
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRare earth dialkyl cations and monoalkyl dications supported by a rigid neutral pincer ligand: synthesis and ethylene polymerization†
Aathith
Vasanthakumar
,
Jeffrey S.
Price
and
David J. H.
Emslie
 *
*
Department of Chemistry, McMaster University, 1280 Main Street West, Hamilton, Ontario L8S 4M1, Canada. E-mail: emslied@mcmaster.ca
First published on 24th July 2023
Abstract
A palladium-catalyzed coupling reaction between 4,5-dibromo-2,7-di-tert-butyl-9,9-dimethylxanthene and 2 equiv. of 1,3-diisopropylimidazolin-2-imine afforded the rigid neutral 2,7-di-tert-butyl-4,5-bis(1,3-diisopropylimidazolin-2-imino)-9,9-dimethylxanthene (XII2) pincer ligand. Reaction of XII2 with YCl3(THF)3.5 provided [(XII2)YCl3] (1). However, compound 1 failed to react cleanly with 3 equiv. of LiCH2SiMe3, and the reaction of XII2 with [Y(CH2SiMe3)3(THF)2] afforded a complex mixture of products. To access group 3 alkyl complexes without the intermediacy of [(XII2)M(CH2SiMe3)3], the XII2 ligand was protonated using [H(OEt2)2][B(C6F5)4] to form [H(XII2)][B(C6F5)4], and subsequent reaction with [M(CH2SiMe3)3(THF)2] (M = Y, Sc) directly afforded the cationic scandium and yttrium dialkyl complexes [(XII2)M(CH2SiMe3)2][B(C6F5)4] {M = Y (2) and Sc (3)}. Reaction of 3 with B(C6F5)3 in C6D5Br afforded dicationic [(XII2)Sc(CH2SiMe2CH2SiMe3)][MeB(C6F5)3][B(C6F5)4] (4) featuring a CH2SiMe2CH2SiMe3 ligand, formed as a result of methyl anion abstraction from silicon, with concomitant migration of the neighbouring CH2SiMe3 group from scandium to silicon. The MeB(C6F5)3 anion in 4 forms a contact ion pair. By contrast, reaction of 1 with [CPh3][B(C6F5)3] in C6D5Br/toluene or o-C6H4F2/toluene afforded dicationic [(XII2)Sc(CH2SiMe3)(ηx-toluene)n][B(C6F5)4]2 (5). Compounds 2–4 showed negligible ethylene polymerization activity, whereas 5 is highly active (up to 870 kg mol−1 h−1 atm−1 in o-C6H4F2/toluene under 1 atm of ethylene at room temperature).
Introduction
Homogeneous rare earth (and transition metal) catalysts for olefin insertion polymerization are primarily monocationic alkyl species, generated by electrophilic abstraction of an alkyl group from a neutral polyalkyl pre-catalyst (which may itself be generated in situ) in arene or alkane solvent.1–3 Dicationic alkyl complexes, particularly those which are monometallic and can provide access to a vacant coordination site, offer new catalyst design possibilities and may afford particularly high polymerization activities due to enhanced electrophilicity of the metal centre. However, such complexes have rarely been described or utilized in catalysis (vide infra).A handful of monometallic rare earth alkyl dications which are stabilized through coordination of THF and/or a crown ether have been reported: [MR(THF)x]2+ (x = 4–6; R = Me,4,5 CH2Ar,6 or CH2SiMe3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 4,7), [M(CH2SiMe3)(THF)x(12-crown-4)]2+ (x = 2 or 3),7 [MR(12-crown-4)2]2+ (R = Me or CH2SiMe3),7–9 and [MMe(L)(THF)x]2+ (L = 15-crown-5 or 18-crown-6; x = 1 or 2),8,9 where M is a rare earth element.10–12 These complexes were generated in THF, and have not been reported to be substantially active ethylene or α-olefin polymerization catalysts, presumably due to a high degree of coordinative saturation. However, highly active ethylene polymerization