
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOrganophotocatalytic carbo-heterofunctionalization of unactivated olefins with pendant nucleophiles†
David M.
Fischer‡
 ,
Manuel
Freis‡
,
Willi M.
Amberg
,
Henry
Lindner
and
Erick M.
Carreira
*
,
Manuel
Freis‡
,
Willi M.
Amberg
,
Henry
Lindner
and
Erick M.
Carreira
*
Laboratory of Organic Chemistry, Department of Chemistry and Applied Biosciences, ETH Zürich, 8093 Zurich, Switzerland. E-mail: carreira@ethz.ch
First published on 14th June 2023
Abstract
We report the difunctionalization of unactivated, terminal olefins through intermolecular addition of α-bromoketones, -esters, and -nitriles followed by formation of 4- to 6-membered heterocycles with pendant nucleophiles. The reaction can be conducted with alcohols, acids, and sulfonamides as nucleophiles furnishing products bearing 1,4 functional group relationships that offer various handles for further manipulation. Salient features of the transformations are the use of 0.5 mol% of a benzothiazinoquinoxaline organophotoredox catalyst and their robustness with respect to air and moisture. Mechanistic investigations are carried out and a catalytic cycle for the reaction is proposed.
Introduction
1,4 Functional group relationships are found as motifs in a range of natural products, pharmaceuticals, and polymers.1 New methods to access these motifs constitute valuable additions to the synthetic toolkit. While 1,3 and 1,5 relationships (consonant) are attainable through transformations such as aldol, Michael, and Mannich reactions, 1,2 and 1,4 relationships (dissonant) require the use of alternative methods.2 Synthetic strategies to generate dissonant relationships include the use of 3-membered rings (epoxide or cyclopropane opening),3 Umpolung reactions,4 homoenolates,5 sulfonium [3,3]-sigmatropic rearrangements,6 and radical additions7 (Scheme 1). Photoredox catalysis has emerged as a convenient way of generating radicals and effecting their subsequent transformation to cations and anions with attendant reactivity.8 Herein, we report the organophotocatalytic carbo-heterofunctionalization of unactivated olefins under visible-light irradiation (Scheme 1). Products readily accessed through this transformation include γ-acyloxy esters, ketones, and nitriles that are akin to homoaldol addition products and the reaction can be conveniently carried out in the presence of oxygen and moisture.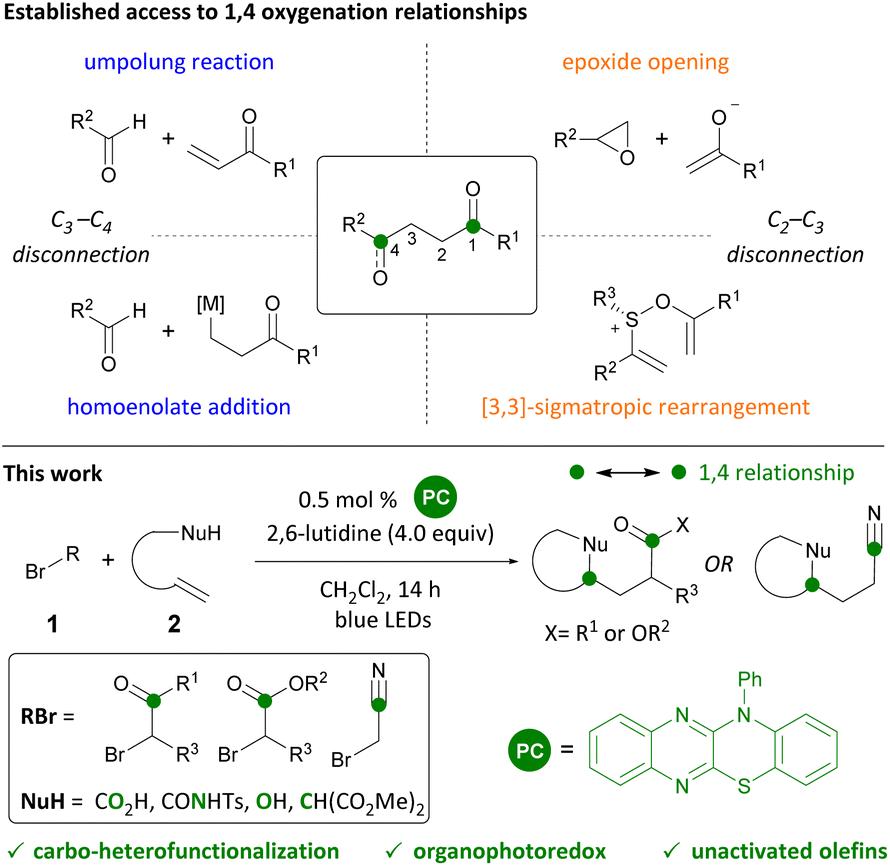 | ||
| Scheme 1 Synthesis of 1,4 oxygenation motifs. Carbo-functionalizations of unactivated olefins. | ||
Carbo-oxygenations of olefins involving activated bromides and O-nucleophiles have been reported in the presence of Pd, Cu, and Fe complexes.9 These transformations have focused on fluorinated alkyl bromides and activated alkenes such as styrenes. With the advent of photoredox catalysis, methods involving Ir photocatalysts have been investigated (Scheme 2).10
