
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDeep removal of trace C2H2 and CO2 from C2H4 by using customized potassium-exchange mordenite†
Hongwei
Chen‡
 a,
Binyu
Wang‡
b,
Bin
Zhang
c,
Jiuhong
Chen
a,
Jiabao
Gui
a,
Xiufeng
Shi
c,
Wenfu
Yan
b,
Jinping
Li
a and
Libo
Li
*a
a,
Binyu
Wang‡
b,
Bin
Zhang
c,
Jiuhong
Chen
a,
Jiabao
Gui
a,
Xiufeng
Shi
c,
Wenfu
Yan
b,
Jinping
Li
a and
Libo
Li
*a
aCollege of Chemical Engineering and Technology, Shanxi Key Laboratory of Gas Energy Efficient and Clean Utilization, Taiyuan University of Technology, Taiyuan 030024, China. E-mail: lilibo@tyut.edu.cn; chenhongwei3197@hotmail.com
bState Key Laboratory of Inorganic Synthesis and Preparative Chemistry, College of Chemistry, Jilin University, 2699 Qianjin Street, Changchun 130012, China. E-mail: 864421855@qq.com
cCollege of Chemistry, Taiyuan University of Technology, Taiyuan 030024, China
First published on 26th May 2023
Abstract
Adsorptive separation using porous materials is a promising approach for separating alkynes/olefins due to its energy efficiency, while the deep removal of trace amounts of C2H2 and CO2 from C2H4 is still very challenging for a commercial adsorbent. Herein, we report a low-cost inorganic metal cation-mediated mordenite (MOR) zeolite with the specific location and distribution of K+ cations acting as a goalkeeper for accurately controlling diffusion channels, as evidence of the experimental and simulation results. Deep purification of C2H4 from ternary CO2/C2H2/C2H4 mixtures was first realized on K–MOR with exceptional results, achieving a remarkable polymer-grade C2H4 productivity of 1742 L kg−1 for the CO2/C2H2/C2H4 mixture. Our approach which only involves adjusting the equilibrium ions, is both promising and cost-effective, and opens up new possibilities for the use of zeolites in the industrial light hydrocarbon adsorption and purification process.
Introduction
Ethylene (C2H4) is an essential petrochemical intermediate and currently the world's most extensively consumed organic compound, with a production capacity that exceeded 200 million tons in 2021.1 C2H4 is primarily obtained through steam cracking or thermal decomposition of either naphtha or ethane (C2H6), which inevitably leads to the presence of acetylene (C2H2) and carbon dioxide (CO2) as downstream byproducts. Purification of C2H4 from ternary CO2/C2H2/C2H4 mixtures is a crucial process to yield polymer-grade C2H4 (C2H4 > 99.9% purity and C2H2 < 40 ppm).2 Currently, in the petrochemical industry, C2H2 is first removed through catalytic hydrogenation using noble-metal catalysts or solvent extraction using organic solvents, and CO2 is subsequently separated from C2H4 by the organic amine adsorption method. These step-by-step purification processes are complex, environmentally unfriendly, and energy-intensive.3 In fact, the total energy used for the purification of ethylene and propene alone accounts for 0.3% of global energy consumption.4 This high energy footprint has led to extensive research aimed at developing more energy-efficient approaches for the purification of C2H4.Physical adsorption is a promising approach that has the potential to significantly reduce the energy consumption of separation processes.5–13 Selective adsorption relies on the specific interaction between olefins and chemically modified adsorbents.14–18 Most adsorbents use kinetic or steric effects to separate alkynes from their corresponding olefins, which requires an additional CO2 removal step. Currently, there are few materials capable of effectively separating C2H4 from CO2/C2H2/C2H4 in a single step, and the majority of these materials are metal–organic frameworks (MOFs) or other porous materials that have been reported in research studies.19–25 Our research focuses on exploring the potential applications of zeolites, which are highly versatile and functional materials. These materials feature robust frameworks, molecule-sized pores, large surface areas, and other tunable properties.26–32 These unique characteristics make zeolites capable of creating suitable pore space and environment to accommodate specific guest molecules. Numerous studies revealed that the adsorption behaviours were governed not only by the valence of the cations and the composition of the skeleton but also by their ionic radii and locations.33–37 These merits are responsible for the successful applications of zeolites in various industrial processes. The material of our study is the MOR zeolite, which has a unique channel structure and adsorption properties that have been extensively studied by scientists. Furthermore, there has been significant progress in exploring the potential of the MOR zeolite in carbon dioxide capture,8,38,39 removal of nitrous oxide,40 and olefin/paraffin separation.41
The fundamental challenge in purifying C2H4 from ternary CO2/C2H2/C2H4 mixtures primarily arises from the physicochemical properties of the respective gas molecules, including their size, boiling point, electrostatic potential distribution (Fig. S8†), kinetic diameters (C2H2: 3.3 Å, C2H4: 4.2 Å, and CO2: 3.3 Å), quadrupole moments (7.2 × 10−26vs. 1.5 × 10−26vs. 4.3 × 10−26 esu cm2), and polarizabilities (39.3 × 10−25vs. 42.52 × 10−25vs. 29.11 × 10−25 cm3). The physicochemical properties of C2H4 are intermediate between those of C2H2 and CO2, making it challenging for most physisorbents to demonstrate high selectivity. Since larger quadrupole moments and more π electrons of C2H2, C2H4, and CO2 gases enable a stronger interaction with cations or highly polar T atoms of the skeleton, deep purification of C2H4 from ternary CO2/C2H2/C2H4 mixtures would require physisorbents to show significantly stronger affinity toward both C2H2 and CO2 over C2H4. To overcome this challenge, a diffusion channel regulation strategy using classic MOR zeolites (Fig. 1) has been proposed. In terms of C2H2/C2H4 separation, porous materials can typically achieve preferential adsorption of C2H2 over C2H4 with relatively high selectivity through pore size fine-tuning, channel environment adjustments, or incorporation of new skeleton components such as heteroatoms in molecular sieves, new groups in MOF ligands and more ways. However, due to the opposite electrostatic potential distributions of CO2 and C2H2, it is nearly impossible for a C2H2-selective zeolite to simultaneously increase both C2H2 and CO2 adsorption capacities. Instead, selectively inhibiting the C2H4-affinity sites in zeolites seems to be a feasible strategy for enhancing C2H4 purification capability without sacrificing the original C2H2 and CO2 adsorption capacity. This special approach not only reduces C2H4 co-adsorption and enhances the recovery during the adsorption process, but also reduces the energy consumption of regeneration. Additionally, the unique CO2 adsorption confinement effect offered by the MOR zeolite can be leveraged to overcome this challenge.42
