
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSurface polarization-induced emission and stability enhancement of CsPbX3 nanocrystals†
Keqiang
Chen
 abcd,
Zixin
Gu
a,
Zhiqing
Wang
b,
Mengyu
Guan
a,
Xiu
Tan
a,
Wanqing
Xu
a,
Xinyu
Ji
a,
Weiqi
Lu
a,
Yueli
Liu
*b and
Guogang
Li
*ac
abcd,
Zixin
Gu
a,
Zhiqing
Wang
b,
Mengyu
Guan
a,
Xiu
Tan
a,
Wanqing
Xu
a,
Xinyu
Ji
a,
Weiqi
Lu
a,
Yueli
Liu
*b and
Guogang
Li
*ac
aFaculty of Materials Science and Chemistry, China University of Geosciences, Wuhan, 430074, P. R. China. E-mail: ggli@cug.edu.cn
bState Key Laboratory of Advanced Technology for Materials Synthesis and Processing, State Key Laboratory of Silicate Materials for Architectures, School of Materials Science and Engineering, Wuhan University of Technology, Wuhan, 430070, P. R. China. E-mail: lylliuwhut@whut.edu.cn
cZhejiang Institute, China University of Geosciences, Hangzhou, 311305, China
dShenzhen Research Institute, China University of Geosciences, Shenzhen, 518052, China
First published on 27th July 2023
Abstract
Recently, the polarization effect has been receiving tremendous attention, as it can result in improved stability and charge transfer efficiency of metal-halide perovskites (MHPs). However, realizing the polarization effect on CsPbX3 NCs still remains a challenge. Here, metal ions with small radii (such as Mg2+, Li+, Ni2+, etc.) are introduced on the surface of CsPbX3 NCs, which facilitate the arising of electric dipole and surface polarization. The surface polarization effect promotes redistribution of the surface electron density, leading to reinforced surface ligand bonding, reduced surface defects, near unity photoluminescence quantum yields (PLQYs), and enhanced stability. Moreover, further introduction of hydroiodic acid results in the in situ formation of tert-butyl iodide (TBI), which facilitates the successful synthesis of pure iodine-based CsPbI3 NCs with high PLQY (95.3%) and stability under ambient conditions. The results of this work provide sufficient evidence to exhibit the crucial role of the surface polarization effect, which promotes the synthesis of high-quality MHPs and their applications in the fields of optoelectronic devices.
Introduction
All-inorganic CsPbX3 (X = Cl, Br, I) perovskite nanocrystals (NCs) represent a flourishing area of research over the past decade, owing to their impressive optoelectronic properties, including strong light-absorption coefficients, photoluminescence (PL) with a narrow full width at half-maximum (FWHM) and high quantum yields, exceptional defect tolerance, and long carrier diffusion lengths. All these properties make them ideal candidates for a wide range of applications such as solar cells, light-emitting diodes (LEDs), lasers, and photodetectors.1–8 However, the poor stability of CsPbX3 NCs greatly limits their wide applications, which should be primarily ascribed to their ionic bonding nature induced highly dynamic surface ligands.9–13 The loss of surface ligands would lead to the formation of halide vacancies, and eventually to the failure of the crystal structure with substantial degradation of photoluminescence quantum yields (PLQYs).14–20 Therefore, much effort has been dedicated to passivate the surface of CsPbX3 NCs by introducing various kinds of ligands with enhanced bonding energy, such as trioctylphosphine (TOP),21 2,2′-iminodibenzoic acid (IDA),22 glutathione,23 4-bromo-butyric acid,24etc.25–32Our previous report suggested that short-chain ligands (such as octanoic acid and octylamine) possess larger polarity in comparison to the commonly used oleylamine (OLA) and oleic acid (OA). The increased polarity leads to stronger adsorption energy, which can result in improved structural stability of CsPbX3 NCs.33 Moreover, it is well known that the polarity is closely related to dielectric properties, that is, a highly polarized material has a larger dielectric constant, which facilitates the charge transfer process.34–37 Recently, Liu et al. suggested that an increase in the dielectric constant could result in a decrease in the electrostatic force between the defects and carriers, which reduced the defect-capture radius.38 In addition, introducing dipolar molecules is known to be an effective way to enhance the stability of perovskite.39–41 Li et al. employed a dipole layer between organic transport layers and inorganic perovskite, leading to reduced energetic mismatch and increased charge extraction.41 Therefore, it is clear from previous studies that increasing the surface polarization of CsPbX3 NCs potentially provides an alternative strategy to strengthen the surface ligand bonding and decrease the defect-capture radius. However, a dipolar-molecule-based dipole layer is most commonly used to achieve the polarization effect in bulk perovskites,42 and this strategy is not suitable for CsPbX3 NCs dispersed in non-polar solvent with a small size (∼10 nm). It is reasonable to suppose that inducing distortion into the intrinsic surface structure of CsPbX3 NCs might be a feasible way to realize the polarization effect.
In this contribution, we develop a co-solvent assisted reprecipitation (CSARP) strategy to synthesize CsPbX3 NCs under ambient conditions. Various kinds of metal halides (such as MgX2, X = Cl, Br, I) with relatively smaller ionic radii (compared with the Pb2+ ion) are employed as halide precursors; sufficient halide ions can passivate the surface halide vacancies. More importantly, the metal ions with small radii would distort the metal-halide octahedron (such as [MgBr6]4−, which prefers to emerge on the surface of CsPbX3 NCs), resulting in the arising of surface polarization. As a result, CsPbX3 NCs with reduced surface defect density and improved surface ligand bonding are achieved, leading to near unity photoluminescence quantum yields (PLQYs) and enhanced stability. This phenomenon is further verified by using metal ions with various ionic radii. Moreover, pure iodine-based CsPbI3 NCs can be directly synthesized by the introduction of hydroiodic acid. Our results provide a facile solution for the preparation of CsPbX3 NCs with optimized optoelectronic properties and enhanced stability.
