF)2]n− system. Gray indicates a proposed, not isolated species. | ||
For example, the measured BDFE of the O–H bond in TEMPOH averages 65.0 ± 1.3 kcal mol−1 in solvents whose dielectric constants differ by 38 units (MeCN vs. hexane). The known Mn(II)OH2 complexes which result from HAT by a {Mn(III)OH} complex for which an O–H BDFE has been reported is limited to those shown in Table 1. The oxidizing power of 3 is comparable to [Mn(III)(SMe2N4(tren))(OH)]+ from the Kovacs group,16 and [Mn(III)(OH)(dpaqH)]+ and its methylated analogue from the Jackson group.15,45 [Mn(III)(OH)(PY5)]2+ from the Stack group is unique among these complexes as it is the only example reported to oxidize C–H bonds.18
| Complex | pKa Mn(III)OH2 | E (V) | Solvent | BDFE (kcal mol−1) | Ref. |
|---|---|---|---|---|---|
| a Potentials vs. Fc+/0. b Estimated in MeCN. c Calculated. d Measured vs. SHE and converted to Fc+/0. e E p,c. f In MeCN. | |||||
| [Mn(III)(SMe2N4(tren))(OH)]+ | 21.2b | −0.6 | MeCN | 70.1b | 16 |
| [Mn(III)(OH)(dpaqH)]+ | 6.8 | −0.73c | MeCN | 79.4 | 39,45 |
| [Mn(III)(OH)(dpaq2me)]+ | 20c | −0.62c | MeCN | 80.9 | 45 |
| [Mn(III)(OH)(PY5)]2+ | 13f | 0.186d | MeCN | 82 | 18 |
| [Mn(OH)(pinF)2]2− | 18.3 ± 0.3 | −0.725e | THF | 67–70 | This work |
Based on the measurements described above and the observed stability of 3 in Et2O under N2 it is unlikely that 3 is responsible for the oxidation of Et2O that results in 2. Because the synthesis of 2 is performed in air, if 3 is formed in solution, it could react with O2 to form a higher valent Mn species capable of oxidizing Et2O. To test this hypothesis a solution of 3 prepared under N2 in either Et2O (solid lines) or MTBE (dotted lines) was cooled to −100 °C in the spectrophotometer and the spectra recorded (Fig. 10, blue traces). Next, excess O2 was added and in both solvents a new chromophore appears with λmax = 550 nm that reaches a maximum absorbance after 50 minutes at −100 °C (Fig. 10, red traces). The solution was then gradually warmed in 20 °C increments. At −80 °C no change was observed, but at −60 °C the chromophore has decayed to a new spectrum that does not match any of the chromophores discussed here (Fig. 10, black traces). Crucially, the chromophore shows much greater decay in MTBE than in Et2O. Further warming to −40 °C did not change the spectrum of this decayed product. Efforts to isolate this intermediate and obtain more indirect evidence via reactivity are underway.
