DOI:
10.1039/D3SC02292G
(Edge Article)
Chem. Sci., 2023,
14, 8524-8530
A dinuclear Rh(−I)/Rh(I) complex bridged by biphilic phosphinine ligands†
Received
5th May 2023
, Accepted 11th July 2023
First published on 25th July 2023
Abstract
Bimetallic complexes have enabled precise control of catalysis by accumulating two discrete metal centres. In these complexes, bridging ligands are essential to combine multiple metals into one molecule. Among some bridging modes, an unsymmetric bridging mode will differentiate the electronic structures of the two metal centres. In this study, a dinuclear Rh(−I)/Rh(I) complex bridged by tridentate phosphine–phosphinine–phosphine ligands was prepared by reduction of the corresponding Rh(I) complex. Single-crystal X-ray analysis and DFT calculations suggest that the phosphinine ligands adopt an unsymmetric bridging mode wherein phosphinine accepts d-electrons from one Rh centre and, at the same time, donates lone pairs to the other Rh centre.
Introduction
Combining multiple metals is an attractive method to accumulate more than one function in one complex enabling precise tuning of the reactivity of each metal centre.1–5 Bridging ligands play a crucial role in constructing a rigid multimetallic skeleton and facilitating electronic communication between metals. The coordination mode of bridging ligands can be classified into the following three types (Fig. 1a). Two electron donors, such as a hydride anion and carbanions, form three-centre two-electron bonding (Type I).6 Anionic ligands with lone pairs, such as halide or dianionic ligands, can donate four electrons via two σ bonds with each metal centre (Type II).6 When a ligand simultaneously has a lone pair and an electron-accepting orbital, the bridging ligand enables an unsymmetric coordination environment where two electrons are donated from a ligand to an electron-poor metal, and two electrons are accepted from an electron-rich metal (Type III). For example, the carbonyl ligand (CO) bridges two or more metal centres, and it has some bridging modes (Fig. 1b);7 Type I with a lone pair on the C atom, Type II where CO donates four electrons by both σ- and π-donation,8 and Type III where the carbon atom coordinates to one metal centre with a σ-type lone pair and accepts two electrons from the other metal centre using the π*-orbital. Since a Type III bridging mode was found in a dinuclear iron carbonyl complex in 1961,9 this bridging mode has been observed in some dinuclear metal carbonyl complexes.10–12
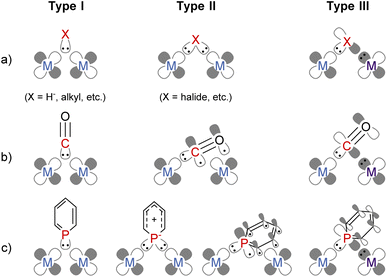 |
| | Fig. 1 Classification of the electronic structures of bridging ligands; (a) general description; (b) carbonyl ligands; (c) phosphinine ligands. | |
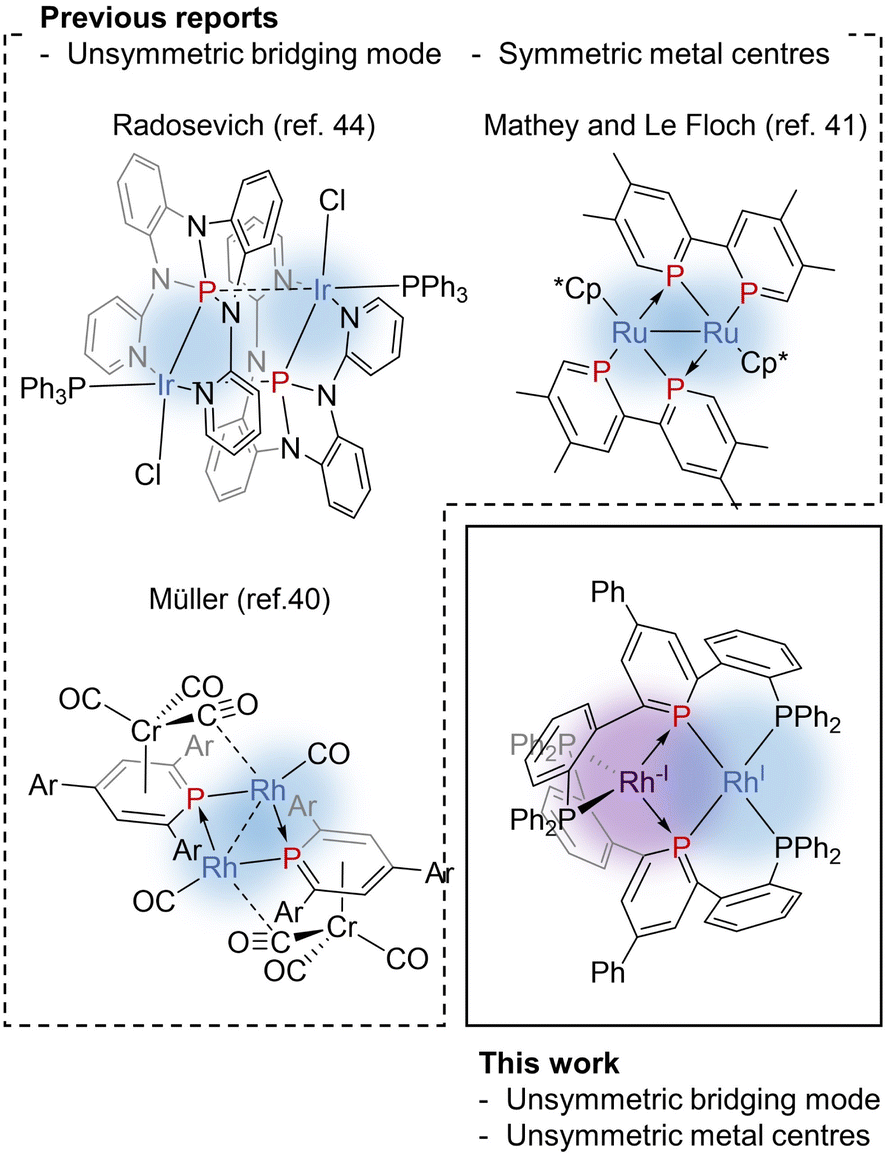
Phosphinine (phosphabenzene) is a well-studied equivalent to carbon monoxide in organometallic chemistry since phosphinine exhibits σ-donating and strong π-accepting properties due to its low-lying π* orbital on the P atom.13–15 To date, numerous phosphinine metal complexes have been synthesized, exhibiting various coordination modes, reactivities, and physical properties, derived from the π-acidity of phosphinine ligands.15–18 Thanks to their strong π-accepting properties, phosphinine ligands often show biphilic reactivity, such as nucleophilic attacks at the P atom19,20 and metal–ligand cooperative C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond cleavage of CO2.21 Phosphinine compounds form multinuclear complexes by the bridging of the P atom like the carbonyl ligand while the use of multidentate ligands also afforded multinuclear complexes.22–26 Phosphinine can exhibit all three bridging modes illustrated in Fig. 1c;27 Type I with a lone pair on the phosphorus atom,28–34 Type II with a phosphide-like resonance structure28,35–37 or 8e donor character where both the lone pair on the P atom and π-electrons of the phosphinine ring are donated,38–40 and Type III utilizing the lone pair and the low-lying π* orbital.40,41 Mathey, Le Floch, and co-workers reported that a dinuclear ruthenium complex with bisphosphinine ligands showed Type III bridging mode (Fig. 2).41 Recently, Müller and co-workers reported that an η6-phosphinine chromium complex acted as a Type III bridging ligand to form dirhodium complexes (Fig. 2).40 Geometrically constrained tricoordinate phosphorus compounds are another class of biphilic phosphorus compounds.42 They exhibited phosphorus-centred bond activation reactions and related catalytic activities, although their coordination chemistry is less explored than that of phosphinine ligands. Radosevich and co-workers reported that geometrically constrained tricoordinate phosphorus ligands show electrophilic reactivity43 and form an iridium dinuclear complex in a Type III manner (Fig. 2).44 Although the acid/base bifunctional character is quite beneficial to construct an unsymmetric multimetallic environment, to the best of our knowledge, there is no example where a phosphinine ligand is employed to bridge two metal centres in different electronic states.