Results and discussion
In the commonly used ligand-assisted reprecipitation (LARP) approach, DMF/DMSO are employed as “good solvents” to dissolve metal halide salts (such as CsBr or PbBr2), and expectedly, these solvents would also lead to the failure of the as-formed perovskite structure.43 Therefore, the strategy adopted in this study is to dissolve metallic and halide precursors separately, which can minimize the negative effect of solvents. Fig. S1† presents a schematic illustration of the synthetic process of CsPbX3 NCs via our co-solvent assisted reprecipitation (CSARP) strategy. Briefly, Cs+ and Pb2+ precursors dissolved in acetic acid are added to a mixture of n-hexane, tert-butanol alcohol (TBA), OLA, and OA to form a homogeneous solution. Successively, a halide precursor (such as MgX2, X = Cl and Br) dissolved in water (HI for MgI2) is injected into the above solution, resulting in the formation of the desired CsPbX3 NCs; detailed information can be found in Experimental section, Fig. S2, Tables S1 and S2.† Here, TBA is employed as a transitionary solvent to balance the huge discrepancy between hexane and water (or hydriodic acid). In the absence of TBA, the immiscibility of hexane and water leads to the failure of the formation of perovskite NCs, as the metal and halide precursors are dissolved in hexane and water (or hydriodic acid), respectively.To start with, MgBr2 is used as a halide precursor to synthesize CsPbBr3 NCs. Fig. 1a shows typical optical images of the as-synthesized CsPbBr3 NCs under room light and a UV lamp (λ = 365 nm), respectively. A strong green emission emerges under a UV lamp, which is quite an expected result. Interestingly, however, prominent PL emission is also observed even under room light, which clearly suggests the remarkable optical properties of the as-prepared CsPbBr3 NCs. The UV-vis absorption spectrum of the as-synthesized CsPbBr3 NCs shown in Fig. 1b exhibits a clear excitonic absorption peak and absorption centered at 2.480 eV. In addition, near-unity PLQY with the emission peak at 2.431 eV is shown. The small Stokes shift (49 meV) suggests a direct exciton recombination process. These results further confirm the near ideal optical properties of the as-synthesized CsPbBr3 NCs. More discussions on the synthesis process can be found in ESI Note 1, Fig. S2, and Table S1.† X-ray diffraction (XRD) patterns and transmission electron microscope (TEM) images shown in Fig. 1c and d verify the pure phase and regular morphology of the as-synthesized CsPbBr3 NCs (Fig. S3†).44,45 Furthermore, the high-angle annular dark-field (HAADF) scanning TEM (STEM) image and the energy-dispersive X-ray spectroscopy (EDX) mappings shown in Fig. 1e reveal that a small amount of Mg2+ ions is uniformly dispersed in the NCs. Quantitative analysis of the EDX results further suggests that the atom ratio between Mg and Pb is about 5.2%. Compared with the XRD pattern of NCs synthesized through CSARP with/without Mg2+ ions and a hot-injection process, the diffraction peaks almost remain unchanged (Fig. 1c). Moreover, the NCs are further in situ etched by argon ion bombardment on the surface for various depths. It is clearly shown in Fig. 1f that the atom ratio of Mg/Pb is 6.6% at the very surface of NCs, which is also well in agreement with the EDX results. With the increase of etching depth, the atom ratio of Mg/Pb decreases rapidly (2–3%). Therefore, it can be inferred that Mg2+ ions should emerge on the surface of CsPbBr3 NCs.46
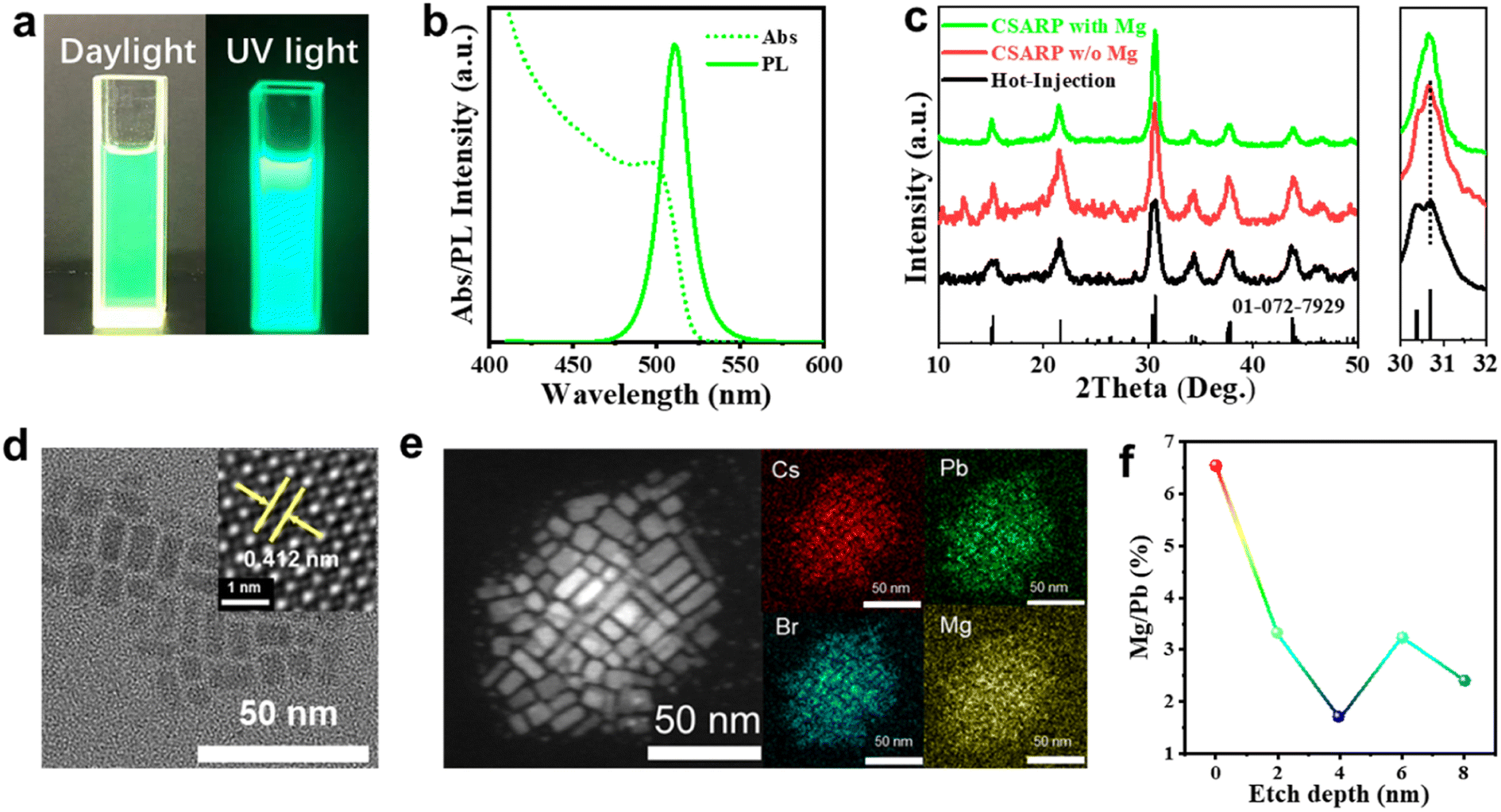 | ||
| Fig. 1 (a) Photographs of CSARP-CsPbBr3 NCs under room light (left) and a UV lamp (right, λ = 365 nm). (b) UV-vis and PL spectra of CSARP-CsPbBr3 NCs. (c) X-ray diffraction pattern of CSARP-CsPbBr3 NCs with/without Mg2+ ions and hot-injection-CsPbBr3, the right inset shows the magnified XRD patterns for the 2θ region of 30–32°. (d) TEM image of the CSARP-CsPbBr3 NCs (inset: HRTEM). (e) EDX mappings of the CSARP-CsPbBr3 NCs. (f) The Mg/Pb ratio in CSARP-CsPbBr3 NCs for various etching depths. | ||
Density functional theory (DFT) calculations are further taken into account to provide a comprehensive understanding on the role of Mg2+ ions in CsPbBr3 NCs. It is known that the adsorption of protonated OLA (H+-OLA) on the surface of CsPbBr3 NCs is crucial for their stability and optoelectronic properties.13 There is no net polarization on the surface of pure CsPbBr3 NCs due to the fact that the Pb atom is almost at the exact center of the [PbBr6]4− octahedron void, as shown in Fig. 2a. In contrast, when the Pb atoms on the surface layer are replaced by Mg atoms, their relatively smaller ionic radii remarkably increase the distortion of [MgBr6]4− octahedra (Fig. 2b). The lattice distortion drives the Mg atoms off the octahedral center, leading to the misalignment of positive and negative charges on the surface of CsPbBr3 NCs. As a result, the microscopic particles form an electric dipole and exhibit polarity. The surface polarization directly promotes the redistribution of the surface electron density, and the Br atom located right above the Mg atom (Fig. 2d, one Pb atom on the surface layer replaced by one Mg atom, marked as 1Mg) has higher electron density compared to that in pure CsPbBr3 (Fig. 2c). This phenomenon is also observed for the completely substituted one (i.e., all the Pb atoms on the surface layer are replaced by Mg atoms, marked as nMg, Fig. 2e). The enhanced electron density facilitates the adsorption of the protonated OLA with increased adsorption energy (Eads), which is found to be 5.457, 5.805, and 6.021 eV for pure CsPbBr3, 1Mg-, and nMg-CsPbBr3, respectively (trans-OLA with negligible polarity is selected for the calculation to exclude the influence of capping ligands). Furthermore, this observation could be also verified by differential charge density (Fig. 2f and g), which exhibits increased net interfacial electron transfer and a reduced Br-OLA bond length as the Mg atoms are introduced. Therefore, it is reasonable to conclude that the excellent optical properties of the as-synthesized CsPbBr3 NCs can be attributed to the reduced halide-vacancy density and enhanced surface ligand bonding.