catalysts were accessed by treatment of toluene solutions of [M(CH2SiMe3)3(THF)2] (M = Tm, Er, Y, Ho, Dy, Tb) with 5 equiv. of [HNMe2Ph][B(C6F5)4] in the presence of excess Al(CH2SiMe3)3 or AliBu3; one equivalent of [HNMe2Ph][B(C6F5)4] was insufficient to achieve high polymerization activities, suggesting that the active species is a coordinatively unsaturated alkyl dication. A similarly active ethylene polymerization catalyst was accessed via the reaction of [Y(CH2SiMe3)2(thf)4][Al(CH2SiMe3)4] with 5 equiv. of [HNMe2Ph][B(C6F5)4] in toluene in the presence of AliBu3. However, neither of the catalytically active species generated in toluene were spectroscopically or crystallographically characterized due to low solubility.4
4,7), [M(CH2SiMe3)(THF)x(12-crown-4)]2+ (x = 2 or 3),7 [MR(12-crown-4)2]2+ (R = Me or CH2SiMe3),7–9 and [MMe(L)(THF)x]2+ (L = 15-crown-5 or 18-crown-6; x = 1 or 2),8,9 where M is a rare earth element.10–12 These complexes were generated in THF, and have not been reported to be substantially active ethylene or α-olefin polymerization catalysts, presumably due to a high degree of coordinative saturation. However, highly active ethylene polymerization catalysts were accessed by treatment of toluene solutions of [M(CH2SiMe3)3(THF)2] (M = Tm, Er, Y, Ho, Dy, Tb) with 5 equiv. of [HNMe2Ph][B(C6F5)4] in the presence of excess Al(CH2SiMe3)3 or AliBu3; one equivalent of [HNMe2Ph][B(C6F5)4] was insufficient to achieve high polymerization activities, suggesting that the active species is a coordinatively unsaturated alkyl dication. A similarly active ethylene polymerization catalyst was accessed via the reaction of [Y(CH2SiMe3)2(thf)4][Al(CH2SiMe3)4] with 5 equiv. of [HNMe2Ph][B(C6F5)4] in toluene in the presence of AliBu3. However, neither of the catalytically active species generated in toluene were spectroscopically or crystallographically characterized due to low solubility.4
Rare earth alkyl dications which are free from oxygen-donor solvent or crown ether ligands are shown in Fig. 1. The scandium alkyl dications (A–B) were generated in situ via reactions of a neutral trialkyl precursor with two equiv. of [CPh3][B(C6F5)4] in chlorobenzene or dichloromethane (for A),13,14 or bromobenzene or toluene (for B),15 and were assigned on the basis of solution NMR studies, and because two equivalents of [CPh3][B(C6F5)4] were required to generate an active α-olefin polymerization catalyst.16 Neodymium complex C can be viewed as an alkyl dication with tight ion pairing of the two HB(C6F5)3− anions. This compound was characterized in solution and by X-ray crystallography, and was found to be a highly active catalyst for butadiene polymerization.17
 | ||
| Fig. 1 Rare earth alkyl dications (S = arene solvent) free from oxygen-donor solvent or crown ether ligands: the scandium complexes (A–B) were characterized in solution, whereas neodymium complex C was characterized in solution and the solid state. | ||
Herein we report the synthesis of a rigid neutral NON-donor pincer ligand, which has enabled the synthesis of scandium and yttrium dialkyl monocations, as well as the monoalkyl dications [(XII2)Sc(CH2SiMe2CH2SiMe3)][MeB(C6F5)3][B(C6F5)4] and [(XII2)Sc(CH2SiMe3)(ηx-toluene)n][B(C6F5)4]2. These dications were characterized by solution NMR spectroscopy, and the latter was found to be highly active for ethylene polymerization.