O bond cleavage of CO2.21 Phosphinine compounds form multinuclear complexes by the bridging of the P atom like the carbonyl ligand while the use of multidentate ligands also afforded multinuclear complexes.22–26 Phosphinine can exhibit all three bridging modes illustrated in Fig. 1c;27 Type I with a lone pair on the phosphorus atom,28–34 Type II with a phosphide-like resonance structure28,35–37 or 8e donor character where both the lone pair on the P atom and π-electrons of the phosphinine ring are donated,38–40 and Type III utilizing the lone pair and the low-lying π* orbital.40,41 Mathey, Le Floch, and co-workers reported that a dinuclear ruthenium complex with bisphosphinine ligands showed Type III bridging mode (Fig. 2).41 Recently, Müller and co-workers reported that an η6-phosphinine chromium complex acted as a Type III bridging ligand to form dirhodium complexes (Fig. 2).40 Geometrically constrained tricoordinate phosphorus compounds are another class of biphilic phosphorus compounds.42 They exhibited phosphorus-centred bond activation reactions and related catalytic activities, although their coordination chemistry is less explored than that of phosphinine ligands. Radosevich and co-workers reported that geometrically constrained tricoordinate phosphorus ligands show electrophilic reactivity43 and form an iridium dinuclear complex in a Type III manner (Fig. 2).44 Although the acid/base bifunctional character is quite beneficial to construct an unsymmetric multimetallic environment, to the best of our knowledge, there is no example where a phosphinine ligand is employed to bridge two metal centres in different electronic states.
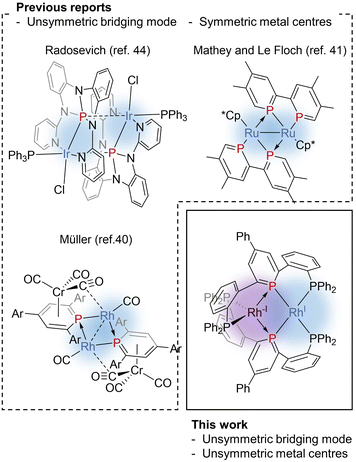 |
| | Fig. 2 Previous reports on unsymmetrically bridging phosphorus ligands and this work, a Rh(−I)/Rh(I) dinuculear complex bridged by phosphinine pincer ligands. | |
Recently, we reported a phosphinine pincer ligand bearing two phosphine pendants.45 The phosphine ligands stabilize the coordination of the phosphinine moiety. The phosphinine ligand formed a dinuclear complex when coordinated to silver centres.46 Here, in this report, we present syntheses and structural analysis of a dinuclear Rh(−I)/Rh(I) complex bridged by phosphinine pincer ligands (Fig. 2). Unlike the precedents shown in Fig. 2, the two rhodium centres are non-equivalent, where one rhodium centre can be regarded as a two electron-accepting Rh(I) centre, and the other can be regarded as a two electron-donating Rh(−I) centre.
Results and discussion
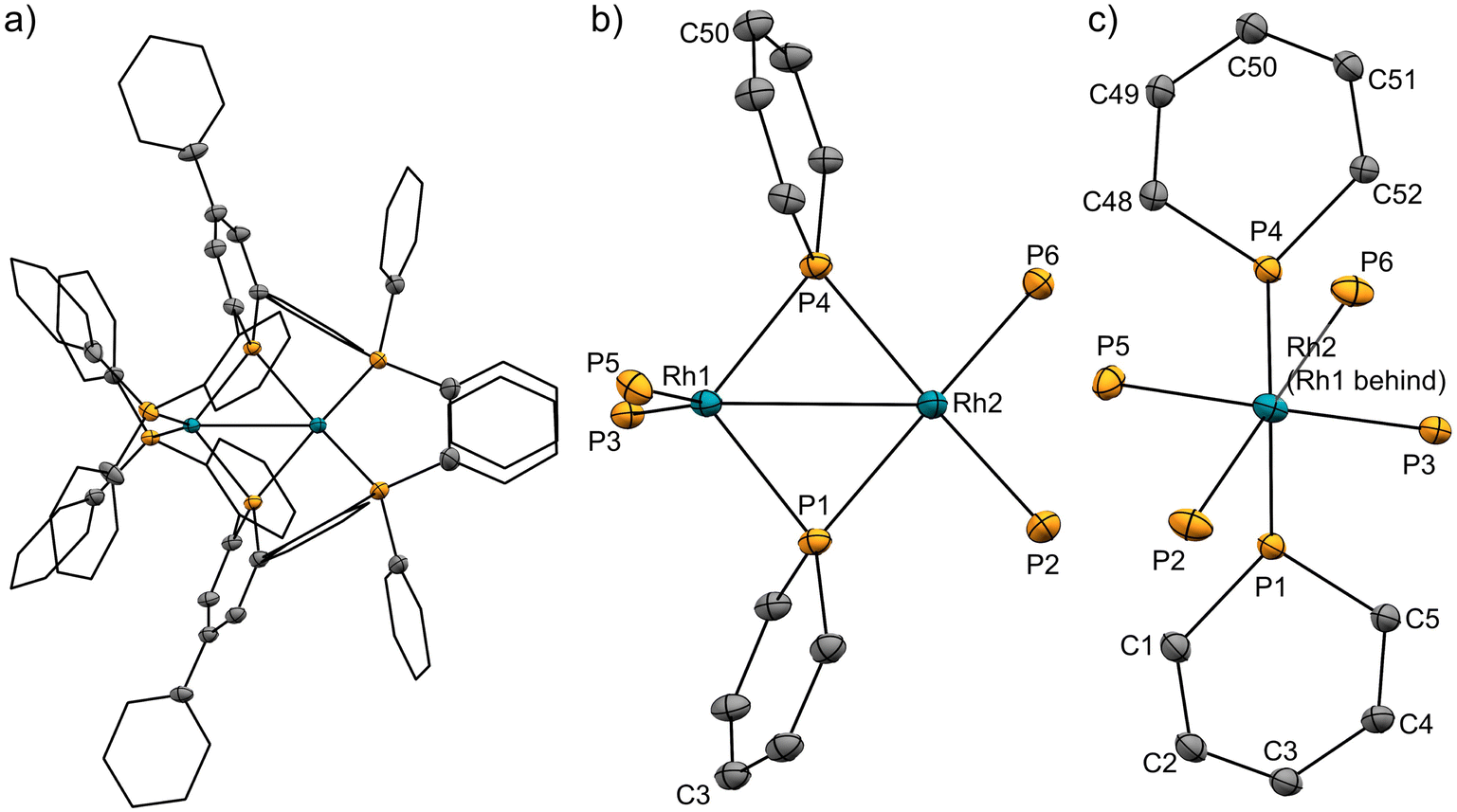
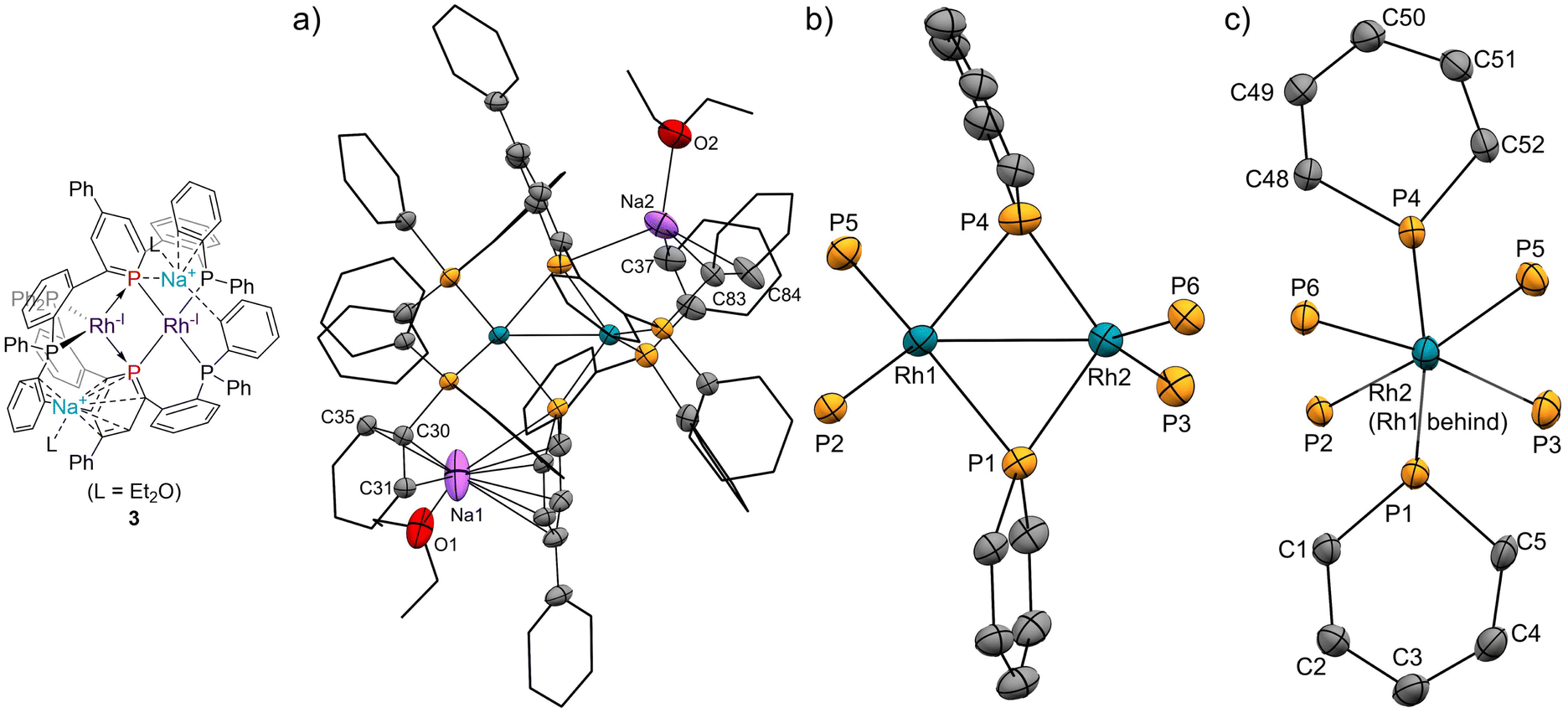
The pincer ligand 1 was synthesized following our previous report.46 By treating ligand 1 with [RhCl(cod)]2, an orange powder was obtained. The powder was insoluble in common solvents, which suggests that a bimetallic or polymetallic structure was formed in the solid. The product was reduced by sodium naphthalenide to afford dinuclear Rh complex 2 in 43% yield (Scheme 1). The structure of complex 2 was determined by single-crystal X-ray crystallographic analysis (Fig. 3). Further reduction to dinuclear Rh(−I)/Rh(−I) complex 3 was observed in the reaction by using an excess amount of metallic sodium (see the ESI† for details). The structure of 3 was determined by X-ray crystallography (Fig. 4) although 3 was too unstable to be fully characterized by spectroscopic methods.