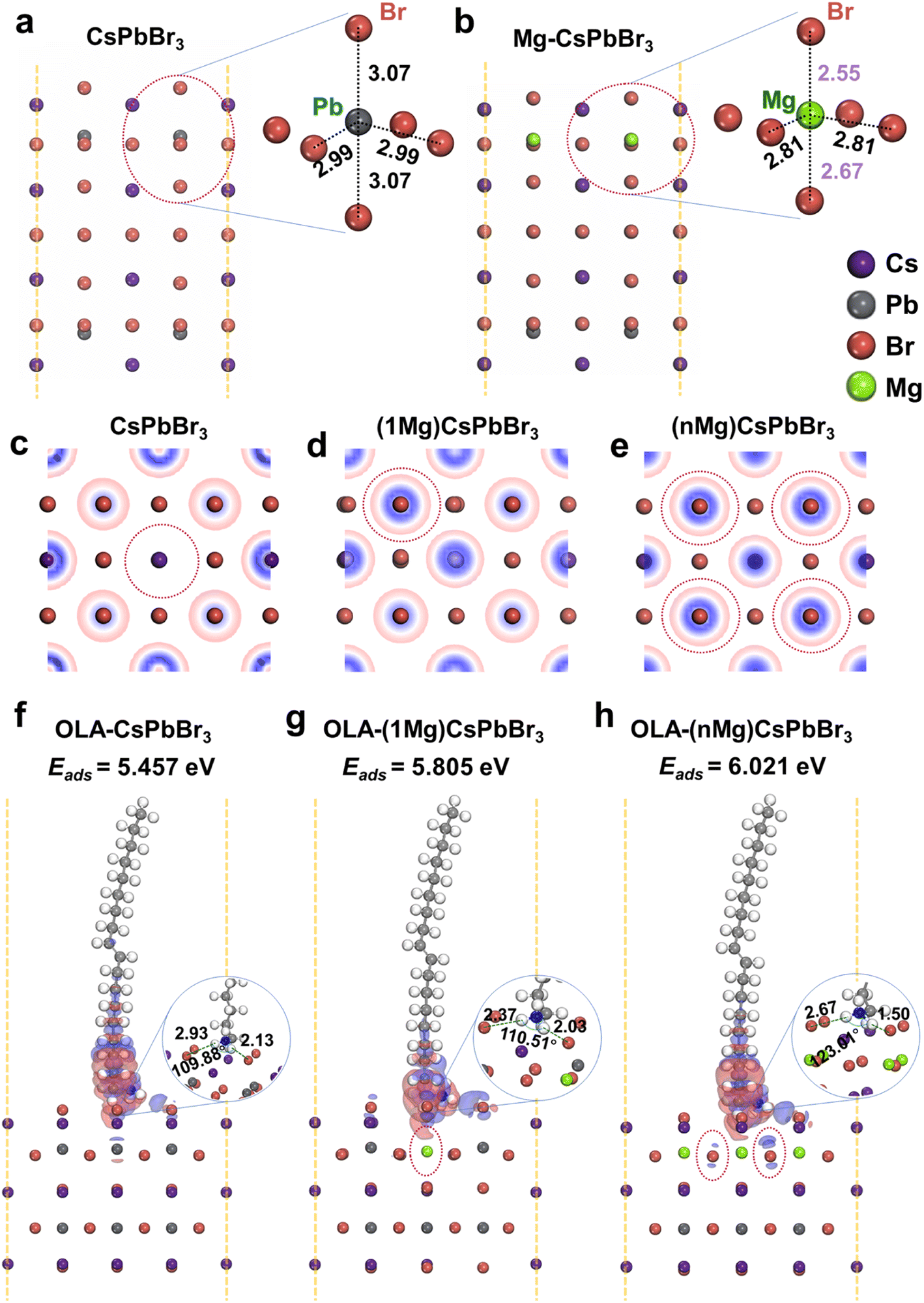 | ||
| Fig. 2 Surface electronic structure of (a) CsPbBr3, and (b) Mg–CsPbBr3. Insets: the geometries of [PbBr6]4− and [MgBr6]4− octahedra with Pb–Br and Mg–Br interatomic distances, respectively. The electron density distribution profiles of (c) CsPbBr3, (d) (1Mg) CsPbBr3, and (e) (nMg) CsPbBr3, respectively, where the blue color indicates an increase in electron density. Differential charge density for (f) OLA-CsPbBr3, (g) OLA-(1Mg)–CsPbBr3, and (h) OLA-(nMg)-CsPbBr3, respectively. Insets: local amplification images of the adsorption configurations. Purple, gray, brown, and green spheres represent Cs, Pb, Br, and Mg atoms, respectively. | ||
In order to have a more comprehensive understanding of the relationship between the polarity and optical properties of perovskite NCs, we further carry out a series of calculations based on various kinds of alkaline-earth metal halides (Be, Mg, Ca, Sr, and Ba). As shown in Fig. 3, destructive lattice distortions of [BeBr6]4− and [BaBr6]4− octahedra can be clearly observed when Pb atoms in the [PbBr6]4− octahedra are replaced by Be and Ba atoms, which should be ascribed to their unsuitable radii (either too small or too large). That is to say, Be2+ and Ba2+ are not suitable for the synthesis of perovskite NCs. In contrast, when MgBr2, CaBr2, and SrBr2 are employed as precursors, all of [MgBr6]4−, [CaBr6]4−, and [SrBr6]4− octahedra can be well maintained. Moreover, [MgBr6]4− and [SrBr6]4− octahedra exhibit the largest (ΔD = 0.12) and smallest (ΔD = 0.06) distortions, respectively. Therefore, we hypothesize that when alkaline-earth metal halides are used as precursors for the synthesis of perovskite NCs via the CSARP strategy, MgBr2, CaBr2, and SrBr2 could result in luminescent phase NCs, and the MgBr2-based one should possess the highest PLQY, while BeBr2 and BaBr2 would lead to non-luminescent phase NCs.
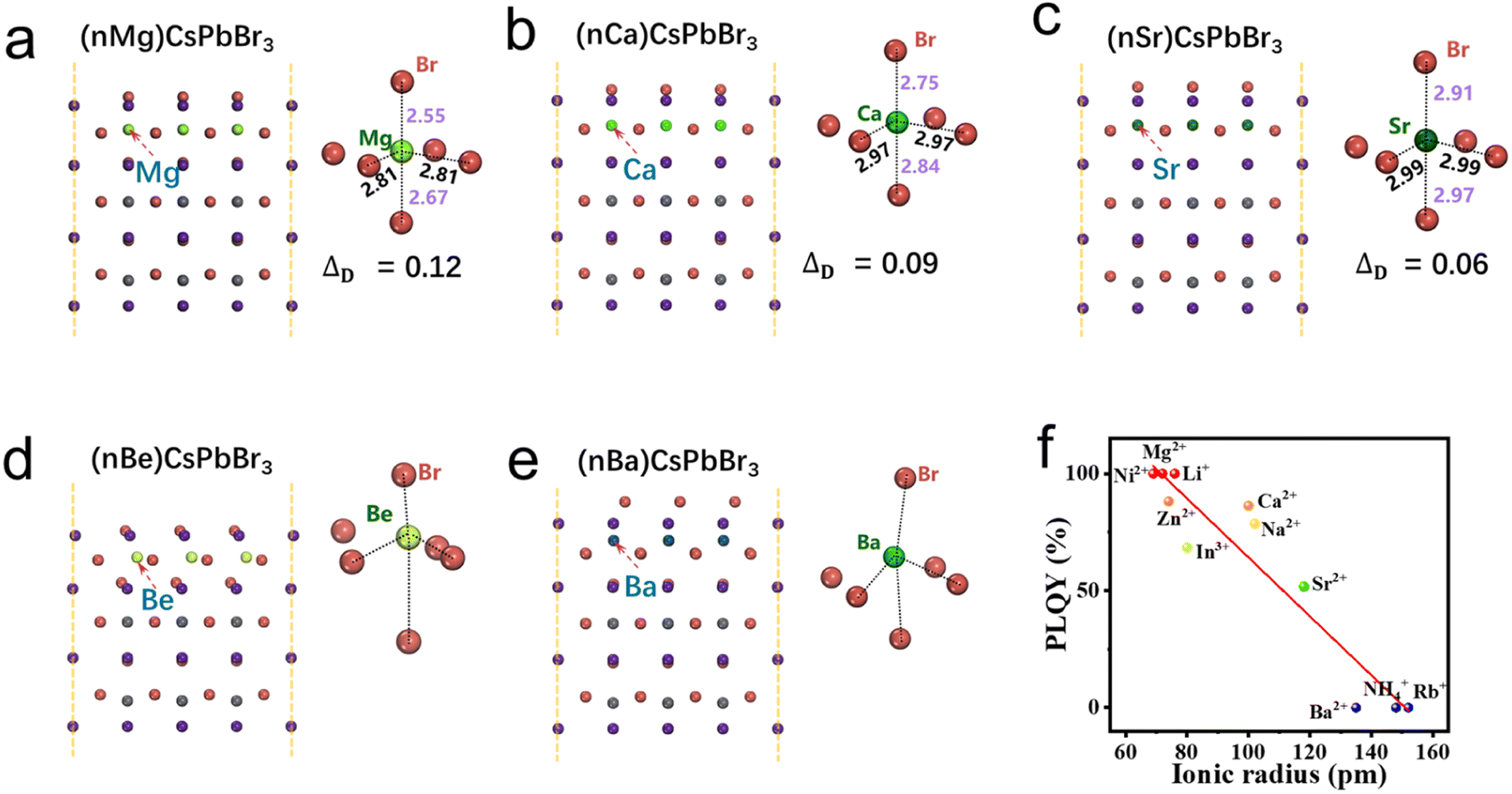 | ||
| Fig. 3 (a)–(e) Surface polarization of CsPbBr3 NCs based on alkaline-earth metal ions. (f) PLQY vs. ionic radius of various metal bromide precursors. | ||
Whereafter, CsPbBr3 NCs are synthesized using alkaline-earth metal halides as precursors via the CSARP process (as BeBr2 is unavailable, there are no corresponding data). From Fig. 3f, one can clearly find that Mg2+-based CsPbBr3 NCs exhibits the champion PLQY (∼100%), while the PLQYs of Ca2+-(86%) and Sr2+-(52%) based ones decrease in turn. Interestingly, the Ba2+-based one leads to the emergence of non-luminescent phase NCs (PLQY = 0%). As the concentration of Br− in these reactions is the same, therefore, the distinct discrepancy of the PLQY should be attributed to the arising of various kinds of cations. These outcomes are entirely consistent with the aforementioned theory calculations, which also provide substantial evidence for the previous supposition.
