
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMechanism landscape in pyrylium induced organic afterglow systems†
Guangming
Wang
,
Xuefeng
Chen
,
Xun
Li
,
Ying
Zeng
and
Kaka
Zhang
 *
*
Key Laboratory of Synthetic and Self-Assembly Chemistry for Organic Functional Molecules, Shanghai Institute of Organic Chemistry, University of Chinese Academy of Sciences, Chinese Academy of Sciences, 345 Lingling Road, Shanghai 200032, People's Republic of China. E-mail: zhangkaka@sioc.ac.cn
First published on 6th July 2023
Abstract
Manipulation of excited states and their dynamics represents a central topic in luminescence systems. We report an unexpected emergence of a high-performance organic afterglow in pyrylium induced photopolymerization systems, as well as the establishment of the mechanism landscape of the afterglow systems as a function of monomer types. In the case of methyl methacrylate, after pyrylium-catalyzed photopolymerization, the obtained materials exhibit a TADF-type organic afterglow with an afterglow efficiency of 70.4%. By using heavy-atom-containing methacrylate, the external heavy atom effect speeds up phosphorescence decay and switches on room-temperature phosphorescence in pyrylium-polymer systems. When 9-vinylcarbazole is used, the resultant materials display organic long persistent luminescence with hour-long durations and emission maxima around 650 nm. The intriguing mechanism landscape reflects the delicate balance of multiple photophysical processes in the pyrylium induced organic afterglow systems, which has been rarely explored in the reported studies.
Introduction
Manipulation of the rate constants of key photophysical processes is of vital importance for fabricating high-performance photofunctional materials.1–4 From the perspective of organic room-temperature phosphorescence (RTP) and afterglow materials,5–10 due to the spin-forbidden nature of triplet formation and transformation, it remains a formidable task to achieve highly efficient and long-lived afterglow systems.11–18 Singlet-to-triplet intersystem crossing and phosphorescence decay represent two of the most important photophysical processes in organic RTP and afterglow systems, with rate constants of kISC and kP, respectively. Pioneering studies exhibited that judicious molecular design, deliberate control of aggregation states, and well-defined supramolecular assembly can increase kISC and kP and thus improve the performance of organic afterglow materials.19–23 Besides, the employment of crystalline and glassy environments to suppress nonradiative decay (knr) and oxygen quenching (kq), dopant-matrix design, thermally activated delayed fluorescence, and retarded charge recombination can enhance afterglow performance.24–36 Among these studies, heavy atom effects and n–π* transition are the most frequently used and reliable strategies to increase both kISC and kP (where kISC and kP are bound),21,37,38 which would boost phosphorescence efficiency (ΦP) but show the severe side effect of significantly shortening phosphorescence lifetimes (τP). For constructing efficient and long-lived organic afterglow materials, we reason that the key is to selectively enhance kISC (to unbound kISC and kP), with other photophysical processes being flexibly adjustable. Consequently, the delicate control of the important photophysical processes would give rise to organic afterglow materials with high performance, desired functions and intriguing features.Here we report a serendipitous finding of high-performance organic afterglow in pyrylium induced photopolymerization systems and the establishment of an intriguing mechanism landscape versus monomer types in the afterglow systems (Fig. 1). The pyrylium-polymer afterglow materials display TADF-type afterglow with 70.4% efficiency, RTP mechanism, and hour-long OLPL in methyl methacrylate, heavy-atom-containing methacrylate, and 9-vinylcarbazole polymerization systems, respectively. In-depth studies reveal that intramolecular and intermolecular charge transfer are responsible for the enhancement of kISC and the formation of the charge-separated state in pyrylium-polymer systems, respectively. Being equally important, the suppression of nonradiative decay and charge recombination in glassy polymer matrices is also crucial for the high-performance organic afterglow. In addition, the obtained OLPL material with emission maxima >600 nm has been coupled with a rare-earth up-conversion material to exhibit near infrared light excitable properties under 2 mm tissue and display promising biological applications.
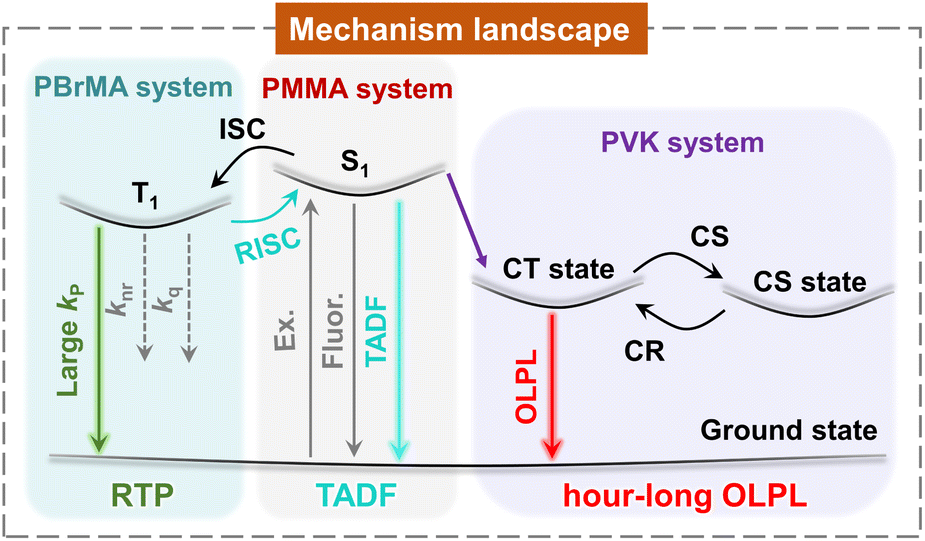 | ||
| Fig. 1 In the poly(methyl methacrylate) (PMMA) system, the pyrylium salt shows a TADF-type afterglow which features a moderate kRISC of 100–101 s−1 to improve afterglow efficiency and maintain afterglow lifetime. In the poly(2-bromoethyl acrylate) (PBrMA) system, the external heavy atom effect greatly increases pyrylium's kP and switches on pyrylium's RTP. In the poly(vinyl carbazole) (PVK) system, the lower-lying intermolecular charge transfer (CT) state of the PVK donor and pyrylium acceptor blocks the population of pyrylium's T1 states, leading to charge separation (CS) states and giving rise to organic long persistent luminescence (OLPL) because of the retarded charge recombination (CR). | ||
Results and discussion
The pyrylium salts (Scheme 1) are synthesized by the cascade reaction developed in our lab39,40 (ESI†). The pyrylium salt, P1, undergoes thorough structural and solution photophysical characterization, with an intense intramolecular charge transfer absorption band at 449 nm (ε = 33![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 800 M−1 cm−1), bright green emission at 499 nm in dichloromethane (PLQY, 90.7%; τF, 3.32 ns), HOMO level of −6.53 eV and LUMO level of −3.95 eV (Fig. S1 and S2†). TD-DFT calculations confirm P1's charge transfer characteristics in S1 states with oscillator strength (fOSC) up to 0.79 (Fig. S3†).