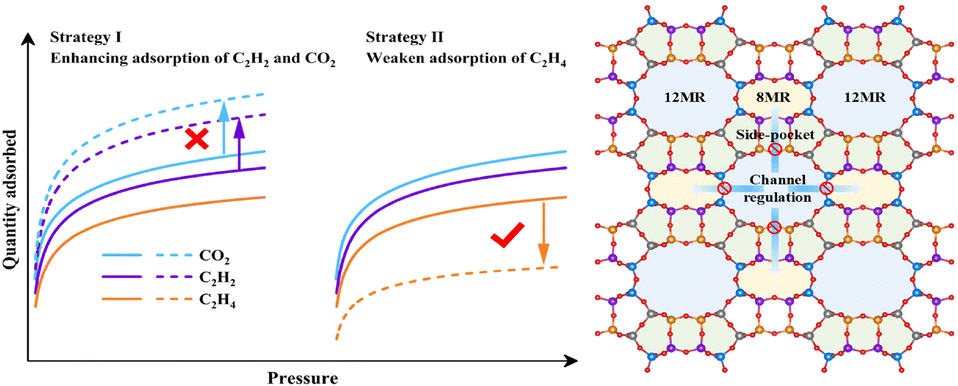 | ||
| Fig. 1 Diffusion channel regulation strategy for CO2/C2H2/C2H4 separation. | ||
It is a mechanism that is often neglected among the many common adsorption and separation mechanisms in zeolites; channel and pore accessibility is another crucial factor in determining adsorption capacity and selectivity. An illustrative example using Na–ZSM-5,43 where DFT calculations were performed, revealed that only the Na+ site located in the 10 MR channel interacts with CO2 as it is the only accessible site to CO2. Conversely, the Na+ located in the 5 MR adjacent to the wall is inaccessible to CO2 due to the kinetic diameter of CO2 being larger than the corresponding pore size (approximately 1.5 Å) through which the cation is accessed.
Zeolites offer the unique capability to regulate the accessibility of their channels through the presence of different cations. To create efficient adsorbent materials customized for targeted separation or purification processes, zeolite products must undergo ion exchange with diverse alkali metals, and alkaline earth, transition, or rare earth elements.44 This customizable process allows the development of the corresponding special adsorbents for different practical application environments. Our study deduced that strategically introducing appropriate cations into the inert channel space of MOR zeolites can effectively impede the diffusion of C2H4 into specific adsorption sites. This mechanism can be combined with the confinement effect of CO2 in MOR to achieve simultaneous separation of C2H2/C2H4 and CO2/C2H4 using a single adsorbent. In this study, we provide a highly feasible reference for deep purification of C2H4 from 1/1/98 CO2/C2H2/C2H4 mixtures, only facilitated through employment of a common zeolite via a traditional and effortless ion exchange process with an impressive C2H4 purification productivity.
Experimental
Synthesis of Na–MOR
All reagents used in the experiment were commercially sourced and used without any purification (Table S1†). Na–MOR (N–MOR) zeolites were synthesized using a template-free method. 0.9475 g NaOH and 0.5075 g NaAlO2 were mixed with 23.53 g deionized water in a plastic beaker and stirred vigorously at room temperature for 30 minutes. Next, 14.18 g of 30 wt% silica sol solutions were added to the mixture and stirred for 4 hours until a homogeneous slurry was formed. The slurry was then transferred to a Teflon-lined stainless-steel autoclave and statically crystallized at 453 K for 3 days. The solid was washed with deionized water and air-dried. Through large-scale synthesis carried out in a 10 L reactor, we were able to produce over 1 kg of N–MOR in a single reaction.Preparation of M–MOR
The M–MOR (M = NH4+, Li+, Na+, Cs+, Mg2+, Ca2+, Sr2+, Ba2+) were prepared via ion-exchange (ionic radius: Mg2+ < Li+ < Na+ < Ca2+ < Sr2+ < K+ < Ba2+ < Cs+ and ionic hydrated radius: Cs+ < K+ < Na+ < Li+ < Ba2+ < Sr2+ < Ca2+ < Mg2+). As an example, the as-synthesized N–MOR was exchanged with NH4Cl solution (1 mol L−1) at a solid-to-liquid ratio of 1 g/50 mL under stirring (298 K, 4 h). The slurry was filtered, washed with deionized water three times, and dried overnight at 373 K; other M–MOR were prepared in the same way. In addition, attempts were made to directly synthesize K–MOR, but only amorphous products were obtained. During the synthesis process, NaOH acted as a crucial mineralizing and structure-guiding agent, playing an essential role in MOR crystallization. It is difficult to obtain K–MOR through a one-step method without any organic template.Structure of MOR
X-ray diffraction (XRD) patterns were obtained using a Bruker D8 ADVANCE powder diffractometer equipped with a copper-sealed target (λ = 0.15418 nm) at room temperature. The measurements were conducted by scanning over the 4–40° (2θ) range at a rate of 7.55 ° min−1. The Na, Si, Al, and K content of the zeolite samples was analyzed using an inductively coupled plasma optical emission spectrometer (Avio™ 200 ICP-OES). The original CIF structure model was sourced from the zeolite database of the Material Studio, and the zeolitic structure was considered rigid. The reported aluminum distribution was used for CIF file modeling,45 and the parameters describing the interaction between the adsorbent and cation were taken from COMPASS II. The sorption simulations were carried out using the Material Studio software.Single-component static adsorption
Isotherms were collected on a Micromeritics ASAP 2020 automatism isothermal adsorption instrument using a calculated amount of adsorbent placed in a sample tube. Prior to the measurements, all the samples were degassed to 10−7 bar at 473 K. Adsorption isotherms were fitted with a double-site Langmuir–Freundlich isotherm model with six parameters. Ideal adsorbed solution theory (IAST) was used to predict binary adsorption isotherms from the single-component adsorption isotherms of C2H2, C2H4, and CO2. Isosteric heat adsorption (Qst) values were calculated from isotherms measured at 298 K, 318 K and 338 K for C2H2, C2H4, and CO2.The porosity of the samples was characterized using N2 sorption isotherms at 77 K on an ASAP 3020 automated isothermal adsorption instrument. The Brunauer–Emmett–Teller method was employed to estimate the specific surface areas (SBET), and the micropore volume was determined simultaneously.
