
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMolecular in situ monitoring of the pH-triggered response in adaptive polymers by two-dimensional Raman micro-correlation-spectroscopy†
Julian
Hniopek
 abc,
Josefine
Meurer
de,
Stefan
Zechel
de,
Michael
Schmitt
bc,
Martin D.
Hager
*de and
Jürgen
Popp
*abce
abc,
Josefine
Meurer
de,
Stefan
Zechel
de,
Michael
Schmitt
bc,
Martin D.
Hager
*de and
Jürgen
Popp
*abce
aDepartment Spectroscopy & Imaging, Leibniz Institute of Photonic Technology, Albert-Einstein-Str. 9, 0775 Jena, Germany. E-mail: juergen.popp@leibniz-ipht.de
bInstitute of Physical Chemistry (IPC), Friedrich Schiller University Jena, Helmholtzweg 4, 07743 Jena, Germany
cAbbe Center of Photonics, Friedrich Schiller University Jena, Albert-Einstein-Str. 6, 07745 Jena, Germany
dLaboratory of Organic and Macromolecular Chemistry (IOMC), Friedrich Schiller University Jena, Humboldtstr. 10, 07743 Jena, Germany. E-mail: martin.hager@uni-jena.de
eJena Center of Soft Matter (JCSM), Friedrich Schiller University Jena, Philosophenweg 7, 07743 Jena, Germany
First published on 6th June 2023
Abstract
Stimuli-responsive polymers can switch specific physical properties in response to a change of the environmental conditions. This behavior offers unique advantages in applications where adaptive materials are needed. To tune the properties of stimuli-responsive polymers, a detailed understanding of the relationship between the applied stimulus and changes in molecular structure as well as the relationship between the latter and macroscopic properties is required, which until now has required laborious methods. Here, we present a straightforward way to investigate the progressing trigger, the change of the chemical composition of the polymer and the macroscopic properties simultaneously. Thereby, the response behavior of the reversible polymer is studied in situ with molecular sensitivity and spatial as well as temporal resolution utilizing Raman micro-spectroscopy. Combined with two-dimensional correlation analysis (2DCOS), this method reveals the stimuli-response on a molecular level and determines the sequence of changes and the diffusion rate inside the polymer. Due to the label-free and non-invasive approach, it is furthermore possible to combine this method with the investigation of macroscopic properties revealing the response of the polymer to the external stimulus on both the molecular and the macroscopic level.
Introduction
Switchable and adaptive polymeric materials, which feature the unique ability to change their physicochemical properties upon application of an external stimulus, have gained much interest over the last few years.1,2 Within this context, different materials have been designed and synthesized, and their properties have been studied in detail.3 The first applications of these switchable materials have already been demonstrated, ranging from self-healing to shape-memory polymers.4,5 One major field of application is the design of switchable surfaces,6 which are able to change their wetting/dewetting behavior by the application of a specific stimulus,7 such as temperature,8 light irradiation,9–11 or pH-value change.12Ideally, these substrates can be designed by polymeric materials featuring a reversible structural unit. The most commonly applied reversible groups are azobenzenes and spiropyrans,10,13,14 which can simply be switched by light irradiation. Furthermore, ferrocene or disulfide containing polymers can be applied for switching the wettability by reversible redox reaction.15,16
To understand and optimize the molecular properties required for the desired switching behavior, it would be desirable to directly monitor the molecular structure of the responsive materials during the application of the external stimulus, which leads to a change of the properties of the material.
However, currently in most cases, the change of molecular structure can only be correlated indirectly via studying the change of certain properties, e.g., wettability determined by contact angle measurements.17 This technique allows the analysis of the interaction of a substrate with water (or oil) over time and during stimulus application. However, a correlation of the molecular change and the interaction of the substrate with the water (oil) droplet is only possible with laborious methods, such as introducing fluorescent particles into the structure.18
A less complex method to monitor the structural changes alongside the macroscopic changes in surface properties would therefore be beneficial in order to optimize such intelligent substrates further and to understand structure–property relationships in more detail. We propose Raman micro-spectroscopy as a powerful tool to monitor the chemical structure of responsive polymers in situ during stimulus application. It is important to note that this technique enables us to measure simultaneously the stimulus progression within the material and the response of the chemical structure of the polymer, and to correlate this with the changed properties (cf.Fig. 1b). Raman spectroscopy is an established tool for the analysis of smart polymers and has been applied, for example, to monitor the structural changes in electro-responsive,19 shape-memory,20 and self-healing polymers,21,22 as well as to monitor chemical processes like diffusion23 or dynamic bond opening in polymers in general.24 Here, the opportunity to combine Raman microspectroscopy with two-dimensional correlation analysis (2DCOS), introduced by Isao