800 M−1 cm−1), bright green emission at 499 nm in dichloromethane (PLQY, 90.7%; τF, 3.32 ns), HOMO level of −6.53 eV and LUMO level of −3.95 eV (Fig. S1 and S2†). TD-DFT calculations confirm P1's charge transfer characteristics in S1 states with oscillator strength (fOSC) up to 0.79 (Fig. S3†).
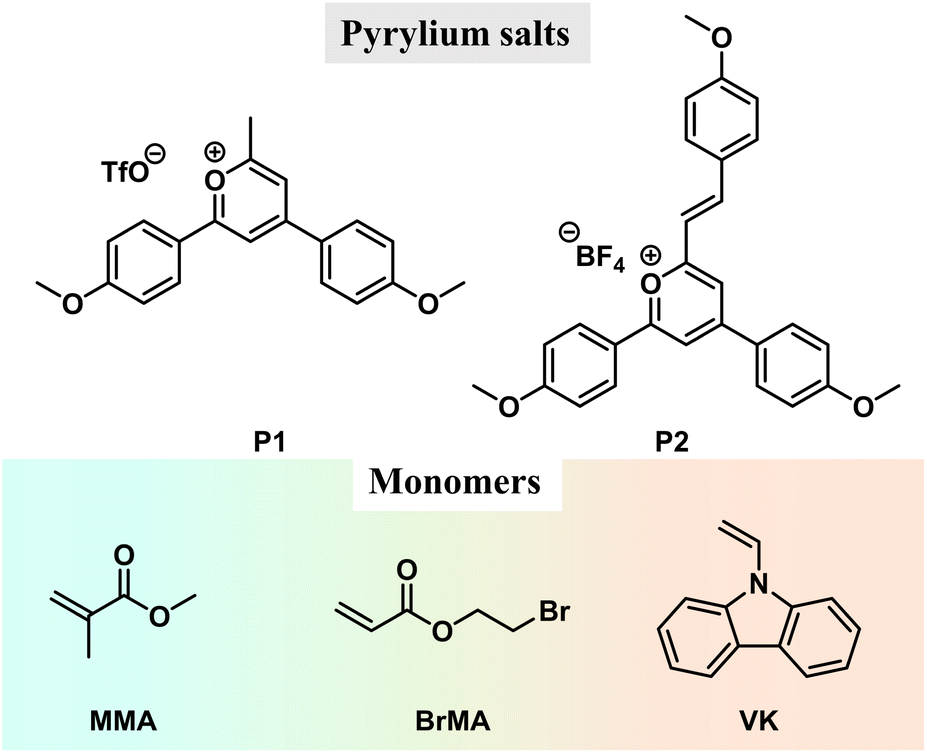 | ||
| Scheme 1 Chemical structures of pyrylium salts and monomers. | ||
The photocatalytic property of pyrylium salts has been reported in small-molecule systems,41,42 with polymerization systems being less explored. When P1 was dissolved in methyl methacrylate (MMA), after standing under ambient light for hours (for example, in the case of 0.05 wt% P1 in MMA, after 10 h ambient light irradiation), a glass-like object was obtained, indicating the formation of poly(methyl methacrylate) (PMMA, Fig. S4;† the formation of PMMA was confirmed by H NMR and GPC, Fig. S5 and S6†); PMMA has a glassy transition temperature of around 110 °C. Unexpectedly, the P1-PMMA material has been found to store energy upon 365 nm UV or 445 nm laser irradiation and then display sky-blue emission in a dark room upon ceasing the excitation (Fig. 2A and S7†). The steady-state emission spectra of P1-PMMA are found to coincide with the delayed emission (1 ms delay) spectra, both of which exhibit 420 nm to 600 nm bands with emission maxima at 484 nm (Fig. 2B). The PLQY has been measured to be 87.1% (Fig. S8†), and from the excited state decay profile (Fig. 2C), the afterglow emission constitutes 80.8% of the total emission (vide infra). The afterglow efficiency can be estimated to be 70.4%, which is among the highest values in the reported afterglow systems (Fig. S9 and S10†). Due to the electronic inertness of the PMMA component, several mechanisms that have been reported to give rise to room-temperature afterglow in two-component systems, such as excited state energy transfer,43–45 donor–acceptor OLPL,23 and matrices' T1 mediation,46 can be ruled out (Text S1†). When P1 was doped into commercial PMMA′, the obtained P1-PMMA′ also showed room-temperature afterglow properties and the coincidence of steady-state and delayed emission spectra (Fig. S11†). This, together with other evidence, can be used to refute the impurity mechanism47 for the emergence of organic afterglow in the present system (Text S2†). Variable temperature delayed emission studies reveal the phosphorescence bands at 539 nm (green afterglow, T1 level, 2.30 eV) at 77 K (Fig. 2D). The delayed emission bands at 487 nm are found to increase with temperature and dominate at room temperature and higher temperatures (Fig. 2D). Because of their identical or very similar emission maxima to the steady-state emission spectra, the 487 nm delayed emission bands can be assigned to delayed fluorescence of P1-PMMA samples with an S1 level of 2.55 eV. P1-PMMA samples with different P1 concentrations (0.001% to 0.1%) have been measured to show similar delayed fluorescence behaviors with a slight red-shift of the delayed emission bands upon increasing P1 concentrations (Fig. S12†). At low P1 concentrations such as 0.001% and 0.01%, the bimolecular triplet–triplet annihilation mechanism that can also give rise to delayed fluorescence would be insignificant. All of this suggests the delayed fluorescence of long-lived emission characteristics originates from thermally activated reverse intersystem crossing (RISC) and the subsequent S1-to-S0 delayed fluorescence of P1 in PMMA glassy matrices. The power-dependent delayed fluorescence intensity measurements exhibit a quasi-linear relationship between excitation power and delayed fluorescence intensity (Fig. S13†), further supporting the TADF afterglow mechanism.