Adsorption kinetics and desorption behaviours
Single composition CO2, C2H2, and C2H4 dynamic adsorption profiles at 298 K were measured on an intelligent gravimetric analyzer (IGA-100, HIDEN). About 80–120 mg of the sample was first loaded into the sample chamber and activated at 473 K under high vacuum, the pressure was increased and decreased with a ramp rate of 100 mbar min−1 during the desorption process, and a fast response furnace was employed for heating.Breakthrough experiments
The setup was designed to measure and analyze the adsorption and separation of gases in real-time, replicating practical conditions for industrial applications, and the outlet gas was detected using an Agilent GC490 gas chromatograph (Table S3†). As an example, the C2H2/C2H4 (1/99, v/v) breakthrough experiment using a dynamic gas breakthrough equipment set up at 298 K was conducted. The experiment was carried out using a stainless-steel column with dimensions of 10 mm inner diameter and 110 mm length. The column was packed with a sample weighing 3.68 g. Prior to each breakthrough experiment, the column was vacuumed at 473 K until no guest molecule was observed, and then purged with Ar at room temperature. The mixed gas (C2H2/C2H4 1/99, v/v) was then introduced at a flow rate of 10 mL min−1. The outlet gas from the column was monitored using gas chromatography (GC-490, Agilent) with a thermal conductivity detector (TCD). After completing the breakthrough experiment, the column was regenerated with an Ar flow at a temperature of 453 K and 573 K. The breakthrough experiments performed in our study included the C2H2/C2H4 (50/50, v/v), C2H2/C2H4/C2H6 (1/90/9, v/v/v), and CO2/C2H2/C2H4 (1/1/98, v/v/v) breakthrough experiments under humid conditions, in addition to the C2H2/C2H4 (1/99, v/v) breakthrough experiment. These experiments were similar in their operating way and were designed to measure the adsorption and separation of mixed gases under various conditions.The dynamic separation selectivity was calculated using 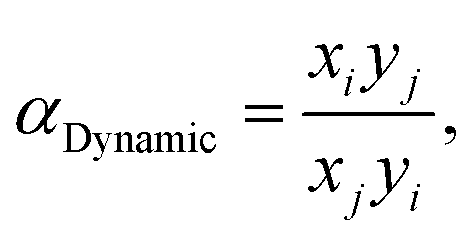 where xi and xj are the mole fractions of components i and j, respectively, in the adsorbed phase, and yi and yj are the mole fractions of components i and j, respectively, in the gas phase.
where xi and xj are the mole fractions of components i and j, respectively, in the adsorbed phase, and yi and yj are the mole fractions of components i and j, respectively, in the gas phase.
Results and discussion
To minimize environmental impact and reduce costs, we utilized a green and classic hydrothermal synthesis route to synthesize MOR zeolites; this approach allowed us to avoid the use of toxic organic reagents and complex synthesis routes. The environmentally friendly and low-cost MOR zeolites served as the study object and parent material with a composition of Na5[Al5Si43O96]. MOR zeolites possess a unique channel system consisting of parallel linear channels. Specifically, the main channel is composed of a 12-membered ring (12 MR) (6.5 × 7.0 Å) and runs along the c-axis. In addition, another channel called a side pocket is present, which consists of an 8 MR (2.6 × 5.7 Å) and runs along the b-axis. Therefore, there are two distinct adsorption spaces available: the 12 MR main channel and the narrow, highly restricted 8 MR side pocket. The larger 12 MR main channel provides a larger adsorption space with weaker interactions with the framework and balancing cations, leading to fewer constraints on guest molecules. Diffusion and adsorption in the 8 MR side pocket, on the other hand, are dominated by the interaction between the guest molecule framework and balancing cations, resulting in significant differences in both thermodynamic and kinetic properties when compared to those in the main channel.The crystalline structure of the MOR zeolite remained intact after ion exchange with NH4+, Ca2+, Mg2+, and K+ ions, as evidenced by the presence of all reflections corresponding to the ion-exchanged zeolites. However, the XRD patterns of Cs–MOR and Ba–MOR exhibited significant differences, and electron microscopy images confirmed multiple structural breakages in Li-, Cs-, Sr-, and Ba–MOR samples (Fig. S1–S3†).
After analyzing the adsorption isotherms of C2H2, C2H2, and CO2 on M–MOR (Fig. S9†), K–MOR and NH4–MOR stand out as the candidate materials. Although NH4–MOR demonstrated exceptional performance during the initial adsorption and desorption, its application was limited due to the instability of NH4+ in MOR and weak interaction with the MOR skeleton. With an increase in the number of cycle test times, NH4+ may convert to NH3 and become detached from MOR during the degassing step, which caused a significant drawback for its practical application.
It is well known that the hydrated ion radius of K+ is much smaller than that of Na+, which makes K+ ions easily exchange with Na+ ions in MOR. The BET and the micropore volume decrease regularly (Fig. 2), which can be can be attributed to the large ionic radius of the K+ ion and its specific occupying effect, which limits the part of the 8 MR side pocket and 8 MR space that the N2 molecule can access and occupy. After undergoing K+ ion exchange, the diffusion channel of MOR was effectively regulated by K+ cations, leading to alterations in the channel-pore environment. C2H2 molecules were still able to access and occupy the side pocket of MOR, while C2H4 molecules were unable to efficiently utilize the adsorption site and space within the side pocket, resulting in a significant decrease in the amount of C2H4 adsorbed. Conversely, due to the intrinsic properties of MOR regarding CO2 confinement in the 8 MR side pocket, the amount of CO2 adsorbed remained relatively constant. Through calculations of the Qst of C2H2, C2H4, and CO2 adsorption, it was found that the Qst of C2H2 and CO2 were higher than that of C2H4 at both zero-coverage and for the whole process, indicating the strong interaction between K–MOR and CO2/C2H2 molecules. Moreover, it was discovered that N–MOR displayed a significant attraction towards C2H2, C2H4, and CO2, with Qst values that were comparatively higher than those of K–MOR. Specifically, N–MOR exhibited a higher heat of adsorption for C2H4 than for CO2, indicating a more significant interaction between N–MOR and C2H4 (Fig. S17 and S18†). In addition, IAST was employed to predict selectivity on M–MOR at 298 K, and the adsorption isotherms were fitted using the dual-site Langmuir–Freundlich adsorption model. K–MOR-1 (K–MOR) exhibited both the highest 1/99 C2H2/C2H4 (28.6) and 1/99 CO2/C2H4 (9.21) IAST selectivity compared to other M–MOR and K–MOR-X (X = 0.01, 0.05, 0.1, 0.5, 1.5, and 2, X represent different ion-exchange concentrations) (Fig. S11–S16†). This exhilarating selectivity makes dynamic breakthrough experiments more desirable. Furthermore, K–MOR exhibited exceptional moisture and pH stability (Fig. S10†), which is advantageous for practical applications in the industry.