Likewise, the crucial relationships between the ionic radius, polarity, and optical properties are further verified by employing various kinds of metal halides (including LiBr, NaBr, RbBr, NH4Br, NiBr2, ZnBr2, and InBr3) as precursors for the synthesis of CsPbBr3 NCs. The PLQYs of NCs based on these precursors are shown in Fig. 3f. A clear tendency, that is, the PLQY having a negative correlation with the cation radius, can be observed. Within a proper range of cation radii, the PLQY can be estimated through the following equation:
| PQLY% = 190.13 − 1.26r | (1) |
For comparison, CsPbBr3 NCs are also prepared via the widely used LARP strategy (denoted as LARP-CsPbBr3 NCs). Optical property characterization studies shown in Fig. 4a and b suggest that the LARP-CsPbBr3 NCs possess a similar PL peak and absorption onset in comparison to those of perovskite NCs synthesized through the CSARP process, while the LARP-CsPbBr3 NCs have lower PLQY (ca. 70%), revealing their higher trap states. Meanwhile, we further calculate the Urbach energies (EU) of these samples by plotting the absorption coefficient as a function of photon energy. The value of EU can be extracted by fitting the exponential part of the Urbach tail according to the following equation:
α(E) = α0![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) exp[σ(T)(E − E0)/(kBT)] exp[σ(T)(E − E0)/(kBT)] | (2) |
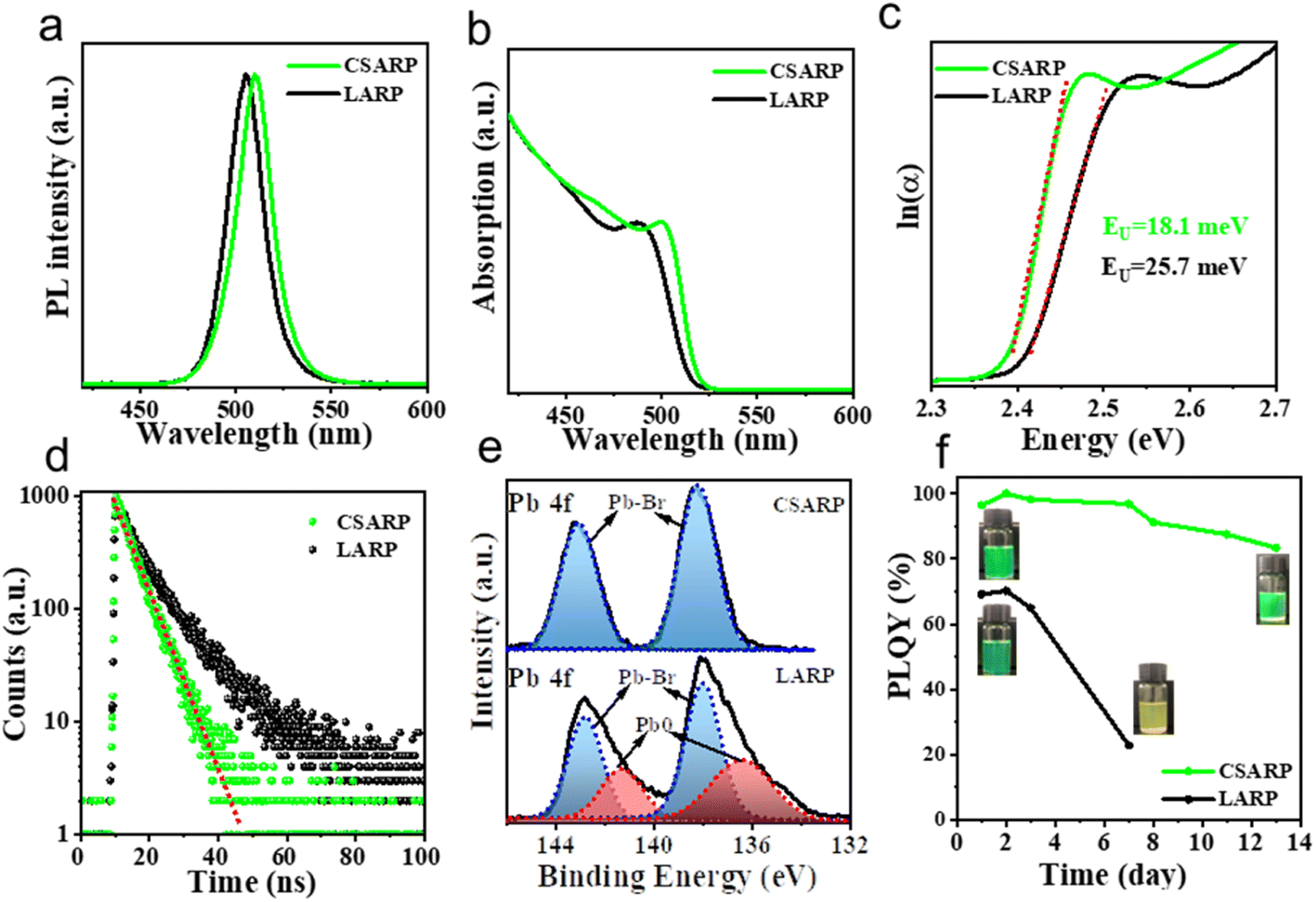 | ||
| Fig. 4 PL spectra (a), UV-vis absorption spectra (b), Urbach energies (EU) (c), time-resolved PL spectra (d), XPS spectra (e), and PL stability (f) of CSARP-CsPbBr3 (green lines) and LARP-CsPbBr3 (black lines). | ||
Furthermore, X-ray photoelectron spectroscopy (XPS) measurement is performed to monitor the chemical state of the CSARP- and LARP-CsPbBr3 NCs (Fig. 4e and S4†). The high-resolution XPS (HR-XPS) spectra of Pb illustrate distinct differences between these two samples, as shown in Fig. 4e. Two sets of peaks are observed for the LARP-CsPbBr3 NCs. One set of peaks at 138.0 (4f7/2) and 142.8 eV (4f5/2) are assigned to the internal Pb–Br bond, and another set of shoulder peaks at lower binding energies of 137.5 (4f7/2) and 142.4 eV (4f5/2) correspond to the Pb0 species on the surface with Br vacancies.49 In contrast, only a single set of peaks located at 138.3 (4f7/2) and 143.2 eV (4f5/2), which are assigned to the internal Pb–Br bond, can be observed for the CSARP-CsPbBr3 NCs. That is to say, the CSARP-CsPbBr3 NCs should have a more ideal structure with lower defect density than the LARP-CsPbBr3 NCs. The long-term stabilities of these samples are further investigated by monitoring the change in their PLQYs with time, as shown in Fig. 4f. One can easily find that the PLQY of the CSARP-CsPbBr3 NCs can be maintained over 90% for 14 days, while the PLQY of LARP-CsPbBr3 NCs shows a drastic decline, and only remains 23% after 7 days. These results clearly confirm the advantage of our synthesis strategy over the LARP process, which can obtain NCs with perfect optical properties, reduced surface defects, and improved stability.