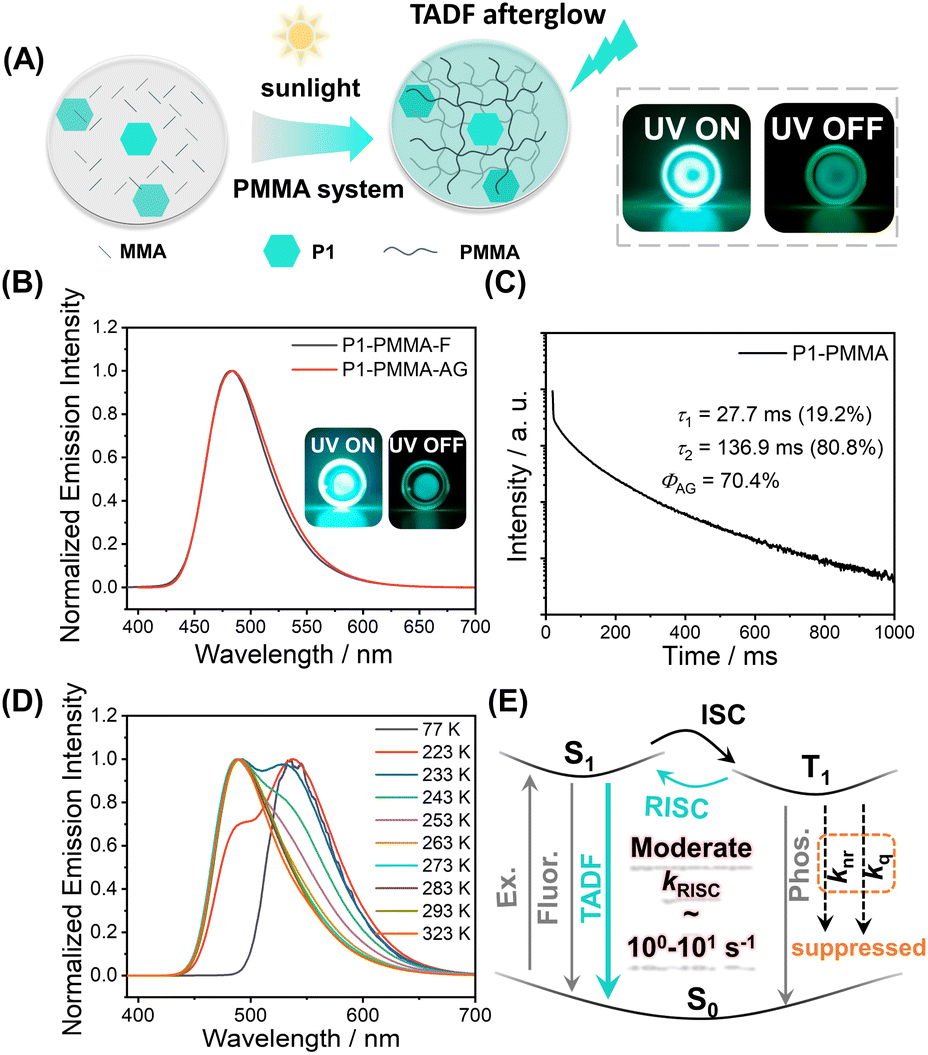 | ||
| Fig. 2 (A) Pyrylium-catalyzed photopolymerization of MMA and the resultant P1-PMMA TADF afterglow materials. (B) Room-temperature steady-state emission and delayed emission (1 ms delay) spectra of P1-PMMA materials at 0.01 wt% P1 concentration (excited at 373 nm). (C) Room-temperature excited state decay profile and its fitting of P1-PMMA materials at 0.1 wt% P1 concentration (excited at 373 nm and monitored at 484 nm). (D) Variable temperature delayed emission spectra (1 ms delay) of P1-PMMA materials at 0.1 wt% P1 concentration (excited at 373 nm). (E) The proposed TADF-type afterglow mechanism in the P1-PMMA system which features moderate kRISC of 100–101 s−1 to improve afterglow efficiency and maintain afterglow lifetimes. | ||
Excited state decay profiles monitored at 484 nm show that the delayed fluorescence lifetime (τ2, which is responsible for the afterglow) is 136.9 ms (Fig. 2C and S14†). The τ1 (27.7 ms, Fig. 2C) can be assigned as prompt fluorescence lifetime, which is greatly overestimated because a microsecond flash lamp is used as the excitation source; the accurate value of the prompt fluorescence lifetime determined by a picosecond pulse laser technique is 3.55 ns (Fig. S14†). Given the RISC process is the rate-determine step for TADF afterglow, the kRISC value of the P1-PMMA system can be estimated to be on the order of 100–101 s−1. Such moderate kRISC values are a distinct feature of TADF-type afterglow systems different from conventional TADF systems for efficient OLED devices (kRISC, 103–106 s−1).3,48–51 In most heavy-atom-free organic systems, the kP values are usually 10−2–103 s−1, so such moderate kRISC values of 100–101 s−1 would be enough to open a TADF pathway to harvest triplet energies and enhance organic afterglow efficiency; here the room-temperature kRISC value would be much larger than the kP value in the P1-PMMA system as can be seen from the room-temperature delayed emission spectra. The molecular design for TADF afterglow emitter is different from TADF OLED emitters. In conventional TADF systems to achieve large kRISC of 103–106 s−1, both small singlet–triplet splitting energy (ΔEST of around 0.2 eV or lower) and large spin–orbit coupling matrix elements (SOCME of around 0.5 cm−1 or above) are considered to be necessary.52,53 In the present system, the P1 molecules possess intramolecular charge transfer characteristics where the HOMO–LUMO separation can reduce ΔEST to some extent. Besides, the multiple electron-donating groups (two anisole groups and one methyl group) can endow P1 molecules with rich excited states, where some Tn states may have different symmetry from S1 states (Fig. S3†). For instance, the T3 state has intramolecular charge transfer character mainly from one anisole group to pyrylium group, while the S1 state has intramolecular charge transfer character mainly from two anisole groups to the pyrylium group. According to the El-Sayed rule, such difference would lead to enhanced intersystem crossing. TD-DFT calculations exhibit that the S1–T2 and S1–T3 channels have SOCME values of 0.31 cm−1 and 0.39 cm−1, respectively, which can mediate intersystem crossing of the present system; S1 and T1 states have similar symmetry of intramolecular charge transfer mainly from two anisole groups to the pyrylium group and thus show S1–T1 SOCME of only 0.09 cm−1. In addition, P1's S1 states with charge transfer characteristics exhibit large dipole moments. Recent studies by us and other groups have revealed that the dipole–dipole interactions between luminescent molecules' S1 states and environments can reduce S1 levels with less effect on T1 levels and thus enhance both ISC and RISC.54,55 Based on the above analyses, it should be understandable that the moderate ΔEST (0.25 eV) and the moderate SOCME (around 0.3 cm−1) can give rise to the moderate kRISC of 100–101 s−1 in the present P1-PMMA system. The molecular design for TADF afterglow here would be less stringent than TADF OLED; organic molecules with large twisted angles are not necessary for TADF afterglow.