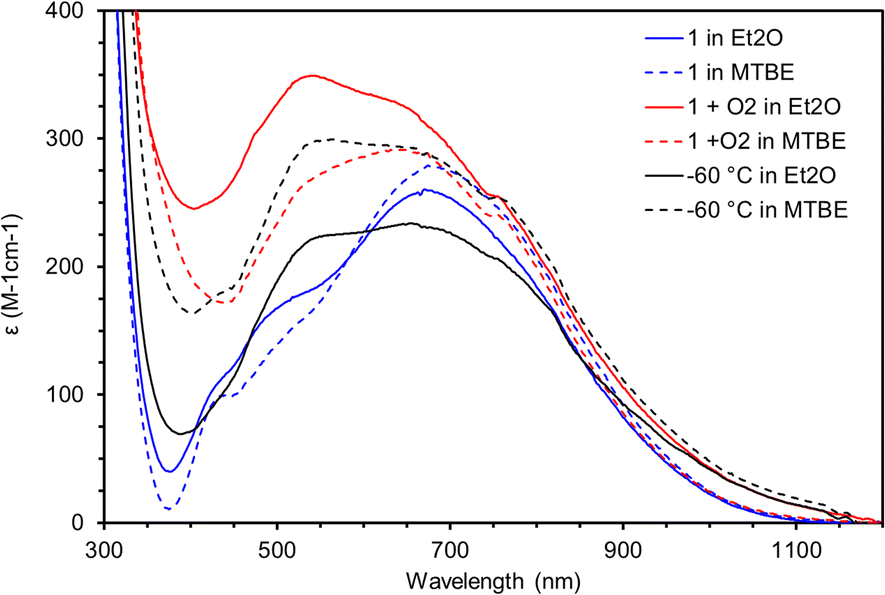 | ||
| Fig. 10 UV-vis spectra for the addition of O2 to 3 (2 mM) at −100 °C in MTBE (dotted) and Et2O (solid). After O2 addition the solution was allowed to rest in the spectrophotometer at −100 °C before warming. | ||
Conclusions
Described here are the synthesis and structural characterization of five Mn complexes supported by the perfluoropinacolate ligand. The synthesis of K[Mn(OH2)(pinF)2], 1, and K(18C6)[Mn(pinF)2], 4, demonstrates the dependence of water ligand lability on cation encapsulation and provides in 4 the first example of a {Mn(III)O4} complex with ∼D4h geometry. The Mn(II) complex (Me4N)2[Mn(pinF)2], 5, was synthesized and revealed by SCXRD to have a ∼Td geometry in contrast to the one electron oxidized product 4. When performed in the presence of excess base, the synthesis of 1 results in K2[Mn(OAc)(pinF)2], 2, with an unexpected acetate ligand proposed to be the result of oxidation of Et2O by an air and water tolerant oxidant. The dependence of this reaction on the amount of base used led us to deprotonate 1 under controlled conditions resulting in K2[Mn(OH)(pinF)2], 3, a rare example of a crystallographically characterized mononuclear {Mn(III)OH} complex, the first with an all O-donor environment and the only anion. The redox properties of 1 and 3 were investigated with cyclic voltammetry which reveals quasi-reversible redox behavior with exceptionally large ΔE that results from a change in geometry and/or composition upon reduction for both complexes. This observation is supported by the differing geometries of 4 and 5 in the solid-state. The pKa of the OH2 ligand in 1 is estimated to be 18.3 ± 0.3 by reaction with stoichiometric amounts of bases. Complex 3 oxidizes the N–H and O–H bonds via HAA. The BDFE of the O–H bond in transient {Mn(II)–OH2} formed when 3 abstracts an H-atom is 67–70 kcal mol−1. While 3 is unable to oxidize Et2O on its own, low temperature addition of O2 to solutions of 3 results in the formation of a new chromophore which decays upon warming. The identity of this proposed Mn(IV) chromophore is still under investigation. This system is the first all O-donor Mn complex shown to oxidize C–H bonds.Data availability
All relevant data are available from the corresponding authors upon reasonable request.Author contributions
S. M. M. – investigation, writing – original draft, C. S. – investigation, J. L. S. – investigation, E. M. L. - investigation, A. L. R. – investigation, L. H. D. – conceptualization, project administration, funding acquisition, writing – review and editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful to the National Science Foundation (NSF, CHE 2102532 to LHD), to the National Institutes of Health (S10OD028585 for SCXR diffractometer), to the Boston University Undergraduate Research Opportunity Program (BU UROP to EML), and to Boston University Post-Doctoral Faculty Fellowship (BU Postdoctoral Faculty Fellowship to JLS) for their support of this work. We would also like to thank Dr Jeffrey Bacon and Dr William Tucker for helpful discussions.Notes and references
- H. Kuhn, S. Banthiya and K. van Leyen, Biochim. Biophys. Acta, 2015, 1851, 308–330 CrossRef CAS PubMed.
- A. Wennman, A. Magnuson, M. Hamberg and E. H. Oliw, J. Lipid Res., 2015, 56, 1606–1615 CrossRef CAS PubMed.
- E. H. Oliw, Prostaglandins Other Lipid Mediators, 2002, 68–69, 313–323 CrossRef CAS PubMed.
