
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA high-field cellular DNP-supported solid-state NMR approach to study proteins with sub-cellular specificity†
David
Beriashvili
 a,
Ru
Yao
b,
Francesca
D'Amico
c,
Michaela
Krafčíková
a,
Andrei
Gurinov
a,
Adil
Safeer
a,
Xinyi
Cai
b,
Monique P. C.
Mulder
c,
Yangping
Liu
b,
Gert E.
Folkers
a and
Marc
Baldus
*a
a,
Ru
Yao
b,
Francesca
D'Amico
c,
Michaela
Krafčíková
a,
Andrei
Gurinov
a,
Adil
Safeer
a,
Xinyi
Cai
b,
Monique P. C.
Mulder
c,
Yangping
Liu
b,
Gert E.
Folkers
a and
Marc
Baldus
*a
aNMR Spectroscopy, Bijvoet Center for Biomolecular Research, Utrecht University, Padualaan 8, 3584 CH Utrecht, The Netherlands. E-mail: m.baldus@uu.nl
bTianjin Key Laboratory on Technologies Enabling Development of Clinical Therapeutics and Diagnostics, School of Pharmacy, Tianjin Medical University, Tianjin 300070, P. R. China
cDepartment of Cell and Chemical Biology, Leiden University Medical Center (LUMC), Einthovenweg 20, 2333 ZC, Leiden, The Netherlands
First published on 5th September 2023
Abstract
Studying the structural aspects of proteins within sub-cellular compartments is of growing interest. Dynamic nuclear polarization supported solid-state NMR (DNP-ssNMR) is uniquely suited to provide such information, but critically lacks the desired sensitivity and resolution. Here we utilize SNAPol-1, a novel biradical, to conduct DNP-ssNMR at high-magnetic fields (800 MHz/527 GHz) inside HeLa cells and isolated cell nuclei electroporated with [13C,15N] labeled ubiquitin. We report that SNAPol-1 passively diffuses and homogenously distributes within whole cells and cell nuclei providing ubiquitin spectra of high sensitivity and remarkably improved spectral resolution. For cell nuclei, physical enrichment facilitates a further 4-fold decrease in measurement time and provides an exclusive structural view of the nuclear ubiquitin pool. Taken together, these advancements enable atomic interrogation of protein conformational plasticity at atomic resolution and with sub-cellular specificity.
Introduction
Organelles are physiochemically distinct microenvironments tuned to efficiently execute biological processes.1–3 Recent technological advancements have made rapid in situ whole-cell and organelle-specific protein interactome mapping with nanometer resolution possible.4–6 The next step in deepening our knowledge of these processes lies in gathering organelle-specific atomic-level structural data.Nuclear magnetic resonance (NMR) provides atomic-resolution data and is amenable for application in situ. Prominently, solution-state NMR has been used to characterize rapidly tumbling or intrinsically disordered biomolecules within intact eukaryotic cells and isolated mitochondria.7–12 To overcome tumbling limitations, magic angle spinning (MAS) solid-state NMR (ssNMR), has been applied to probe abundant globular proteins,13 soluble as well membrane-embedded proteins in bacteria,14,15 and protein-enriched eukaryotic membrane vesicles.16
Overcoming sensitivity limitations in ssNMR is increasingly possible by application of dynamic nuclear polarization (DNP) based-ssNMR (DNP-ssNMR),17 which greatly increases sensitivity by transferring electron polarization from carefully chosen biradical polarization agents (PAs) to NMR active nuclei. It has already allowed to determine the supramolecular structure of native diatom biosilica as well as to visualize structural elements of and binding events to supra-protein complexes embedded in native-membranes derived from both bacteria and human cells.16,18,19 Moreover, DNP-ssNMR has been applied to cell lysates,20,21 eukaryotic cells grown as 2D or 3D cultures,22–24 and to soluble proteins in bacteria15 and within eukaryotic cells at near endogenous (μM) concentrations.25 In spite of these advancements, our current knowledge regarding line-broadening effects under low temperature-DNP conditions stems from in vitro preparations where inhomogeneous broadening usually dominates and correlates with local protein dynamics,26–29 resulting in narrow line widths for rigid protein segments and broader distributions for mobile or surface exposed residues.26,27,30,31
However, all these applications have been limited to DNP setups operating at comparatively low magnetic field (400 MHz/600 MHz).20,22,24,25 Extending such experiments to higher magnetic fields has been complicated by a lack of suitable PAs.22,23 Secondly, obtaining detailed information regarding the penetration and distribution of PAs within whole cells is crucial for correlating in-cell DNP-ssNMR readouts with sub-cellular localization. Lastly, further complications may arise in cells where the surrounding physio–chemical environment varies in space and time (see, e.g. ref. 32 and 33) leading to additional unfavorable broadening mechanisms that may prohibit a detailed conformational analysis of the protein of interest at low magnetic fields.
Here, we set out to address these issues and to determine whether SNAPol-1, a PA with demonstrated exceptional high-field performance in vitro,34,35 could be efficiently utilized in the context of high-field in cell DNP-ssNMR. Through confocal z-stack microscopy we investigated the distribution of SNAPol-1 in whole cells and cell nuclei. In line with DNP ssNMR data, our findings suggest a homogeneous distribution of PA in both preparations. To confirm confocal data and further probe potential SNAPol-1-related sensitivity and resolution improvements, we conducted high-field (800 MHz) DNP-ssNMR experiments on HeLa cells and isolated nuclei containing [13C,15N] ubiquitin (Ub), a post-translational modifier that regulates a host of cellular functions,36 delivered by electroporation.25 Comparison of our high-field in-cell and in nuclei data with in-cell data acquired at 400 MHz and on in vitro Ub revealed significant improvement in spectral sensitivity and resolution at 800 MHz DNP conditions thus providing an avenue for broadly studying the conformational landscape of proteins in cells at atomic resolution and with sub-cellular precision.