 | ||
| Fig. 2 C2H2, C2H4, and CO2 adsorption isotherms of (a) N–MOR and (b) K–MOR. (c) IAST selectivity of K–MOR for mixtures of C2H2/C2H4 (1/99) and CO2/C2H4 (1/99). (d) Isosteric heats of C2H2, C2H4, and CO2 adsorption on K–MOR. (e) N2 sorption isotherms at 77 K on K–MOR with different K salt concentrations. (f) Comparison of ion-exchange concentration, specific surface areas (SBET) and micropore volume in K–MOR. | ||
Adsorption kinetics and desorption behaviours (Fig. 3) are critical parameters to consider when evaluating the separation efficiency of a material for industrial application. A key finding of this research was the enhanced desorption kinetics of CO2 and C2H4 on K–MOR, which became more easier compared to that on N–MOR. Notably, only vacuum conditions at room temperature were sufficient for achieving desorption on K–MOR, demonstrating a significant energy consumption advantage over N–MOR. In contrast, desorbing adsorbents for C2H2, C2H4, and CO2 from N–MOR through vacuuming proved to be difficult and time-consuming, requiring additional heating operations and energy consumption. While a small amount of C2H2 heating was necessary to completely desorb from K–MOR, this result suggests that K–MOR retains its strong interaction force with C2H2 while interactions with C2H4 weakened. As for adsorption, the C2H2 fractional uptake Qt/Qe exhibited a relatively slow increase with time for both K–MOR and N–MOR during the adsorption process, possibly due to the confinement effect of MOR and the strong interaction of the cations in MOR to reach the adsorption equilibrium. As for C2H4, the equilibrium time of C2H4 for N–MOR was significantly longer than that of K–MOR, possibly attributed to the fact that C2H4 molecules are limited by K+ ions, which makes it difficult for the C2H4 molecules to access and be retained in the 8 MR side-pocket as easily as C2H2 and CO2, and mostly only diffuse and adsorb in the space of the 12 MR of MOR, which accelerates the process of C2H4 reaching the adsorption equilibrium and also contributes to the desorption of C2H4 in K–MOR.
 | ||
| Fig. 3 Kinetics of adsorption and desorption behaviours for CO2, C2H2, and C2H4 on (a and b) N–MOR and (c and d) K–MOR. | ||
The outstanding C2H2/C2H4 separation performance of K–MOR is further confirmed dynamically in a fixed bed (Fig. 4). The breakthrough curves are plotted with time (min g−1), which is already adjusted for the dead volume obtained from the blank experiment. As expected, the C2H2/C2H4 mixture was effectively separated using K–MOR. C2H4 was the first to elute through the bed, and then the outlet gas quickly reached pure grade with no detectable C2H2 (below the detection limit of the experimental setup), whereas the solid adsorbent retained C2H2 for a remarkable time before the breakthrough of C2H2, which is in line with the sorption experiments. Through conservation calculations, the co-adsorbed C2H4 can be partially replaced by C2H2, which enhances the dynamic separation performance to a certain extent, and makes it possible to remove C2H2 from C2H4 without losing any valuable C2H4. The amount of polymer-grade C2H4 purified from the C2H2/C2H4 (1/99, v/v) mixture was as high as 2029 L kg−1 (C2H2 < 10 ppm), the recovery rate was up to 97.61%, the amount of C2H2 adsorbed from the mixture was up to 20.35 cm3 g−1, and a remarkable C2H2/C2H4 dynamic selectivity of 54.4 was achieved, compared with that of reported state-of-the-art sorbents (Table S4†). Moreover, regeneration of K–MOR under an Ar flow at 453 K demonstrated complete recovery of the adsorbed gas within 60 min g−1 at 10 mL min−1, which is significantly faster than the Ag-doped adsorbent (which can take up to thousands of minutes).46 These results demonstrate that C2H2 removal can be effectively addressed under mild conditions (1–2 bar and 298 K), consistent with the realistic pressure of cracked gas steam leaving the thermal cracking furnace (ca. 1.55 bar).47 The cycling C2H2/C2H4 (1/99, v/v) breakthrough tests were conducted on K–MOR under the aforementioned operating conditions, and the results showed the same retention time as the initial experiment (Fig. S21†).