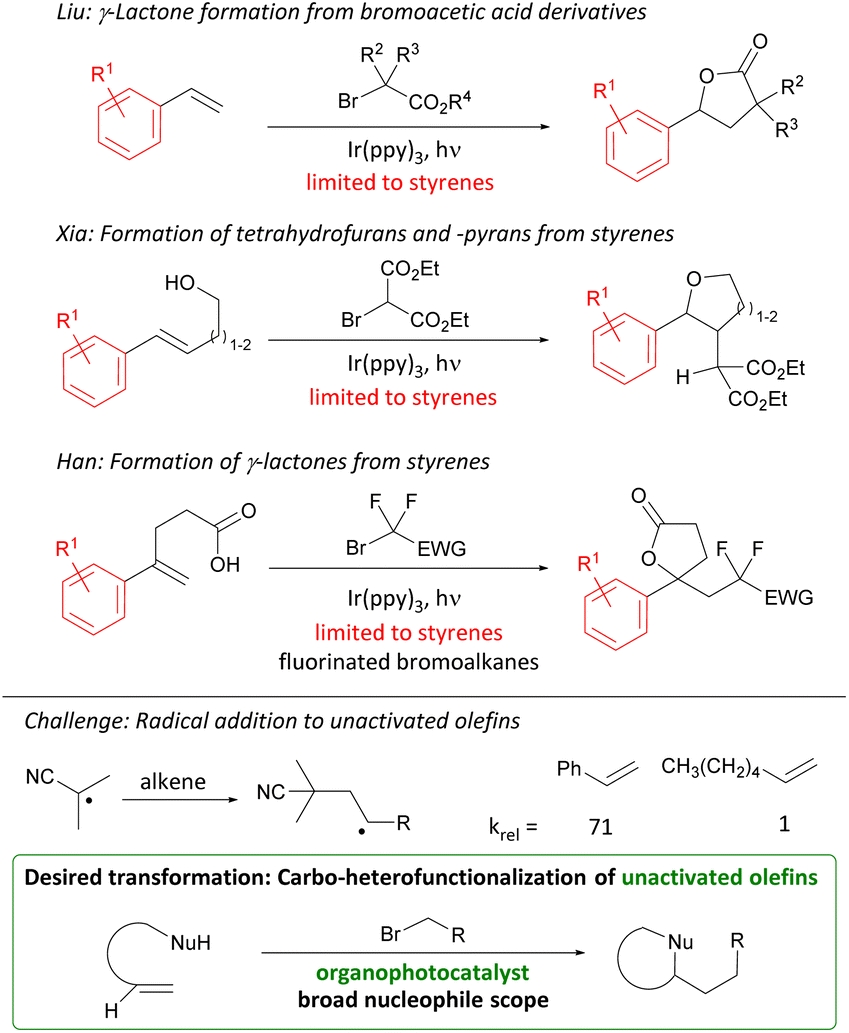 | ||
| Scheme 2 Previous photocatalytic alkene difunctionalization reactions, challenge, and targeted transformation. | ||
Early studies by Liu focused on the addition of bromoacetic acid esters over styrenes furnishing γ-lactones. Later work by Xia and Han employed styrenes and doubly activated alkyl bromides to furnish tetrahydrofurans, -pyrans, and lactones. The direct photocatalytic carbofunctionalization of unactivated olefins with alkyl bromides stands out as a desirable transformation for investigation. However, the challenge of using unactivated olefins is their attenuated reactivity as illustrated by the rate of radical addition to styrenes vs. terminal olefins (Scheme 2).11
Results and discussion
Guided by our interest in olefin functionalizations and, more recently, organophotocatalysis, we examined pent-4-enoic acid as part of a study on olefin cyclopropanation reactions.8d When pent-4-enoic acid was subjected to blue-light irradiation in the presence of benzothiazinoquinoxaline catalyst PC, 2,6-lutidine, LiBF4 and diethyl bromomalonate in sulfolane, no cyclopropane product was isolated. Instead, γ-lactone 3a was isolated along with a second related product 3a′ (Scheme 3). Intrigued by this result, we set out to investigate the carbo-heterofunctionalization of unactivated olefins under organophotocatalytic conditions to access lactone 3a and related products.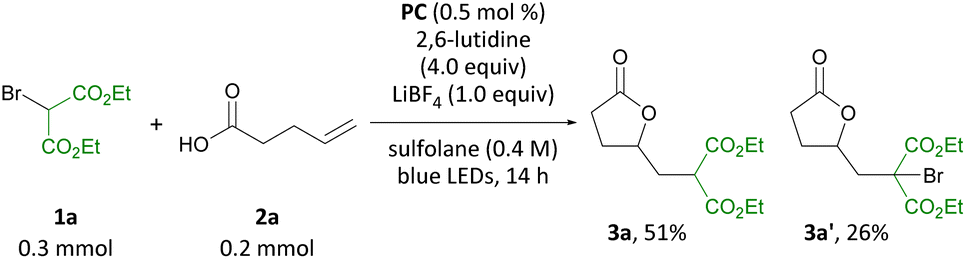 | ||
| Scheme 3 Initial result. | ||
On the basis of our earlier mechanistic work, we hypothesize side product 3a′ is formed through bromide exchange between 3a and excess bromomalonate 1a in the presence of Li+.8d Gratifyingly, in the absence of LiBF4 under otherwise identical conditions formation of 3a′ was not observed, and 3a was produced in 82% yield (Table 1, entry 2). Further optimization studies (see ESI†) revealed that a combination of 0.5 mol% PC, 4.0 equiv. 2,6-lutidine, and 1.5 equiv. diethyl bromomalonate in CH2Cl2 is optimal for the transformation reported (entry 3). Related photocatalysts PC-2 and PC-3 were also able to effect the transformation, albeit in lower yields (entries 4 and 5). In the absence of photocatalyst or light, no reaction was observed, and only 7% product were obtained in the absence of 2,6-lutidine (entries 6–8). No benefit was observed for the reaction carried out under argon atmosphere and in degassed, anhydrous solvent (entry 9). Indeed, an outstanding feature of this transformation is its air and moisture tolerance, which is in contrast to previous methods carried out under argon.10a,c An identical yield was obtained when the reaction was irradiated using a commercial Kessil light (see ESI†).