It is worth mentioning that polymer-based organic afterglow materials display excellent mechanical properties, which would be necessary for their application in diverse circumstances. For the preparation of polymer-based afterglow materials, solution casting and melt casting are the most used techniques which require the use of solvents or high processing temperatures. Here the pyrylium induced photopolymerization provides a solvent-free method for the fabrication of polymer-based afterglow materials under mild conditions.
We also test the photocatalytic property of P1 using other monomers, such as 2-bromoethyl acrylate and 9-vinylcarbazole (BrMA and VK, Scheme 1). In the P1-BrMA system, photopolymerization has been found to form materials with fluorescence maxima at 484 nm and phosphorescence maxima at 533 nm at 77 K (Fig. 3B). At room temperature, the delayed emission spectra exhibit a red-shift emission band when compared to the steady-state emission spectra (Fig. 3C). After peak separation, the delayed emission band has been found to show a 26.1% delayed fluorescence component and 73.9% phosphorescence component (Fig. 3D). The RTP of P1-PBrMA materials has an emission lifetime of 1.54 ms (Fig. 3E). When compared to the P1-PMMA system, these observations indicate the presence of an external heavy atom effect in the P1-PBrMA system. P1's kP values can be greatly increased by PBrMA's heavy atoms to compete with P1's kRISC, leading to a drastic change in the delayed emission spectra and delayed emission lifetimes. It is interesting to find that the simple variation of monomers significantly changes the photophysical behaviors and mechanism in the resultant P1-polymer system, that is, the reversal of the TADF mechanism and RTP mechanism.
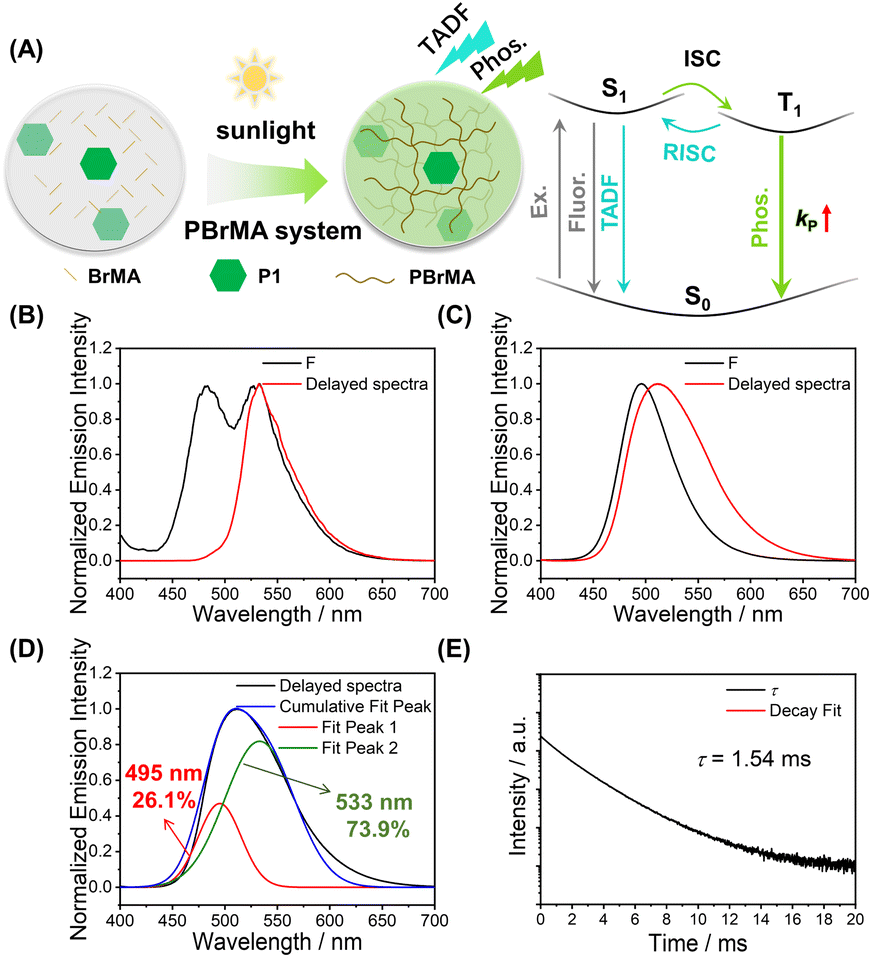 | ||
| Fig. 3 (A) Pyrylium-catalyzed photopolymerization of BrMA and the proposed RTP plus TADF mechanism in P1-PBrMA materials. (B and C) Steady-state emission and delayed emission (1 ms delay) spectra of 0.1%-P1-PBrMA materials at 77 K (excited at 380 nm) (B) and at room temperature (C). (D) Room-temperature delayed emission (1 ms delay) spectra of 0.1%-P1-PBrMA materials after peak separation (excited at 380 nm). (E) Excited state decay profile of 0.1%-P1-PBrMA materials at room temperature (excited at 380 nm and monitored at 506 nm). | ||
More interestingly, in the case of P1-VK, the obtained materials after photopolymerization (the formation of PVK was confirmed by HNMR and GPC, Fig. S16 and S17†) have been found to exhibit orange-red afterglow with hour-long durations and emission maxima at 607 nm under ambient conditions (Fig. 4A). Room-temperature afterglow emission spectra as a function of time (excited at 442 nm) show that the afterglow intensity follows a power law decay, that is, the afterglow intensity is proportional to t−m, where the index m can be fitted as 1.08 (Fig. 4B). The hour-long duration and power law decay suggest that the P1-PVK system has an OLPL mechanism caused by retarded charge recombination in glassy PVK matrices formed. VK solids, PVK polymers and P1-VK materials show insignificant room-temperature afterglow, so the carbazole isomeric mechanism is not related to the OLPL in the present study (Fig. S18†). In addition, the P1-PVK' materials obtained by doping P1 into commercial PVK' can only show room-temperature afterglow properties under degassed conditions, which further confirms the advantages of in situ photopolymerization (Fig. S19†). Upon photoirradiation, PVK (HOMO, −5.29 eV; LUMO, −1.80 eV) and P1 (HOMO, −6.53 eV; LUMO, −3.95 eV) form an exciplex with emission maxima at 607 nm in the steady-state emission spectra, which is significantly longer than those in the steady-state spectra of both the P1-PMMA sample and P1 solution in dichloromethane, as well as PVK solution (Fig. S20†). The delayed emission spectra at 77 K (610 nm, 2.03 eV) show similar emission maxima to the steady-state emission spectra at room temperature (607 nm, 2.04 eV). These observations agrees with the photophysical behavior of reported exciplex systems.33 Exciplex systems are assumed to possess nearly degenerate S1 and T1 levels with intermolecular charge transfer characteristics. Such S1 and T1 levels, lower than P1's T1 levels (2.30 eV), can block the pathway of energy