Results and discussion
Palladium-catalyzed coupling of 4,5-dibromo-2,7-di-tert-butyl-9,9-dimethylxanthene with 2 equiv. of 1,3-diisopropylimidazolin-2-imine afforded the neutral XII2 ligand (2,7-di-tert-butyl-4,5-bis(1,3-diisopropylimidazolin-2-imino)-9,9-dimethylxanthene; Scheme 1), in which the ether donor of the xanthene backbone is flanked by imidazolin-2-imine donors. Two resonance structures for the XII2 ligand are depicted in Scheme 1; the zwitterionic resonance structure is responsible for the high donor ability of the imidazolin-2-imine groups, and also provides a means for the imidazole rings to lie perpendicular to the plane of the xanthene backbone.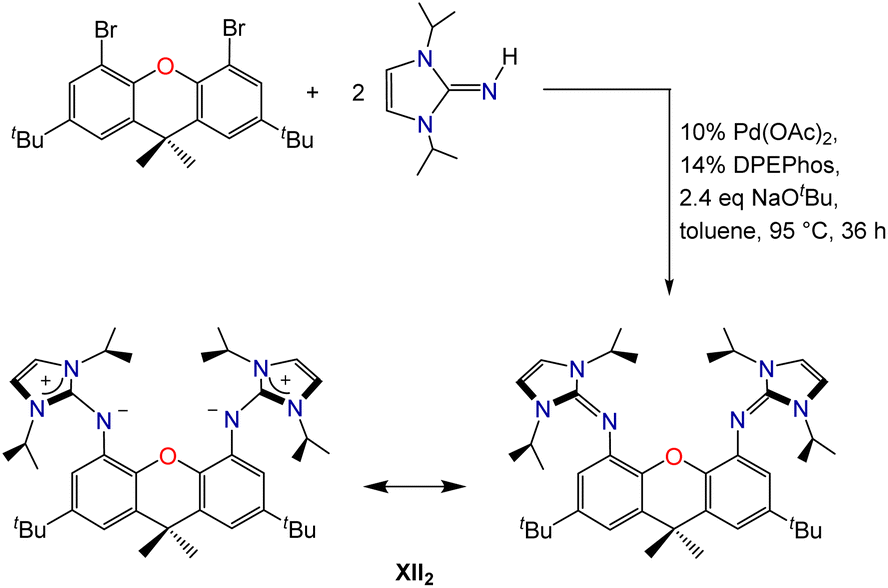 | ||
| Scheme 1 Synthesis and resonance structures of the neutral XII2 pincer ligand. | ||
The XII2 ligand is a neutral analogue of our recently reported AII2 monoanion (4,5-bis(1,3-diisopropylimidazolin-2-imino)-2,7,9,9-tetramethylacridanide; Fig. 2).18 It is also a close steric analogue of the dianionic XA2 ligand (4,5-bis(2,6-diisopropylanilido)-2,7-di-tert-butyl-9,9-dimethylxanthene; Fig. 2), which we reported in 2007.19 This dianionic pincer ligand, and relatives with alternative aryl substituents on nitrogen, have been employed for the synthesis of a broad range of highly reactive early transition metal,20 rare earth,21,22 actinide,19,23–30 and main group31–45 species.