Noda,25 makes it possible to mathematically retrieve relationships between stimulus progression and molecular changes inside the polymer. This enables the derivation of structure–property relationships in a straightforward and unbiased manner. Furthermore, Raman spectroscopy offers the distinct advantage that it directly probes the molecular structure of the sample via its characteristic molecular vibrations and is thus label-free.26 As a consequence, there is no need for expensive and laborious modifications of the polymers to enable the observation of structural changes, which moreover might influence the properties of the polymer. Furthermore, water does not show strong Raman signals, due to its highly polar nature and resulting comparatively low scattering cross-section.27,28 Therefore, Raman spectroscopy can be combined with methods like contact angle measurements or water-based triggers (e.g., changes of the pH-value) in a straightforward manner without significant contribution of the water to the spectrum.29,30
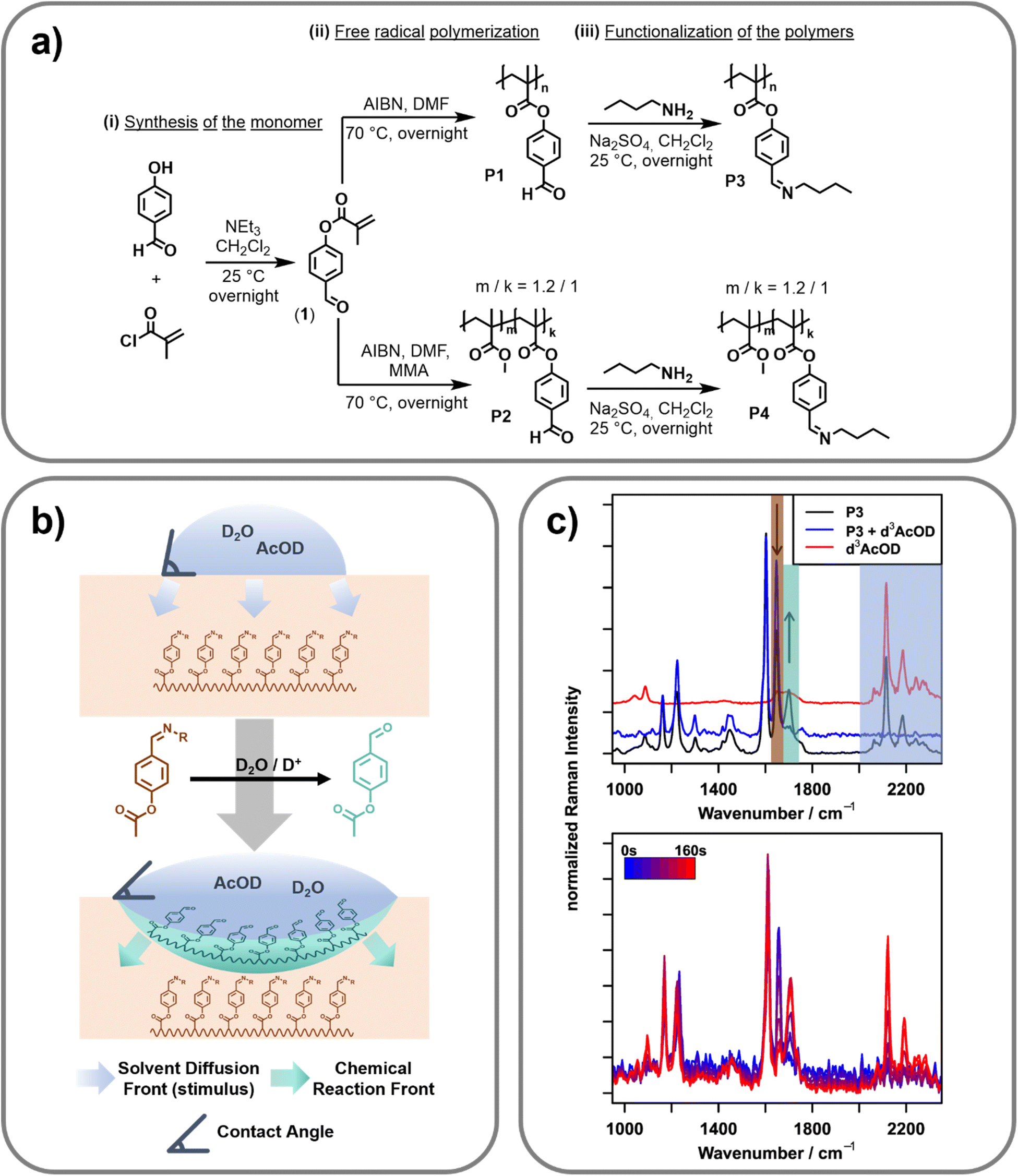 | ||
| Fig. 1 (a) Reaction scheme for the synthesis of (i) 4-formyl phenyl methacrylate (1), (ii) the homopolymer P1 and the copolymer P2via free radical polymerization and (iii) the imine bond containing polymers P3 (homopolymer) and P4 (copolymer) via the functionalization with n-butyl amine. (b) Schematic overview of the design principle of the surface active polymer. Upon application of a stimulus (D2O/AcOD, blue), the imine side-groups of the polymer (P3/P4, dark-orange) get hydrolyzed to aldehydes (mint green). After the cleavage, solvent (activating mixture) diffuses further into the polymer, leading to a delay between the chemical reaction front (at the interface) and the solvent diffusion front. (c) FT-Raman spectra (top, λexc = 1064 nm) of P3 (bottom, black), P3 with d3-AcOD (blue, middle) and pure d3-AcOD (red, top). Shaded regions indicate the relevant band areas to monitor the pristine polymer (orange), the hydrolyzed polymer (mint green) and d3-AcOD (blue). Bottom: Raman spectra (λexc = 514 nm) of a solid sample of P4 recorded 12.5 μm below the surface, 0 (blue) to 160 s (red) after placing a drop of D2O/d3-AcOD onto the surface. | ||
Results and discussion
Synthesis of the polymers
In order to synthesize a pH-triggerable polymer, imine bonds were integrated as side chains into the polymeric structure. These bonds can be activated by a slight decrease of the pH-value and, thus, were already utilized for the design of pH-responsive functional materials.31 Herein, the imine bonds should be integrated into the polymer side chain. Therefore, 4-formylphenyl-methacrylate (1) was synthesized by adopting literature procedures.32,33 Afterwards, this monomer was utilized in free radical polymerization resulting in the homopolymer (P1) as well as the copolymer (P2) utilizing methyl methacrylate (MAA) as the second monomer (feed ratio of the two monomers: MMA![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 = 1:1). 2,2-Azobis(2-methylpropionitrile) (AIBN) was utilized as initiator. Molar masses (Mn) of 26.9 and 35.6 kDa with dispersities of 5.95 and 3.78 were reached respectively. The composition of the copolymer was determined by 1H NMR spectroscopic measurements. The ratio of MMA to 4-formylphenyl-methacrylate was found to be 1.2 to 1. Since the two synthesized polymers feature the 4-formylphenyl functionality in their side chain, the imine bonds were simply introduced via the reaction of the aldehyde groups with n-butyl amine. This functionalization step yielded the imine containing polymers P3 (homopolymer derived from P1) and P4 (copolymer derived from P2). These polymers were further utilized for all investigations. A reaction scheme of the synthesis is presented in Fig. 1a. Additional information regarding the synthetic procedure and the characterization is provided in the ESI.†
:1 = 1:1). 2,2-Azobis(2-methylpropionitrile) (AIBN) was utilized as initiator. Molar masses (Mn) of 26.9 and 35.6 kDa with dispersities of 5.95 and 3.78 were reached respectively. The composition of the copolymer was determined by 1H NMR spectroscopic measurements. The ratio of MMA to 4-formylphenyl-methacrylate was found to be 1.2 to 1. Since the two synthesized polymers feature the 4-formylphenyl functionality in their side chain, the imine bonds were simply introduced via the reaction of the aldehyde groups with n-butyl amine. This functionalization step yielded the imine containing polymers P3 (homopolymer derived from P1) and P4 (copolymer derived from P2). These polymers were further utilized for all investigations. A reaction scheme of the synthesis is presented in Fig. 1a. Additional information regarding the synthetic procedure and the characterization is provided in the ESI.†
Raman reference measurements
To investigate the spectroscopic changes in the surface-active polymer upon activation by pH-change reference experiments, Fourier transform (FT) Raman spectroscopy at a 1064 nm excitation wavelength was carried out (cf.Fig. 1c, top). An overview of the bands relevant to this work as well as their assignment can be found in Table 1.| Wavenumber position/cm−1 | Assignment |
|---|---|
| 1600 |
ν(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C), aromatic ring; polymer C), aromatic ring; polymer |
| 1656 |
ν(CN), imine; native polymer |
| 1702 |
ν(CO), imine; hydrolyzed polymer |
| 2110, 2180, 2250, 2280 | ν(C–D), d3-AcOD |