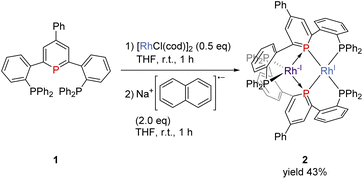 |
| | Scheme 1 Formation of complex 2. | |
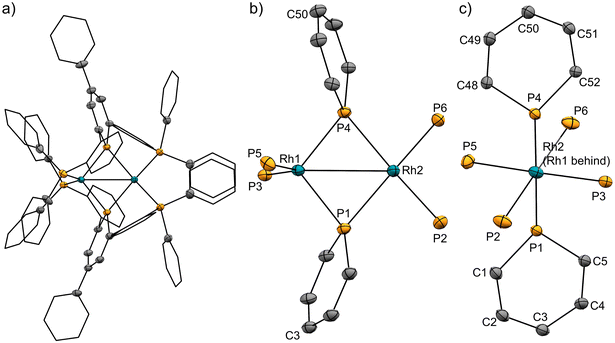 |
| | Fig. 3 (a) X-ray crystal structure of complex 2 (thermal ellipsoids are drawn with 50% probability; the hydrogen atoms are omitted, and the phenyl groups and the phenylene linkers are simplified for clarity); (b) and (c) geometries around rhodium and phosphorus atoms. Selected bond lengths [Å] and bond angles [deg]: Rh1⋯Rh2, 2.8493(3); Rh1–P1, 2.2262(8); Rh1–P4, 2.2182(8); Rh1–P3, 2.2848(8); Rh1–P5, 2.2840(8); Rh2–P1, 2.2731(8); Rh2–P4, 2.2884(8); Rh2–P2, 2.2554(9); Rh2–P6, 2.2569(9); P1–C1, 1.758(3); P1–C5, 1.763(3); C1–C2, 1.387(5); C2–C3, 1.411(5); C3–C4, 1.408(4); C4–C5, 1.384(4); P4–C48, 1.767(3); P4–C52, 1.758(3); C48–C49, 1.377(4); C49–C50, 1.399(4); C50–C51, 1.408(4); C51–C52, 1.392(4); P1–Rh1–P5, 112.40(3); P3–Rh1–P5, 130.89(3); P1–Rh2–P6, 152.26(3); P2–Rh2–P4, 152.65(3). | |
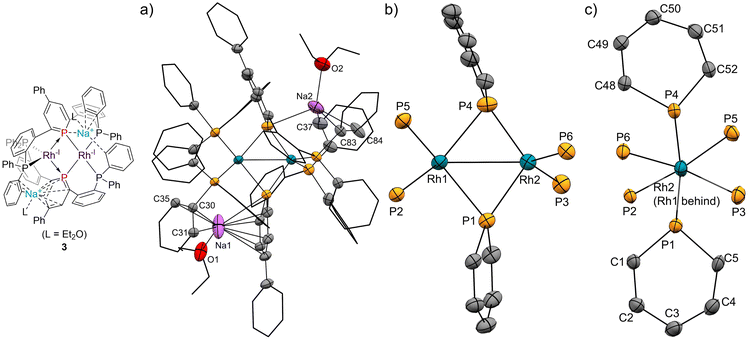 |
| | Fig. 4 (a) X-ray crystal structure of complex 3 (thermal ellipsoids are drawn with 50% probability; the hydrogen atoms are omitted, and the phenyl groups and the phenylene linkers are simplified for clarity); (b) and (c) geometries around rhodium and phosphorus atoms. Selected bond lengths [Å] and bond angles [deg]: Rh1⋯Rh2, 2.7436(4); Rh1–P1, 2.3335(10); Rh1–P4, 2.3666(11); Rh1–P2, 2.2375(10); Rh1–P5, 2.2785(10); Rh2–P1, 2.2278(10); Rh2–P4, 2.2275(11); Rh2–P3, 2.2575(11); Rh2–P6, 2.2440(10); P1–C1, 1.802(4); P1–C5, 1.804(4); C1–C2, 1.377(5); C2–C3, 1.426(6); C3–C4, 1.403(6); C4–C5, 1.384(6); P4–C48, 1.799(4); P4–C52, 1.793(4); C48–C49, 1.372(6); C49–C50, 1.409(6); C50–C51, 1.391(6); C51–C52, 1.398(5); P1–Na1, 3.149(3); Na1–C1, 2.993(5); Na1–C2, 2.947(5); Na1–C3, 2.911(5); Na1–C4, 2.731(5); Na1–C5, 2.708(5); Na1–C30, 2.732; Na1–C31, 3.015(5); Na1–C35, 2.793(5); P4–Na2, 2.976(3); Na2–C37, 2.848(6); Na2–C83, 2.987(5); Na2–C84, 2.818(8); P1–Rh1–P4, 101.59(4); P2–Rh1–P5, 113.64(4); P1–Rh2–P4, 109.67(4); P3–Rh2–P6, 119.85(4). | |
Complex 2 has an unsymmetric structure where both phosphinine rings tilt to one Rh centre (∠C3–P1–P4 = 160.9° and ∠C50–P4–P1 = 165.0°). One Rh centre (Rh1) adopts tetrahedral geometry (τ4 = 0.83), whereas the other Rh centre (Rh2) adopts distorted square planar geometry (τ4 = 0.39)47 (vide infra for detailed discussions on the unsymmetric structure of complex 2). Complex 3 also has an unsymmetric structure where one phosphinine ring tilts to one Rh centre (∠C50–P4–P1 = 156.9°) and the other phosphinine ring shows symmetric coordination (∠C3–P1–P4 = 178.0°). In complex 3, both Rh centres adopt tetrahedral geometry (τ4 = 0.67 and 0.78). DFT calculations (calculated at the PBE0-D3/def2-SVP level) suggest that the unsymmetric coordination of phosphinine is due to the perturbation by the contact of the Na cations. The calculated structure of complex 3 matched with the experimental result (Fig. S10†). When the geometry optimization of complex 3 was conducted without the contacting Na cations, the resulting structure showed symmetric coordination of both phosphinine rings (Fig. S11†). These results suggest that the unsymmetric coordination in complex 3 is mainly derived from the contacting Na cations.