Results and discussion
It is well established that a PA's DNP performance is highly reliant on it being in close proximity to the molecular species of interest.24–26,37 To assess SNAPol's suitability for cellular applications we first investigated its ability to passively enter and distribute within mammalian cells. Confocal z-stack microscopy of HeLa cells treated with fluorescein isothiocyanate (FITC) tagged-SNAPol-1 (Fig. S1†) dissolved in “DNP Juice” (6![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3:1 d8 glycerol:D2O:H2O) revealed that SNAPol-1 passively entered cells distributing homogenously within the luminal space, similar to PyPOL (Fig. 1A and S2†).25 Due to its large size (1.6 kDa), we postulate that glycerol-induced membrane-permeability plays a role in facilitating SNAPol-1 entry into cells.38
:3:1 d8 glycerol:D2O:H2O) revealed that SNAPol-1 passively entered cells distributing homogenously within the luminal space, similar to PyPOL (Fig. 1A and S2†).25 Due to its large size (1.6 kDa), we postulate that glycerol-induced membrane-permeability plays a role in facilitating SNAPol-1 entry into cells.38
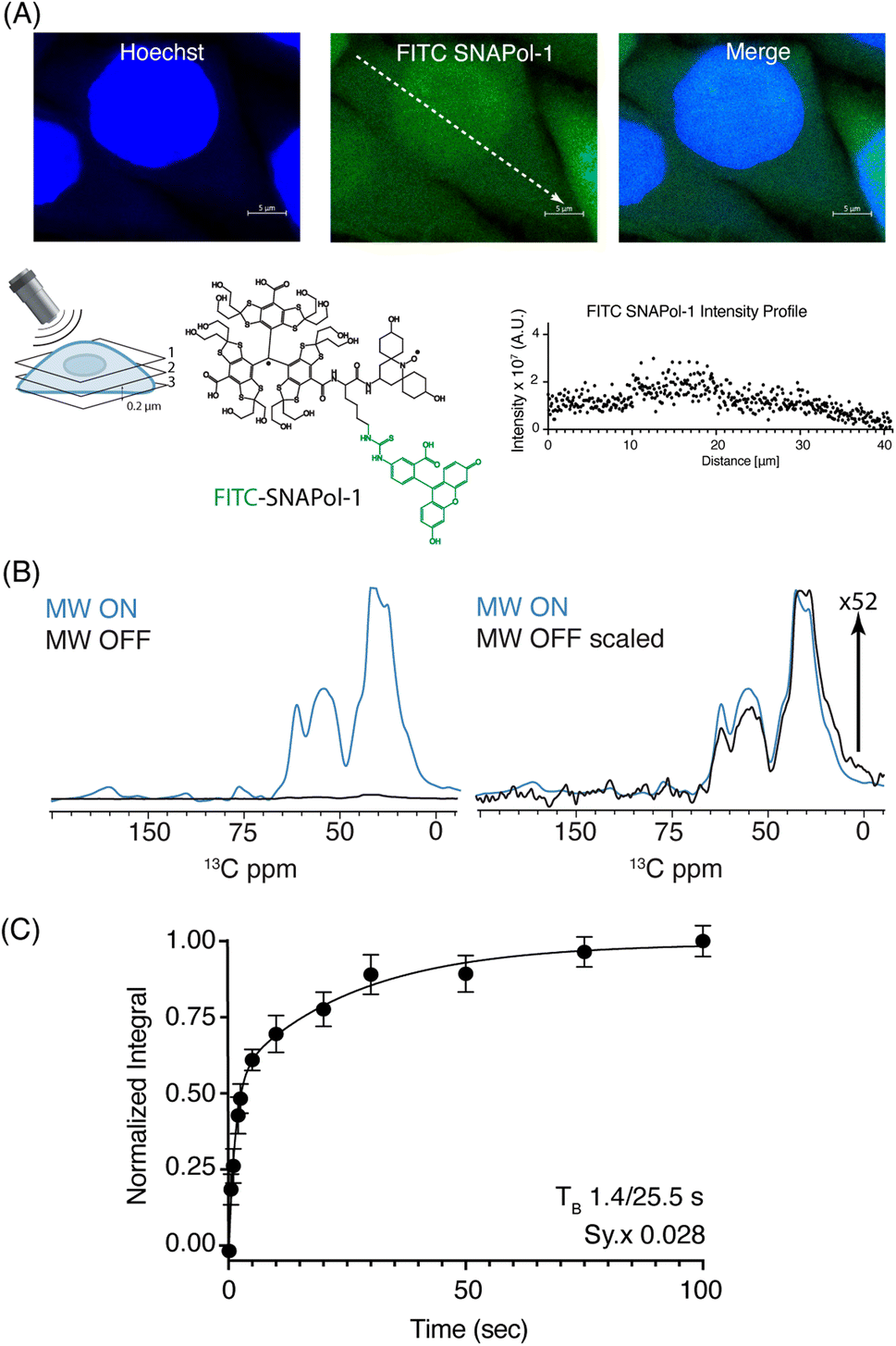 | ||
| Fig. 1 (A): Confocal z-stack of FITC-SNAPol-1 (green) treated HeLa cells counterstained with Hoechst (blue) along with an illustrative FITC-SNAPol-1 intensity profile taken along the arrow depicted in the green panel. (B/C): SNAPol-1 DNP enhancement on 8 million HeLa cells electroporated with 1.2 mM [13C,15N] Ub treated with 30 mM SNAPol-1 embedded in “DNP Juice” (6:4 d8, 12C3-enriched glycerol:D2O with 1 × Hank's buffered salt solution HBSS) as previously described in ref. 25. In (C), polarization buildup (TB) on the carbonyl signal was best fit by a biexponential with error bars denoting the standard deviation (s.d.) of the signal to noise ratio (S/N) and Sy.x the s.d. residual of the fit. See ESI† for more details on error analysis. | ||
To validate our confocal data, we initially conducted DNP-ssNMR experiments on HeLa cells and later isolated nuclei electroporated with 1.2 mM [13C,15N] Ub. For both type of preparations, we also performed solution-state NMR control experiments (Fig. S3†). These experiments showed signals exclusively for residues in Ub's dynamic C-terminal tail within intact cells in full agreement with published in-cell solution-state NMR data11,25,39 We also note that these results obtained in at least two different human cell lines are at odds with a recent report from Kadavath et al.,40 possibly due to their use of a non-human cell line. Notably, we did not observe any solution-state NMR correlations for Ub within the intact nuclei (Fig. S3†) in line with a high degree of conjugation of the nuclear Ub pool.41
We conducted adiabatic 1H–13C cross polarization (CP) and saturation recovery experiments (Fig. 1B/C) on HeLa cells electroporated with 1.2 mM [13C,15N] Ub, which equates to 40 μM of delivered protein.25 Doping with 30 mM SNAPol-1 revealed a DNP enhancement of 52 (Table 1), characterized by a biexponential polarization buildup time (TB) of 1.40/25.5 s with the former accounting for 57% of the buildup.34 These results are in good agreement with our recent findings for a bioresistant variant (Stapol-1)35 and suggested the presence of two PA populations. We attribute the fast buildup rate (1.40 s) to lumen-localized SNAPol-1 with the longer (25.5 s) likely arising from SNAPol-1 outside or at the cell's periphery. Comparison of absolute enhancements (Σ) at 800 MHz/527 GHz for proline, revealed that the weighted average Σ for in-cell SNAPol-1 is an order of magnitude greater than reported for AMUPol which was previously used for cellular DNP-ssNMR.22,24,25 Notably and in line with earlier observations for complex biological systems,16,22,34 in-cell enhancements decreased compared to in vitro SNAPol-1 values recorded on proline.