Due to the FT based detection approach and the use of an excitation wavelength relatively far in the near infrared region, FT-Raman is ideally suited to acquire reference spectra free of fluorescence with very high spectral precision. It can therefore serve as a gold standard tool to identify artifacts introduced by fluorescence or other optical effects in the following confocal Raman microscopy measurements. To acquire the spectra, powdered polymer P3 was placed in an aluminium pot and a drop of heavy water/deuterated acetic acid (D2O/d3-AcOD, 9:1 v/v) was added. Due to the higher atomic mass of deuterium compared to protium, deuterated species lead to a characteristic shift of the molecular vibrations involving hydrogen atoms (C–D/O–D) to lower wavenumbers. This shift is especially pronounced for the respective stretching vibrations, which are shifted from over 3000 cm−1 to the “silent” region of the spectrum (2200–2500 cm−1). In this spectral region, only very few moieties show vibrational bands, which enables straightforward separation of the signals arising from the activating mixture (D2O/d3-AcOD) and the signals of the polymer.
Besides the obvious appearance of the d3-AcOD related signals in this silent region, adding the activating mixture also changes the spectral region under 1750 cm−1 (decreasing the intensity at 1656 cm−1 and increasing the intensity at 1702 cm−1). These bands do not correspond with bands of the d3-AcOD and must, therefore, stem from changes of the polymer upon addition of the stimulus, i.e., the acid. Indeed, the positions of these bands correspond well to the CN stretching vibration of an imine (1656 cm−1), and the CO stretching vibration of an aldehyde (1702 cm−1),27 indicating the stimulus-response of the imine bond by hydrolysing the imine to an aldehyde. Together with the well separated signals of the d3-AcOD, this enables orthogonal investigation of the presence of acetic acid (the stimulus) and the onset of molecular changes within the polymer (the response).
Raman microscopy
After collecting the reference spectra, the same measurement principle was transferred to a microscopic setup, enabling spatially resolved Raman spectroscopy of the polymer film in order to study the change of the surface properties in correlation with the stimulus progression and the molecular change of the polymeric structure. For this purpose, a confocal Raman microscope with an excitation wavelength of 514 nm that achieves diffraction limited lateral resolution and axial resolutions in the range of 1 μm was utilized. The reference Raman spectra collected on the FT-NIR instrument could be reproduced here using an integration time of 150 s (a comparison of the full spectra can be found in Fig. S6 in the ESI†). The intrinsically higher Raman intensity when exciting at 514 nm compared to 1064 nm (ca. 18× due to ω4 (ω: frequency of excitation light) dependency) furthermore shortens the necessary integration time for following the stimuli response to 2 s per point,34 making temporally resolved measurements possible. Utilizing this setup, it should therefore be possible to monitor the progression of the stimulus (d3-AcOD) and the response of the polymer in a spatially and temporally resolved manner. To carry out this investigation, a hot-pressed sample of polymer P4 was embedded into an epoxy matrix to prevent bending of the sample due to changes of the molecular structure during stimulus application. Furthermore, the surface was polished to ensure surface flatness. This sample was placed under the Raman microscope and the initial focus was set to the surface using a 100×/0.8 NA objective. A scan of the surface from 0 to 50 μm depth in 12.5 μm steps was performed. At each depth, two points laterally separated by 10 μm were recorded with an integration time of two seconds and subsequently averaged to minimize lateral variations.Afterwards, a drop of a 9:1 (v/v) mixture of D2O/d3-AcOD was placed directly on top of the focused points and the scan was started to afford a spatially and temporally resolved data set. The right panel of Fig. 1c exemplarily shows a snippet of the time series recorded 12.5 μm below the surface. As determined via the reference measurements (cf.Fig. 1c, left), hydrolyzation of the imine (decreasing the intensity at 1650 cm−1 and increasing the intensity at 1700 cm−1), as well as the influx of the deuterated acetic acid (appearance of bands between 2100 and 2300 cm−1) can be seen. Naturally, as the reaction progresses, both the degree of hydrolyzation (response) and the amount of deuterated species (progressing stimulus) increase with time. Looking more closely at the traces, the influx of solvent and the hydrolyzation seem to happen on slightly different timescales, as hydrolysed polymer bands can be seen already before acetic acid bands can be detected (first signals at 60 s). This indicates that the reaction mainly happens on the interface between the activating mixture and the polymer. Consequently, before switching the polymer, only a minute amount of the activation mixture (and therefore d3-AcOD) is present inside the focal volume of the confocal Raman microscope. While this minute amount is large enough to cause hydrolyzation of the polymer, its concentration stays below the detection limit of the measurement setup. After the hydrolyzation, the polymer changes from being more hydrophobic (caused by the butyl groups of the imine) to being hydrophilic (free aldehyde), which allows the bulk of the (water-based) activation mixture to penetrate deeper into the polymer.