The geometries of complex 2 can be rationalized as follows. The distorted square planar geometry of Rh2 suggests that it has Rh(I) character. The unsymmetric coordination mode of the phosphinine ligands suggests that the phosphinine ligands behave as Z-type ligands on the Rh1 centre. In comparison with the computed structure of a model ML2Z2-type Rh(−I) complex, [Rh(PMe3)2(BMe3)2]− (Fig. S12†), the geometry around Rh1 is consistent with Rh(−I) character. The P–Rh–P angles (∠P3–Rh1–P5 = 130.9° for complex 2 and ∠P–Rh–P = 138.6° for [Rh(PMe3)2(BMe3)2]−) and the geometry indices (τ4 = 0.83 for Rh1 in complex 2, τ4 = 0.76 for [Rh(PMe3)2(BMe3)2]−) resemble each other (see the ESI† for further discussion). Consequently, the electronic structure of complex 2 is an unsymmetric Rh(−I)/Rh(I) dinuclear structure, as shown in Scheme 1. This description is also supported by the bond lengths between Rh centres and phosphorus atoms. Rh1 had shorter distances from the P atoms on the phosphinine rings (Rh1–P1 = 2.2262 Å and Rh1–P4 = 2.2182 Å) than Rh2 (Rh2–P1 = 2.2731 Å and Rh2–P4 = 2.2884 Å) whereas Rh1 had longer distances from P atoms on the phosphine moiety (Rh1–P3 = 2.2848 Å and Rh1–P5 = 2.2840 Å) than Rh2 (Rh2–P2 = 2.2554 Å and Rh2–P6 = 2.2569 Å). Phosphinine ligands act as a strong π-acceptor and weak σ-donor, so P atoms on the phosphinine rings have stronger interaction with the Rh(−I) centre, which acts as a strong electron-donor.
In addition, the X-ray photoelectron spectroscopy (XPS) analysis of complex 2 was conducted to obtain more information on the electronic states of the Rh centres (Fig. 5). The XPS spectrum showed that complex 2 contains both Rh(I) species (311.7 and 307.0 eV) and Rh(−I) species (310.8 and 306.1 eV). For the binding energy of Rh 3d5/2, the higher value (307.0 eV) is slightly lower than that of referential Rh(I) species ([RhCl(C2H4)2]2: 307.3 eV; RhCl(PPh3)3: 307.4 eV).48 The lower value (306.1 eV) is lower than that of metallic Rh (307.0 eV)49 and a reported Rh(−I) species, [K(18-crown-6)][Rh(CO)(PPh3)3]50 (306.8 eV, see Fig. S8† for the XPS spectrum measured in this work). These results strongly support that the Rh centres in complex 2 have unsymmetric Rh(−I)/Rh(I) character.
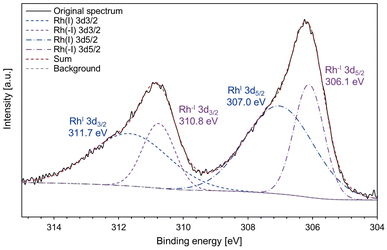 |
| | Fig. 5 The XPS spectrum of complex 2. The continuous black line indicates the original spectrum; the blue and purple dashed lines indicate the deconvoluted signals; the red dashed line is the sum of the deconvoluted signals; the grey dashed line is the background signal. | |
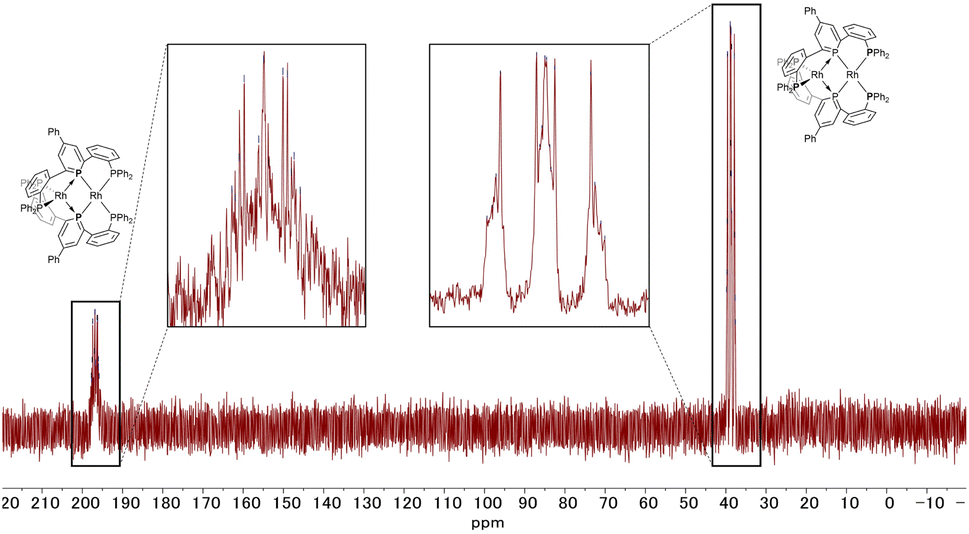
The 31P NMR spectrum of complex 2 (Fig. 6) showed two peaks with complex coupling patterns by JPP and JPRh, which accords with the dinuclear structure observed in the solid state. The 1H, 13C, and 31P NMR spectra suggest that all the PPh2 moieties are chemically equivalent, possibly due to the flipping of the phosphinine rings that is faster than the timescale of NMR (vide infra for the calculation of the stability of the unsymmetric structure). Under more concentrated conditions, 31P NMR showed complicated NMR spectra, probably due to aggregation (see Fig. S6 in the ESI† for details).