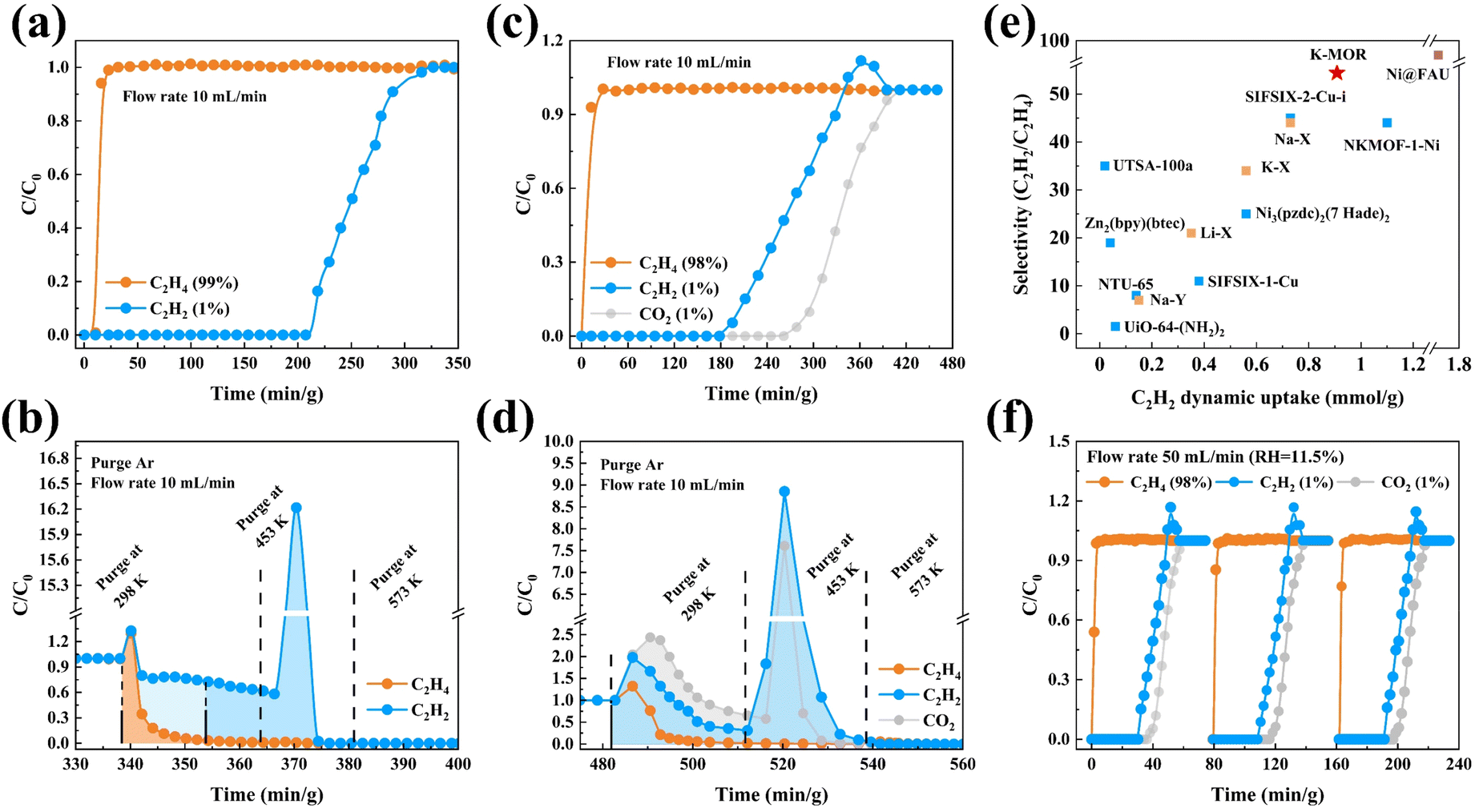 | ||
| Fig. 4 (a and b) C2H2/C2H4 (1/99, v/v) and (c and d) CO2/C2H2/C2H4 (1/1/98, v/v/v) breakthrough experiments at 298 K. (e) Comparation of C2H2/C2H4 dynamic selectivity against C2H2 dynamic uptake under ambient conditions with a state-of-the art adsorbent. (f) Cycling breakthrough tests on K–MOR under humid conditions (RH = 11.5%). | ||
Due to the MOR zeolite nature structure properties,42 K–MOR shows excellent CO2 capture ability at extremely low CO2 concentrations and room temperature as N–MOR. And the 1/1/98 CO2/C2H2/C2H4 breakthrough experiment conducted on K–MOR showed that it is capable of capturing both CO2 and C2H2 simultaneously, while producing highly purified polymer-grade C2H4 with a productivity of up to 1742 L kg−1 (CO2 < 100 ppm and C2H2 < 10 ppm). The dynamic uptake of C2H2 was up to 27.15 cm3 g−1, while that of CO2 was up to 32.61 cm3 g−1 when adsorption equilibrium was reached. The desorption process revealed that the majority of adsorbed CO2 was rapidly released during argon purging, with only a small portion requiring temperature desorption. Similarly, a fraction of adsorbed C2H2 was swiftly desorbed, while another portion required temperature desorption. In contrast, C2H4 showed significant replacement during the adsorption process, as confirmed by conservation calculations. During the desorption process, the majority of adsorbed C2H4 was directly purged and released at room temperature.
Typically, the cracking gas contains some H2O and the adsorbent operates at higher flow rates. Breakthrough experiments were carried out at different relative humidities and RH levels of 75%, 43.5%, and 11.5%. The separation performance of K–MOR was largely maintained, with the polymer-grade C2H4 production productivity reaching 1074 L kg−1 under humid conditions (RH = 75%). Furthermore, breakthrough cycling experiments were conducted on 1/1/98 CO2/C2H2/C2H4 with a 50 mL min−1 flow (RH = 11.5%) (Fig. S22 and S23†), indicating that this material has great potential and feasibility for challenging separation of the C2H4 component in actual industrial cracked gas streams.
To explore the diffusion behaviours and adsorption density distribution of C2H2, C2H4, and CO2 molecules in MOR, we integrate practical separation experiments with computer simulation techniques. While it is possible to determine the mean structure and chemical composition of a zeolite using established methods such as X-ray diffraction and elemental analysis, determining the specific location of aluminum atoms within the zeolite framework is challenging using conventional approaches. Although the determination of the mean structure and the chemical composition of a zeolite is well established, it is still very hard to get a direct determination of aluminum siting by the conventional methods. Many researchers have investigated the Al distribution in the MOR zeolite in both experimental48–54 and simulated45,54,55 ways, confirmed that Al atoms preferentially occupy tetrahedral positions in the four-membered ring (T3 and T4 sites) of the MOR zeolite structure and the proportion of Al atoms in the main channels relatively decreased with an increase in the Si/Al ratio of the MOR zeolite, and indicate that the order of occupancy of the sites obtained by many simulation methods is the same: T3 > T4 > T1 > T2. Our model was based on an up-to-date and reliable molecular simulation method reported by Jeffroy and co-workers45 (Table S6†) to determine the Al distribution for the CIF file modification; this simulation result is not only suiting the case for the MOR zeolite, but also for several other materials studied by simulation belonging to the GOO and ANA families.45,49–52,54
Adsorption properties of aluminosilicate zeolites are related to the location of cations; however, the location and distribution of aluminum atoms can strongly influence the cation location.55–58 As a result of the atomic charge difference between silicon and aluminum atoms, the repulsive forces exerted on cations are expected to be smaller for aluminum atoms. This means that cations are likely to be located close to aluminum atoms, with the number of aluminum atoms close to a cation dependent on the Si/Al ratio. In general, a higher Si/Al ratio is expected to result in a smaller number of aluminum atoms surrounding a cation. However, the local Si/Al ratio around a cation can be lower than the global Si/Al ratio, and this can lead to the cation being more sensitive to the presence of aluminum atoms and being located closer to them. Understanding the relationship between aluminum atoms, cations, and Si/Al ratios is important in predicting and controlling the adsorption properties of zeolites for specific gas separation applications.