|
|
||
|---|---|---|
| Entry | Change from standard conditions | Yieldb (%) |
| a Reaction temperature rises to 40 °C. b Yields obtained by 1H NMR (1,3,5-mesitylene as internal standard). | ||
| 1 | Sulfolane instead of CH2Cl2 1.0 equiv. LiBF4 | 51 |
| 2 | Sulfolane instead of CH2Cl2 | 82 |
| 3 | — | 93 |
| 4 | PC-2 | 80 |
| 5 | PC-3 | 83 |
| 6 | In the dark, 40 °C | 0 |
| 7 | Without photocatalyst | 0 |
| 8 | Without 2,6-lutidine | 7 |
| 9 | Under Ar; degassed, dry solvent | 90 |
|
||
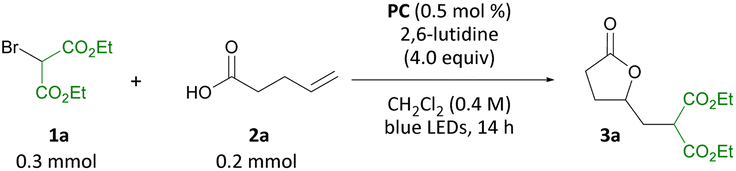
With the optimized conditions in hand, the substrate scope of the carbo-lactonization of pent-4-enoic acid was investigated (Scheme 4).12 The transformation was amenable to a range of activated alkyl bromides. Acetoacetate derived γ-lactones 3b to 3d were accessed from the corresponding bromides 1b to 1d in 70–80% yield. Substituted α-bromo-β-ketoesters were also tolerated with Et and i-Pr derivatives 1e and 1f furnishing 3e and 3f in 84 and 85% yield, respectively. Phenethyl ketone 1g provided lactone 3g in 74% yield and tert-butyl ketone 1h produced 3h in 38% yield. The 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 d.r. observed in this transformation can be attributed to epimerization of Cα-H of the β-ketoester group, which we have previously shown to exchange in the presence of 2,6-lutidine.8d 3-Bromobutanone 1i gave ketone 3i in 66% yield. α-Bromo ethyl acetate and α-bromo acetonitrile were shown to be suitable coupling partners in the reaction giving access to ester 3j and nitrile 3k in 68 and 85% yield, respectively. A variety of substituted bromoacetophenones bearing electron-withdrawing and electron-donating aryl substituents were well tolerated and provided γ-lactones 3l–3o in 65–85% yield. Thus, it was shown that in this system a single activating group suffices for the reaction to proceed. Finally, it was demonstrated that the reaction may also be carried out on 2.00 mmol scale in comparable yield (3a formed in 77% yield). Notably, the use of activated alkyl bromides in this transformation constitutes a valuable example of a bifunctional reagent in synthesis.13
:1 d.r. observed in this transformation can be attributed to epimerization of Cα-H of the β-ketoester group, which we have previously shown to exchange in the presence of 2,6-lutidine.8d 3-Bromobutanone 1i gave ketone 3i in 66% yield. α-Bromo ethyl acetate and α-bromo acetonitrile were shown to be suitable coupling partners in the reaction giving access to ester 3j and nitrile 3k in 68 and 85% yield, respectively. A variety of substituted bromoacetophenones bearing electron-withdrawing and electron-donating aryl substituents were well tolerated and provided γ-lactones 3l–3o in 65–85% yield. Thus, it was shown that in this system a single activating group suffices for the reaction to proceed. Finally, it was demonstrated that the reaction may also be carried out on 2.00 mmol scale in comparable yield (3a formed in 77% yield). Notably, the use of activated alkyl bromides in this transformation constitutes a valuable example of a bifunctional reagent in synthesis.13
 | ||
| Scheme 4 Substrate scope for the olefin difunctionalization of pent-4-enoic acid with activated alkyl bromides. aReaction carried out on 2.00 mmol scale. bd.r. = 1:1 by 1H NMR analysis of the unpurified reaction mixture. c1.00 mmol of bromide 1i was used. | ||
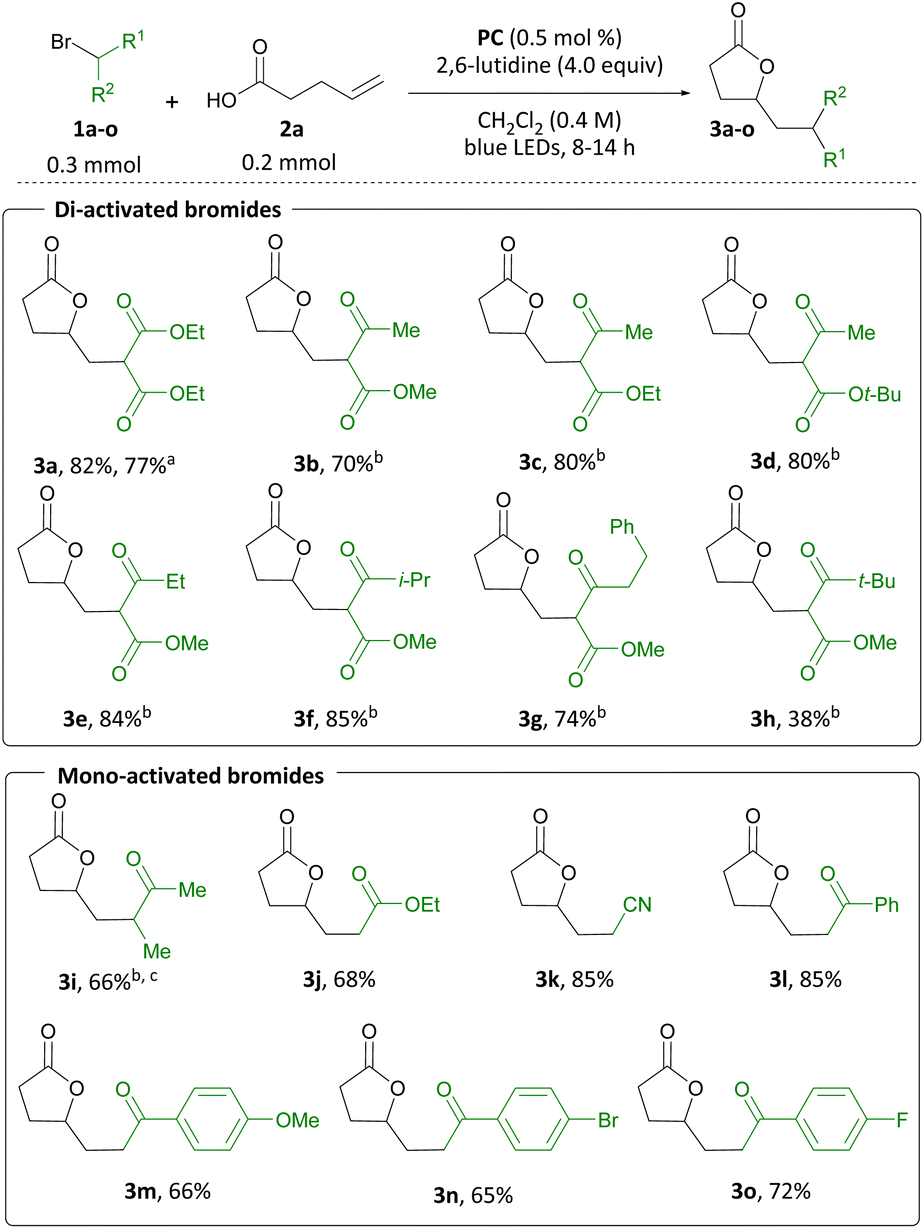
Having established the feasibility and generality of the transformation with pent-4-enoic acid, we sought to expand the substrate scope to include more highly substituted carboxylic acids (Scheme 5). A range of α-substituted carboxylic acids were suitable for use in this transformation, furnishing N-Boc amine 4b, spirocyclic ether 4c and spiro[3.4]lactone 4d in 94, 80, and 70% yield, respectively. N-Boc-protected amino acid 2e produced 4e in 93% yield and 1.2:1 d.r. Finally, δ-lactone 4f was accessed from the corresponding carboxylic acid in 85% yield.