Besides the use of toxic polar solvents with high boiling points, the inability to synthesize pure iodine-based perovskite NCs is another drawback of the LARP strategy. In comparison, we successfully realize the synthesis of pure CsPbI3 with a high PLQY of 95.3% through the CSARP technique. Impressively, obvious PL can be also observed under room light, as shown in Fig. 5a. In addition, the as-synthesized CsPbI3 NCs have uniform size distribution and good crystallization, as shown in Fig. 5c. Stability tracking results show that the PL spectra of the as-prepared CsPbI3 NCs can maintain 90% of their initial intensity after 28 days of being kept under ambient conditions (Fig. 5b), which is even better than the stability of previously reported CsPbI3 NCs synthesized by the hot-injection process.22,50 It should be noted here that MgI2 dissolved in HI rather than water is selected as the iodine precursor for the synthesis of CsPbI3 NCs, as the latter resulted in the formation of the non-luminescent yellow phase (γ-CsPbI3). It is well known that a unimolecular nucleophilic substitution (SN1) chemical reaction can take place between alcohol and halogenated acid,51 leading to the formation of halohydrocarbon. Therefore, it is hypothesized that the in situ formed tert-butyl iodide (TBI) obtained from eqn (3) should be crucial for the successful synthesis of black phase CsPbI3 NCs (Fig. S5†).52
| TBA + HI → TBI + H2O | (3) |
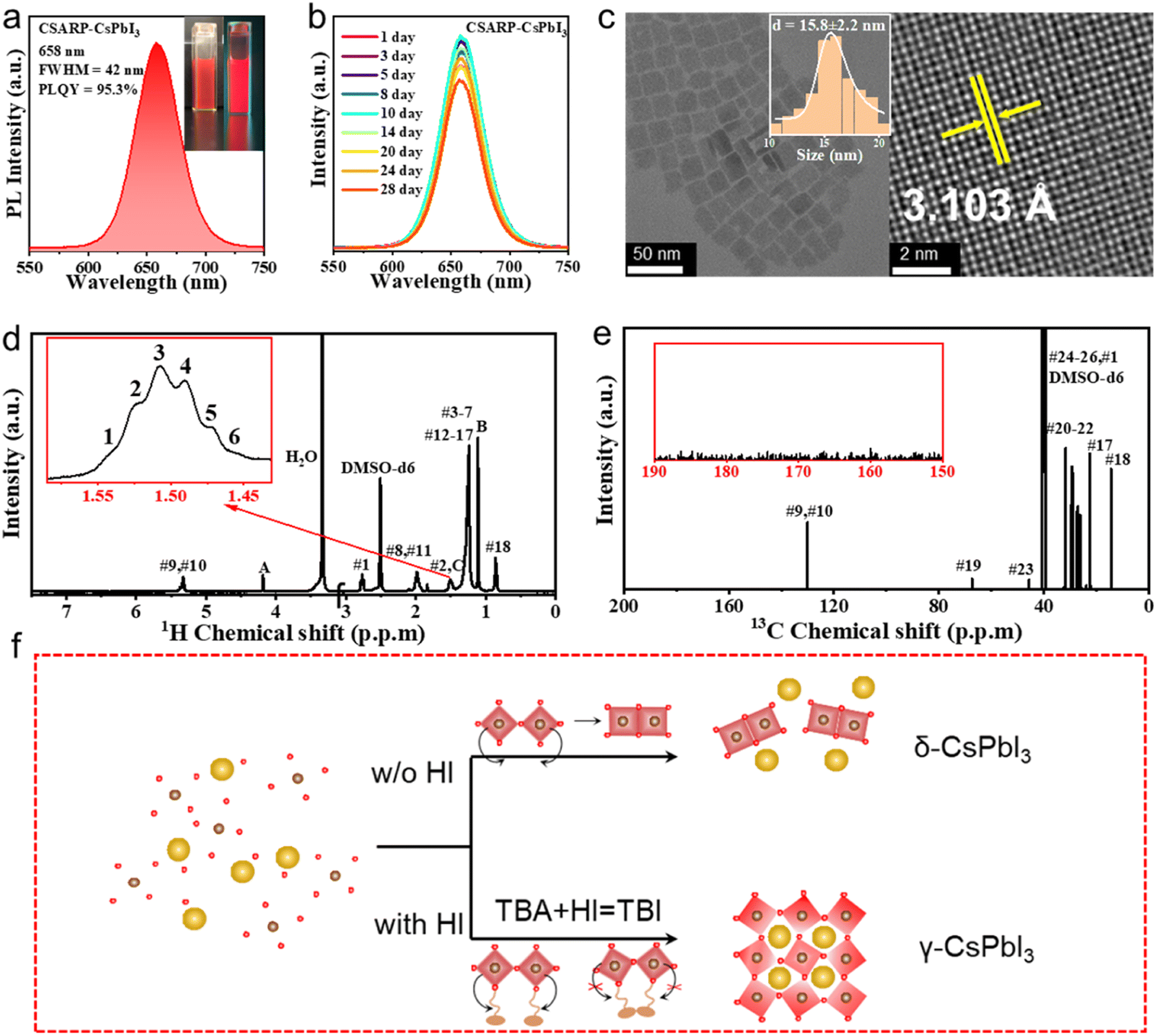 | ||
| Fig. 5 (a) PL spectra of CSARP-CsPbI3. The insets show photographs under room light (left) and a UV lamp (right, λ = 365 nm). (b) PL spectra of CSARP-CsPbI3 kept under ambient conditions for different times. (c) TEM and HRTEM images of the CSARP-CsPbI3 NCs. 1H NMR (d) and 13C NMR (e) spectra (dispersed in DMSO-d6, at 298 K, with 400 MHz) of CSARP-CsPbI3 NCs. (f) Mechanism diagram of CSPbI3 synthesis by the CSARP strategy at room temperature. | ||
A series of experiments are carried out to verify this supposition, as shown in Fig. S6 and Video S1–4.† It is clearly seen that the use of a MgI2–HI mixture results in the formation of a black phase, while the yellow phase which arises for the MgI2–H2O one at the beginning, is subsequently turned into a mixture of black and yellow phases, and completely transforms into the yellow phase finally. In contrast, when TBI is pre-injected into the solution (containing hexane, TBA, OLA, and OA), the NCs based on the MgI2–H2O precursor can maintain the black phase. That is to say, TBI plays a significant role in the formation of the black phase. The change in color for the MgI2–H2O one without pre-injected TBI should be ascribed to the formation of a trace amount of TBI induced by the hydrolysis of MgI2. Furthermore, when MgI2–H2O is injected into the solution without TBA (containing hexane, OLA, and OA), the yellow phase emerges for the entire duration of the reaction. In addition, nuclear magnetic resonance (NMR) measurement is performed to further confirm the type of organic ligands adhered on the surface of the as-synthesized CsPbI3 NCs. The characteristic resonance peak of TBI can be observed at 1.50 ppm from the 1H NMR spectrum shown in Fig. 5d (Fig. S7†). Moreover, this can be further verified by the 13C NMR spectrum (Fig. 5e), where the resonance peaks at 40.0 and 45.6 ppm can be assigned to TBI. Based on these results, it is reasonable to conclude that the formation of black phase CsPbI3 NCs via the CSARP technique is dominated by the in situ formed TBI, which can also passivate the surface and enhance the stability of the as-synthesized CsPbI3 NCs (Fig. 5f).
Similarly, a series of CsPbX3 NCs with various optical properties are achieved by adjusting their composition (ratio of halides in the mixed halide NCs), as shown in Fig. 6a and Table S1.† Distinct PL emission with various colors of these NCs can be see under a UV lamp and room light (Fig. 6b), revealing their outstanding optical properties. CsPbBrxCl3−x NCs with blue emission is believed to have great potential in blue LEDs; however, the low stability induced by phase segregation is one of the biggest restrictions. Here, we successfully realized the synthesis of CsPbBrxCl3−x NCs with near unity PLQY in the blue region (465 nm) (Fig. 6c). More importantly, the optical properties of CSARP-CsPbBrxCl3−x NCs can be well maintained even after 28 days under ambient conditions, as shown in Fig. 6d. For comparison, serious phase segregation and PL decline are observed for the LARP-CsPbBrxCl3−x NCs (Fig. 6e). That is to say, the CSARP-CsPbBrxCl3−x NCs exhibit a much slower phase segregation rate and improved stability than LARP-CsPbBrxCl3−x NCs, which should be attributed to the reduced surface defect density and suppressed ion migration.