From our simulation result (Fig. 5), the K+ cations tend to be distributed near the center of the 8 MR side pocket which could adjust the connection channel of the 12 MR main channel and side pocket; the Na+ cations tend to be distributed in the 8 MR, which indicated that K–MOR could block some bigger molecules from diffusing into the 8 MR side pocket and the 8 MR of the MOR zeolite, and influence the adsorption environment of the 12 MR, providing special adsorption and separation properties to K–MOR. The high electro-positivity and large ionic size of K+ cations regulate the diffusion channel and alter the channel environment. As a result, C2H4 molecules cannot easily diffuse into the 8 MR side-pocket and 8 MR, and the majority of C2H4 can only compete with CO2 and C2H2 for adsorption sites in the 12 MR. Additionally, K+ cations weaken the interaction between the host and the guest, which significantly reduces the adsorption capacity and strength of C2H4. This is consistent with the results of static adsorption performance and breakthrough experiments, and both C2H4 adsorption and desorption behaviours of K–MOR.
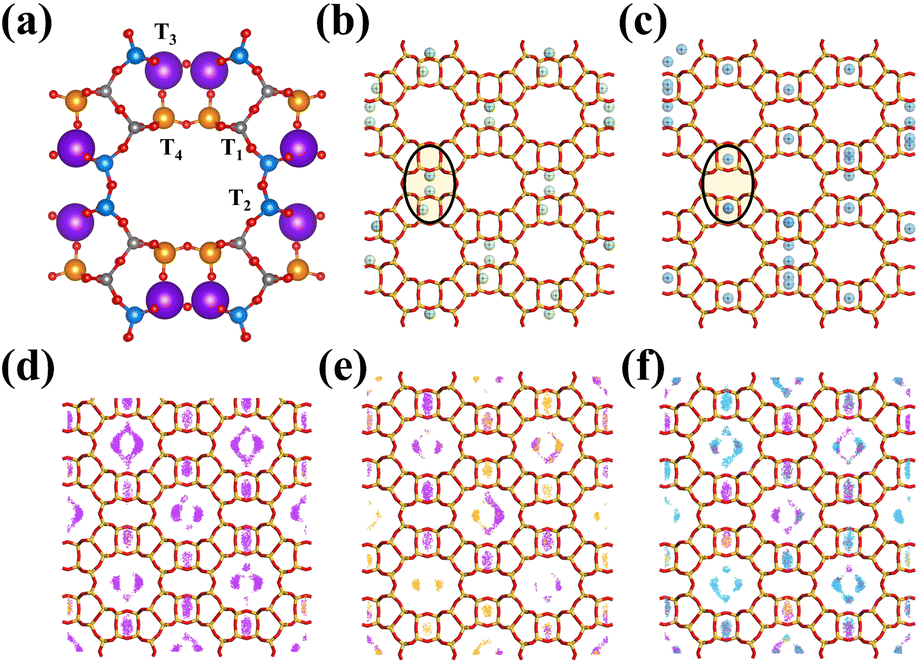 | ||
| Fig. 5 (a) Aluminium distribution in different T sites (T1, T2, T3, and T4) of mordenite used for simulation, and the sphere diameter is proportional to the aluminium population in the corresponding site. The skeleton and cation distribution of (b) N–MOR and (c) K–MOR. Density distribution of C2H2 (purple), C2H4 (orange), CO2 (blue) under the loading calculated from (d) C2H2/C2H4 (50/50, v/v), (e) C2H2/C2H4 (1/99, v/v) and (f) CO2/C2H2/C2H4 (1/1/98, v/v/v) separation on K–MOR at 298 K. | ||
To better understand the selective adsorption behaviours of C2H2 and CO2 over C2H4, GCMC simulation was conducted using the sorption module of Material Studio to calculate the adsorption probability distribution of C2H2, C2H4, and CO2 in N–MOR and K–MOR at 298 K and 1.0 bar. This simulation allows us to gain insights into the molecular-level mechanisms that govern the preferential adsorption of certain gases over others in these zeolite materials. To ensure a fair comparison, a super unit cell (222) containing 40 Al atoms based on the experimental Si/Al ratio was built, and 40 cations were used as the equilibrium cations. Sorption simulations were carried out for the same fixed loading of C2H2, C2H4, and CO2 in N–MOR as in the blank control (Fig. S24†). The sorption calculation results based on the breakthrough experiment revealed that the density distribution of CO2 and C2H2 was high in both the 8 MR side pocket and the 12 MR of K–MOR. The main potential adsorption site density of C2H4 was primarily distributed in the 12 MR of K–MOR and was rare in the 8 MR side pocket. These findings are consistent with our predicted adsorption behaviours and practical separation experiments, providing a better comprehension of the actual separation mechanism and guest molecular distribution in K–MOR.
Conclusion
The K–MOR zeolite was synthesized by a feasible and template-free green method. The special channel regulation of K+ cations poses a challenge for C2H4 to access and occupy the 8 MR side pocket as easily as C2H2 and CO2, which reduces the accessible space for adsorption of C2H4, and meanwhile weakens the host–guest interaction. Breakthrough experiments reveal that K–MOR exhibits exceptional separation performance, achieving a remarkable polymer-grade C2H4 productivity of up to 2029 L kg−1 for the separation of the 1/99 C2H2/C2H4 mixture and 1742 L kg−1 for the 1/1/98 CO2/C2H2/C2H4 mixture. Deep purification of C2H4 from CO2/C2H2/C2H4 with high purity and productivity was achieved in this customized zeolite, while operating in an energy-efficient and cost-effective way. We believe that further research will lead to the discovery of more exceptional zeolites with potential applications in the adsorption and separation field, addressing some critical tasks in complex industrial processes.Author contributions
All the experiments were conducted by H. C., B. W., B. Z., J. C., J. G., and X. S. with the supervision of W. Y., J. L., and L. L. All authors discussed the results and co-wrote the manuscript.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
We acknowledge the financial support from the National Key Research and Development Program of China (2022YFB3806800, 2021YFA1500401, and 2021YFA1501202) and the National Natural Science Foundation of China (22090062, 22288101, U1967215, and 22178244).References
- Y. Jiang, Y. Hu, B. Luan, L. Wang, R. Krishna, H. Ni, X. Hu and Y. Zhang, Nat. Commun., 2023, 14, 401 CrossRef PubMed
.