 | ||
| Scheme 5 Substrate scope for the olefin difunctionalization reaction with diethyl bromomalonate. | ||
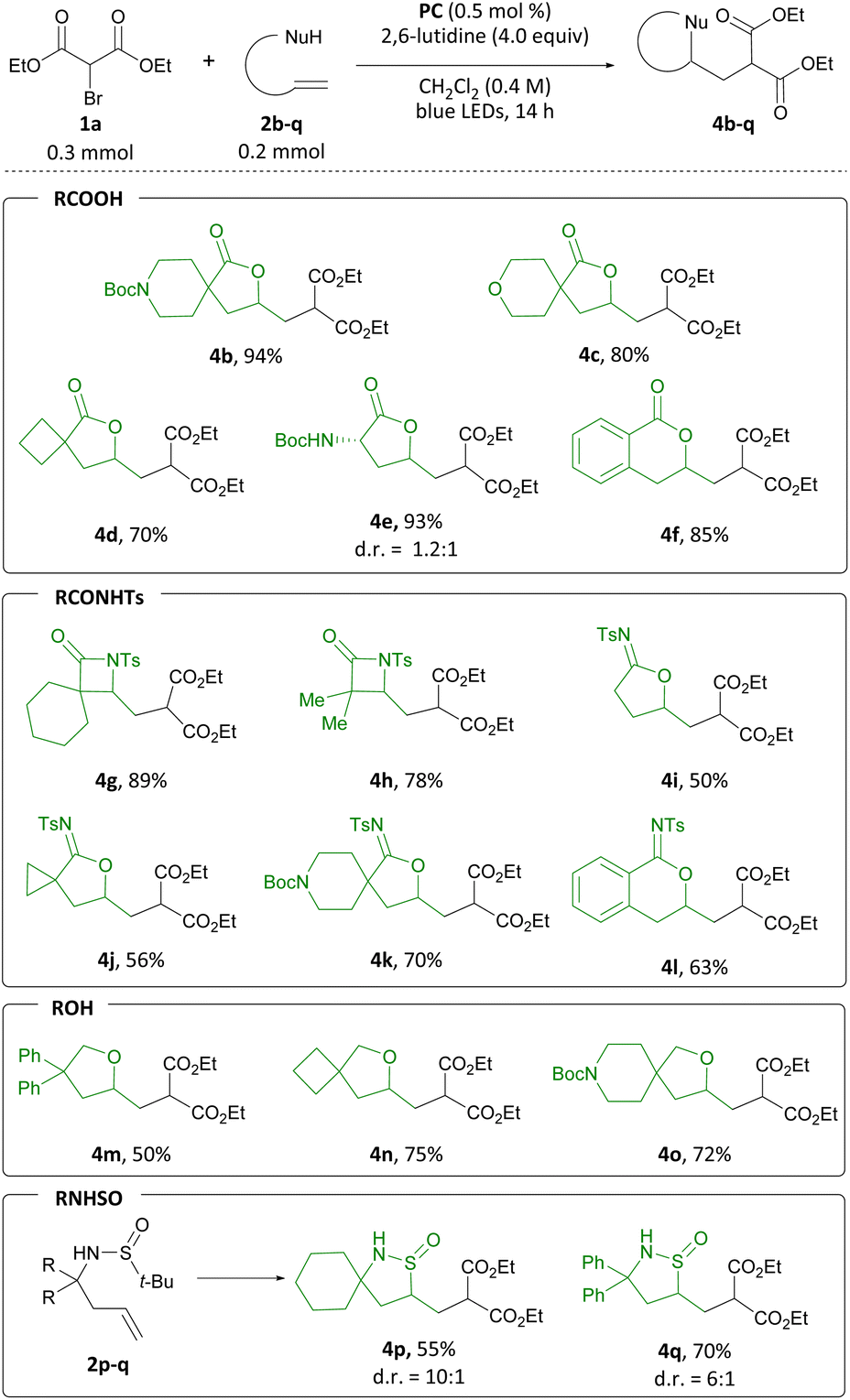
To broaden the range of heterocycles accessible through this transformation, we examined additional pendant nucleophiles. Guided by the similar pKa of carboxylic acids and N-acyl sulfonamides, the latter were considered as nucleophiles in the difunctionalization reaction.14 We examined β,γ-unsaturated N-acylsulfonamides first, and when 2g was subjected to the reaction conditions, β-lactam 4g was isolated in 89% yield. Analogously, β-lactam 4h was produced from 2h in 78% yield. When γ,δ- and δ,ε-unsaturated alkenyl N-acyl sulfonamides were employed, N-sulfonyl imidates 4i to 4l were formed in 50 to 70% yield. Aiming to probe whether this transformation was amenable to less acidic nucleophiles, we considered alcohols next.15 To this end, tetrahydrofuran 4m was produced in 50% yield from alcohol 2m. Spirocyclic ethers 4n and 4o were accessed from 2n and 2o in 75% and 72% yield, respectively. Finally, inspired by Zhu's work on olefin sulfinylation employing CF3 radicals,16 we investigated sulfinamides as potential substrates. Gratifyingly, we could access two heterocyclic isothiazolidine S-oxides, 4p and 4q, in 55 and 70% yield, respectively. Derivatives of this class of heterocycles have found application in drug discovery research.17
Interested in whether this method could be extended to carbon nucleophiles, we subjected dimethyl allyl malonate to a combination of 0.5 mol% PC, 2.0 equiv. of bromoacetonitrile, 1.0 equiv. LiCl and 4.00 equiv. of 2,6-lutidine in CH2Cl2 under blue-light irradiation. Gratifyingly, formation of cyclopropane 4r was observed in 65% yield (Scheme 6). LiCl was added to effect soft enolization of the malonate group.