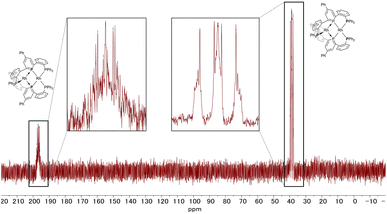 |
| | Fig. 6
31P NMR spectrum of complex 2 (202 MHz, THF-d8). | |
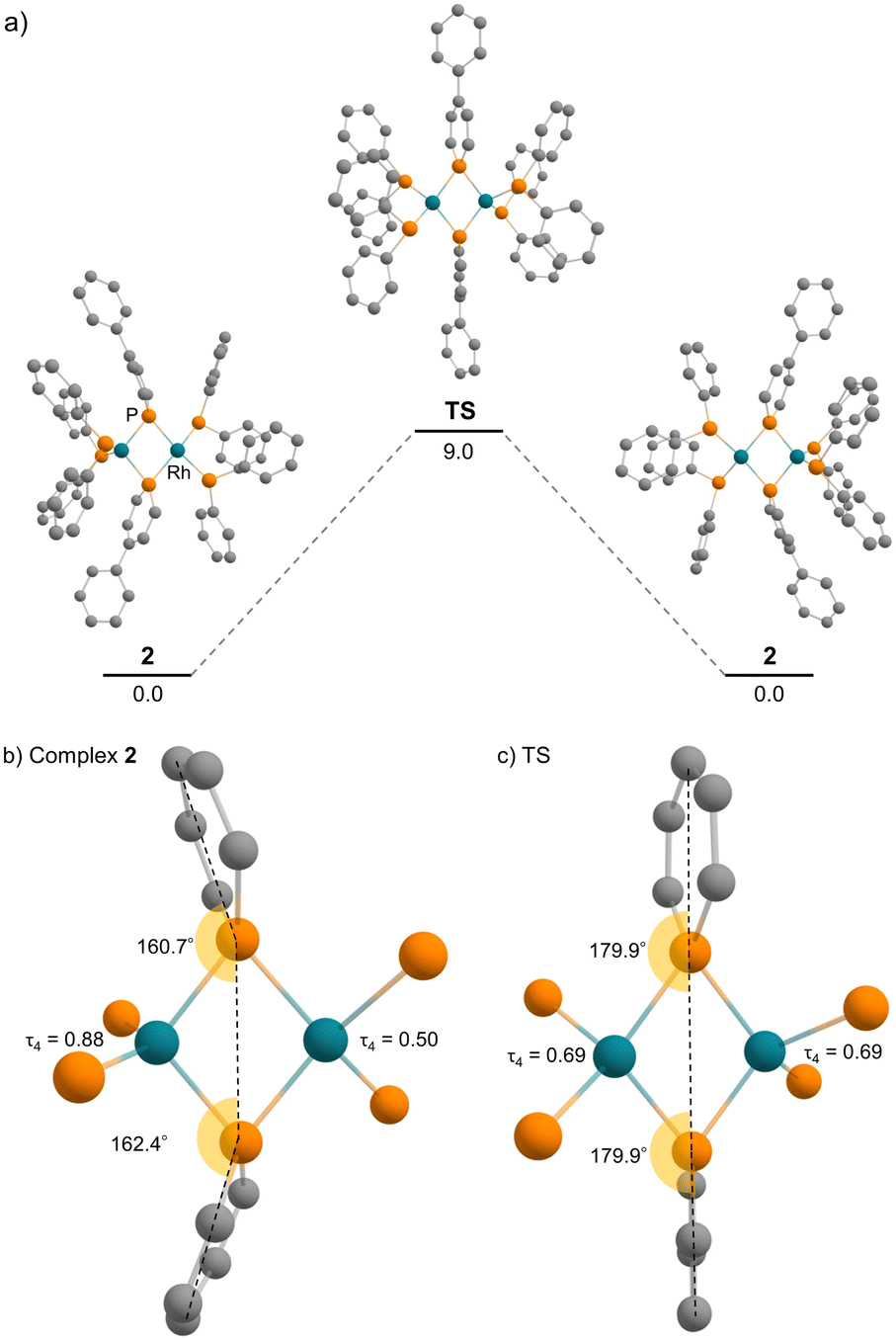
DFT calculations were employed to obtain deeper insight into the electron donation from the Rh(−I) centre to the phosphinine ring in complex 2. The calculation in the gas phase reproduced the unsymmetric structure of complex 2, which was observed in the solid state. In addition, a symmetric structure could not be located as a stable intermediate but exists as a transition state of the flipping of the phosphinine rings (Fig. 7). The activation energy is 9.0 kcal mol−1 (calculated at the ωB97XD/def2-TZVP//PBE0-D3/def2-SVP level), which supports the discussion that fast flipping of the phosphinine rings occurred in the solution state. This result suggests that the unsymmetric structure of complex 2 is favourable compared with the symmetric structure. Therefore, the unsymmetric structure was considered for further discussion.