- Y. Umena, K. Kawakami, J.-R. Shen and N. Kamiya, Nature, 2011, 473, 55–60 CrossRef CAS PubMed.
- K. N. Ferreira, T. M. Iverson, K. Maghlaoui, J. Barber and S. Iwata, Science, 2004, 303, 1831 CrossRef CAS PubMed.
- J. Azadmanesh and G. E. O. Borgstahl, Antioxidants, 2018, 7(2), 25–41 CrossRef.
- H. Isobe, M. Shoji, T. Suzuki, J.-R. Shen and K. Yamaguchi, J. Photochem. Photobiol., A, 2021, 405, 112905 CrossRef CAS.
- D. B. Rice, A. A. Massie and T. A. Jackson, Acc. Chem. Res., 2017, 50, 2706–2717 CrossRef CAS PubMed.
- A. S. Borovik, Chem. Soc. Rev., 2011, 40, 1870–1874 RSC.
- Y.-R. Luo, Handbook of Bond Dissociation Energies in Organic Compounds, CRC Press LLC, 2002 Search PubMed.
- M.-H. Baik, M. Newcomb, R. A. Friesner and S. J. Lippard, Chem. Rev., 2003, 103, 2385–2420 CrossRef CAS PubMed.
- Z. Shirin, B. S. Hammes, V. G. Young and A. S. Borovik, J. Am. Chem. Soc., 2000, 122, 1836–1837 CrossRef CAS.
- Z. Shirin, B. S. Hammes, V. G. Young and A. S. Borovik, J. Am. Chem. Soc., 2000, 122(8), 1836–1837 CrossRef CAS.
- D. C. Cummins, J. G. Alvarado, J. P. T. Zaragoza, M. Q. Effendy Mubarak, Y.-T. Lin, S. P. de Visser and D. P. Goldberg, Inorg. Chem., 2020, 59, 16053–16064 CrossRef CAS PubMed.
- G. B. Wijeratne, B. Corzine, V. W. Day and T. A. Jackson, Inorg. Chem., 2014, 53, 7622–7634 CrossRef CAS PubMed.
- M. K. Coggins, L. M. Brines and J. A. Kovacs, Inorg. Chem., 2013, 52, 12383–12393 CrossRef CAS.
- X. Wu, M. S. Seo, K. M. Davis, Y.-M. Lee, J. Chen, K.-B. Cho, Y. N. Pushkar and W. Nam, J. Am. Chem. Soc., 2011, 133, 20088–20091 CrossRef CAS PubMed.
- C. R. Goldsmith, A. P. Cole and T. D. P. Stack, J. Am. Chem. Soc., 2005, 127, 9904–9912 CrossRef CAS PubMed.
- R. L. Halbach, D. Gygi, E. D. Bloch, B. L. Anderson and D. G. Nocera, Chem. Sci., 2018, 9, 4524–4528 RSC.
- K.-B. Cho, S. Shaik and W. Nam, J. Phys. Chem. Lett., 2012, 3, 2851–2856 CrossRef CAS.
- A. Grass, D. Wannipurage, R. L. Lord and S. Groysman, Coord. Chem. Rev., 2019, 400, 213044 CrossRef CAS.
- A. Nandy and H. J. Kulik, ACS Catal., 2020, 10, 15033–15047 CrossRef CAS.
- J. K. Elinburg and L. H. Doerrer, Polyhedron, 2020, 190, 114765 CrossRef CAS.
- J. L. Steele, L. Tahsini, C. Sun, J. K. Elinburg, C. M. Kotyk, J. McNeely, S. A. Stoian, A. Dragulescu-Andrasi, A. Ozarowski, M. Ozerov, J. Krzystek, J. Telser, J. W. Bacon, J. A. Golen, A. L. Rheingold and L. H. Doerrer, Chem. Commun., 2018, 54, 12045–12048 RSC.
- L. Tahsini, S. E. Specht, J. S. Lum, J. J. M. Nelson, A. F. Long, J. A. Golen, A. L. Rheingold and L. H. Doerrer, Inorg. Chem., 2013, 52, 14050–14063 CrossRef CAS PubMed.