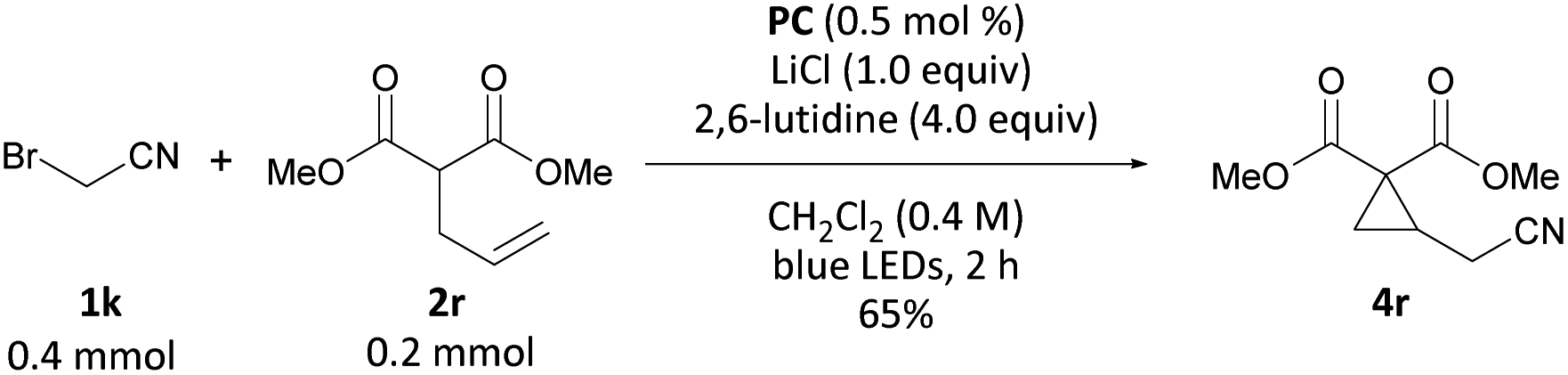 | ||
| Scheme 6 Reaction of dimethyl allyl malonate with bromoacetonitrile. | ||
We set out to examine the mechanism of the transformation reported herein next (Scheme 7). To illuminate the photocatalytic part of the transformation, Stern–Volmer quenching studies were conducted, which revealed strong quenching of the photocatalyst excited state by bromides 1l and 1a (Scheme 7A and ESI†). Interestingly, weak Stern–Volmer quenching was also observed for carboxylic acid 2a, however no quenching was observed for alcohol 2n (see ESI†). Thus, we suggest that quenching of the photocatalyst by the carboxylic acid group in 2a is inconsequential for the mechanism of this transformation.
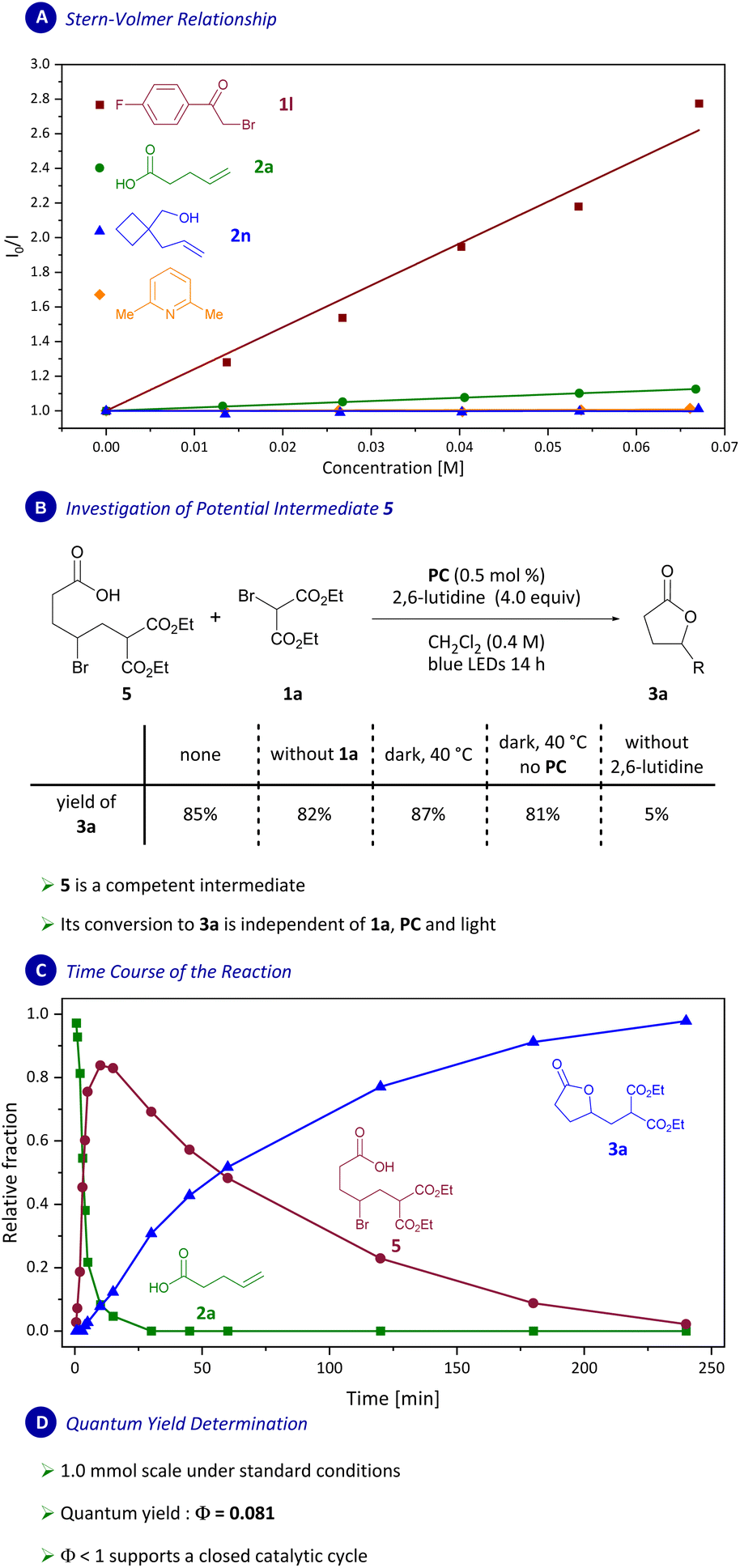 | ||
| Scheme 7 Mechanistic investigations: (A) Stern–Volmer relationship study between PC and reagents. (B) Reaction of putative intermediate 5 under reaction conditions and in the absence of light, photocatalyst, base or 1a. Yields were determined by analysis of 1H NMR spectra of the unpurified reaction mixture with 1,3,5-mesitylene as internal standard. R = CH2CH(CO2Et)2. (C) Time course of the reaction (ratio determined by 1H NMR). (D) Quantum yield determination. | ||
In difunctionalization reactions of styrenes catalyzed by Ir(ppy)3 as photocatalyst, an intermediate benzylic carbocation has been speculated to directly undergo intramolecular trapping.10a,b Based on our previous work,8d we considered the feasibility of an additional pathway proceeding via an n-alkyl bromide intermediate. To distinguish between these two options, we designed a set of mechanistic experiments. Consequently, bromide 5 was independently synthesized to assess if it is an intermediate in the transformation reported herein (Scheme 7B and ESI†). When 5 was subjected to the reaction conditions (0.5 mol% PC, 4.0 equiv. 2,6-lutidine in CH2Cl2 under blue-light irradiation) product 3a was formed in 85% yield as determined by analysis of 1H NMR spectra (Scheme 7B).