Quantification of molecular changes
The areas of the bands corresponding to hydrolyzation (CO vibration at 1702 cm−1) and the influx of the activation mixture (C–D vibration at 2110 cm−1) were integrated for each measurement in order to investigate this behavior in more detail. These integrated band areas directly relate to the amount of the respective species present in the focal volume. Plotting of the integrated values against time and depth therefore illustrates the progress of both the hydrolyzation reaction (i.e. molecular changes) and the macroscopic progression of the stimulus (deuterated acetic acid) (cf.Fig. 2, left). For all depths, the hydrolyzation (blue shading in the figure) happens earlier than the influx of acid (yellow to red shading), confirming the previously discussed behavior. Furthermore, it is apparent that the band area for both the hydrolyzed polymer and the acetic acid signal does not strictly increase but decreases again after a certain time. Presumably, this is caused by a slight swelling of the polymer due to the diffusion of water, which causes optical distortions in the swollen part and thereby lowers the efficiency of the Raman signal collection. Swelling in aqueous media is known in the literature for pure PMMA, and is expected to also be present in the PMMA based polymers in this study.35,36 To confirm this assumption, a swelling experiment was performed by swelling P3 and P4 in a mixture of water:acetic acid (9:1, v/v), which showed a swelling degree of 1310% (P3) and 234% (P4) (for further information see the ESI†). Additionally, it can be noticed that the temporal width of the signals broadens with increasing depth. This behavior is caused by the slightly different time constants for hydrolyzation and diffusion across the polymer multiplied by increasing depth. These variations in time constants are caused on the one hand by the fact that reaction and diffusion are statistical processes for which the time constants only hold true on average, not for a specific sample in an ensemble, and on the other hand by the slight variations in polymer composition.
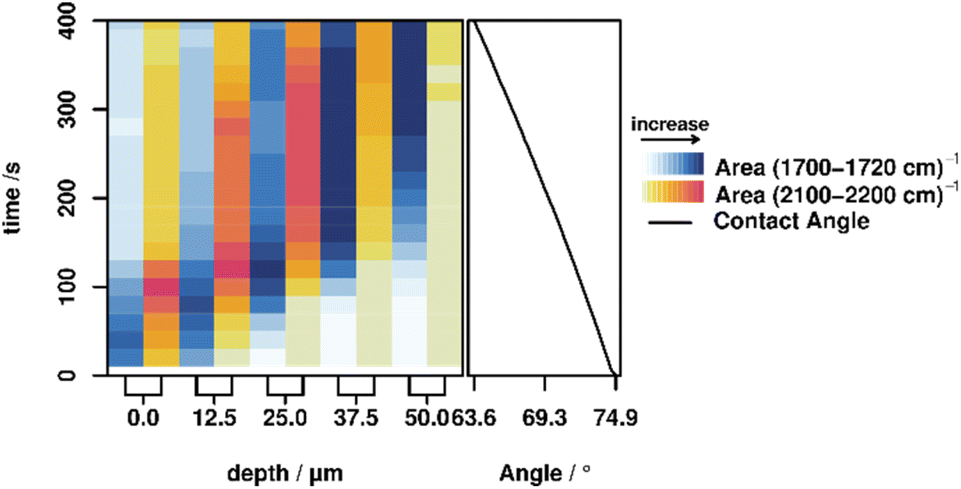 | ||
| Fig. 2 Left: Integrated band areas of the CO vibration of the hydrolyzed polymer P4 (1700–1720 cm−1, blue) and the C–D vibration of the acetic acid (2100–2200 cm−1, yellow–red), at depths between 0 and 50 μm from 0 to 400 s after adding a drop of the activation mixture (D2O/d3-AcOD, 9:1 v/v). Right: Contact angle of the droplet over the same 0 to 400 s time span, corresponding to an increase in hydrophilicity. | ||
As a method with molecular specificity, Raman spectroscopy enables the amount of hydrolyzed imines to be monitored over the activation process.
For this, the band area of the imine was integrated and compared with the band area of a C–C backbone vibration (1220 cm−1), revealing a constant level during the FT-Raman reference measurements. By monitoring this ratio with respect to the ratio between the two bands in a pristine sample (AImine/AC–C = 1.63), the hydrolyzation ratio can be calculated. Here, for all depths a hydrolyzation ratio of 65–81% could be found before the aforementioned effect of polymer swelling set in (for calculation details see the ESI†). To further investigate the changes in the data, we employed two-dimensional correlation analysis (2DCOS), which has been described in detail elsewhere.25,37,38 In short, 2DCOS recovers the cross-correlation function between spectral variables (bands) under the external perturbation, thereby revealing information about their relationship (direction and especially order of changes). Since 2DCOS uses a well-defined mathematical approach to get this information it is less susceptible to subjective interpretation as, e.g., plotting kinetic traces or difference spectra and can also deal with more complex response (e.g., not strictly decreasing) functions that are not easily analyzed visually. 2DCOS is a well-established method to analyze sets of spectra recorded under external perturbation (in this case time after addition of the activation mixture) in general,39–42 and in the study of smart polymers in particular.21,22 2DCOS results are usually displayed in the form of contour plots, that depict the real (synchronous, Φ) and imaginary (asynchronous, Ψ) parts of the complex correlation function. These maps visualize if spectral changes within a data set are correlated (Φ) and if a phase-shift is present between the changes (Ψ), respectively. Rules for interpreting these maps can be derived from the underlying maths, the so-called Noda Rules.38
(1) A positive correlation signal in Φ shows that changes at the respective x and y positions are correlated and they change in the same direction.