- T. Ren, M. Patel and K. Blok, Energy, 2006, 31, 425–451 CrossRef CAS
- S. M. Sadrameli, Fuel, 2016, 173, 285–297 CrossRef CAS
- D. S. Sholl and R. P. Lively, Nature, 2016, 532, 435–437 CrossRef PubMed
- Y. Chai, X. Han, W. Li, S. Liu, S. Yao, C. Wang, W. Shi, I. da-Silva, P. Manuel, Y. Cheng, L. D. Daemen, A. J. Ramirez-Cuesta, C. C. Tang, L. Jiang, S. Yang, N. Guan and L. Li, Science, 2020, 368, 1002–1006 CrossRef CAS PubMed
- J. Li, Z. R. Gao, Q.-F. Lin, C. Liu, F. Gao, C. Lin, S. Zhang, H. Deng, A. Mayoral, W. Fan, S. Luo, X. Chen, H. He, M. A. Camblor, F.-J. Chen and J. Yu, Science, 2023, 379, 283–287 CrossRef CAS PubMed
- X. Cui, K. Chen, H. Xing, Q. Yang, R. Krishna, Z. Bao, H. Wu, W. Zhou, X. Dong, Y. Han, B. Li, Q. Ren, M. J. Zaworotko and B. Chen, Science, 2016, 353, 141–144 CrossRef CAS PubMed
- Y. Zhou, J. Zhang, L. Wang, X. Cui, X. Liu, S. S. Wong, H. An, N. Yan, J. Xie, C. Yu, P. Zhang, Y. Du, S. Xi, L. Zheng, X. Cao, Y. Wu, Y. Wang, C. Wang, H. Wen, L. Chen, H. Xing and J. Wang, Science, 2021, 373, 315–320 CrossRef CAS PubMed
- Z. Chen, P. Li, R. Anderson, X. Wang, X. Zhang, L. Robison, L. R. Redfern, S. Moribe, T. Islamoglu, D. A. Gomez-Gualdron, T. Yildirim, J. F. Stoddart and O. K. Farha, Science, 2020, 368, 297–303 CrossRef CAS PubMed
- L. Li, R. B. Lin, R. Krishna, H. Li, S. Xiang, H. Wu, J. Li, W. Zhou and B. Chen, Science, 2018, 362, 443–446 CrossRef CAS PubMed
- B. Zhu, J. W. Cao, S. Mukherjee, T. Pham, T. Zhang, T. Wang, X. Jiang, K. A. Forrest, M. J. Zaworotko and K. J. Chen, J. Am. Chem. Soc., 2021, 143, 1485–1492 CrossRef CAS PubMed
- Y. Li, H. Chen, C. Wang, Y. Ye, L. Li, X. Song and J. Yu, Chem. Sci., 2022, 13, 5687–5692 RSC
- X. Wang, Y. Wang, K. Lu, W. Jiang and F. Dai, Chin. Chem. Lett., 2021, 32, 1169–1172 CrossRef CAS
- C. Gu, N. Hosono, J. J. Zheng, Y. Sato, S. Kusaka, S. Sakaki and S. Kitagawa, Science, 2019, 363, 387–391 CrossRef CAS PubMed
- P. Zhang, Y. Zhong, Y. Zhang, Z. Zhu, Y. Liu, Y. Su, J. Chen, S. Chen, Z. Zeng, H. Xing, S. Deng and J. Wang, Sci. Adv., 2022, 8, eabn9231 CrossRef CAS PubMed
- H. Furukawa, K. E. Cordova, M. O'Keeffe and O. M. Yaghi, Science, 2013, 341, 1230444 CrossRef PubMed
- S. Mukherjee, N. Kumar, A. A. Bezrukov, K. Tan, T. Pham, K. A. Forrest, K. A. Oyekan, O. T. Qazvini, D. G. Madden, B. Space and M. J. Zaworotko, Angew. Chem., Int. Ed., 2021, 60, 10902–10909 CrossRef CAS PubMed
- C. He, H. Yang and L. Fu, Chin. Chem. Lett., 2023, 34, 107581 CrossRef CAS
- O. T. Qazvini, R. Babarao and S. G. Telfer, Chem. Mater., 2019, 31, 4919–4926 CrossRef CAS
- Q.-L. Qian, X.-W. Gu, J. Pei, H.-M. Wen, H. Wu, W. Zhou, B. Li and G. Qian, J. Mater. Chem. A, 2021, 9, 9248–9255 RSC
- Y. Wang, C. Hao, W. Fan, M. Fu, X. Wang, Z. Wang, L. Zhu, Y. Li, X. Lu, F. Dai, Z. Kang, R. Wang, W. Guo, S. Hu and D. Sun, Angew. Chem., Int. Ed., 2021, 60, 11350–11358 CrossRef CAS PubMed
- J. W. Cao, S. Mukherjee, T. Pham, Y. Wang, T. Wang, T. Zhang, X. Jiang, H. J. Tang, K. A. Forrest, B. Space, M. J. Zaworotko and K. J. Chen, Nat. Commun., 2021, 12, 6507 CrossRef CAS PubMed
- Y. Yang, L. Li, R. B. Lin, Y. Ye, Z. Yao, L. Yang, F. Xiang, S. Chen, Z. Zhang, S. Xiang and B. Chen, Nat. Chem., 2021, 13, 933–939 CrossRef CAS PubMed
- X.-H. Xiong, L. Zhang, W. Wang, N.-X. Zhu, L.-Z. Qin, H.-F. Huang, L.-L. Meng, Y.-Y. Xiong, M. Barboiu and D. Fenske, ACS Appl. Mater. Interfaces, 2022, 14, 32105–32111 CrossRef CAS PubMed
- J.-W. Cao, S. Mukherjee, T. Pham, Y. Wang, T. Wang, T. Zhang, X. Jiang, H.-J. Tang, K. A. Forrest, B. Space, M. J. Zaworotko and K.-J. Chen, Nat. Commun., 2021, 12, 6507 CrossRef CAS PubMed
- T. De Baerdemaeker and D. De Vos, Nat. Chem., 2013, 5, 89–90 CrossRef CAS PubMed
- A. J. W. Physick, D. J. Wales, S. H. R. Owens, J. Shang, P. A. Webley, T. J. Mays and V. P. Ting, Chem. Eng. J., 2016, 288, 161–168 CrossRef CAS
- H. J. Choi, D. Jo, J. G. Min and S. B. Hong, Angew. Chem., Int. Ed., 2021, 60, 4307–4314 CrossRef CAS PubMed