We further demonstrated that the conversion of intermediate 5 to 3a is independent of irradiation, photocatalyst or bromomalonate 1a but relies on the presence of 2,6-lutidine. 13C NMR experiments were carried out and are consistent with 2a being predominantly present in its deprotonated form (RCOO−) under the reaction conditions (4 equiv. of 2,6-lutidine). Thus, we show that 5 could be a relevant intermediate in a catalytic cycle.
To determine if 5 is, in fact, a kinetically competent intermediate, time-series experiments were carried out next (Scheme 7C). In the experiments we observed rapid accumulation of a species early on (t < 30 min) which was then consumed during the remainder of the reaction time with concomitant formation of product 3a. The initially formed species was isolated and identified as 5, providing support for its role as an intermediate in this transformation.
Certain photocatalytic radical additions to olefins have been postulated to proceed via radical-chain mechanisms.18 To distinguish between such a mechanism and one that involves a closed catalytic cycle, the quantum yield for the transformation was measured and determined as Φ = 0.081 (Scheme 7D and ESI†). A low quantum yield of 8.1% supports a closed catalytic cycle and leads us to disfavor mechanisms involving a radical-chain process.18,19
Together, all of these experiments have guided us to propose a catalytic cycle (Scheme 8). At the outset, PC absorbs a photon in the blue-light range (λmax,abs = 420 nm, Step A) leading to the photocatalyst in the excited state (PC*). Reduction of bromomalonate 1a by PC* forms a stabilized α-malonyl radical 6 (Step B).8c,d,15 Radical addition to the unactivated olefin prefentially takes place to furnish the more substituted, and thus more stable, secondary carbon radical 7 (Step C). Subsequent oxidation of 7 (ref. 20) by oxidized photocatalyst PC˙+ leads to formation of secondary carbocation 8 which is trapped by Br− (Steps D and E).12,21 SN2 displacement of the alkyl bromide in 5 by the pendant nucleophile leads to formation of product 3a (Step F). It is worth noting that the accumulation of intermediate 5 identifies SN2 displacement as the rate-determining step in the carbo-functionalization reaction.