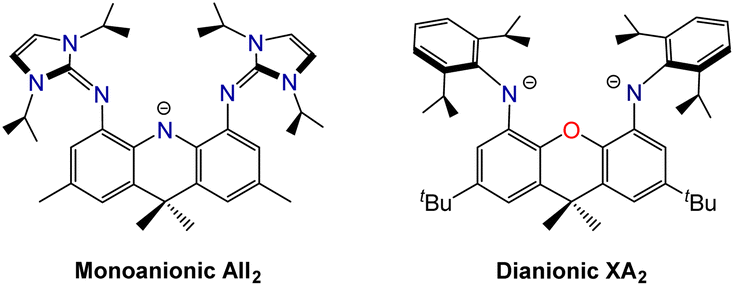 | ||
| Fig. 2 Monoanionic AII2 and dianionic XA2 pincer ligands which are structurally related to the neutral XII2 ligand in this work. | ||
Reaction of XII2 with YCl3(THF)3.5 cleanly generated [(XII2)YCl3] (1; Scheme 2), which was isolated in 82% yield, and gave rise to 1H and 13C NMR spectra corresponding to the expected C2v symmetry. However, compound 1 failed to react cleanly with 3 equiv. of LiCH2SiMe3 to afford [(XII2)Y(CH2SiMe3)3]. Furthermore, the reaction of XII2 with [Y(CH2SiMe3)3(THF)2] afforded a complex mixture of products. Based on these results, we reasoned that [(XII2)Y(CH2SiMe3)3] may be unstable as a consequence of steric crowding. A related, albeit less-pronounced, situation was previously observed in the uranium chemistry of the dianionic XA2 ligand, which shares a similar steric profile to XII2: [(XA2)U(CH2SiMe3)2] is thermally stable in solution at room temperature, whereas [Li(THF)x][(XA2)U(CH2SiMe3)3] decomposed over several days to afford SiMe4 and unidentified paramagnetic products.26
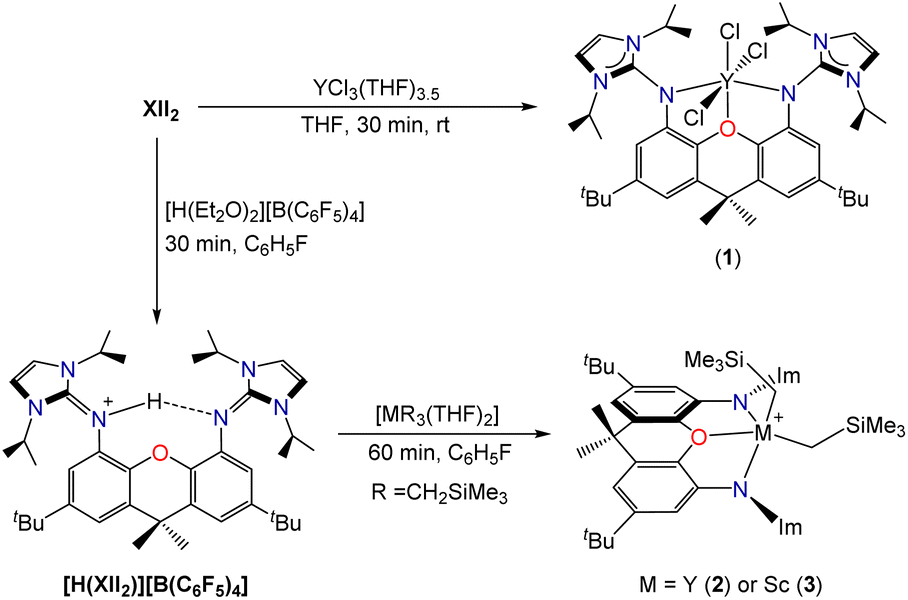 | ||
| Scheme 2 Synthesis of complexes 1–3 (Im = 1,3-diisopropylimidazol-2-ylidene). | ||
To bypass potentially unstable [(XII2)Y(CH2SiMe3)3], an alternative ligand attachment strategy was pursued (Scheme 2). Reaction of the XII2 ligand with [H(Et2O)2][B(C6F5)4] in fluorobenzene, followed by the addition of hexanes provided [H(XII2)][B(C6F5)4]·0.5 hexane as a white solid in 86% yield, and subsequent reaction with [M(CH2SiMe3)3(THF)2] (M = Y or Sc) afforded [(XII2)M(CH2SiMe3)2][B(C6F5)4] [M = Y (2) or Sc (3)] in greater than 90% yield. The room temperature 1H and 13C NMR spectra of 2 are consistent with apparent C2v symmetry, with the MCH2 signal at −0.77 ppm (1JC,H 102 Hz) in the 1H NMR spectrum and 37.2 ppm (d, 1JC,Y 41 Hz) in the 13C{1H} NMR spectrum. The 1H and 13C NMR spectra of scandium complex 3 are similar, but with broadened signals from atoms located outside of the plane of the ligand backbone, indicative of a fluxional process involving migration of the alkyl groups between coordination sites within and above/below the plane of the ligand backbone (with concomitant flexing of the ligand backbone). Consistent with this explanation, a sharper ScCH21H NMR signal was observed at 48 °C (−0.33 ppm), whereas upon cooling to −33 °C, this signal decoalesced to afford two broad peaks at 0.22 and −0.87 ppm. A similar fluxional process is presumably operative for 2, but would be expected to be more rapid due to reduced steric hindrance around the larger metal ion.