:3:1 glycerol:D2O:H2O). εon/off, εabs, and Σ as per convention.34,43,44 Avg. denotes weighted average Σ (by percent of the fit)
| Sample | ε on/off | ε abs | T B (s) | Σ |
|---|---|---|---|---|
| a Data taken from ref. 34. See ESI for further information. | ||||
| SNAPol-1 whole cell (30 mM) | 52 | 45.26 | 1.4/25.5 | 340 ± 10/77 ± 7 avg. 229 ± 9 |
| SNAPol-1 nuclei (30 mM) | 60 | 54 | 1.4 | 406 ± 19 |
| SNAPol-1 proline (10 mM)a | 133 | 116 | 4.2/3.15 | 505 ± 7/544 ± 10 |
| AMUPol proline (10 mM)a | 35 | 19 | 5.0 | 76 ± 8 |
| AMUPol proline (30 mM)a | 43 | 23.22 | 8.2 | 72 ± 11 |
Next, we investigated improvements in ssNMR resolution at 800 MHz DNP conditions on cells electroporated with 1.2 mM [13C,15N] Ub by recording 3D double-quantum, single-quantum, single-quantum 13C–13C–13C (DQSQSQ)42 experiments (see ESI for details) using 30 mM SNAPol-1. To examine resolution improvements, we compared these data to Ub in-cell DNP-ssNMR experiments conducted at 400 MHz (30 mM AMUPol, Fig. 2A).
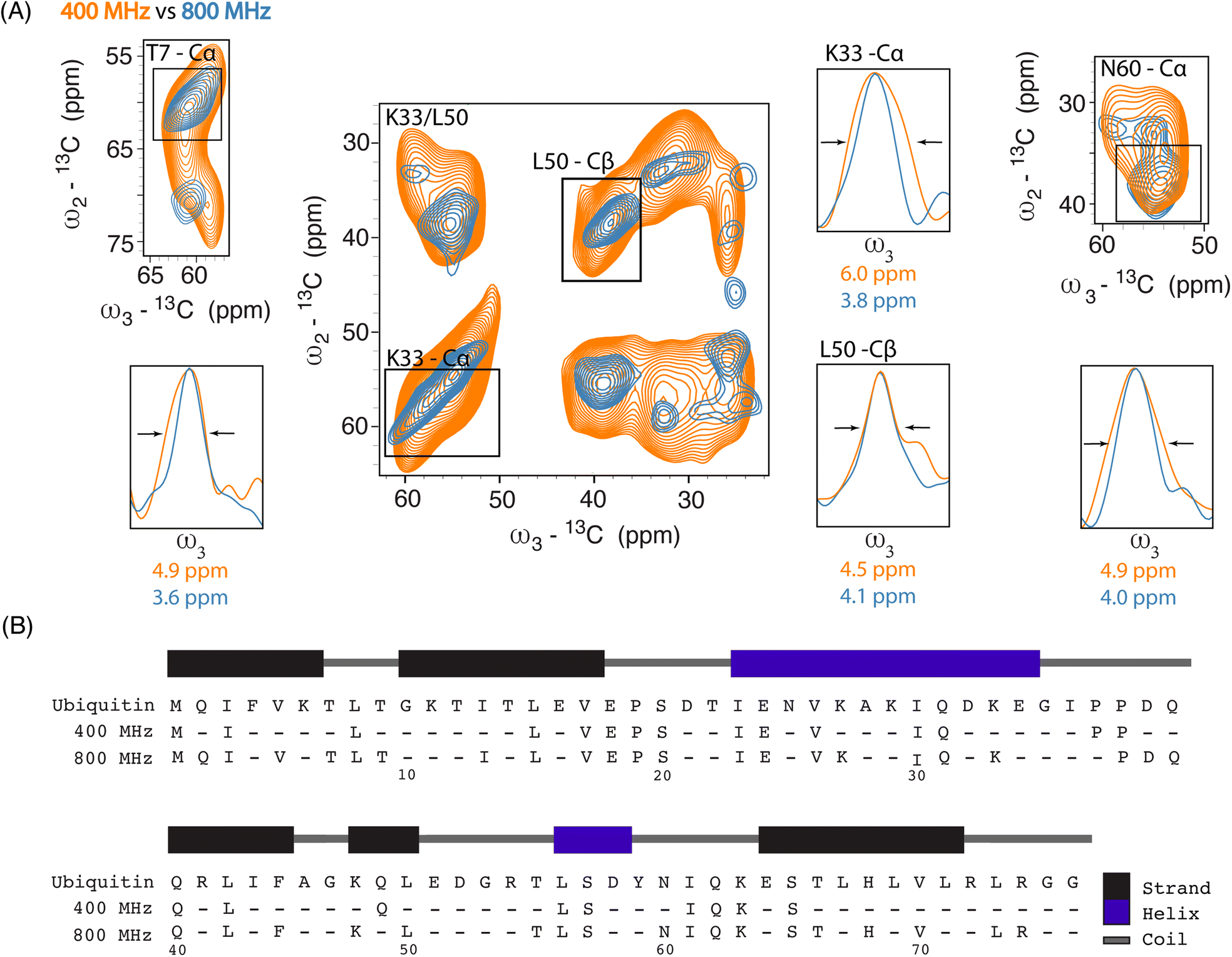 | ||
| Fig. 2 (A) Overlay of 2D SQSQ planes extracted from 13C–13C–13C DQSQSQ 3D experiments recorded on 8 million HeLa cells electroporated with 1.2 mM [13C,15N] Ub and doped with 30 mM AMUPol, at 400 MHz (orange) and with 30 mM SNAPol-1 at 800 MHz (blue). 1D projections detail linewidth improvement (full width at half maximum (FWHM)). (B) Ubiquitin sequence showing the increase in assignments between Ub in-cell samples measured at 400 (25 residues) and 800 MHz (41 residues) (assignment required 2 or more resolved correlations). | ||
Linewidth analysis of Cα and Cβ correlations corresponding to residues located in various secondary structural elements showed a reduction in linewidth from about 10% (L50) up to 35% for residues such as T7, K33, N60, most likely reflecting different relative contributions of homogeneous and inhomogeneous line broadening.31,43,45 Cumulatively, the improvement in resolution seen in our 3D data sets enabled the assignments of an additional 16 residues (leading to 41 assigned residues at 800 MHz) (Fig. 2B) and in a few cases allowed to discern between residues with overlapping chemical shifts (e.g., P37 and P38). Further, comparison of in cell 800 MHz line widths with those obtained on microcrystalline Ub (also recorded at 800 MHz) revealed that while in cell line widths are broader (Fig. S4†), differences varied significantly between residues (36% for T7 and 60% in the case of K33) in line with the dynamic nature of Ub previously found in vitro (see, e.g., ref. 46–49). It is important to highlight that variation in line widths could arise from differences in measurement temperatures, chemical exchange (100 K whole cells vs. 278 K microcrystals), and that the microcrystals are solely reflective of Ub's free monomeric state. Correspondingly, this comparison serves as a qualitative measure.