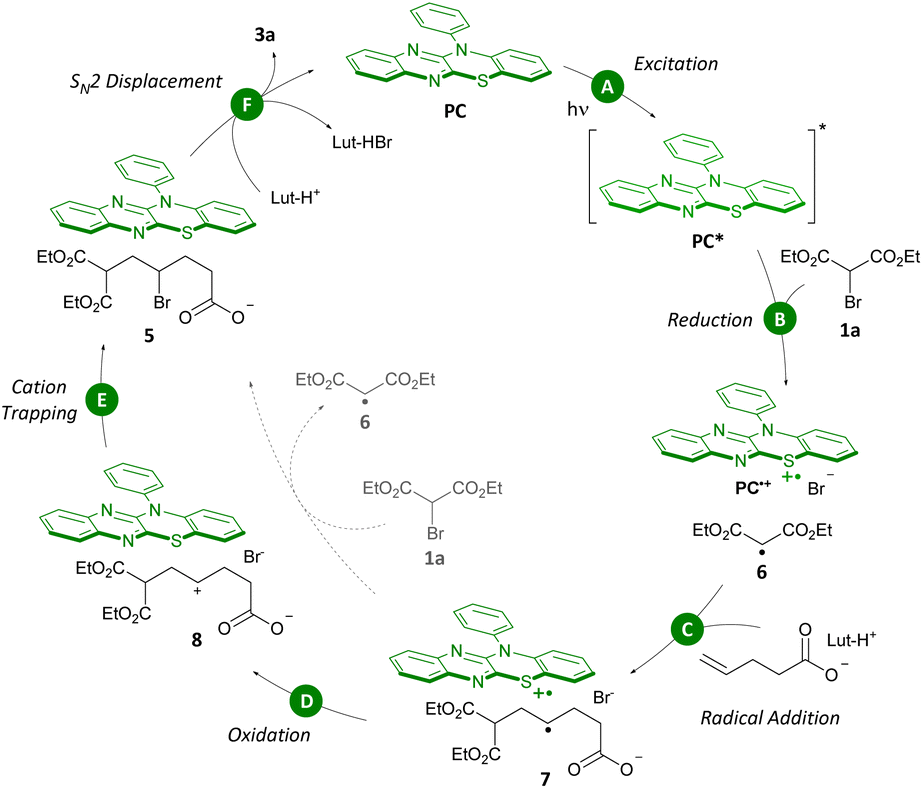 | ||
| Scheme 8 Proposed catalytic cycle. Lut = 2,6-lutidine. | ||
Conclusion
We have developed a carbo-heterofunctionalization reaction of unactivated olefins under organophotocatalytic conditions. In addition to the formation of a new C–C bond, the transformation gives access to a variety of saturated heterocycles including tetrahydrofurans, lactones, and cyclic imidates, all displaying 1,4 functional group relationships. The method was then extended to include cyclization via nitrogen and sulfur groups to furnish products that feature β-lactams and isothiazolidine S-oxides. Mechanistic investigations were carried out, which identified key intermediates and the rate-determining step. In employing a benzothiazinoquinoxaline we highlight the utility of these photocatalysts. Investigations into further transformations unlocked by this class of catalysts may expand the synthetic toolkit and provide access to novel and exciting synthetic building blocks.Data availability
Data supporting this article have been uploaded as ESI.†Author contributions
D. M. F. and E. M. C. conceived the project. D. M. F., M. F., W. M. A., and H. L. conducted the investigations under supervision of E. M. C., D. M. F. and M. F. contributed equally. D. M. F., W. M. A., H. L., and E. M. C. wrote the manuscript. All authors contributed to the revision of the manuscript and have approved the final version.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was funded by the European Research Council (OLECAT, Grant ID 833540). We are grateful to Dr M.-O. Ebert, R. Frankenstein, S. Burkhardt and R. Arnold for support with NMR experiments, Dr E. Meister for support with fluorescence spectroscopy, C. A. Bärtschi, C. Marro, and P. Trüssel for the maintenance and construction of the photoreactor (all ETH Zurich). W. M. Amberg is grateful for partial support with funding from the SSCI (Scholarship Fund of the Swiss Chemical Industry). We thank Yuxuan Li for early investigations into preparation of 4r.Notes and references
- M. Lemmerer, M. Schupp, D. Kaiser and N. Maulide, Nat. Synth., 2022, 1, 923–935 CrossRef.
- D. A. Evans and G. C. Andrews, Acc. Chem. Res., 1974, 7, 147–155 CrossRef CAS.
- (a) G. H. Posner, J. P. Maxwell and M. Kahraman, J. Org. Chem., 2003, 68, 3049–3054 CrossRef CAS PubMed; (b) W. E. Grigsby, J. Hind, J. Chanley and F. Westheimer, J. Am. Chem. Soc., 1942, 64, 2606–2610 CrossRef CAS; (c) S. K. Taylor, Tetrahedron, 2000, 56, 1149–1163 CrossRef CAS.
- (a) A. Biju, S. Yetra and A. Patra, Synthesis, 2015, 47, 1357–1378 CrossRef; (b) M. M. Heravi, V. Zadsirjan, K. Kafshdarzadeh and Z. Amiri, Asian J. Org. Chem., 2020, 9, 1999–2034 CrossRef CAS; (c) H. Stetter, Angew. Chem., Int. Ed., 1976, 15, 639–647 CrossRef; (d) D. Enders, K. Breuer, J. Runsink and J. H. Teles, Helv. Chim. Acta, 1996, 79, 1899–1902 CrossRef CAS; (e) C. Burstein and F. Glorius, Angew. Chem., Int. Ed., 2004, 43, 6205–6208 CrossRef CAS PubMed; (f) Q. Liu, S. Perreault and T. Rovis, J. Am. Chem. Soc., 2008, 130, 14066–14067 CrossRef CAS PubMed; (g) S. Barik, S. Shee, S. Das, R. G. Gonnade, G. Jindal, S. Mukherjee and A. T. Biju, Angew. Chem., Int. Ed., 2021, 60, 12264–12268 CrossRef CAS PubMed; (h) M. Rezazadeh Khalkhali, M. M. D. Wilde and M. Gravel, Org. Lett., 2021, 23, 155–159 CrossRef CAS PubMed.
- L. R. Mills and S. A. L. Rousseaux, Eur. J. Org. Chem., 2019, 2019, 8–26 CrossRef CAS.
- (a) D. Kaldre, I. Klose and N. Maulide, Science, 2018, 361, 664–667 CrossRef CAS PubMed; (b) W. Zhou and A. Voituriez, Org. Lett., 2021, 23, 247–252 CrossRef CAS PubMed.
- (a) B. Giese and S. Lachhein, Angew. Chem., Int. Ed., 1981, 20, 967 CrossRef; (b) T. Morack, C. Mück-Lichtenfeld and R. Gilmour, Angew. Chem., Int. Ed., 2019, 58, 1208–1212 CrossRef CAS PubMed; (c) Y. Y. Cheng, J. X. Yu, T. Lei, H. Y. Hou, B. Chen, C. H. Tung and L. Z. Wu, Angew. Chem., Int. Ed., 2021, 60, 26822–26828 CrossRef CAS PubMed; (d) C. Che, Z. Qian, M. Wu, Y. Zhao and G. Zhu, J. Org. Chem., 2018, 83, 5665–5673 CrossRef CAS PubMed.