transfer from the P1-PVK exciplex to P1's T1 states.56 Consequently, the P1-PVK exciplex undergoes charge separation to form P1 radical and PVK radical cations. Fig. S22† shows the time-integrated transient absorption spectrum from 0 to 130 μs, where the broad band from 700 nm to 1400 nm supports the presence of PVK radical cations.57,58 We also illustrate the transient absorption spectra collected at delay times from 20 ns to 100 μs in Fig. 4C, as well as the decay profiles monitored at different wavelengths (Fig. S23†). The long-lived characteristics of PVK radical cations as can be seen from the decay profiles (Fig. S23†) further support the charge separation and retarded charge recombination processes for OLPL emergence. Here the OLPL mechanism also receives support from Adachi's recent study on small-molecule pyrylium-donor systems.33 It should be noted that here the pyrylium salts are synthesized via our cascade reaction, the OLPL materials are polymer-based, and the photopolymerization and mechanism landscape are unexpected findings; these are the distinct differences between the present study and Adachi's study.
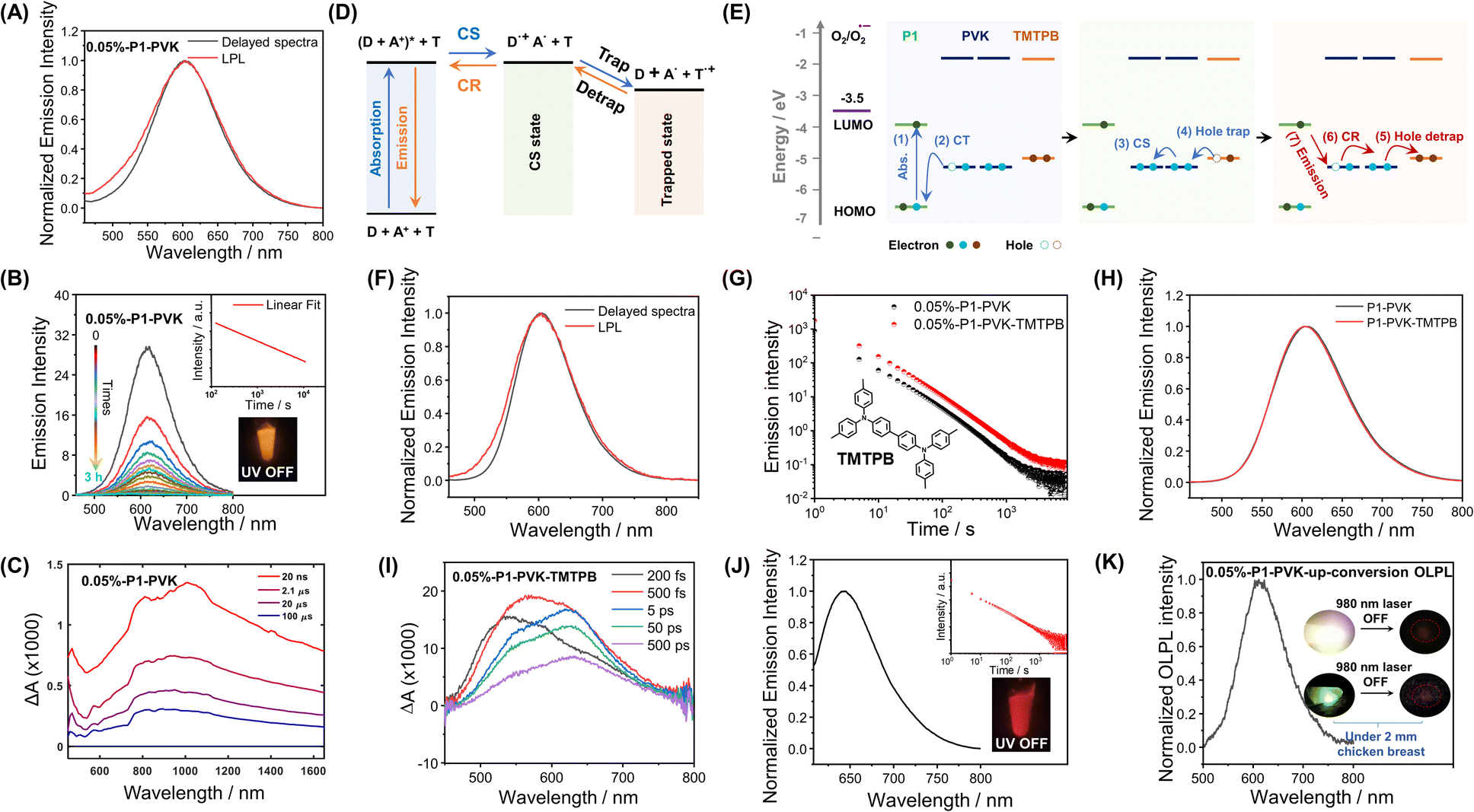 | ||
| Fig. 4 (A) Room-temperature delayed emission (1 ms delay) spectra and emission spectra 10 s after photoexcitation (OLPL) of the 0.05%-P1-PVK materials (excited at 442 nm). (B) OLPL spectra of the 0.05%-P1-PVK materials as a function of delay times (excited at 442 nm). The inset shows a logarithmic plot of the OLPL decay profile of 0.05%-P1-PVK materials, monitored at a peak wavelength of 606 nm. (C) Transient absorption spectra of the 0.05%-P1-PVK materials (excited by a 355 nm pump laser) collected at delay times from 20 ns to 100 μs. (D) The proposed charge separation, retarded charge recombination and hole trapping mechanism in the pyrylium-PVK OLPL system. (E) Proposed emission mechanism of the pyrylium-PVK OLPL system with relevant HOMO–LUMO energy level diagrams of materials and reduction potential of oxygen. (F) Room-temperature delayed emission (1 ms delay) spectra and emission spectra 10 s after photoexcitation (OLPL) of the 0.05%-P1-PVK-TMTPB materials (excited at 442 nm). (G) Room-temperature emission decay (1 ms delay) of 0.05%-P1-PVK and 0.05%-P1-PVK-TMTPB materials (excited at 442 nm and monitored at 606 nm). (H) Room-temperature delayed emission (1 ms delay) spectra of the 0.05%-P1-PVK materials and 0.05%-P1-PVK-TMTPB materials. (I) Transient absorption spectra of the 0.05%-P1-PVK-TMTPB materials (excited by a 390 nm pump laser) collected at delay times from 200 fs to 500 ps. (J) Room-temperature delayed emission (1 ms delay) spectra of 0.1%-P2-PVK materials with a red afterglow and emission maxima around 650 nm (excited at 588 nm). Inset shows the logarithmic plot of their emission decay (1 ms delay), monitored at a peak wavelength of 650 nm. (K) Room-temperature OLPL spectra (10 s delay) of the 0.05%-P1-PVK-up-conversion materials after photoexcitation by a 980 nm laser (500 mW). The inset shows the photographs of 0.05%-P1-PVK-up-conversion orange-red afterglow materials after ceasing 980 nm NIR excitation (top row, without 2 mm chicken breast; bottom row, the afterglow materials and the 980 NIR excitation source are separated by 2 mm chicken breast). | ||
To enhance the brightness of the OLPL system, we screen various compounds with higher HOMO levels than PVK to serve as hole trap (Fig. 4D and E). It has been found that, when 0.05 wt% N,N,N′,N′-tetrakis(4-methylphenyl)-benzidine (TMTPB) was incorporated into the P1-VK system, the obtained three-component materials after photopolymerization exhibit the enhancement of orange-red afterglow intensity by around 2 times with the delayed emission maxima nearly unchanged when compared to the P1-PVK two-component materials (Fig. 4F–H). In the case of P1-PVK-TMTPB materials, the transient absorption spectra (excited by a 390 nm pump laser) collected at delay times from 200 fs to 500 ps show a broad band from 500 nm to 750 nm (Fig. 4I and S24†), which can be mainly attributed to P1's excitons59–63 and possibly the minor contribution of P1's radicals.62–64 At such an early stage, the contribution of P1's radicals for the transient absorption spectra should be small, because the extent of intermolecular charge transfer from PVK to P1 