Considering the respectable performance of SNAPol-1 at 800 MHz, we proceeded to investigate whether conducting organelle specific DNP-ssNMR was technically feasible. We settled on investigating cell nuclei, isolated via a near quantitative detergent nucleus enrichment protocol known to retain transcriptional activity.50 The rapid nature of isolation afforded by this method would also minimize stress-induced ubiquitinome alterations.51 Similar to whole cells, confocal z-stack microscopy of FITC-SNAPol-1 treated isolated cell nuclei showed SNAPol-1 entry and homogenous distribution (Fig. 3A and S5†). In addition, we examined whether Ub electroporated into whole cells would be retained upon nuclei isolation. Therefore, we conducted an analogous confocal z-stack microscopy experiment with N-terminal 5-carboxytetramethylrhodamine-tagged Ub (TAMRA-UB), which showed that cell nuclei isolated from whole-cells, electroporated with TAMRA-Ub (Fig. S6†), retained morphology and Ub upon fractionation (Fig. 3B). The lack of TAMRA-Ub in certain locations of the cell nuclei mirrored previously reported results further purporting that the isolation procedure was minimally invasive and retained sub-cellular morphology.25,52,53 In addition, we tested the suitability of isolated nuclei for DNP ssNMR (i.e., MAS (8 kHz), 60% glycerol, and cryogenic temperature, Fig. S7†) by brightfield microscopy. No adverse effects on morphology were observed in line with earlier studies54,55 suggesting that transcriptional activity and morphology of isolated nuclei are maintained if stored in high glycerol concentrations (>60%) and at cryogenic temperatures.
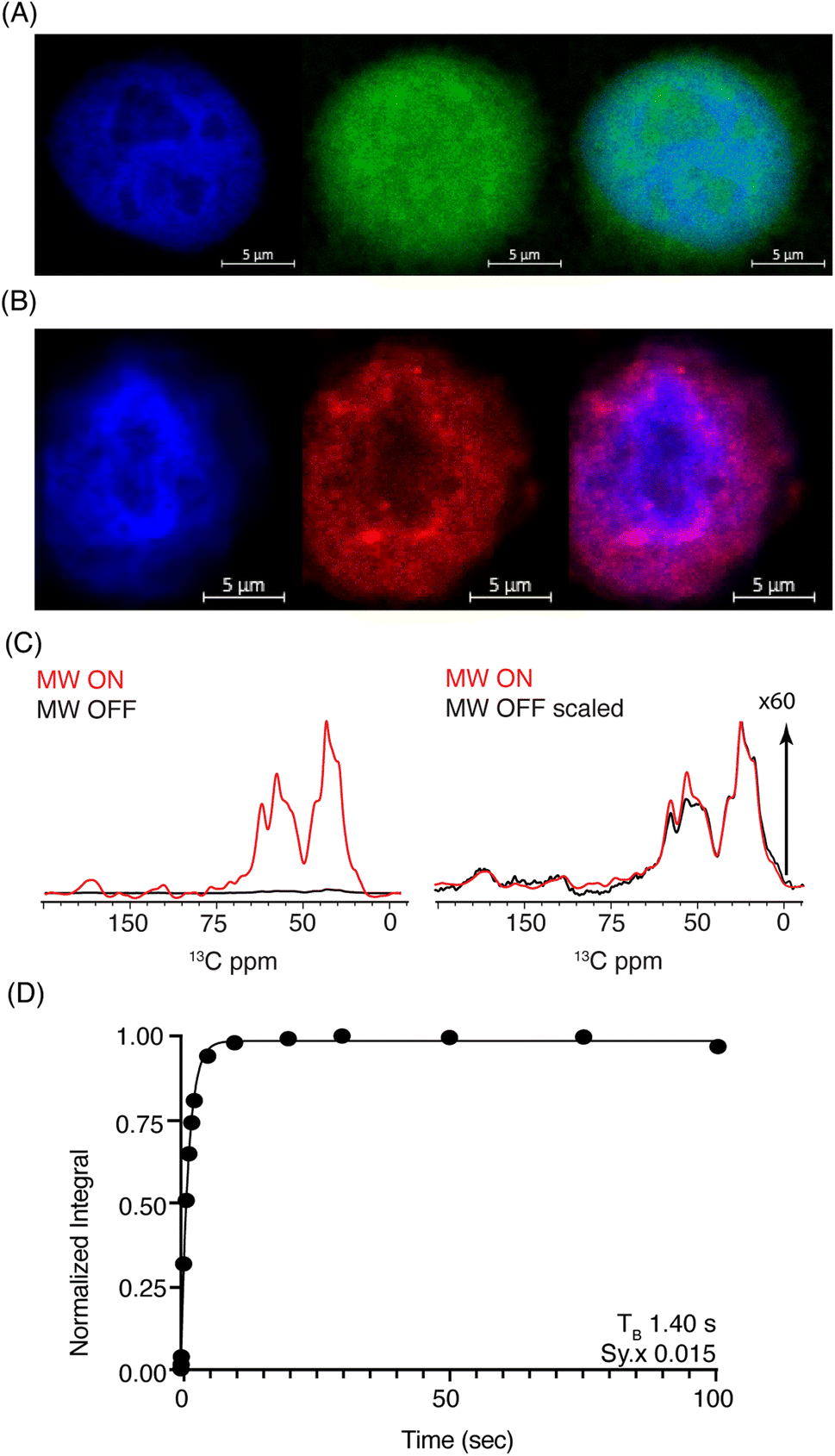 | ||
| Fig. 3 (A) Confocal z-stack of cell nuclei stained with Hoechst (blue) treated FITC-SNAPol-1 (green). (B) Confocal z-stack of cell nuclei isolated from HeLa cells electroporated with 1.2 mM of TAMRA-Ub (red) stained with Hoechst (blue). Panel (C/D) SNAPol-1 DNP enhancement (judged on Ub carbonyl signal 170 to 185 ppm) on cell nuclei isolated from 16 million HeLa cells electroporated with 1.2 mM [13C,15N] Ub and treated with 30 mM SNAPol-1 dissolved in “DNP Juice” (6:4 d8, 12C3-enriched glycerol:D2O with 1 × HBSS) as previously described in ref. 25. Polarization buildup (TB) on the carbonyl signal was best fit by a mono-exponential with Sy.x denoting the residual s.d. of the fit. Error bars are not visible due to high S/N ratio. See also Table S1.† | ||
DNP-ssNMR adiabatic 1H–13C cross polarization (CP) and saturation recovery experiments conducted on nuclei isolated from HeLa cells electroporated with 1.2 mM [13C,15N] Ub (Fig. 3C/D) and then doped with 30 mM SNAPol-1, exhibited a DNP enhancement of 60, characterized by a short mono-exponential polarization buildup time (TB) of 1.40 s, confirming that SNAPol-1 was in the vicinity of Ub. In line with this notion, we observed an in-nuclei DNP enhancement Σ (Table 1) of 406 compared to 229 in whole cells, approaching values obtained with SNAPol-1 in proline.