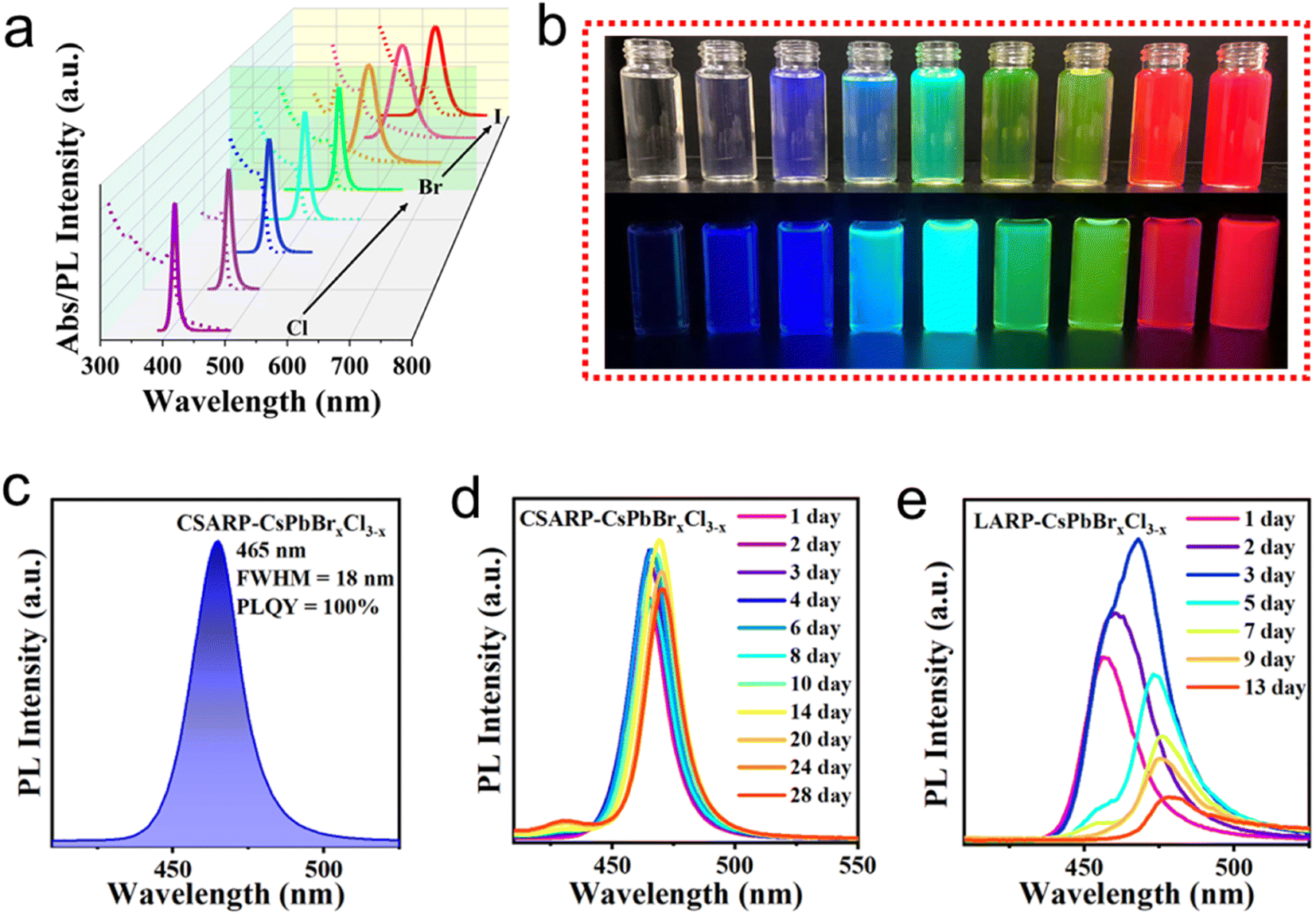 | ||
| Fig. 6 (a) PL spectra of CSARP-CsPbX3 NCs. (b) Photograph of CsPbX3 NCs with different emission colors under room light (top) and a UV lamp (bottom, λ = 365 nm). (c) PL spectra of CSARP-CsPbBrxCl3−x. Insets: photographs under room light (left) and a UV lamp (right, λ = 365 nm). PL spectra of CSARP-CsPbBrxCl3−x (d) and LARP-CsPbBrxCl3−x (e) NCs kept under ambient conditions for different times. | ||
Conclusion
In summary, this work provides a simple and environment-friendly strategy to synthesize CsPbX3 NCs with near ideal optical properties and enhanced stabilities. It is found that polarization effects induced by metal ions with small radii significantly enhance the adsorption energy of ligands, which eventually inhibits the formation of surface defects and results in CsPbX3 NCs with near-unity PLQY. Interestingly, the synthesis of black phase CsPbI3 NCs is successfully realized via the CSARP approach, which is a serious limitation of the traditional LARP process. Further studies reveal that the in situ formed TBI dominates the formation of black phase CsPbI3 NCs, which can also passivate the surface and enhance the stability of CsPbI3 NCs. Moreover, the CSARP strategy can also suppress the ion migration of mixed halide CsPbX3 NCs, which further leads to inhibited phase segregation and enhanced stability.Experimental section
Chemicals
Lead acetate trihydrate (PbAc2·3H2O, 99.99%), cesium acetate (CsAc, 99.9%), lead chloride (PbCl2, 99.99%), lead bromide (PbBr2, 99.999%), magnesium chloride (MgCl2, 99.9%), magnesium bromide (MgBr2, 99.99%), magnesium iodide (MgI2, 98%), zinc bromide (ZnBr2, 99.9%), sodium bromide (NaBr, 99.99%), ammonium bromide (NH4Br, 99.99%), lithium bromide (LiBr, 99.9%), rubidium bromide (RbBr, 99.5%), nickel bromide (NiBr2, 98%), calcium bromide (CaBr2, 98%), strontium bromide (SrBr2, 99%), barium bromide (BaBr2, 99%), indium bromide (InBr3, 99%), hydrobromic acid (HBr, 48%, ACS), hydriodic acid (HI, 45–50%), oleylamine (C18H37N, OLA, 80–90%), oleic acid (C18H34O2, OA, AR), tert-butanol alcohol (C4H10O, TBA, AR), n-hexane (C6H14, HEX, AR), acetic acid (C2H4O2, HAc, AR), isopropyl alcohol (C3H8O, IPA, AR), methyl acetate (C3H6O2, MeOAc, AR), ethyl acetate (C4H8O2, EA, AR), N,N-dimethylformamide (C3H7NO, DMF, AR), 1-octadecene (C18H36, ODE, >90%), dimethyl sulfoxide-d6 (C2D6SO, DMSO-d6, 99.9%), and tert-butyl iodide (C4H9I, TBI, > 95.0%) were purchased from Aladdin Industrial Corporation, China. Toluene (C7H8, AR) was purchased from Sinopharm Chemical Reagent Co., Ltd. All of the reagents were commercial and used without further purification. Deionized water was used in all experiments.Preparations of precursor solutions
Synthesis of CSARP-CsPbX3 NCs
HEX and TBA were mixed as reaction solvent, 200 μL OA and 200 μL OLA were used as ligands and added into the above mixture under intense stirring to form a homogeneous solution. Then, 50 μL Precursor 1 was injected and stirred evenly for 5 min. After that, 50 μL Precursor 2 (Precursor 3 was used for iodide-contained reactions) was added and stirred continuously for 30–120 min to obtain desired CsPbX3 NCs. All the reactions were carried out at room temperature under ambient conditions. The volume ratio of HEX to TBA is important for this strategy, which was tuned from 1:4 to 4:1. The detailed volume used for various NCs can be found in Table S2.† The crude solution was centrifuged at 16000 rpm for 10 min, and the precipitate was dispersed in 5 mL hexane after discarding the supernatant. Whereafter, another centrifugation process was carried out (8000 rpm for 5 min) to remove oversized NCs. Finally, the supernatant containing CsPbX3 NCs was saved.