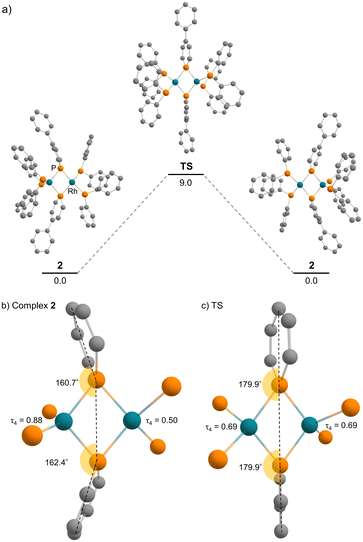 |
| | Fig. 7 (a) Energy diagram of the flipping of the phosphinine rings (unit: kcal mol−1); (b) calculated geometries around rhodium and phosphorus atoms of complex 2; (c) calculated geometries around rhodium and phosphorus atoms of the transition state. | |
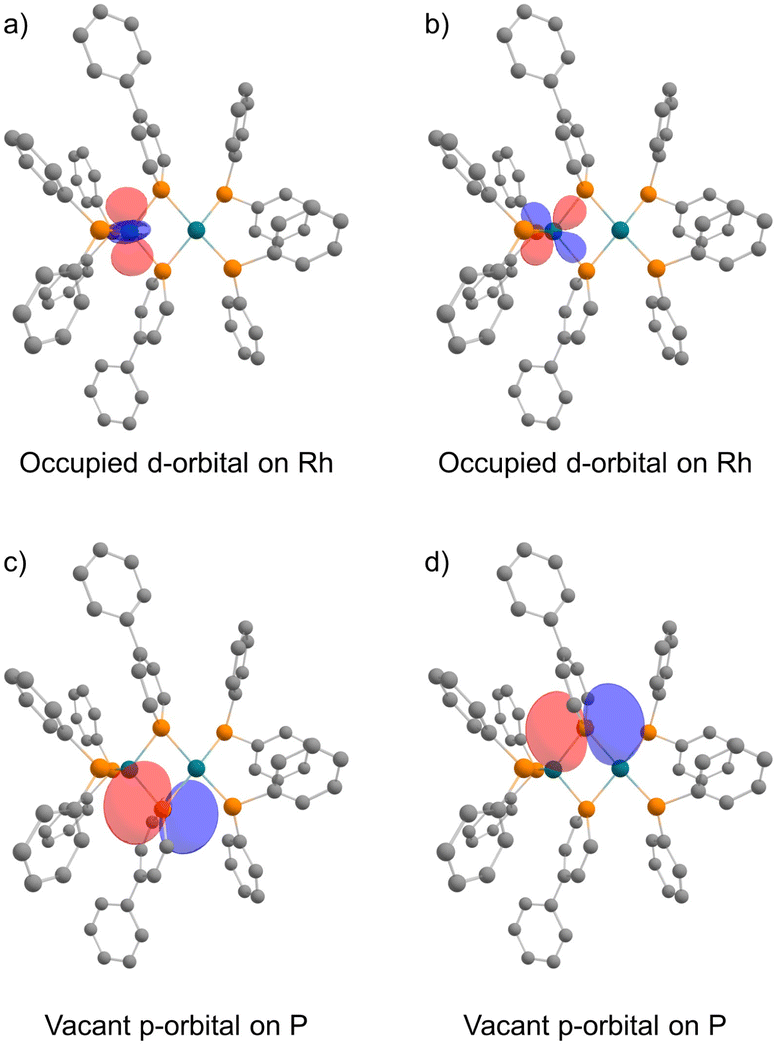
In the NBO analysis of complex 2 shown in Fig. 8 (calculated at the PBE0-D3/def2-TZVP//PBE0-D3/def2-SVP level), the vacant p-orbital of the phosphorus atom on phosphinine (Fig. 8a and b) and the occupied d-orbital of the Rh centre are overlapping each other (Fig. 8c and d). Second-order perturbation analysis suggested strong donation from the occupied d-orbital of the Rh centre to the vacant p-orbital of the P atoms on the phosphinine rings (see the ESI† for details). Interaction of these orbitals can be found in HOMO−40, HOMO−39, the HOMO, and the LUMO (Fig. 9). In HOMO−40 and HOMO−39, the bonding interaction of the square planar rhodium centre with the P atoms on the phosphinine rings also has some contribution. The existence of these orbitals accords with the electronic structure shown in Scheme 1, where the Rh(−I) centre donates d-electrons to the P atoms on the phosphinine rings and the lone pairs on the P atom are donated to the Rh(I) centre.
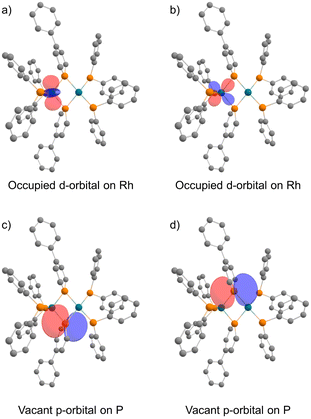 |
| | Fig. 8 Calculated NBOs of complex 2: (a) and (b) occupied d-orbitals on the Rh atom; (c) and (d) vacant p-orbitals on P atoms (hydrogens and phenylene linkers are omitted for clarity). | |
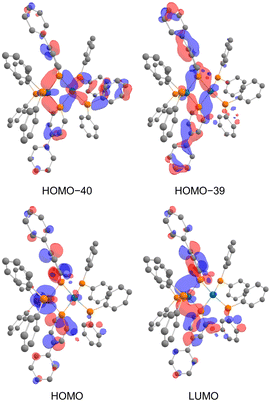 |
| | Fig. 9 Calculated molecular orbitals of complex 2 (hydrogens and phenylene linkers are omitted for clarity). | |
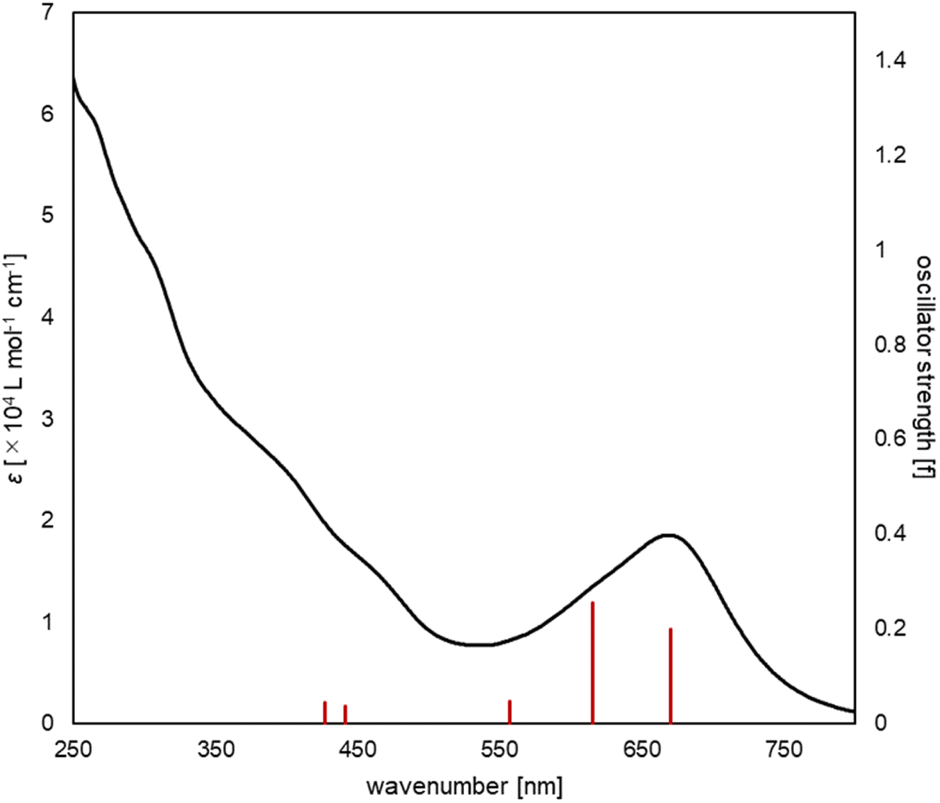
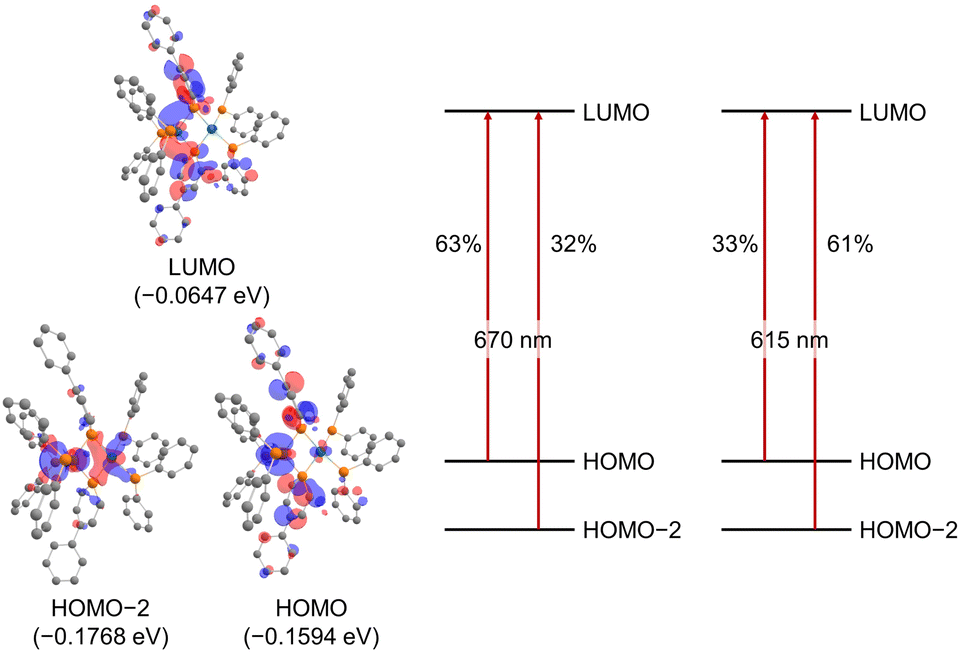
The UV/vis absorption spectrum of complex 2 is shown in Fig. 10. The absorption spectrum of complex 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) (1 × 10−5 mol L−1) showed maximum absorption at 669 nm. TD-DFT calculations (calculated at the PBE0-D3/def2-TZVP//PBE0-D3/def2-SVP level) showed that the absorption of complex 2 at 669 nm can be assigned to the HOMO-to-LUMO and HOMO−2-to-LUMO transition (λcalc = 670 nm (fcalc = 0.1999) and λcalc = 615 nm (fcalc = 0.2548), Fig. 11). As shown in Fig. 11, this transition involves a d–d transition process (HOMO → LUMO), as well as an MLCT process (HOMO−2 → LUMO) like the previous reports on the photochemical properties of phosphinine metal complexes.32,51–54
(1 × 10−5 mol L−1) showed maximum absorption at 669 nm. TD-DFT calculations (calculated at the PBE0-D3/def2-TZVP//PBE0-D3/def2-SVP level) showed that the absorption of complex 2 at 669 nm can be assigned to the HOMO-to-LUMO and HOMO−2-to-LUMO transition (λcalc = 670 nm (fcalc = 0.1999) and λcalc = 615 nm (fcalc = 0.2548), Fig. 11). As shown in Fig. 11, this transition involves a d–d transition process (HOMO → LUMO), as well as an MLCT process (HOMO−2 → LUMO) like the previous reports on the photochemical properties of phosphinine metal complexes.32,51–54
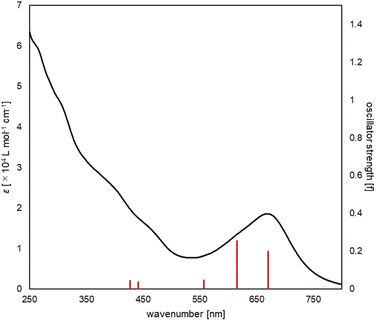 |
| | Fig. 10 UV/vis spectrum of complex 2 (black line, experimental, 1 × 10−5 M in THF), along with the oscillator strengths of complex 2 (red bars) obtained by the TD-DFT calculations. | |
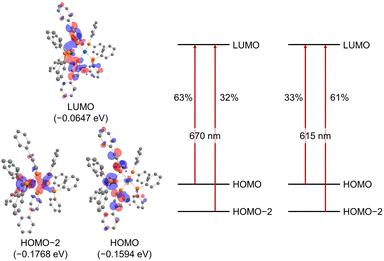 |