Next, we performed 3D DQSQSQ 13C–13C–13C experiments to visualize Ub's nuclear pool at atomic resolution. A cumulative 1D projection, derived from the SQSQ dimension, showed a 2 fold increase in S/N for both carbonyl and aliphatic signals in-nuclei (Fig. S8†) which we attribute to physical sample enrichment (allowing for increased nuclei packing) and more efficient SNAPol-1 entry into nuclei, possibly via the nuclear pore complex and also through detergent-induced membrane perturbations.56
Analysis of individual residues (Fig. 4A) confirmed the global increase in S/N, revealed the appearance of novel correlations for previously assigned residues (e.g., T66 – Cγ), and increased the number of assigned residues to a total of 49 (Fig. 4B). It is evident from Fig. 4A that residues exhibited variations in Cα intensity (in cell versus in nuclei) and varying degrees of chemical shift (CS) perturbations (e.g., E18) which suggested the presence of multiple Ub conformers. The observed variation in Ub's linewidth in cell versus microcrystals further substantiates this hypothesis. For example, while the (Cα,Cβ) cross peak correlation for L50 was dominated by α-helical conformations, we observed a broader distribution of (Cα, Cβ) correlations for T66 matching both β-sheet and random coil secondary structure values (Fig. 4C). The latter observation agrees with previous in vitro solution-state NMR studies suggesting that T66 adopts both β-sheet and random coil secondary structure.48 Taken together these observations suggest that the increased spectral resolution and sensitivity seen in our 800 MHz DNP-ssNMR in cell and in nuclei data not only facilitate structural analysis, but also enable the study of conformational heterogeneity, that so far was mainly studied by in vitro solid-state NMR.58,59
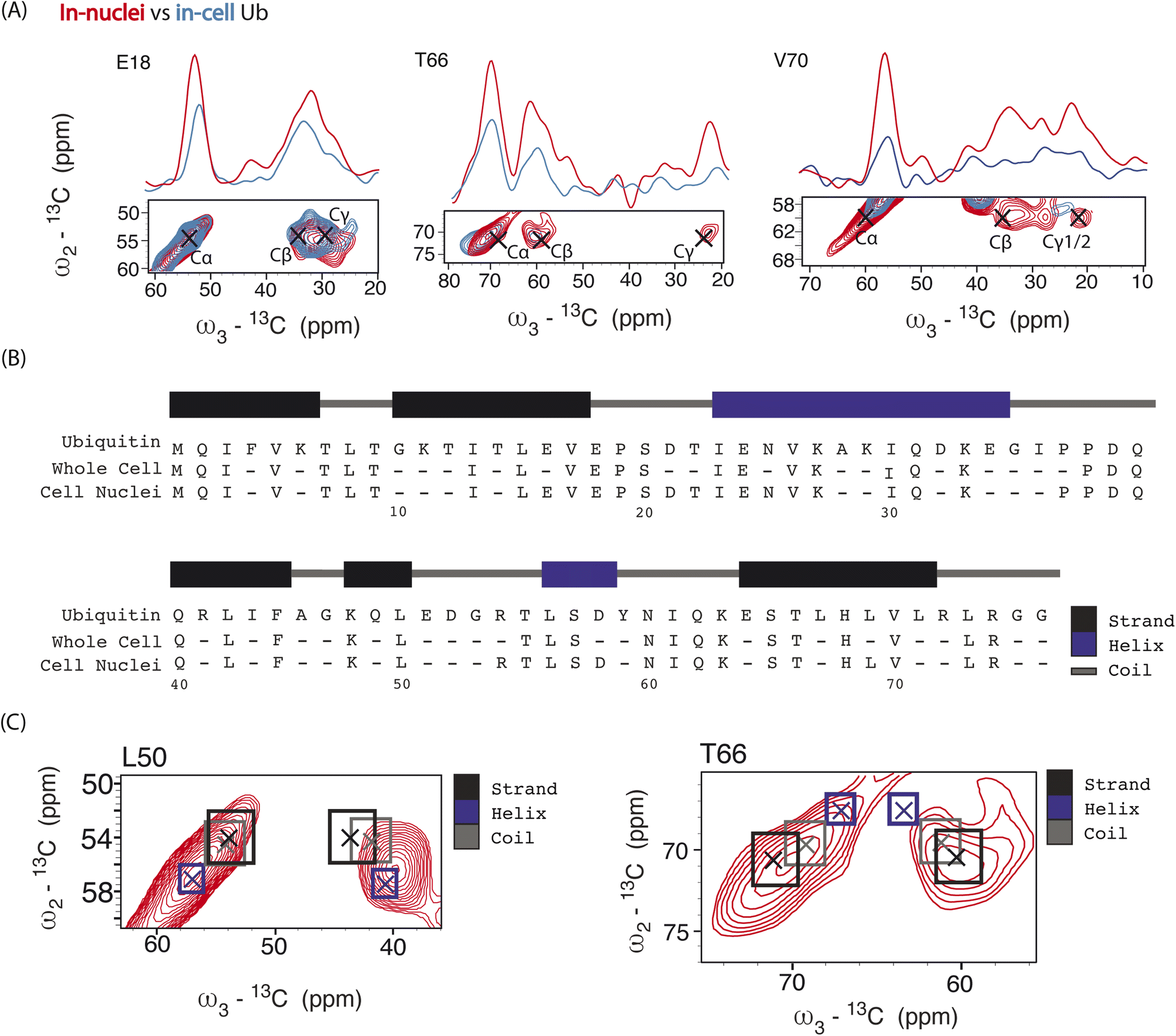 | ||
| Fig. 4 Panel (A) overlay of aliphatic 2D projections and 1D slices extracted from 13C–13C–13C DQSQSQ 3D experiment conducted at 800 MHz on 8 million HeLa cells (blue) electroporated with 1.2 mM [13C,15N]-Ub and on 1.6 × 107 isolated cell nuclei (red), both doped with 30 mM SNAPol-1. Black crosses denote literature assignments (BMRB ID: 15410 and 7111). Panel (B) sequence showing the assigned Ub residues at 800 MHz in isolated cell nuclei (49 residues) versus whole cells (41 resides). Panel (C) comparison of experimental 2D projections for L50 and T66 Cα/β overlayed with literature secondary chemical-shift values (with crosses denoting the average secondary structure chemical-shift) taken from ref. 57. | ||
Conclusion
Both sensitivity and resolution are critical for increasing the wide-spread use of in-cell DNP-ssNMR. Here we have shown that SNAPol-1 enables high field DNP-ssNMR for cellular applications not only with superior sensitivity but also leads to enhanced spectral resolution in a complex cellular setting and for a protein known to engage a multitude of intermolecular interactions.60 A combination of microscopy and in-cell ssNMR data showed that SNAPol-1 readily enters and homogeneously distributes inside whole cells and isolated cell nuclei. By using HeLa cells containing near physiological concentrations of [13C,15N] Ub and doped with SNAPol-1, we observed significant improvements in resolution that approach data quality seen on in vitro microcrystalline biological preparations. Further, we have expanded the scope of DNP-ssNMR to conduct experiments on intact nuclei. The physical removal of the cytoplasm facilitated improved SNAPol-1 entry and enabled data acquisition on the nuclear Ub pool with a 4-fold reduction in measurement time. A preliminary comparison of the resulting ssNMR correlations (Fig. 4) between in cell and in nuclei Ub spectra suggests that a multitude of Ub conformers can exist inside cells. Importantly, unlike in cell solution-state NMR which probes mostly monomeric conformers,11,25,39 our presented approach probes all species,41,61 regardless of molecular size, thus laying a foundation to comprehensively probe conformational protein states with sub-cellular selectivity.Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.Author contributions
D. B., G. E. F., and M. B. designed experiments, analysed data, and wrote the manuscript. D. B. produced protein, samples, conducted confocal microscopy, and NMR experiments. M. K. assisted in confocal microscopy acquisition and analysis. A. G. maintained DNP instrumentation and A. S. assisted in Ub production. R. Y., X. C. and Y. L. produced synthetic SNAPol-1 and FITC-SNAPol-1. F. D., and M. P. C. M. produced TAMRA-Ub. All authors reviewed the manuscript and agreed to its publication.Conflicts of interest
The authors have no conflicts to declare.Acknowledgements
We thank Johan van der Zwan for technical support, Prof. Dr A. J. J. M. Bonvin for helpful discussions, and Dr Ilya Grigoriev of the Biology Imaging Center (BIC), Department of Biology, Utrecht University for confocal microscopy access and training. This work was supported by the Dutch Science Foundation NWO (grants 175.010.2009.002, 718.015.001 and 711.018.008 to M. B.) and by uNMR-NL-grid, a distributed state-of-the-art Magnetic Resonance Facility for the Netherlands (grant 184.035.002). In addition, our work was funded in part by iNEXT-Discovery (grant number 871037), a Horizon 2020 program of the European Commission, and the National Natural Science Foundation of China (No. 21871210, 22174099, and 21572161 to Y. P. L). F. D. acknowledges support from the European Union's Horizon 2020 research and innovation programme under the Marie Skłodowska-Curie grant agreement no. 813599. And M. P. C. M. acknowledges support from NWO (VIDI Grant VI. 213.110). The graphical abstract was prepared with Bio-render software.Notes and references
- P. G. Heytler and W. W. Prichard, Biochem. Biophys. Res. Commun., 1962, 7, 272–275 CrossRef CAS.
- J. Llopis, J. M. McCaffery, A. Miyawaki, M. G. Farquhar and R. Y. Tsien, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 6803–6808 CrossRef CAS PubMed.
- J. Davoust, J. Gruenberg and K. E. Howell, EMBO J., 1987, 6, 3601–3609 CrossRef CAS PubMed.
- N. H. Cho, K. C. Cheveralls, A.-D. Brunner, K. Kim, A. C. Michaelis, P. Raghavan, H. Kobayashi, L. Savy, J. Y. Li, H. Canaj, J. Y. S. Kim, E. M. Stewart, C. Gnann, F. McCarthy, J. P. Cabrera, R. M. Brunetti, B. B. Chhun, G. Dingle, M. Y. Hein, B. Huang, S. B. Mehta, J. S. Weissman, R. Gómez-Sjöberg, D. N. Itzhak, L. A. Royer, M. Mann and M. D. Leonetti, Science, 2022, 375, eabi6983 CrossRef CAS.
- E. Lundberg and G. H. H. Borner, Nat. Rev. Mol. Cell Biol., 2019, 20, 285–302 CrossRef CAS PubMed.
- M. Mattiazzi Usaj, E. B. Styles, A. J. Verster, H. Friesen, C. Boone and B. J. Andrews, Trends Cell Biol., 2016, 26, 598–611 CrossRef CAS PubMed.
- F.-X. Theillet, A. Binolfi, B. Bekei, A. Martorana, H. M. Rose, M. Stuiver, S. Verzini, D. Lorenz, M. van Rossum, D. Goldfarb and P. Selenko, Nature, 2016, 530, 45–50 CrossRef CAS PubMed.
- L. Barbieri, E. Luchinat and L. Banci, Biochim. Biophys. Acta, 2014, 1843, 2492–2496 CrossRef CAS PubMed.
- R. Hänsel, L. M. Luh, I. Corbeski, L. Trantirek and V. Dötsch, Angew Chem. Int. Ed. Engl., 2014, 53, 10300–10314 CrossRef PubMed.