Synthesis of CSARP-CsPbBr3 NCs using various kinds of metal bromide precursors
All the steps were the same as in the above process except usage of various metal bromides for the preparation of Precursor 2.Synthesis of LARP-CsPbX3 NCs
0.4 mmol PbX2 and 0.4 mmol CsX were dissolved in 10 mL DMF, 1 mL OA and 0.5 mL OLA were added to stabilize this precursor solution. Then, 1 mL of the precursor solution was quickly added to 10 mL toluene under vigorous stirring to obtain CsPbX3 NCs.Characterization
Powder XRD patterns were recorded using a Bruker D8 Advance X-ray diffractometer (Cu Kα, λ = 1.5406 Å). TEM analysis was carried out with an FEI Talos F200X microscope at an operating voltage of 200 kV. UV-vis absorption spectra were obtained using a Specord 200 Plus spectrophotometer. PL, PLQYs, and PL lifetimes were obtained by using an EI-FLS1000 fluorescence spectrometer (Edinburgh Instruments) equipped with an integrating sphere. XPS measurements were performed using an ESCALAB Xi+ (Thermo Fischer), monoatomic Ar+ ions were employed for XPS etching with various depths (the etching rate was estimated through Ta2O5, the data were collected at each 2 nm depth); all the XPS data are calibrated by using the C 1s core level. A Bruker AVANCE III (400 MHz) was used to record the NMR data.Computational methods
Electronic structure calculations were performed systematically within the framework of the density functional theory (DFT) formalism as implemented in Materials Studio (MS) to explore the electronic properties of pure CsPbBr3, 1Mg-, and nMg-CsPbBr3. General gradient approximation (GGA) in the form of a Perdew–Burke–Ernzerhof (PBE) exchange-correlation functional was employed for optimizing the geometrical structure. Each (001) CsPbBr3 slab consisted of 7 single layers, and a large vacuum spacing of >20 Å was used to prevent inter-slab interactions. Ligands and the top four layers of the CsPbBr3 slab were fully relaxed until the structure satisfied the following relaxation criteria: the energy cutoff used throughout the calculations was set at 700 eV, while the first Brillouin zone integrations were sampled using a 7 × 7 × 1 Monkhorst pack for the ionic relaxation of the system. The convergence thresholds were set at 0.002 Hartree per Å for the maximum force, 0.005 Å for the maximum displacement, and 1.0 × 10−5 Hartree for the energy change.Data availability
The data supporting this study are available within the main text and the ESI.†Author contributions
K. C. and Z. G. conceived the study; K. C., Z. G., Y. L., and G. L. designed the experiments and supervised the research. K. C., Z. G., and Z. W. performed the experiments and analyzed the data; M. G., X. T., W. X., X. J., and W. L. helped with the data collection; K. C., Z. G., Y. L., and G. L. wrote and revised the manuscript; all authors discussed the results and approved the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (NSFC No. 52072349 and 12174298), the Natural Science Foundation of Zhejiang Province (No. LZ22A040005 and LR22E020004), the Natural Science Foundation of Hubei Province (No. 2022CFB654), the Guangdong Basic and Applied Basic Research Foundation (No. 2023A1515030021), the Shenzhen Science and Technology Program (No. JCYJ20220530162405012 and JCYJ20210324135002007), the Hainan Provincial Joint Project of Sanya Yazhou Bay Science and Technology City (No. 520LH053), the 111 Project (No. B18038), the Fundamental Research Funds for the Central Universities (No. 2021Szvup104), the State Key Laboratory of Advanced Technology for Materials Synthesis and Processing (Wuhan University of Technology) (No. 2022-KF-30), and the State Key Laboratory of Silicate Materials for Architectures (Wuhan University of Technology) (No. SYSJJ2022-11).References
- A. Kojima, K. Teshima, Y. Shirai and T. Miyasaka, J. Am. Chem. Soc., 2009, 131, 6050–6051 CrossRef CAS PubMed.
- L. Protesescu, S. Yakunin, M. I. Bodnarchuk, F. Krieg, R. Caputo, C. H. Hendon, R. X. Yang, A. Walsh and M. V. Kovalenko, Nano Lett., 2015, 15, 3692–3696 CrossRef CAS PubMed.
- A. Swarnkar, A. R. Marshall, E. M. Sanehira, B. D. Chernomordik, D. T. Moore, J. A. Christians, T. Chakrabarti and J. M. Luther, Science, 2016, 354, 92–95 CrossRef CAS PubMed.
- Y. Dong, Y.-K. Wang, F. Yuan, A. Johnston, Y. Liu, D. Ma, M.-J. Choi, B. Chen, M. Chekini, S.-W. Baek, L. K. Sagar, J. Fan, Y. Hou, M. Wu, S. Lee, B. Sun, S. Hoogland, R. Quintero-Bermudez, H. Ebe, P. Todorovic, F. Dinic, P. Li, H. T. Kung, M. I. Saidaminov, E. Kumacheva, E. Spiecker, L.-S. Liao, O. Voznyy, Z.-H. Lu and E. H. Sargent, Nat. Nanotechnol., 2020, 15, 668–674 CrossRef CAS PubMed.
- K. Chen, W. Jin, Y. Zhang, T. Yang, P. Reiss, Q. Zhong, U. Bach, Q. Li, Y. Wang, H. Zhang, Q. Bao and Y. Liu, J. Am. Chem. Soc., 2020, 142, 3775–3783 CrossRef CAS PubMed.
- K. Chen, Y. Wang, J. Liu, J. Kang, Y. Ge, W. Huang, Z. Lin, Z. Guo, Y. Zhang and H. Zhang, Nanoscale, 2019, 11, 16852–16859 RSC.
- J. S. Yao, J. Ge, K. H. Wang, G. Zhang, B. S. Zhu, C. Chen, Q. Zhang, Y. Luo, S. H. Yu and H. B. Yao, J. Am. Chem. Soc., 2019, 141, 2069–2079 CrossRef CAS PubMed.
- C.-T. Wang, K. Chen, P. Xu, F. Yeung, H.-S. Kwok and G. Li, Adv. Funct. Mater., 2019, 29, 1903155 CrossRef.
- Q. Zeng, X. Zhang, Q. Bing, Y. Xiong, F. Yang, H. Liu, J.-y. Liu, H. Zhang, W. Zheng, A. L. Rogach and B. Yang, ACS Energy Lett., 2022, 7, 1963–1970 CrossRef CAS.
- J. Ye, Z. Li, D. J. Kubicki, Y. Zhang, L. Dai, C. Otero-Martinez, M. A. Reus, R. Arul, K. R. Dudipala, Z. Andaji-Garmaroudi, Y. T. Huang, Z. Li, Z. Chen, P. Muller-Buschbaum, H. L. Yip, S. D. Stranks, C. P. Grey, J. J. Baumberg, N. C. Greenham, L. Polavarapu, A. Rao and R. L. Z. Hoye, J. Am. Chem. Soc., 2022, 144, 12102–12115 CrossRef CAS PubMed.
- Z. L. Tseng, L. C. Chen, L. W. Chao, M. J. Tsai, D. Luo, N. R. Al Amin, S. W. Liu and K. T. Wong, Adv. Mater., 2022, 34, 2109785 CrossRef CAS PubMed.
- D. Luo, X. Li, A. Dumont, H. Yu and Z. H. Lu, Adv. Mater., 2021, 33, e2006004 CrossRef PubMed.
- J. De Roo, M. Ibáñez, P. Geiregat, G. Nedelcu, W. Walravens, J. Maes, J. C. Martins, I. Van Driessche, M. V. Kovalenko and Z. Hens, ACS Nano, 2016, 10, 2071–2081 CrossRef CAS PubMed.
- K. Chen, C. Wang, Z. Peng, K. Qi, Z. Guo, Y. Zhang and H. Zhang, Coord. Chem. Rev., 2020, 418, 213333 CrossRef CAS.
- J. Shamsi, A. S. Urban, M. Imran, L. De Trizio and L. Manna, Chem. Rev., 2019, 119, 3296–3348 CrossRef CAS PubMed.
- Q. Zhao, R. Han, A. R. Marshall, S. Wang, B. M. Wieliczka, J. Ni, J. Zhang, J. Yuan, J. M. Luther, A. Hazarika and G. R. Li, Adv. Mater., 2022, 34, e2107888 CrossRef PubMed.
- L. Liu, A. Najar, K. Wang, M. Du and S. F. Liu, Adv. Sci., 2022, 9, 2104577 CrossRef CAS PubMed.
- Y. Wang, K. Chen, H. Hao, G. Yu, B. Zeng, H. Wang, F. Zhang, L. Wu, J. Li, S. Xiao, J. He, Y. Zhang and H. Zhang, Nanoscale, 2019, 11, 2637–2643 RSC.
- Y. Wang, J. Yuan, X. Zhang, X. Ling, B. W. Larson, Q. Zhao, Y. Yang, Y. Shi, J. M. Luther and W. Ma, Adv. Mater., 2020, 32, 2000449 CrossRef CAS.
- H. Wang, F. Ye, J. Sun, Z. Wang, C. Zhang, J. Qian, X. Zhang, W. C. H. Choy, X. W. Sun, K. Wang and W. Zhao, ACS Energy Lett., 2022, 7, 1137–1145 CrossRef CAS.