X-ray quality crystals of 2·2hexane were grown from flurobenzene/hexanes at −29 °C (Fig. 3). The XII2 ligand is κ3NON-coordinated, with a ligand backbone bend (the angle between the planes of the two aryl rings of the ligand backbone) of 38° (cf. 29° in [(AII2)Y(CH2SiMe3)2],18 and 25° in [(XA2)Y(CH2SiMe3)(THF)]21).46 One alkyl ligand is located above the plane of the XII2 ligand backbone, whereas the other is located approximately in the plane, affording a distorted square pyramidal geometry.
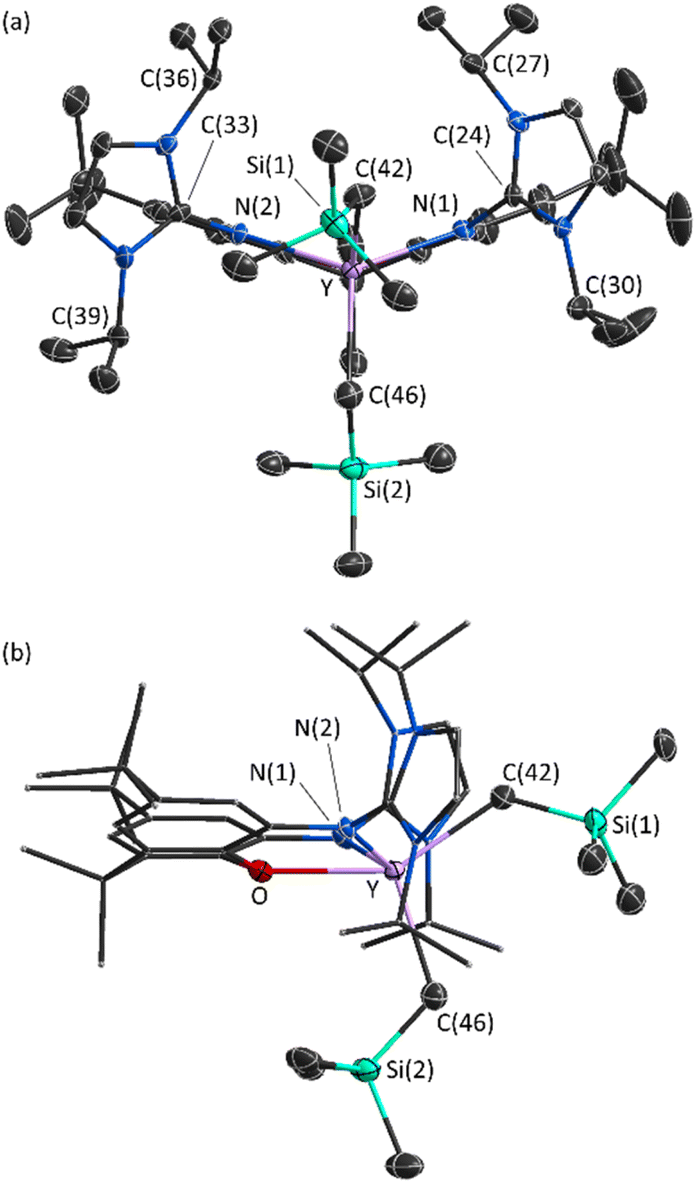 | ||
| Fig. 3 Front (a) and side (b) views of the cationic portion of the X-ray crystal structure of [(XII2)Y(CH2SiMe3)2][B(C6F5)4]·2hexane (2·2hexane). Ellipsoids are set to 30% probability, and hydrogen atoms are omitted for clarity. Both CMe3 groups and one SiMe3 group (attached to C(42)) are disordered, and only one orientation is shown. In view (b), all atoms of the XII2 ligand, except for O, N(1) and N(2) are shown in wireframe. Selected bond lengths (Å) and angles (°): Y–N(1) 2.336(4), Y–N(2) 2.330(3), Y–O 2.425(3), Y–C(42) 2.393(5), Y–C(46) 2.385(5), N(1)–C(24) 1.367(5), N(2)–C(33) 1.378(5), C(27)⋯C(36) 4.80, C(30)⋯C(39) 8.43, N(1)–Y–N(2) 122.1(1), O–Y–C(42) 145.5(2), O–Y–C(46) 107.4(2), C(42)–Y–C(46) 107.0(2), Y–C(42)–Si(1) 126.3(4), Y–C(42)–Si(1A) 122.8(7), Y–C(46)–Si(2) 119.6(2). | ||
The flanking imidazole rings are oriented approximately perpendicular to the plane of the ligand backbone (with angles of 80° and 87° between the plane of each imidazole ring and the adjacent aryl ring). Furthermore, the imine N(1)–C(24) and N(2)–C(33) distances {1.367(5) and 1.378(5) Å} are significantly longer than those in free imidazolin-2-imines such as (NHC)![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N{C6H4(OMe)-p} (1.308(2) Å; NHC = 1,3-diisopropyl-4,5-dimethylimidazol-2-ylidene)47 and [{κ2-2,6-C5H3N(CH2N{NHC})2}FeCl2] (1.294(3) Å; NHC = 1,3-di-tert-butylimidazol-2-ylidene).48 These observations are consistent with a substantial contribution from the zwitterionic resonance form of the imidazolin-2-imine donors.
N{C6H4(OMe)-p} (1.308(2) Å; NHC = 1,3-diisopropyl-4,5-dimethylimidazol-2-ylidene)47 and [{κ2-2,6-C5H3N(CH2N{NHC})2}FeCl2] (1.294(3) Å; NHC = 1,3-di-tert-butylimidazol-2-ylidene).48 These observations are consistent with a substantial contribution from the zwitterionic resonance form of the imidazolin-2-imine donors.
Cationic 2 is qualitatively isostructural with neutral [(AII2)Y(CH2SiMe3)2].18 However, the Y–C distances of 2.385(5) and 2.393(5) Å in 2 are considerably shorter than those in [(AII2)Y(CH2SiMe3)2] (2.434(2) and 2.482(2) Å). Similarly, the Y–N(1) and Y–N(2) distances of 2.330(3) Å and 2.336(4) Å in 2 are notably shorter than the Y–Nimidazolin-2-imine distances in [(AII2)Y(CH2SiMe3)2] (2.394(2), 2.421(2) Å).49 These bond metrics are indicative of a significantly more electron deficient yttrium center in cationic 2 than in neutral [(AII2)Y(CH2SiMe3)2].
X-ray quality crystals of 3·2PhF were also obtained from fluorobenzene/hexanes at −29 °C (Fig. S44†). The structure of 3 is analogous to that of 2, but with M–C and M–N bonds which are shorter by 0.14–0.18 Å, primarily reflecting the lower ionic radius of ScIII (0.745 Å) versus YIII (0.90 Å).50 The ligand backbone bend angle is slightly more obtuse (41°) than that in 2, and the N(1)–C(24) and N(2)–C(33) distances of 1.369(4) Å (for both) are equal within error to those in 2.