(2) A negative correlation signal in Φ shows that changes at the respective x and y positions are correlated and they change in different directions.
(3) If the signs of the signals in Φ and Ψ are the same for a combination of spectral positions x and y, the change at x happens before the change at y.
(4) If the signs of the signals in Φ and Ψ are different for a combination of spectral positions x and y, the change at y happens before the change at x.
A plot with the synchronous (Φ) and asynchronous (Ψ) parts of the 2DCOS at 25 μm depth can be seen in Fig. 3. It can be derived that the bands of acetic acid (2100–2200 cm−1) are positively correlated (positive signal in Φ → 1st Noda Rule, change in the same direction) with the signal for the hydrolyzed polymer at 1702 cm−1 and negatively correlated (negative signal in Φ → change in different directions, 2nd Noda Rule) with the CN signal of the imine signal in the unhydrolyzed polymer. This once again confirms the observation that addition of the activation mixture causes the polymer to hydrolyze. Furthermore, by combining the signals in Φ and Ψ the temporal relationship of the changes to the polymer structure and the influx of acetic acid can be elucidated.
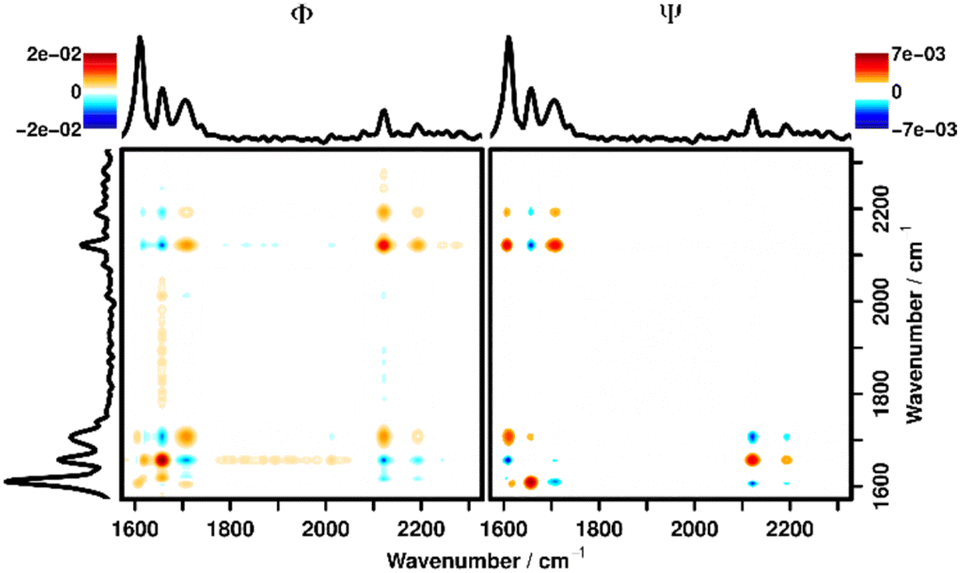 | ||
| Fig. 3 Synchronous (left) and asynchronous (right) correlation spectrum of the layer at z = 25 μm in the timeframe between 0 and 400 s after adding the activation mixture (D2O/d3-AcOD, 9:1 v/v) to P4. | ||
To interpret the signals in Ψ the 3rd and 4th Noda Rules must be applied, which state that the change at the respective wavenumber position plotted on the x-axis happens before the change at the y-position if the signs of a signal are the same in Φ and Ψ and vice versa. Since the signs of the signal at (1702, 2110), (1656, 2110) and (1656, 2180) cm−1 are all equal in Φ and Ψ, it is confirmed that the diffusion of acetic acid always happens after the polymer has been hydrolyzed. The same plots, focused on this area of interest for all depths can be found in the ESI (Fig. S7–S11†) showing the same behavior in all depths. Additionally, in Fig. 3, correlation signals with the aromatic vibration at 1600 cm−1 can be seen, which stand in for generally observable correlation signals with all polymer bands outside of the shown wavenumber region. These correlation signals are caused by the aforementioned swelling of the polymer and subsequent loss of Raman intensity after the liquid has diffused into the polymer. As can be seen from the differing signs of all correlation signals along the (1600, y) cm−1 line in Φ and Ψ these changes are strictly happening later than the hydrolyzation as well as the diffusion of liquid.
The power of 2DCOS to extract small, systematic changes from a series of spectra could be utilized to directly visualize the correlation of the aldehyde/imine bonds with the influx of D2O as well. For this purpose, the O–D stretching vibration located between 2500 and 2600 cm−1 could be used. While this band shows very low Raman intensity, which makes integrating the band impossible, using 2DCOS it was still possible to find correlations with the CO/N stretching bands (cf. Fig. S12–S16 in the ESI†). These maps show similar results as shown in Fig. 3 and S7–S11.† While the correlation intensities are lower and the asynchronous correlation is therefore only clearly visible for the aldehyde CO stretching vibration at 1702 cm−1, they also indicate bulk influx of the D2O only after hydrolyzation of the imine. Together, these 2DCOS observations further support the mechanism proposed in the previous section: the hydrolyzation reaction is mediated by minute amounts of the activation mixture without significant penetration of the imine-containing polymer. Only after the opening of the imine bond and a resulting change in hydrophilicity can the majority of the mixture penetrate the polymer, leading to the observed lags in their Raman signals.