- P. Broft, S. Dzatko, M. Krafcikova, A. Wacker, R. Hänsel-Hertsch, V. Dötsch, L. Trantirek and H. Schwalbe, Angew. Chem., Int. Ed., 2021, 60, 865–872 CrossRef CAS PubMed.
- K. Inomata, A. Ohno, H. Tochio, S. Isogai, T. Tenno, I. Nakase, T. Takeuchi, S. Futaki, Y. Ito, H. Hiroaki and M. Shirakawa, Nature, 2009, 458, 106–109 CrossRef CAS PubMed.
- B. M. Burmann, J. A. Gerez, I. Matečko-Burmann, S. Campioni, P. Kumari, D. Ghosh, A. Mazur, E. E. Aspholm, D. Šulskis, M. Wawrzyniuk, A. Schmidt, S. G. D. Rüdiger, R. Riek and S. Hiller, Nature, 2020, 577, 127–132 CrossRef CAS PubMed.
- S. Reckel, J. J. Lopez, F. Löhr, C. Glaubitz and V. Dötsch, ChemBioChem, 2012, 13, 534–537 CrossRef CAS PubMed.
- L. A. Baker, T. Sinnige, P. Schellenberger, J. de Keyzer, C. A. Siebert, A. J. M. Driessen, M. Baldus and K. Grünewald, Structure, 2018, 26, 161–170 CrossRef CAS PubMed.
- S. Chordia, S. Narasimhan, A. Lucini Paioni, M. Baldus and G. Roelfes, Angew. Chem., Int. Ed., 2021, 60, 5913–5920 CrossRef CAS PubMed.
- M. Kaplan, S. Narasimhan, C. de Heus, D. Mance, S. van Doorn, K. Houben, D. Popov-Čeleketić, R. Damman, E. A. Katrukha, P. Jain, W. J. C. Geerts, A. J. R. Heck, G. E. Folkers, L. C. Kapitein, S. Lemeer, P. M. P. van Bergen En Henegouwen and M. Baldus, Cell, 2016, 167, 1241–1251 CrossRef CAS PubMed.
- Q. Z. Ni, E. Daviso, T. V. Can, E. Markhasin, S. K. Jawla, T. M. Swager, R. J. Temkin, J. Herzfeld and R. G. Griffin, Acc. Chem. Res., 2013, 46, 1933–1941 CrossRef CAS PubMed.
- M. Kaplan, A. Cukkemane, G. C. P. van Zundert, S. Narasimhan, M. Daniëls, D. Mance, G. Waksman, A. M. J. J. Bonvin, R. Fronzes, G. E. Folkers and M. Baldus, Nat. Methods, 2015, 12, 649–652 CrossRef CAS PubMed.
- A. Jantschke, E. Koers, D. Mance, M. Weingarth, E. Brunner and M. Baldus, Angew. Chem., Int. Ed., 2015, 54, 15069–15073 CrossRef CAS PubMed.
- K. K. Frederick, V. K. Michaelis, B. Corzilius, T.-C. Ong, A. C. Jacavone, R. G. Griffin and S. Lindquist, Cell, 2015, 163, 620–628 CrossRef CAS PubMed.
- T. Viennet, A. Viegas, A. Kuepper, S. Arens, V. Gelev, O. Petrov, T. N. Grossmann, H. Heise and M. Etzkorn, Angew. Chem., Int. Ed., 2016, 55, 10746–10750 CrossRef CAS PubMed.
- B. J. Albert, C. Gao, E. L. Sesti, E. P. Saliba, N. Alaniva, F. J. Scott, S. Th. Sigurdsson and A. B. Barnes, Biochemistry, 2018, 57, 4741–4746 CrossRef CAS.
- R. Damman, A. Lucini Paioni, K. T. Xenaki, I. Beltrán Hernández, P. M. P. van Bergen En Henegouwen and M. Baldus, J. Biomol. NMR, 2020, 74, 401–412 CrossRef CAS PubMed.
- R. Ghosh, Y. Xiao, J. Kragelj and K. K. Frederick, J. Am. Chem. Soc., 2021, 143, 18454–18466 CrossRef CAS PubMed.
- S. Narasimhan, S. Scherpe, A. Lucini Paioni, J. van der Zwan, G. E. Folkers, H. Ovaa and M. Baldus, Angew. Chem., Int. Ed., 2019, 58, 12969–12973 CrossRef CAS PubMed.
- E. J. Koers, E. A. W. van der Cruijsen, M. Rosay, M. Weingarth, A. Prokofyev, C. Sauvée, O. Ouari, J. van der Zwan, O. Pongs, P. Tordo, W. E. Maas and M. Baldus, J. Biomol. NMR, 2014, 60, 157–168 CrossRef CAS PubMed.
- R. Gupta, H. Zhang, M. Lu, G. Hou, M. Caporini, M. Rosay, W. Maas, J. Struppe, J. Ahn, I.-J. L. Byeon, H. Oschkinat, K. Jaudzems, E. Barbet-Massin, L. Emsley, G. Pintacuda, A. Lesage, A. M. Gronenborn and T. Polenova, J. Phys. Chem. B, 2019, 123, 5048–5058 CrossRef CAS PubMed.
- M. Lu, M. Wang, I. V. Sergeyev, C. M. Quinn, J. Struppe, M. Rosay, W. Maas, A. M. Gronenborn and T. Polenova, J. Am. Chem. Soc., 2019, 141, 5681–5691 CrossRef CAS.
- B. Uluca, T. Viennet, D. Petrović, H. Shaykhalishahi, F. Weirich, A. Gönülalan, B. Strodel, M. Etzkorn, W. Hoyer and H. Heise, Biophys. J., 2018, 114, 1614–1623 CrossRef CAS PubMed.
- I. V. Sergeyev, B. Itin, R. Rogawski, L. A. Day and A. E. McDermott, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 5171–5176 CrossRef CAS.
- P. Fricke, D. Mance, V. Chevelkov, K. Giller, S. Becker, M. Baldus and A. Lange, J. Biomol. NMR, 2016, 65, 121–126 CrossRef CAS PubMed.
- W. Liu, Y. Wang, L. H. M. Bozi, P. Fischer, M. P. Jedrychowski, H. Xiao, T. Wu, N. Darabedian, X. He, E. L. Mills, N. Burger, S. Shin, A. Reddy, H.-G. Sprenger, N. Tran, S. Winther, S. M. Hinshaw, J. Shen, H.-S. Seo, K. Song, A. Z. Xu, L. Sebastian, J. Zhao, S. Dhe-Paganon, J. Che, S. P. Gygi, H. Arthanari and E. T. Chouchani, Nature, 2023, 1–3 Search PubMed.