- F. Liu, Y. Zhang, C. Ding, S. Kobayashi, T. Izuishi, N. Nakazawa, T. Toyoda, T. Ohta, S. Hayase, T. Minemoto, K. Yoshino, S. Dai and Q. Shen, ACS Nano, 2017, 11, 10373–10383 CrossRef CAS PubMed.
- J. Pan, Y. Shang, J. Yin, M. De Bastiani, W. Peng, I. Dursun, L. Sinatra, A. M. El-Zohry, M. N. Hedhili, A.-H. Emwas, O. F. Mohammed, Z. Ning and O. M. Bakr, J. Am. Chem. Soc., 2018, 140, 562–565 CrossRef CAS PubMed.
- Y. Hassan, J. H. Park, M. L. Crawford, A. Sadhanala, J. Lee, J. C. Sadighian, E. Mosconi, R. Shivanna, E. Radicchi, M. Jeong, C. Yang, H. Choi, S. H. Park, M. H. Song, F. De Angelis, C. Y. Wong, R. H. Friend, B. R. Lee and H. J. Snaith, Nature, 2021, 591, 72–77 CrossRef CAS PubMed.
- H. Zhu, Y. Pan, C. Peng, H. Lian and J. Lin, Angew. Chem., Int. Ed., 2022, 61, e202116702 CAS.
- Q. A. Akkerman, T. P. T. Nguyen, S. C. Boehme, F. Montanarella, D. N. Dirin, P. Wechsler, F. Beiglböck, G. Rainò, R. Erni, C. Katan, J. Even and M. V. Kovalenko, Science, 2022, 377, 1406–1412 CrossRef CAS PubMed.
- W. Yin, M. Li, W. Dong, X. Zhang and W. Zheng, Angew. Chem., Int. Ed. Engl., 2023, 135, e202303462 CrossRef.
- F. Li, X. Zhang, J. Shi, L. Jin, J. Qiao, J. Guo, H. Yin, Y. Li, J. Yuan and W. Ma, Adv. Funct. Mater., 2023 DOI:10.1002/adfm.202302542.
- J. N. Yang, Z. Y. Ma, J. D. Luo, J. J. Wang, C. Ye, Y. Zhou, Y. C. Yin, X. C. Ru, T. Chen, L. Y. Li, L. Z. Feng, K. H. Song, J. Ge, Q. Zhang and H. B. Yao, Nano Lett., 2023, 23, 3385–3393 CrossRef CAS PubMed.
- H. Zhu, G. Tong, J. Li, E. Xu, X. Tao, Y. Sheng, J. Tang and Y. Jiang, Adv. Mater., 2022, 34, 2205092 CrossRef CAS PubMed.
- W. Sun, R. Yun, Y. Liu, X. Zhang, M. Yuan, L. Zhang and X. Li, Small, 2023, 19, e2205950 CrossRef PubMed.
- N. Fiuza-Maneiro, K. Sun, I. López-Fernández, S. Gómez-Graña, P. Müller-Buschbaum and L. Polavarapu, ACS Energy Lett., 2023, 8, 1152–1191 CrossRef CAS.
- L. De Trizio, I. Infante and L. Manna, Acc. Chem. Res., 2023, 56, 1815–1825 CrossRef CAS PubMed.
- K. Chen, Q. Zhong, W. Chen, B. Sang, Y. Wang, T. Yang, Y. Liu, Y. Zhang and H. Zhang, Adv. Funct. Mater., 2019, 29, 1900991 CrossRef.
- J. Wang, J. Li, Y. Zhou, C. Yu, Y. Hua, Y. Yu, R. Li, X. Lin, R. Chen, H. Wu, H. Xia and H. L. Wang, J. Am. Chem. Soc., 2021, 143, 7759–7768 CrossRef CAS PubMed.
- M. Zhang, Q. Chen, R. Xue, Y. Zhan, C. Wang, J. Lai, J. Yang, H. Lin, J. Yao, Y. Li, L. Chen and Y. Li, Nat. Commun., 2019, 10, 4593 CrossRef PubMed.
- H. Liu, Z. Lu, W. Zhang, J. Wang, Z. Lu, Q. Dai, X. Qi, Y. Shi, Y. Hua, R. Chen, T. Shi, H. Xia and H. L. Wang, Adv. Sci., 2022, 9, 2203640 CrossRef CAS PubMed.
- T. Yang, W. Zhao, X. Liu and S. Liu, Adv. Energy Mater., 2023, 13, 2204192 CrossRef CAS.
- J. Zhang, B. Che, W. Zhao, Y. Fang, R. Han, Y. Yang, J. Liu, T. Yang, T. Chen, N. Yuan, J. Ding and S. Liu, Adv. Mater., 2022, 34, 2202735 CrossRef CAS PubMed.
- B. Guo, R. Lai, S. Jiang, L. Zhou, Z. Ren, Y. Lian, P. Li, X. Cao, S. Xing, Y. Wang, W. Li, C. Zou, M. Chen, Z. Hong, C. Li, B. Zhao and D. Di, Nat. Photonics, 2022, 16, 637–643 CrossRef CAS.
- G. Li, Z. Su, L. Canil, D. Hughes, M. H. Aldamasy, J. Dagar, S. Trofimov, L. Wang, W. Zuo, J. J. Jerónimo-Rendon, M. M. Byranvand, C. Wang, R. Zhu, Z. Zhang, F. Yang, G. Nasti, B. Naydenov, W. C. Tsoi, Z. Li, X. Gao, Z. Wang, Y. Jia, E. Unger, M. Saliba, M. Li and A. Abate, Science, 2023, 379, 399–403 CrossRef CAS PubMed.
- T. Li, J. Xu, R. Lin, S. Teale, H. Li, Z. Liu, C. Duan, Q. Zhao, K. Xiao, P. Wu, B. Chen, S. Jiang, S. Xiong, H. Luo, S. Wan, L. Li, Q. Bao, Y. Tian, X. Gao, J. Xie, E. H. Sargent and H. Tan, Nat. Energy, 2023, 8, 610–620 CrossRef CAS.
- Q. Chen, C. Wang, Y. Li and L. Chen, J. Am. Chem. Soc., 2020, 142, 18281–18292 CrossRef CAS PubMed.
- X. Li, Y. Wu, S. Zhang, B. Cai, Y. Gu, J. Song and H. Zeng, Adv. Funct. Mater., 2016, 26, 2435–2445 CrossRef CAS.
- Y. Yang, C. Hou and T. X. Liang, Phys. Chem. Chem. Phys., 2021, 23, 7145–7152 RSC.
- S. Jeon, M. C. Jung, J. Ahn, H. K. Woo, J. Bang, D. Kim, S. Y. Lee, H. Y. Woo, J. Jeon, M. J. Han, T. Paik and S. J. Oh, Nanoscale Horiz., 2020, 5, 960–970 RSC.
- J.-K. Chen, J.-P. Ma, S.-Q. Guo, Y.-M. Chen, Q. Zhao, B.-B. Zhang, Z.-Y. Li, Y. Zhou, J. Hou, Y. Kuroiwa, C. Moriyoshi, O. M. Bakr, J. Zhang and H.-T. Sun, Chem. Mater., 2019, 31, 3974–3983 CrossRef CAS.
- C. Zheng, C. Bi, F. Huang, D. Binks and J. Tian, ACS Appl. Mater. Interfaces, 2019, 11, 25410–25416 CrossRef CAS PubMed.
- W. J. Mir, A. Alamoudi, J. Yin, K. E. Yorov, P. Maity, R. Naphade, B. Shao, J. Wang, M. N. Lintangpradipto, S. Nematulloev, A. H. Emwas, A. Genovese, O. F. Mohammed and O. M. Bakr, J. Am. Chem. Soc., 2022, 144, 13302–13310 CrossRef CAS PubMed.
- Y. Cai, H. Wang, Y. Li, L. Wang, Y. Lv, X. Yang and R.-J. Xie, Chem. Mater., 2019, 31, 881–889 CrossRef CAS.
- Y. Tang, A. Lesage and P. Schall, J. Mater. Chem. C, 2020, 8, 17139–17156 RSC.
- R. Bruckner, Organic mechanisms, Springer, 2010 Search PubMed.
- D. Jia, J. Chen, X. Mei, W. Fan, S. Luo, M. Yu, J. Liu and X. Zhang, Energy Environ. Sci., 2021, 14, 4599–4609 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sc02109b |
| This journal is © The Royal Society of Chemistry 2023 |