Furthermore, recovering the correlation function between two signals allows for a direct way to measure the delay between the two processes. The average phase angle between the signals in the Fourier domain is encoded in the 2DCOS, which directly relates to the delay between the two signals in the time domain.43 Using the parameters of the Fourier Transform, it is possible to transform those phase angles back to a value in the time domain, which reveals delay times between hydrolyzation and influx of acetic acid ranging from 30 to 55 s (cf.Table 2, for details on the calculation see the ESI†). There is no increase or decrease of the delay time with increasing depth, indicating statistical fluctuations around a constant delay time between hydrolyzation and acetic acid influx. This delay time is governed by the diffusion rate of the activation mixture in the switched polymer, which can be calculated as 0.27–0.3 μm s−1 from the delay times and spacing of the measurement points. Consequently, the diffusion coefficients are in the range of (16.9–18.9) × 10−9 cm2 s−1.
| Depth | 0 μm | 12.5 μm | 25 μm | 37.5 μm | 50 μm |
|---|---|---|---|---|---|
| a Correlating molecular behavior to macroscopic properties. | |||||
| Delay time/s | 43 ± 4Exp | 46 ± 4Exp | 42 ± 4Exp | 41 ± 4Exp | 44 ± 5 |
| Diffusion rate/nm s−1 | 293 ± 3Exp | 270 ± 3Exp | 301 ± 3Exp | 303 ± 3Exp | 282 ± 3 |
| Diffusion coefficient/10−9 cm2 s−1 | 18.3 ± 1.4Exp | 16.9 ± 1.4Exp | 18.8 ± 1.4Exp | 18.9 ± 1.4Exp | 17.6 ± 1.6 |
Comparing these values to the literature values obtained for the diffusion of acetic acid in an epoxy resin (rate: 37 nm s−1)44 and pure PMMA (coefficient: 1.2–3.2 × 10−9 cm2 s−1 depending on the diffusion model),45 these values are roughly an order of magnitude higher. This fits the expectation, as the diffusion of a very polar electrolyte is measured and free aldehydes are more polar compared to the polyether structures present in epoxy resins as well as the ester moieties in PMMA, thereby underpinning the validity of the measurement.
Correlating molecular behavior to macroscopic properties
Contact angle measurements are often utilized as an ‘indirect’ method to study changes on the molecular level via changes in the wetting behaviour. Consequently, we also investigated the correlation between changes on the molecular level, the stimulus progression and the macroscopic property change by simultaneously measuring all parameters, elucidated using Raman microscopy, and macroscopic changes of the polymer properties, elucidated by sessile-drop method contact angle measurements performed on the same polymer over the same time span (cf.Fig. 2, right). A list of the contact angles can be found in the ESI.† These contact angle measurements show a steady decrease in contact angle, corresponding to an increase in hydrophilicity caused by the hydrolyzation of the imine to the relatively hydrophilic free aldehyde. These results are consistent with the observed continuous diffusion of the activating mixture into the polymer, demonstrating that with this method it is possible to correlate the changes at the molecular level with the macroscopic changes of a functional polymer.Conclusion
To conclude, utilizing Raman micro-spectroscopy and 2D correlation analysis, we could, to the best of our knowledge, for the first time demonstrate a method that delivers spatially resolved measurements with molecular specificity inside smart polymers. This method allows not only the investigation of macroscopic properties, but it also furthermore provides invaluable information on the molecular moieties responsible for the functionality. Consequently, this new technique enables the measurement of the stimulus progression, the change of the molecular structure and the change of the macroscopic properties simultaneously. Through the combination of this molecularly resolved method with methods to investigate the macroscopic properties, e.g. hydrophilicity, it is therefore possible to derive a structure–function relationship that will allow tuning of the desired properties of the smart polymer by modifying the structure. For example, in the present case it was demonstrated that the switching-reaction is mainly limited to the interface between the activating mixture and polymer, while the macroscopic influx of the mixture only happens after the stimuli-response. Therefore, the transport (and the spatial progression of stimuli-response) is limited by the diffusion rate of the stimulus (d3-AcOD/D2O) inside the switched phase and therefore by its physical properties. This reveals a clear relationship between the molecular structure (the diffusion coefficient is governed by the molecular structure of the polymer) and the functionality (the speed of property change).As a vibrational spectroscopic method, Raman micro-spectroscopy is broadly applicable to many functional groups and thus presents a technique transferable to different stimuli-responsive systems. The technique is in principle only limited by the acquisition speed and the separability of signals of the activating species and polymer. The technique applied here, isotope labelling, is also broadly applicable and in the case of deuterium labelling usually comparatively easy and cheap to carry out. Using non-linear Raman techniques or resonance enhancement, it is furthermore possible to increase the acquisition speeds to the order of milliseconds, thus enabling the use of the same basic technique also for faster processes.
Experimental section
The synthesis of monomer 1 and polymers P1–P4 (cf.Fig. 1a) was adopted from literature procedures.32,33 Detailed information about the synthesis and characterization of the monomer and polymers is provided in the ESI.†All chemicals were used as received from TCI (Eschborn, Germany), Sigma Aldrich (Darmstadt, Germany), Alfa Aesar (Kandel, Germany), Thermo Fisher Scientific (Geel, Belgium) and Acros Organics (Geel, Belgium) if not otherwise stated. All solvents were dried over a molecular sieve under a nitrogen atmosphere. The stabilizer in the used liquid monomer methyl methacrylate (MMA) was removed over a short aluminium oxide (AlOx) column (neutral AlOx, obtained from Molecula, Darlington, UK).
Nuclear magnetic resonance (NMR) spectra were measured using a Bruker AC 300 (300 MHz) spectrometer (Billerica, MA, USA) at 298 K if not stated differently. The chemical shift is given in parts per million (ppm on δ scale) related to deuterated solvent.