- J. Han and K. Burgess, Chem. Rev., 2010, 110, 2709–2728 CrossRef CAS PubMed.
- X. Cai, A. Lucini Paioni, A. Adler, R. Yao, W. Zhang, D. Beriashvili, A. Safeer, A. Gurinov, A. Rockenbauer, Y. Song, M. Baldus and Y. Liu, Chem.–Eur. J., 2021, 27, 12758–12762 CrossRef CAS PubMed.
- R. Yao, D. Beriashvili, W. Zhang, S. Li, A. Safeer, A. Gurinov, A. Rockenbauer, Y. Yang, Y. Song, M. Baldus and Y. Liu, Chem. Sci., 2022, 13, 14157–14164 RSC.
- J. Liwocha, D. T. Krist, G. J. van der Heden van Noort, F. M. Hansen, V. H. Truong, O. Karayel, N. Purser, D. Houston, N. Burton, M. J. Bostock, M. Sattler, M. Mann, J. S. Harrison, G. Kleiger, H. Ovaa and B. A. Schulman, Nat. Chem. Biol., 2021, 17, 272–279 CrossRef CAS PubMed.
- P. C. A. van der Wel, K.-N. Hu, J. Lewandowski and R. G. Griffin, J. Am. Chem. Soc., 2006, 128, 10840–10846 CrossRef CAS PubMed.
- A. M. Vian and A. Z. Higgins, Cryobiology, 2014, 68, 35–42 CrossRef CAS PubMed.
- W. Zhu, A. J. Guseman, F. Bhinderwala, M. Lu, X.-C. Su and A. M. Gronenborn, Angew. Chem., Int. Ed., 2022, 61, e202201097 CrossRef CAS PubMed.
- H. Kadavath, N. C. Prymaczok, C. Eichmann, R. Riek and J. A. Gerez, Angew. Chem., Int. Ed., 2023, 62, e202213976 CrossRef CAS PubMed.
- S. E. Kaiser, B. E. Riley, T. A. Shaler, R. S. Trevino, C. H. Becker, H. Schulman and R. R. Kopito, Nat. Methods, 2011, 8, 691–696 CrossRef CAS PubMed.
- H. Heise, K. Seidel, M. Etzkorn, S. Becker and M. Baldus, J. Magn. Reson., 2005, 173, 64–74 CrossRef CAS.
- A. B. Barnes, B. Corzilius, M. L. Mak-Jurkauskas, L. B. Andreas, V. S. Bajaj, Y. Matsuki, M. L. Belenky, J. Lugtenburg, J. R. Sirigiri, R. J. Temkin, J. Herzfeld and R. G. Griffin, Phys. Chem. Chem. Phys., 2010, 12, 5861–5867 RSC.
- B. Corzilius, L. B. Andreas, A. A. Smith, Q. Z. Ni and R. G. Griffin, J. Magn. Reson., 2014, 240, 113–123 CrossRef CAS PubMed.
- A. H. Linden, W. T. Franks, Ü. Akbey, S. Lange, B.-J. van Rossum and H. Oschkinat, J. Biomol. NMR, 2011, 51, 283 CrossRef CAS PubMed.
- C. A. Castañeda, E. K. Dixon, O. Walker, A. Chaturvedi, M. A. Nakasone, J. E. Curtis, M. R. Reed, S. Krueger, T. A. Cropp and D. Fushman, Structure, 2016, 24, 423–436 CrossRef PubMed.
- M. K. Hospenthal, S. M. V. Freund and D. Komander, Nat. Struct. Mol. Biol., 2013, 20, 555–565 CrossRef CAS PubMed.
- C. Gladkova, A. F. Schubert, J. L. Wagstaff, J. N. Pruneda, S. M. Freund and D. Komander, EMBO J., 2017, 36, 3555–3572 CrossRef CAS PubMed.
- O. F. Lange, N.-A. Lakomek, C. Farès, G. F. Schröder, K. F. A. Walter, S. Becker, J. Meiler, H. Grubmüller, C. Griesinger and B. L. de Groot, Science, 2008, 320, 1471–1475 CrossRef CAS PubMed.
- L. Sardo, A. Lin, S. Khakhina, L. Beckman, L. Ricon, W. Elbezanti, T. Jaison, H. Vishwasrao, H. Shroff, C. Janetopoulos and Z. A. Klase, J. Cell Sci., 2017, 130, 2926–2940 CAS.
- B. A. Maxwell, Y. Gwon, A. Mishra, J. Peng, H. Nakamura, K. Zhang, H. J. Kim and J. P. Taylor, Science, 2021, 372, eabc3593 CrossRef CAS PubMed.
- K. Juenemann, A. H. P. Jansen, L. van Riel, R. Merkx, M. P. C. Mulder, H. An, A. Statsyuk, J. Kirstein, H. Ovaa and E. A. Reits, Sci. Rep., 2018, 8, 1405 CrossRef PubMed.
- N. P. Dantuma, T. A. M. Groothuis, F. A. Salomons and J. Neefjes, J. Cell Biol., 2006, 173, 19–26 CrossRef CAS PubMed.
- E. Wist and H. Krokan, Exp. Cell Res., 1978, 116, 313–316 CrossRef CAS PubMed.
- R. S. D. Read and C. M. Mauritzen, Can. J. Biochem., 1970, 48, 559–565 CrossRef CAS PubMed.
- K. E. Knockenhauer and T. U. Schwartz, Cell, 2016, 164, 1162–1171 CrossRef CAS PubMed.
- Y. Wang and O. Jardetzky, Protein Sci., 2002, 11, 852–861 CrossRef CAS PubMed.
- H. Heise, S. Luca, B. L. de Groot, H. Grubmüller and M. Baldus, Biophys. J., 2005, 89, 2113–2120 CrossRef CAS PubMed.
- R. H. Havlin and R. Tycko, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 3284–3289 CrossRef CAS PubMed.
- I. Dikic and B. A. Schulman, Nat. Rev. Mol. Cell Biol., 2022, 1–15 Search PubMed.
- K. N. Swatek, J. L. Usher, A. F. Kueck, C. Gladkova, T. E. T. Mevissen, J. N. Pruneda, T. Skern and D. Komander, Nature, 2019, 572, 533–537 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sc02117c |
| This journal is © The Royal Society of Chemistry 2023 |