| | Fig. 11 Molecular orbitals of complex 2 (hydrogens and phenylene linkers are omitted for clarity) and major contributions to the absorption at 670 nm and 615 nm based on TD-DFT calculations (in THF). | |
Conclusions
This study presented the syntheses and structural analysis of dinuclear Rh(−I)/Rh(I) complex 2 bridged by phosphinine pincer ligands. The single-crystal X-ray crystallographic analysis showed an unsymmetric structure of complex 2 where the phosphinine ligands bridged two Rh centres in an unsymmetric mode. This suggests that complex 2 consists of a Rh(−I) centre and a Rh(I) centre. This electronic structure was supported by the DFT calculations, where back-donation from the tetrahedral Rh centre to the P atoms on the phosphinine rings was observed in the NBO analysis. As shown in this study, the phosphinine ligand gave rise to the unsymmetric electronic structure of a dinuclear metal complex. It will enable more precise control of the catalysis of dinuclear metal complexes.
Data availability
The data generated in this study are available in the main text or the ESI. Crystallographic data has been deposited at the joint Cambridge Crystallographic Data Centre (CCDC: 2260653 (2), 2260654 (3)). The NMR spectra and the cartesian coordinates of DFT optimized structure are provided in the ESI.†
Author contributions
K. M. and K. O. performed the synthesis and NMR experiments. K. M. and S. K. performed the crystallographic measurements. K. M. performed the DFT calculations. All authors analysed and discussed the results. K. M. prepared the manuscript with feedback from all authors. K. N. supervised the project.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
Part of this work was supported by “Advanced Research Infrastructure for Materials and Nanotechnology in Japan (ARIM)” of the Ministry of Education, Culture, Sports, Science and Technology (MEXT), (Grant Number JPMXP1222UT0194.). The computations were performed at the Research Center for Computational Science, Okazaki, Japan (Project: 23-IMS-C014). This work was supported by Grant-in-Aid for Challenging Research (Pioneering) JP22K19021, Noguchi institute research fund. K. M. is grateful to the Program for Leading Graduate Schools (MERIT-WINGS) and to the JSPS for a fellowship for young scientists.
Notes and references
- E. K. van den Beuken and B. L. Feringa, Tetrahedron, 1998, 54, 12985–13011 CrossRef CAS.
- J. I. van der Vlugt, Eur. J. Inorg. Chem., 2012, 2012, 363–375 CrossRef CAS.
- I. Bratko and M. Gómez, Dalton Trans., 2013, 42, 10664–10681 RSC.
- J. A. Mata, F. Ekkehardt Hahn and E. Peris, Chem. Sci., 2014, 5, 1723–1732 RSC.
- X. Huo, G. Li, X. Wang and W. Zhang, Angew. Chem., Int. Ed., 2022, 61, e202210086 CAS.
-
R. H. Crabtree, The Organometallic Chemistry of the Transition Metals, Wiley, 2014 Search PubMed.
- R. Colton and M. J. McCormick, Coord. Chem. Rev., 1980, 31, 1–52 CrossRef CAS.
- A. L. Sargent and M. B. Hall, J. Am. Chem. Soc., 1989, 111, 1563–1569 CrossRef CAS.
- A. A. Hock and O. S. Mills, Acta Crystallogr., 1961, 14, 139–148 CrossRef CAS.
- F. A. Cotton, B. A. Frenz and L. Kruczynski, J. Am. Chem. Soc., 1973, 95, 951–952 CrossRef CAS.
- F. A. Cotton and J. M. Troup, J. Am. Chem. Soc., 1974, 96, 1233–1234 CrossRef CAS.
- P. Macchi, L. Garlaschelli and A. Sironi, J. Am. Chem. Soc., 2002, 124, 14173–14184 CrossRef CAS.
- P. D. Burrow, A. J. Ashe III, D. J. Bellville and K. D. Jordan, J. Am. Chem. Soc., 1982, 104, 425–429 CrossRef CAS.