Size exclusion chromatography (SEC) measurements were performed using the following setup: a Shimadzu instrument with a CBM-20A (system controller), DGU-14A (degasser), LC-20AD (pump), SIL-20AHT (auto sampler), CTO-10AC vp (oven), SPD-20A (UV detector), RID-10A (RI detector), PSS SDV guard/1000 Å/1000000 Å (5 μm particle size) chloroform/isopropanol/triethyl-amine [94/2/4] at 1 mL min−1 at 40 °C, and poly(methyl methacrylate) (standard).
Raman-spectroscopic measurements were performed on a MultiRAM near-infrared Fourier-transform Raman spectrometer (Bruker Corporation, Billerica, Massachusetts, United States of America) in the range between 100 and 4000 cm−1 with a spectral resolution of 4 cm−1. The Raman excitation light at 1064 nm was provided by a Nd:YAG laser (Klastech DeniCAFC-LC-3/40, Dortmund, Germany). The laser power was set to 1000 mW at the sample plane and 128 scans were accumulated for each sample.
Raman microscopy was performed on an alpha300 Raman microscope (WiTec GmbH, Ulm, Germany) equipped with a 600 line per mm grating. The excitation source was a continuous-wave diode-pumped solid-state laser (CoboltTM, Solna, Sweden) at 514 nm and 30 mW power in the sample plane. Light was focused onto the sample using a Leica Fluotar long-working distance objective (100×/0.6 NA). Spectra were recorded in the wavenumber region between 0 and 4000 cm−1. A depth scan with 2 × 5 points spread 10 μm laterally and 50 μm axially was performed using an integration time of 2 s per point (for an illustration of the measurement scheme, see Fig. S1 in the ESI†).
All pre-processing of the Raman spectroscopic data and subsequent analysis, including 2D correlation analysis, were performed using GNU R, version 4.2.2 with in-house developed scripts.46 A detailed description of the analysis steps can be found in the ESI.†
Contact angle measurements were performed on an OCA 30 (DataPhysics, Filderstadt, Germany) using the SCA 20 software in dynamic tracking mode.
Data availability
The data supporting the findings of this study are openly available on Zenodo at https://doi.org/10.5281/zenodo.8026026.Author contributions
Synthesis: J. M.; SEC, NMR, CA: J. M.; Raman measurements: J. H.; two-dimensional correlation analysis: J. H.; writing – original draft: J. H., J. M., S. Z.; interpretation of measurement data: J. H., S. Z., M.S.; supervision: S. Z., M. S., M. D. H., J. P.; conceptualization: J. H., S. Z., M. S., M. D. H., J. P.; writing – review & editing: M. D. H., M. S., J. P., funding acquisition: M. D. H., J. P.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the German Research Foundation (DFG, Deutsche Forschungsgemeinschaft) for funding in the framework of FOR5301 “FuncHeal” (projects P3 and P6) as well as SFB/TRR 234 CataLight, project: C2.References
- M. A. C. Stuart, W. T. S. Huck, J. Genzer, M. Müller, C. Ober, M. Stamm, G. B. Sukhorukov, I. Szleifer, V. V. Tsukruk, M. Urban, F. Winnik, S. Zauscher, I. Luzinov and S. Minko, Nat. Mater., 2010, 9, 101–113 CrossRef PubMed.
- Z. P. Zhang, M. Z. Rong and M. Q. Zhang, Prog. Polym. Sci., 2018, 80, 39–93 CrossRef CAS.
- M. Wei, Y. Gao, X. Li and M. J. Serpe, Polym. Chem., 2017, 8, 127–143 RSC.
- S. Wang and M. W. Urban, Nat. Rev. Mat., 2020, 5, 562–583 CrossRef CAS.
- M. D. Hager, S. Bode, C. Weber and U. S. Schubert, Prog. Polym. Sci., 2015, 49–50, 3–33 CrossRef CAS.
- X. Li, L. Zhang, Y. Feng, Y. Zheng, Z. Wu, X. Zhang, N. Wang, D. Wang and F. Zhou, Adv. Funct. Mater., 2021, 31, 2010220 CrossRef CAS.
- M. Motornov, S. Minko, K.-J. Eichhorn, M. Nitschke, F. Simon and M. Stamm, Langmuir, 2003, 19, 8077–8085 CrossRef CAS.
- Z. Rashid, I. Atay, S. Soydan, M. B. Yagci, A. Jonáš, E. Yilgor, A. Kiraz and I. Yilgor, Appl. Surf. Sci., 2018, 441, 841–852 CrossRef CAS.
- P. Giusto, B. Kumru, J. Zhang, R. Rothe and M. Antonietti, Chem. Mater., 2020, 32, 7284–7291 CrossRef CAS.
- N. Wagner and P. Theato, Polymer, 2014, 55, 3436–3453 CrossRef CAS.
- S. Abrakhi, S. Péralta, O. Fichet, D. Teyssié and S. Cantin, Langmuir, 2013, 29, 9499–9509 CrossRef CAS PubMed.
- Q. Zhang, F. Xia, T. Sun, W. Song, T. Zhao, M. Liu and L. Jiang, Chem. Commun., 2008, 1199–1201, 10.1039/B716681H.
- D. Wang, P. Jiao, J. Wang, Q. Zhang, L. Feng and Z. Yang, J. Appl. Polym. Sci., 2012, 125, 870–875 CrossRef CAS.
- A. Athanassiou, M. I. Lygeraki, D. Pisignano, K. Lakiotaki, M. Varda, E. Mele, C. Fotakis, R. Cingolani and S. H. Anastasiadis, Langmuir, 2006, 22, 2329–2333 CrossRef CAS PubMed.