- (a) L. Furst, B. S. Matsuura, J. M. R. Narayanam, J. W. Tucker and C. R. J. Stephenson, Org. Lett., 2010, 12, 3104–3107 CrossRef CAS PubMed; (b) J. D. Nguyen, J. W. Tucker, M. D. Konieczynska and C. R. J. Stephenson, J. Am. Chem. Soc., 2011, 133, 4160–4163 CrossRef CAS PubMed; (c) T. Kodo, K. Nagao and H. Ohmiya, Nat. Commun., 2022, 13, 2684 CrossRef CAS PubMed; (d) D. M. Fischer, H. Lindner, W. M. Amberg and E. M. Carreira, J. Am. Chem. Soc., 2023, 145, 774–780 CrossRef CAS PubMed; (e) R. Wang, C. Zhou, X. Huang, J.-Y. Wu and X. Zhang, ACS Sustainable Chem. Eng., 2022, 10, 4650–4659 CrossRef CAS; (f) M. A. Zeller, M. Riener and D. A. Nicewicz, Org. Lett., 2014, 16, 4810–4813 CrossRef CAS PubMed.
- (a) Y. Da, S. Han, X. Du, S. Liu, L. Liu and J. Li, Org. Lett., 2018, 20, 5149–5152 CrossRef CAS PubMed; (b) T. L. Buchanan, S. N. Gockel, A. M. Veatch, Y.-N. Wang and K. L. Hull, Org. Lett., 2021, 23, 4538–4542 CrossRef CAS PubMed; (c) F. Yuan, S. Zhou, Y. Yang, M. Guo, X. Tang and G. Wang, Org. Chem. Front., 2018, 5, 3306–3309 RSC; (d) F. Xiao, F. Wu, X. Yang, Y. Shen and X. Shi, J. Fluorine Chem., 2005, 126, 319–323 CrossRef CAS; (e) M. Zhang, W. Li, Y. Duan, P. Xu, S. Zhang and C. Zhu, Org. Lett., 2016, 18, 3266–3269 CrossRef CAS PubMed.
- (a) R. Lin, H. Sun, C. Yang, W. Shen and W. Xia, Chem. Commun., 2015, 51, 399–401 RSC ; a singular example of a monoactivated bromide reacting with a styrene was examined in this work; (b) W. Sha, W. Zhang, S. Ni, H. Mei, J. Han and Y. Pan, J. Org. Chem., 2017, 82, 9824–9831 CrossRef CAS PubMed; (c) X.-J. Wei, D.-T. Yang, L. Wang, T. Song, L.-Z. Wu and Q. Liu, Org. Lett., 2013, 15, 6054–6057 CrossRef CAS PubMed.
- (a) K. Münger and H. Fischer, Int. J. Chem. Kinet., 1985, 17, 809–829 CrossRef; (b) K. Hėberger and H. Fischer, Int. J. Chem. Kinet., 1993, 25, 249–263 CrossRef.
- For an overview of unsuccessful substrates see ESI.13.† H.-M. Huang, P. Bellotti, J. Ma, T. Dalton and F. Glorius, Nat. Rev. Chem., 2021, 5, 301–321 CrossRef CAS PubMed.
- H. M. Huang, P. Bellotti, J. Ma, T. Dalton and F. Glorius, Nat. Rev. Chem., 2021, 5, 301–321 CrossRef CAS PubMed.
- D. M. Fischer, M. Balkenhohl and E. M. Carreira, JACS Au, 2022, 2, 1071–1077 CrossRef CAS PubMed.
- C.-J. Wallentin, J. D. Nguyen, P. Finkbeiner and C. R. J. Stephenson, J. Am. Chem. Soc., 2012, 134, 8875–8884 CrossRef CAS PubMed . Stephenson observed formation of a tetrahydrofuran as a byproduct (10%, singular example) during a study on the addition of activated bromides over olefins under metal photoredox catalysis..
- Y. Chen, X. Wu, S. Yang and C. Zhu, Angew. Chem., Int. Ed., 2022, 61, e202201027 CAS.
- J.-H. Lee, C.-Y. Hong, T.-S. Park, J.-H. Kim, S.-H. Choi, S.-K. Yoon, H.-H. Chung, S.-W. Jeong, K.-Y. Hwang and D.-K. Shin, US Pat., US6620831B2, 2003 Search PubMed.
- (a) J. W. Tucker and C. R. J. Stephenson, J. Org. Chem., 2012, 77, 1617–1622 CrossRef CAS PubMed; (b) M. D. Kärkäs, B. S. Matsuura and C. R. Stephenson, Science, 2015, 349, 1285–1286 CrossRef PubMed.
- (a) J. V. Burykina, A. D. Kobelev, N. S. Shlapakov, A. Y. Kostyukovich, A. N. Fakhrutdinov, B. König and V. P. Ananikov, Angew. Chem., Int. Ed., 2022, 61, e2021168 CrossRef PubMed; (b) M. A. Cismesia and T. P. Yoon, Chem. Sci., 2015, 6, 5426–5434 RSC.
- D. Wayner and A. Houmam, Acta Chem. Scand., 1998, 52, 377–384 CrossRef CAS.
- (a) M. Nakagawa, Y. Matsuki, K. Nagao and H. Ohmiya, J. Am. Chem. Soc., 2022, 144, 7953–7959 CrossRef CAS PubMed . Stabilization of carbocation intermediate 8 might be aided by formation of a thiazinium adduct as previously suggested by Ohmiya for HAT functionalization in the presence of a benzophenothiazine catalyst; (b) Although the low quantum yield supports a closed catalytic cycle, a radical-chain process may not be categorically ruled out as has been pointed out by Yoon (see ref. 19b)..
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sc02250a |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2023 |