- J. S. Lum, L. Tahsini, J. A. Golen, C. Moore, A. L. Rheingold and L. H. Doerrer, Chem.–Eur. J., 2013, 19, 6374–6384 CrossRef CAS PubMed.
- S. A. Cantalupo, S. R. Fiedler, M. P. Shores, A. L. Rheingold and L. H. Doerrer, Angew. Chem., Int. Ed., 2012, 51, 1000–1005 CrossRef CAS PubMed.
- S. A. Cantalupo, H. E. Ferreira, E. Bataineh, A. J. King, M. V. Petersen, T. Wojtasiewicz, A. G. DiPasquale, A. L. Rheingold and L. H. Doerrer, Inorg. Chem., 2011, 50, 6584–6596 CrossRef CAS.
- S. A. Cantalupo, J. S. Lum, M. C. Buzzeo, C. Moore, A. G. Di Pasquale, A. L. Rheingold and L. H. Doerrer, Dalton Trans., 2010, 39, 374–383 RSC.
- H. Hirao, L. Que Jr., W. Nam and S. Shaik, Chem.–Eur. J., 2008, 14, 1740–1756 CrossRef CAS PubMed.
- J. K. Elinburg, S. L. Carter, J. J. M. Nelson, D. G. Fraser, M. P. Crockett, A. B. Beeler, E. Nordlander, A. L. Rheingold and L. H. Doerrer, Inorg. Chem., 2020, 59, 16500–16513 CrossRef CAS PubMed.
- C. J. Willis, Coord. Chem. Rev., 1988, 88, 133–202 CrossRef CAS.
- S. E. N. Brazeau and L. H. Doerrer, Dalton Trans., 2019, 48, 4759–4768 RSC.
- S. Hong, Y.-M. Lee, M. Sankaralingam, A. K. Vardhaman, Y. J. Park, K.-B. Cho, T. Ogura, R. Sarangi, S. Fukuzumi and W. Nam, J. Am. Chem. Soc., 2016, 138, 8523–8532 CrossRef CAS PubMed.
- H. M. Neu, R. A. Baglia and D. P. Goldberg, Acc. Chem. Res., 2015, 48, 2754–2764 CrossRef CAS PubMed.
- M. Mondal and U. Bora, RSC Adv., 2013, 3, 18716–18754 RSC.
- T. J. Hubin, J. M. McCormick, N. W. Alcock and D. H. Busch, Inorg. Chem., 2001, 40, 435–444 CrossRef CAS PubMed.
- J. P. T. Zaragoza, M. A. Siegler and D. P. Goldberg, J. Am. Chem. Soc., 2018, 140, 4380–4390 CrossRef CAS.
- M. Sankaralingham, Y-M. Lee, D. G. Karmalkar, W. Nam and S. Fukuzumi, J. Am. Chem. Soc., 2018, 140(40), 12695–12699 CrossRef PubMed.
- W. H. Harman and C. J. Chang, J. Am. Chem. Soc., 2007, 129, 15128–15129 CrossRef CAS PubMed.
- T. H. Parsell, M.-Y. Yang and A. S. Borovik, J. Am. Chem. Soc., 2009, 131, 2762–2763 CrossRef CAS PubMed.
- S. K. Barman, J. R. Jones, C. Sun, E. A. Hill, J. W. Ziller and A. S. Borovik, J. Am. Chem. Soc., 2019, 141, 11142–11150 CrossRef CAS.
- R. G. Agarwal, S. C. Coste, B. D. Groff, A. M. Heuer, H. Noh, G. A. Parada, C. F. Wise, E. M. Nichols, J. J. Warren and J. M. Mayer, Chem. Rev., 2022, 122, 1–49 CrossRef CAS.
- F. G. Bordwell, J. P. Cheng and J. A. Harrelson, J. Am. Chem. Soc., 1988, 110, 1229–1231 CrossRef CAS.
- D. B. Rice, G. B. Wijeratne, A. D. Burr, J. D. Parham, V. W. Day and T. A. Jackson, Inorg. Chem., 2016, 55, 8110–8120 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2246386–2246390. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3sc01971c |
| This journal is © The Royal Society of Chemistry 2023 |