- J. Elbert, M. Gallei, C. Rüttiger, A. Brunsen, H. Didzoleit, B. Stühn and M. Rehahn, Organometallics, 2013, 32, 5873–5878 CrossRef CAS.
- G. Godeau, T. Darmanin and F. Guittard, React. Funct. Polym., 2015, 96, 44–49 CrossRef CAS.
- M. Morra, E. Occhiello and F. Garbassi, Adv. Colloid Interface Sci., 1990, 32, 79–116 CrossRef CAS.
- X. Zhao, A. Best, W. Liu, K. Koynov, H.-J. Butt and C. Schönecker, Phys. Rev. Fluids, 2021, 6, 054004 CrossRef.
- X. Cheng, W. Yang, Y. Zhang, Y. Kang, Y. Ding, Z. Jiao and L. Cheng, Polym. Test., 2018, 65, 90–96 CrossRef CAS.
- J. Meurer, T. Bätz, J. Hniopek, S. Zechel, M. Schmitt, J. Popp, M. D. Hager and U. S. Schubert, J. Mater. Chem. A, 2021, 9, 15051–15058 RSC.
- R. Geitner, J. Kötteritzsch, M. Siegmann, T. W. Bocklitz, M. D. Hager, U. S. Schubert, S. Gräfe, B. Dietzek, M. Schmitt and J. Popp, Phys. Chem. Chem. Phys., 2015, 17, 22587–22595 RSC.
- R. Geitner, J. Kötteritzsch, M. Siegmann, R. Fritzsch, T. W. Bocklitz, M. D. Hager, U. S. Schubert, S. Gräfe, B. Dietzek, M. Schmitt and J. Popp, Phys. Chem. Chem. Phys., 2016, 18, 17973–17982 RSC.
- H. Kubota, K. Sakamoto and T. Matsui, Sci. Rep., 2020, 10, 16426 CrossRef PubMed.
- J. L. Koenig and J. P. Bobiak, Macromol. Mater. Eng., 2007, 292, 801–816 CrossRef CAS.
- I. Noda, Appl. Spectrosc., 1993, 47, 1329–1336 CrossRef CAS.
- H. G. M. Edwards, A. F. Johnson and I. R. Lewis, J. Raman Spectrosc., 1993, 24, 475–483 CrossRef CAS.
- G. Socrates, Infrared and Raman Characteristic Group Frequencies: Tables and Charts, John Wiley & Sons Ltd., Chichester, United Kingdom, 2001 Search PubMed.
- R. Petry, M. Schmitt and J. Popp, ChemPhysChem, 2003, 4, 14–30 CrossRef CAS PubMed.
- I. Lynch, I. A. Blute, B. Zhmud, P. MacArtain, M. Tosetto, L. T. Allen, H. J. Byrne, G. F. Farrell, A. K. Keenan, W. M. Gallagher and K. A. Dawson, Chem. Mater., 2005, 17, 3889–3898 CrossRef CAS.
- H. Tian, J. Fan, Z. Yu, Q. Liu and X. Lu, Minerals, 2023, 13, 164 CrossRef CAS.
- Y. Li, L. Yang, Y. Zeng, Y. Wu, Y. Wei and L. Tao, Chem. Mater., 2019, 31, 5576–5583 CrossRef CAS.
- N. Kuhl, S. Bode, R. K. Bose, J. Vitz, A. Seifert, S. Hoeppener, S. J. Garcia, S. Spange, S. van der Zwaag, M. D. Hager and U. S. Schubert, Adv. Funct. Mater., 2015, 25, 3295–3301 CrossRef CAS.
- B. García-Acosta, F. García, J. M. García, R. Martínez-Máñez, F. Sancenón, N. San-José and J. Soto, Org. Lett., 2007, 9, 2429–2432 CrossRef PubMed.
- A. C. Albrecht and M. C. Hutley, J. Chem. Phys., 1971, 55, 4438–4443 CrossRef CAS.
- K. Tanaka, Y. Fujii, H. Atarashi, K.-i. Akabori, M. Hino and T. Nagamura, Langmuir, 2008, 24, 296–301 CrossRef CAS PubMed.
- M. N'Diaye, F. Pascaretti-Grizon, P. Massin, M. F. Baslé and D. Chappard, Langmuir, 2012, 28, 11609–11614 CrossRef PubMed.
- I. Noda, J. Mol. Struct., 2016, 1124, 29–41 CrossRef CAS.
- I. Noda, A. E. Dowrey, C. Marcott, G. M. Story and Y. Ozaki, Appl. Spectrosc., 2000, 54, 236A–248A CrossRef CAS.
- Y. Park, S. Jin, I. Noda and Y. M. Jung, Spectrochim. Acta, Part A, 2023, 284, 121636 CrossRef CAS PubMed.
- Y. Park, S. Jin, I. Noda and Y. M. Jung, Spectrochim. Acta, Part A, 2023, 284, 121750 CrossRef CAS PubMed.
- Y. Park, S. Jin, I. Noda and Y. M. Jung, J. Mol. Struct., 2020, 1217, 128405 CrossRef CAS.
- Y. Park, S. Jin, I. Noda and Y. M. Jung, Spectrochim. Acta, Part A, 2022, 281, 121573 CrossRef CAS PubMed.
- S.-I. Morita, Y. Ozaki and I. Noda, Appl. Spectrosc., 2001, 55, 1618–1621 CrossRef CAS.
- Y. Dutheillet, M. Mantle, D. Vesely and L. Gladden, J. Polym. Sci., Part B: Polym. Phys., 1999, 37, 3328–3336 CrossRef CAS.
- M. C. Santos, B. Bendiksen and Y. A. Elabd, Ind. Eng. Chem. Res., 2017, 56, 3464–3476 CrossRef CAS.
- R Core Team, R: A Language and Environment for Statistical Computing, R Foundation for Statistical Computing, 2022 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sc01455j |
| This journal is © The Royal Society of Chemistry 2023 |