should be small. Similarly, the signals of PVK radical cations at lower-energy regions should also be insignificant. The hour-long duration and power-law decay still support the OLPL afterglow mechanism in the P1-PVK-TMTPB system. It is proposed that the charge recombination between P1 radicals and the radical cations are suppressed by the glassy PVK matrices, leading to the emergence of hour-long OLPL due to the hole diffusion mechanism in the present study. Other pyrylium salts, such as P2 (Scheme 1), have also been used to catalyze the polymerization of VK monomers. The resultant P2-PVK materials have also been found to exhibit red OLPL properties with emission maxima around 650 nm and hour-long durations (Fig. 4J). In RTP and TADF afterglow systems, due to the energy gap law, it is very challenging to achieve long-lived red afterglow materials.5–18 The present study provides a robust pathway to address this issue. In addition, rare-earth up-conversion materials have also been incorporated into the pyrylium-VK systems. The obtained three-component materials under 2 mm chicken breast have been found to be excitable by 980 nm lasers to emit OLPL under ambient conditions (Fig. 4K), while Fig. S25† proposes a schematic illustration of the OLPL afterglow from the obtained three-component materials. 980 nm near infrared (NIR) lights have deep penetration in biological tissues and show negligible damage to biological systems. In addition, the OLPL materials can escape from the autofluorescence interference in the afterglow imaging mode. The OLPL system with the hole diffusion mechanism is relatively stable against oxygen when compared to the electron diffusion mechanism. These advantages shed light on their promising biological applications by using the obtained NIR excitable OLPL materials.
Conclusion
In conclusion, from an unexpected finding, we establish the mechanism landscape in pyrylium induced afterglow systems as illustrated in Fig. 1. These intriguing afterglow mechanisms originate from the different electronic properties of the resultant polymers after pyrylium induced photopolymerization. The pyrylium salts with intramolecular charge transfer characteristics and multi-donor design show moderate ΔEST. In the pyrylium-PMMA system, with the aid of dipole–dipole interactions, the materials exhibit TADF-type afterglow behaviors, which feature moderate kRISC of 100–101 s−1 to harvest triplet energies and show afterglow efficiency as high as 70.4%. In the pyrylium-PBrMA system, we find the reversal of the TADF afterglow and RTP mechanism because of the involvement of an external heavy atom effect. In the case of pyrylium-PVK, the formation of an exciplex drastically changes the photophysical pathway. Specifically, it blocks the population of pyrylium's T1 states because of the lower energy levels of the exciplex and opens the charge separation plus retarded charge recombination pathway, giving rise to hour-long OLPL properties of the hole diffusion mechanism against the influence of oxygen under ambient conditions. With the incorporation of third components, we achieve brighter OLPL properties and NIR excitable systems under ambient conditions.The present study demonstrates that the control of the rate constants of key photophysical processes and the manipulation of excited state energy levels can give rise to an intriguing mechanism landscape in very simple organic systems. The variation of the photophysical mechanism gives a very significant optical response, through which afterglow materials with desired functions and smart features would be constructed. It is noted that, although the individual afterglow mechanism (RTP, TADF or OLPL) has been frequently reported in previous studies, the establishment of the mechanism landscape in an organic system, such as the pyrylium induced afterglow system here, has been rarely explored (Text S3†). The present study also provides a straightforward and robust pathway for fabricating highly efficient and long-lived polymer-based afterglow materials from luminescent compounds and simple monomers. This study would have a significant impact in the field and further study would be needed for the extension of the concept to other molecular systems and the exploration of the applications of the obtained afterglow materials.