- E. B. Clatworthy, A. A. Paecklar, E. Dib, M. Debost, N. Barrier, P. Boullay, J.-P. Gilson, N. Nesterenko and S. Mintova, ACS Appl. Energy Mater., 2022, 5, 6032–6042 CrossRef CAS
- E. Perez-Botella, S. Valencia and F. Rey, Chem. Rev., 2022, 122, 17647–17695 CrossRef CAS PubMed
- S. J. Datta, C. Khumnoon, Z. H. Lee, W. K. Moon, S. Docao, T. H. Nguyen, I. C. Hwang, D. Moon, P. Oleynikov, O. Terasaki and K. B. Yoon, Science, 2015, 350, 302–306 CrossRef CAS PubMed
- T. Tian, Z. Zeng, D. Vulpe, M. E. Casco, G. Divitini, P. A. Midgley, J. Silvestre-Albero, J. C. Tan, P. Z. Moghadam and D. Fairen-Jimenez, Nat. Mater., 2018, 17, 174–179 CrossRef CAS PubMed
- M. Palomino, A. Corma, J. L. Jorda, F. Rey and S. Valencia, ChemComm, 2012, 48, 215–217 RSC
- J. Shang, G. Li, R. Singh, Q. Gu, K. M. Nairn, T. J. Bastow, N. Medhekar, C. M. Doherty, A. J. Hill, J. Z. Liu and P. A. Webley, J. Am. Chem. Soc., 2012, 134, 19246–19253 CrossRef CAS PubMed
- M. M. Lozinska, J. P. S. Mowat, P. A. Wright, S. P. Thompson, J. L. Jorda, M. Palomino, S. Valencia and F. Rey, Chem. Mater., 2014, 26, 2052–2061 CrossRef CAS
- B. Zhang, G. He, Y. Shan and H. He, Catal. Today, 2019, 327, 177–181 CrossRef CAS
- X. Wang, N. Yan, M. Xie, P. Liu, P. Bai, H. Su, B. Wang, Y. Wang, L. Li, T. Cheng, P. Guo, W. Yan and J. Yu, Chem. Sci., 2021, 12, 8803–8810 RSC
- Y. Li, H. Yi, X. Tang, F. Li and Q. Yuan, Chem. Eng. J., 2013, 229, 50–56 CrossRef CAS
- D.-i. Kwon, M. Numan, J. Kim, M. Yilmaz, S.-E. Park, H. Ihee and C. Jo, J. CO2 Util., 2022, 62, 102064 CrossRef CAS
- K. Yamashita, Z. Liu, K. Iyoki, C. T. Chen, S. Miyagi, Y. Yanaba, Y. Yamauchi, T. Okubo and T. Wakihara, ChemComm, 2021, 57, 1312–1315 RSC
- H. S. Lee, N. S. Kim, D. I. Kwon, S. K. Lee, M. Numan, T. Jung, K. Cho, M. Mazur, H. S. Cho and C. Jo, Adv. Mater., 2021, 33, e2105398 CrossRef PubMed
- D. Fu, Y. Park and M. E. Davis, Proc. Natl. Acad. Sci. U. S. A., 2022, 119, e2211544119 CrossRef CAS PubMed
- H. Chen, Y. J. Zhang, P. Y. He and C. J. Li, Energy, 2019, 179, 422–430 CrossRef CAS
-
S. Kulprathipanja, Zeolites in industrial separation and catalysis, John Wiley & Sons, Weinheim, 2010 Search PubMed
- M. Jeffroy, C. Nieto-Draghi and A. Boutin, Chem. Mater., 2017, 29, 513–523 CrossRef CAS
- B. Li, Y. Zhang, R. Krishna, K. Yao, Y. Han, Z. Wu, D. Ma, Z. Shi, T. Pham, B. Space, J. Liu, P. K. Thallapally, J. Liu, M. Chrzanowski and S. Ma, J. Am. Chem. Soc., 2014, 136, 8654–8660 CrossRef CAS PubMed
- S. M. Sadrameli, Fuel, 2015, 140, 102–115 CrossRef CAS
- P. Bodart, J. B. Nagy, G. Debras, Z. Gabelica and P. A. Jacobs, J. Phys. Chem. C, 2002, 90, 5183–5190 CrossRef
- Y. Li, M. Yu, K. Cai, M. Wang, J. Lv, R. F. Howe, S. Huang and X. Ma, Phys. Chem. Chem. Phys., 2020, 22, 11374–11381 RSC
- S. Calero, D. Dubbeldam, R. Krishna, B. Smit, T. J. Vlugt, J. F. Denayer, J. A. Martens and T. L. Maesen, J. Am. Chem. Soc., 2004, 126, 11377–11386 CrossRef CAS PubMed
- M. Kato, K. Itabashi, A. Matsumoto and K. Tsutsumi, J. Phys. Chem. B, 2003, 107, 1788–1797 CrossRef CAS
- J. L. Schlenker, J. J. Pluth and J. V. Smith, Mater. Res. Bull., 1979, 14, 751–758 CrossRef CAS
- K. Shiokawa, M. Ito and K. Itabashi, Zeolites, 1989, 9, 170–176 CrossRef CAS
- A. Vjunov, J. L. Fulton, T. Huthwelker, S. Pin, D. Mei, G. K. Schenter, N. Govind, D. M. Camaioni, J. Z. Hu and J. A. Lercher, J. Am. Chem. Soc., 2014, 136, 8296–8306 CrossRef CAS PubMed
- B. Liu, E. García-Pérez, D. Dubbeldam, B. Smit and S. Calero, J. Phys. Chem. C, 2007, 111, 10419–10426 CrossRef CAS
- E. Garcia-Perez, D. Dubbeldam, B. Liu, B. Smit and S. Calero, Angew. Chem., Int. Ed., 2007, 46, 276–278 CrossRef CAS PubMed
- C. Mellot-Draznieks, S. Buttefey, A. Boutin and A. H. Fuchs, ChemComm, 2001, 2200–2201 RSC
- H. Fang, A. Kulkarni, P. Kamakoti, R. Awati, P. I. Ravikovitch and D. S. Sholl, Chem. Mater., 2016, 28, 3887–3896 CrossRef CAS
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sc02147e |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2023 |