- M. Doux, A. Moores, N. Mézailles, L. Ricard, Y. Jean and P. L. Floch, J. Organomet. Chem., 2005, 690, 2407–2415 CrossRef CAS.
- C. Müller, L. E. E. Broeckx, I. de Krom and J. J. M. Weemers, Eur. J. Inorg. Chem., 2013, 2013, 187–202 CrossRef.
- P. Le Floch and F. Mathey, Coord. Chem. Rev., 1998, 178–180, 771–791 CrossRef CAS.
- P. L. Floch, Coord. Chem. Rev., 2006, 250, 627–681 CrossRef.
- N. T. Coles, A. Sofie Abels, J. Leitl, R. Wolf, H. Grützmacher and C. Müller, Coord. Chem. Rev., 2021, 433, 213729 CrossRef CAS.
- M. Doux, N. Mézailles, L. Ricard and P. Le Floch, Eur. J. Inorg. Chem., 2003, 2003, 3878–3894 CrossRef.
- A. Campos-Carrasco, L. E. E. Broeckx, J. J. M. Weemers, E. A. Pidko, M. Lutz, A. M. Masdeu-Bultó, D. Vogt and C. Müller, Chem.–Eur. J., 2011, 17, 2510–2517 CrossRef CAS.
- J. Leitl, M. Marquardt, P. Coburger, D. J. Scott, V. Streitferdt, R. M. Gschwind, C. Müller and R. Wolf, Angew. Chem., Int. Ed., 2019, 58, 15407–15411 CrossRef CAS PubMed.
- P. Le Floch, D. Carmichael, L. Ricard and F. Mathey, J. Am. Chem. Soc., 1993, 115, 10665–10670 CrossRef CAS.
- K. Waschbüch, P. Le Floch, L. Ricard and F. Mathey, Chem. Ber., 1997, 130, 843–849 CrossRef.
- N. Mézailles, P. Le Floch, K. Waschbüsch, L. Ricard, F. Mathey and C. P. Kubiak, J. Organomet. Chem., 1997, 541, 277–283 CrossRef.
- R. J. Newland, J. M. Lynam and S. M. Mansell, Chem. Commun., 2018, 54, 5482–5485 RSC.
- C. Tan, H. Tinnermann, V. Wee, S. Tan, S. Sung, Q. Wang and R. D. Young, J. Organomet. Chem., 2023, 986, 122617 CrossRef CAS.
- For a Mn2 complex reported in the following reference, the phosphinine ligands are regarded as 3e donors, and the Mn2 complex cannot be classified into the bridging modes shown in Fig. 1c: C. Elschenbroich, J. Six and K. Harms, Chem. Commun., 2006, 3429–3431 RSC.
- Y. Hou, Z. Li, Y. Li, P. Liu, C.-Y. Su, F. Puschmann and H. Grützmacher, Chem. Sci., 2019, 10, 3168–3180 RSC.
- B. Schmid, L. M. Venanzi, T. Gerfin, V. Gramlich and F. Mathey, Inorg. Chem., 1992, 31, 5117–5122 CrossRef CAS.
- M. T. Reetz, E. Bohres, R. Goddard, M. C. Holthausen and W. Thiel, Chem.–Eur. J., 1999, 5, 2101–2108 CrossRef CAS.
- Y. Mao, K. M. H. Lim, Y. Li, R. Ganguly and F. Mathey, Organometallics, 2013, 32, 3562–3565 CrossRef CAS.
- X. Chen, Z. Li, F. Yanan and H. Grützmacher, Eur. J. Inorg. Chem., 2016, 2016, 633–638 CrossRef CAS.
- M. H. Habicht, F. Wossidlo, T. Bens, E. A. Pidko and C. Müller, Chem.–Eur. J., 2018, 24, 944–952 CrossRef CAS PubMed.
- J. Leitl, P. Coburger, D. J. Scott, C. G. P. Ziegler, G. Hierlmeier, R. Wolf, N. P. van Leest, B. de Bruin, G. Hörner and C. Müller, Inorg. Chem., 2020, 59, 9951–9961 CrossRef CAS PubMed.
- M. J. Bakker, F. W. Vergeer, F. Hartl, K. Goubitz, J. Fraanje, P. Rosa and P. Le Floch, Eur. J. Inorg. Chem., 2000, 2000, 843–845 CrossRef.
- M. J. Bakker, F. W. Vergeer, F. Hartl, P. Rosa, L. Ricard, P. Le Floch and M. J. Calhorda, Chem.–Eur. J., 2002, 8, 1741–1752 CrossRef CAS.
- S. Giese, K. Klimov, A. Mikeházi, Z. Kelemen, D. S. Frost, S. Steinhauer, P. Müller, L. Nyulászi and C. Müller, Angew. Chem., Int. Ed., 2021, 60, 3581–3586 CrossRef CAS.
- F. Nief, C. Charrier, F. Mathey and M. Simalty, J. Organomet. Chem., 1980, 187, 277–285 CrossRef CAS.
- H. Lehmkuhl, R. Paul and R. Mynott, Liebigs Ann. Chem., 1981, 1981, 1139–1146 CrossRef.
- E. Yue, A. Petrov, D. S. Frost, L. Dettling, L. Conrad, F. Wossidlo, N. T. Coles, M. Weber and C. Müller, Chem. Commun., 2022, 58, 6184–6187 RSC.
- P. Rosa, L. Ricard, F. Mathey and P. Le Floch, Organometallics, 2000, 19, 5247–5250 CrossRef CAS.
- J. Abbenseth and J. M. Goicoechea, Chem. Sci., 2020, 11, 9728–9740 RSC.
- S. J. Hwang, A. Tanushi and A. T. Radosevich, J. Am. Chem. Soc., 2020, 142, 21285–21291 CrossRef CAS PubMed.
- M. J. Drance, A. Tanushi and A. T. Radosevich, J. Am. Chem. Soc., 2022, 144, 20243–20248 CrossRef CAS PubMed.
- K. Masada, S. Kusumoto and K. Nozaki, Angew. Chem., Int. Ed., 2022, 61, e202117096 CAS.
- K. Masada, S. Kusumoto and K. Nozaki, Organometallics, 2023, 42, 971–981 CrossRef CAS.
- L. Yang, D. R. Powell and R. P. Houser, Dalton Trans., 2007, 955–964 RSC.
- M. Carvalho, L. F. Wieserman and D. M. Hercules, Appl. Spectrosc., 1982, 36, 290–296 CrossRef CAS.
- J. P. Contour, G. Mouvier, M. Hoogewys and C. Leclere, J. Catal., 1977, 48, 217–228 CrossRef CAS.
- H. Kameo, Y. Hashimoto and H. Nakazawa, Organometallics, 2012, 31, 3155–3162 CrossRef CAS.
- J. Moussa, T. Cheminel, G. R. Freeman, L.-M. Chamoreau, J. A. G. Williams and H. Amouri, Dalton Trans., 2014, 43, 8162–8165 RSC.
- J. Moussa, L. M. Chamoreau and H. Amouri, RSC Adv., 2014, 4, 11539–11542 RSC.
- P. Roesch, J. Nitsch, M. Lutz, J. Wiecko, A. Steffen and C. Müller, Inorg. Chem., 2014, 53, 9855–9859 CrossRef CAS PubMed.
- Y. Li, Z. Li, Y. Hou, Y.-N. Fan and C.-Y. Su, Inorg. Chem., 2018, 57, 13235–13245 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence ,
Kiyosumi
Okabe
,
Kiyosumi
Okabe