Data availability
Further details of the experimental procedures, the computational studies, and the characterization data for the new compounds are available in the ESI.† All relevant data are available from the corresponding authors upon request.Author contributions
G. W. carried out the experiments and analyzed the results. X. C., X. L. and Y. Z. provided assistance for the photophysical measurements and the design of graphical abstract. G. W. and K. Z. wrote and edited the original manuscript. K. Z. supervised the project.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are thankful for the financial support from the National Natural Science Foundation of China (22175194), Shanghai Scientific and Technological Innovation Project (20QA1411600, 20ZR1469200), and Hundred Talents Program from Shanghai Institute of Organic Chemistry (Y121078). We thank the staff of Magnitude Instruments for their assistance with transient absorption measurements.Notes and references
- V. W.-W. Yam, V. K.-M. Au and S. Y.-L. Leung, Chem. Rev., 2015, 115, 7589–7728 CrossRef CAS PubMed.
- J. Mei, N. L. Leung, R. T. Kwok, J. W. Lam and B. Z. Tang, Chem. Rev., 2015, 115, 11718–11940 CrossRef CAS PubMed.
- H. Uoyama, K. Goushi, K. Shizu, H. Nomura and C. Adachi, Nature, 2012, 492, 234–238 CrossRef CAS PubMed.
- K. Y. Zhang, Q. Yu, H. Wei, S. Liu, Q. Zhao and W. Huang, Chem. Rev., 2018, 118, 1770–1839 CrossRef CAS PubMed.
- W. Zhao, Z. He and B. Z. Tang, Nat. Rev. Mater., 2020, 5, 869–885 CrossRef CAS.
- X. Ma, J. Wang and H. Tian, Acc. Chem. Res., 2019, 52, 738–748 CrossRef CAS PubMed.
- S. Hirata, Adv. Opt. Mater., 2017, 5, 1700116 CrossRef.
- A. Forni, E. Lucenti, C. Botta and E. Cariati, J. Mater. Chem. C, 2018, 6, 4603–4626 RSC.
- C. C. Kenry and B. Liu, Nat. Commun., 2019, 10, 2111 CrossRef PubMed.
- N. Gan, H. Shi, Z. An and W. Huang, Adv. Funct. Mater., 2018, 28, 1802657 CrossRef.
- T. Zhang, X. Ma, H. Wu, L. Zhu, Y. Zhao and H. Tian, Angew. Chem., Int. Ed., 2020, 59, 11206–11216 CrossRef CAS PubMed.
- J. Li, G. Wang, X. Chen, X. Li, M. Wu, S. Yuan, Y. Zou, X. Wang and K. Zhang, Chem. –Eur. J., 2022, 28, e202200852 CAS.
- S. Guo, W. Dai, X. Chen, Y. Lei, J. Shi, B. Tong, Z. Cai and Y. Dong, ACS Mater. Lett., 2021, 3, 379–397 CrossRef CAS.
- S. Hirata, Appl. Phys. Rev., 2022, 9, 011304 CAS.
- X. Yan, H. Peng, Y. Xiang, J. Wang, L. Yu, Y. Tao, H. Li, W. Huang and R. Chen, Small, 2022, 18, 2104073 CrossRef CAS PubMed.
- H. Gao and X. Ma, Aggregate, 2021, 2, e38 CAS.
- Q. Li and Z. Li, Acc. Chem. Res., 2020, 53, 962–973 CrossRef CAS PubMed.
- M. Singh, K. Liu, S. Qu, H. Ma, H. Shi, Z. An and W. Huang, Adv. Opt. Mater., 2021, 9, 2002197 CrossRef CAS.
- G. Zhang, G. M. Palmer, M. W. Dewhirst and C. L. Fraser, Nat. Mater., 2009, 8, 747–751 CrossRef CAS PubMed.
- W. Z. Yuan, X. Y. Shen, H. Zhao, J. W. Y. Lam, L. Tang, P. Lu, C. Wang, Y. Liu, Z. Wang, Q. Zheng, J. Z. Sun, Y. Ma and B. Z. Tang, J. Phys. Chem. C, 2010, 114, 6090–6099 CrossRef CAS.
- O. Bolton, K. Lee, H.-J. Kim, K. Y. Lin and J. Kim, Nat. Chem., 2011, 3, 205–210 CrossRef CAS.
- S. Hirata, K. Totani, J. Zhang, T. Yamashita, H. Kaji, S. R. Marder, T. Watanabe and C. Adachi, Adv. Funct. Mater., 2013, 23, 3386–3397 CrossRef CAS.
- R. Kabe and C. Adachi, Nature, 2017, 550, 384–387 CrossRef CAS.
- I. Bhattacharjee and S. Hirata, Adv. Mater., 2020, 32, 2001348 CrossRef CAS.
- Y. Wang, J. Yang, M. Fang, Y. Yu, B. Zou, L. Wang, Y. Tian, J. Cheng, B. Z. Tang and Z. Li, Matter, 2020, 3, 449–463 CrossRef.
- Y. Zhang, Y. Su, H. Wu, Z. Wang, C. Wang, Y. Zheng, X. Zheng, L. Gao, Q. Zhou, Y. Yang, X. Chen, C. Yang and Y. Zhao, J. Am. Chem. Soc., 2021, 143, 13675–13685 CrossRef CAS.
- X. Wang, Y. Sun, G. Wang, J. Li, X. Li and K. Zhang, Angew. Chem., Int. Ed., 2021, 60, 17138–17147 CrossRef CAS PubMed.
- M. Louis, H. Thomas, M. Gmelch, A. Haft, F. Fries and S. Reineke, Adv. Mater., 2019, 31, 1807887 CrossRef.
- J. Jin, H. Jiang, Q. Yang, L. Tang, Y. Tao, Y. Li, R. Chen, C. Zheng, Q. Fan, K. Y. Zhang, Q. Zhao and W. Huang, Nat. Commun., 2020, 11, 842 CrossRef CAS PubMed.
- Z. Yang, C. Xu, W. Li, Z. Mao, X. Ge, Q. Huang, H. Deng, J. Zhao, F. L. Gu, Y. Zhang and Z. Chi, Angew. Chem., Int. Ed., 2020, 59, 17451–17455 CrossRef CAS PubMed.
- X.-F. Wang, H. Xiao, P.-Z. Chen, Q.-Z. Yang, B. Chen, C.-H. Tung, Y.-Z. Chen and L.-Z. Wu, J. Am. Chem. Soc., 2019, 141, 5045–5050 CrossRef CAS PubMed.
- Z. He, H. Gao, S. Zhang, S. Zheng, Y. Wang, Z. Zhao, D. Ding, B. Yang, Y. Zhang and W. Z. Yuan, Adv. Mater., 2019, 31, 1807222 CrossRef.
- K. Jinnai, R. Kabe, Z. Lin and C. Adachi, Nat. Mater., 2022, 21, 338–344 CrossRef CAS PubMed.
- W. Li, Z. Li, C. Si, M. Y. Wong, K. Jinnai, A. K. Gupta, R. Kabe, C. Adachi, W. Huang, E. Zysman-Colman and I. D. W. Samuel, Adv. Mater., 2020, 32, 2003911 CrossRef CAS PubMed.
- B. Chen, W. Huang, X. Nie, F. Liao, H. Miao, X. Zhang and G. Zhang, Angew. Chem., Int. Ed., 2021, 60, 16970–16973 CrossRef CAS.
- X.-K. Ma and Y. Liu, Acc. Chem. Res., 2021, 54, 3403–3414 CrossRef CAS PubMed.
- W. Zhao, Z. He, J. W. Y. Lam, Q. Peng, H. Ma, Z. Shuai, G. Bai, J. Hao and B. Z. Tang, Chem, 2016, 1, 592–602 CAS.
- H. Ma, Q. Peng, Z. An, W. Huang and Z. Shuai, J. Am. Chem. Soc., 2019, 141, 1010–1015 CrossRef CAS.
- G. Wang, X. Li, X. Wang and K. Zhang, New J. Chem., 2021, 45, 12305–12310 RSC.
- G. Wang, J. Li, X. Li, X. Wang, Y. Sun, J. Liu and K. Zhang, Chem. Eng. J., 2022, 431, 134197 CrossRef CAS.
- N. A. Romero and D. A. Nicewicz, Chem. Rev., 2016, 116, 10075 CrossRef CAS.
- D. A. Nicewicz and T. M. Nguyen, ACS Catal., 2014, 4, 355 CrossRef CAS.
- J.-X. Wang, H. Zhang, L.-Y. Niu, X. Zhu, Y.-F. Kang, R. Boulatov and Q.-Z. Yang, CCS Chem., 2020, 2, 1391–1398 CrossRef CAS.
- S. Xu, W. Wang, H. Li, J. Zhang, R. Chen, S. Wang, C. Zheng, G. Xing, C. Song and W. Huang, Nat. Commun., 2020, 11, 4802 CrossRef CAS.
- S. Kuila and S. J. George, Angew. Chem., Int. Ed., 2020, 59, 9393 CrossRef CAS PubMed.
- Y. Lei, W. Dai, J. Guan, S. Guo, F. Ren, Y. Zhou, J. Shi, B. Tong, Z. Cai, J. Zheng and Y. Dong, Angew. Chem., Int. Ed., 2020, 59, 16054–16060 CrossRef CAS PubMed.
- C. Chen, Z. Chi, K. C. Chong, A. S. Batsanov, Z. Yang, Z. Mao, Z. Yang and B. Liu, Nat. Mater., 2020, 20, 175–180 CrossRef PubMed.
- Y. Tao, K. Yuan, T. Chen, P. Xu, H. Li, R. Chen, C. Zheng, L. Zhang and W. Huang, Adv. Mater., 2014, 26, 7931–7958 CrossRef CAS.
- M. Y. Wong and E. Zysman-Colman, Adv. Mater., 2017, 29, 1605444 CrossRef PubMed.
- Y. Liu, C. Li, Z. Ren, S. Yan and M. R. Bryce, Nat. Rev. Mater., 2018, 3, 18020 CrossRef CAS.
- Y. Im, M. Kim, Y. J. Cho, J.-A. Seo, K. S. Yook and J. Y. Lee, Chem. Mater., 2017, 29, 1946–1963 CrossRef CAS.
- H. Noda, X. Chen, H. Nakanotani, T. Hosokai, M. Miyajima, N. Notsuka, Y. Kashima, J. Bredas and C. Adachi, Nat. Mater., 2019, 18, 1084–1090 CrossRef CAS PubMed.
- P. K. Samanta, D. Kim, V. Coropceanu and J. L. Bredas, J. Am. Chem. Soc., 2017, 139, 4042–4051 CrossRef CAS PubMed.
- Y. Sun, J. Liu, J. Li, X. Li, X. Wang, G. Wang and K. Zhang, Adv. Opt. Mater., 2021, 10, 2101909 CrossRef.
- A. J. Gillett, A. Pershin, R. Pandya, S. Feldmann, A. J. Sneyd, A. M. Alvertis, E. W. Evans, T. H. Thomas, L.-S. Cui, B. H. Drummond, G. D. Scholes, Y. Olivier, A. Rao, R. H. Friend and D. Beljonne, Nat. Mater., 2022, 21, 1150–1157 CrossRef CAS PubMed.
- N. Nishimura, Z. Lin, K. Jinnai, R. Kabe and C. Adachi, Adv. Funct. Mater., 2020, 30, 2000795 CrossRef CAS.
- Y. Tsujii, A. Tsuchida, Y. Onogi and M. Yamamoto, Macromolecules, 1990, 23, 4019–4023 CrossRef CAS.
- Y. Tsujii, A. Tsuchida, M. Yamamoto and Y. Nishijima, Macromolecules, 1988, 21, 665–670 CrossRef CAS.
- W. G. Santos, D. S. Budkina, S. H. Santagneli, A. N. Tarnovsky, J. Zukerman-Schpector and S. J. L. Ribeiro, J. Phys. Chem. A, 2019, 123, 7374–7383 CrossRef CAS PubMed.
- L. E. Rosch, M. R. Crawley, R. M. O'Donnell, T. N. Rohrabaugh Jr, T. R. Ensley, T. A. Sobiech and T. R. Cook, Organometallics, 2022, 41, 2301–2316 CrossRef CAS.
- I. S. Banu and P. Ramamurthy, J. Photochem. Photobiol., A, 2009, 201, 175–182 CrossRef CAS.
- P. Montes-Navajas and H. Garcia, J. Colloid Interface Sci., 2013, 410, 111–115 CrossRef CAS PubMed.
- M. A. Miranda and H. Garcia, Chem. Rev., 1994, 94, 1063–1089 CrossRef CAS.
- T. Karatsu, K. Kanayama, M. Takahashi, N. Ishigohoka, K. Fukui and A. Kitamura, Heteroat. Chem., 2001, 12, 269–275 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sc01500a |
| This journal is © The Royal Society of Chemistry 2023 |