Reactions of compound 2 with B(C6F5)3 or [CPh3][B(C6F5)4] in bromobenzene or toluene/bromobenzene afforded mixtures of unidentified products accompanied by SiMe4. By contrast, the reaction of 3 with B(C6F5)3 in bromobenzene (Scheme 3) cleanly produced a new Cs symmetric product with a 1H NMR spectrum featuring SiMex signals at −0.12 and −0.31 ppm, integrating to 9H and 6H respectively, accompanied by two CH2 signals (2H each) at 0.85 and −0.66 ppm, and a broad singlet (3H) at 1.51 ppm (Fig. 4). These data point to the formation of dicationic [(XII2)Sc(CH2SiMe2CH2SiMe3)][MeB(C6F5)3][B(C6F5)4] (4), resulting from methyl anion abstraction from silicon, with concomitant migration of the neighbouring CH2SiMe3 group from scandium to silicon.51
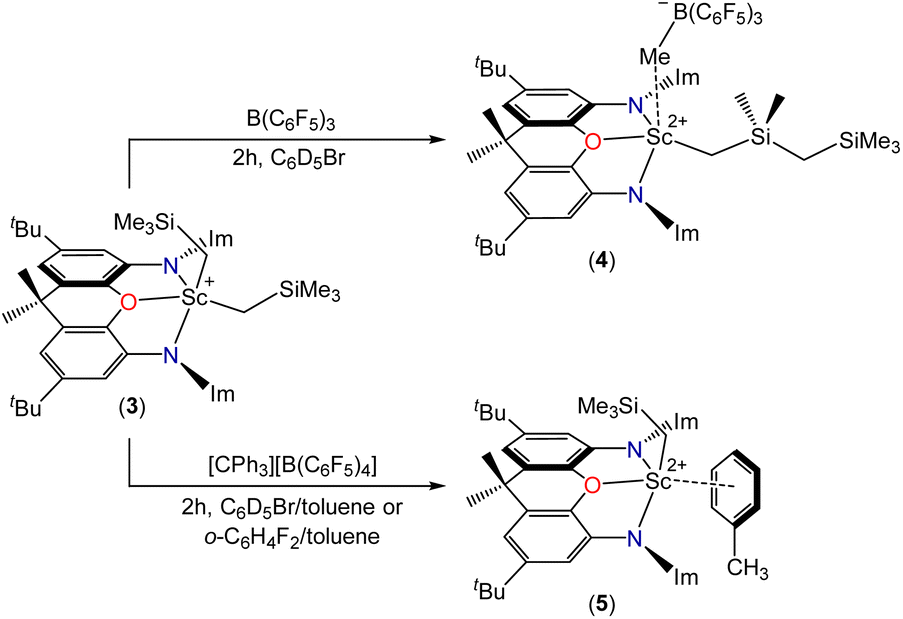 | ||
| Scheme 3 Synthesis of dications 4 and 5 (Im = 1,3-diisopropylimidazol-2-ylidene). In the structures of 4 and 5, the alkyl groups may be coordinated within or above the plane of the ligand backbone, and only one possibility is shown. The hapticity of toluene coordination in 5 is unknown (toluene coordination is proposed because attempted syntheses of 5 in the absence of toluene did not afford a stable product). | ||
 | ||
| Fig. 4 Alkyl region of the 1H NMR spectra of (a) [(XII2)Sc(CH2SiMe3)2][B(C6F5)4] (3), (b) [(XII2)Sc(CH2SiMe2CH2SiMe3)][MeB(C6F5)3][B(C6F5)4] (4) generated in situ, and (c) [(XII2)Sc(CH2SiMe3)(ηx-toluene)n][B(C6F5)4]2 (5) generated in situ in the presence of 5 equiv. of toluene. All spectra are in C6D5Br. The spectra of 3 and 5 are at 298 K, whereas the spectrum of 4 is at 252 K. | ||
The 1H–13C and 1H–29Si HMBC NMR spectra provide further support the formation of a CH2SiMe2CH2SiMe3 ligand. For example, in the 1H–13C HMBC (Fig. S24†), the γ-CH2 carbon signal couples to the proton signals of both the SiMe2 and SiMe3 groups, and the SiMe2 proton signal couples to both the α-CH2 and γ-CH2 carbon signals. In the 1H–29Si HMBC (Fig. S27†), the α-CH2 and SiMe2 protons couple to just one silicon center (SiMe2), the SiMe3 protons couple to a different silicon center (SiMe3), and the γ-CH2 group appears to couple to both silicon centers, although this is harder to distinguish due to overlapping cross-peaks.
The 19F and 11B NMR spectra are also consistent with the proposed structure of 4. For example, the 11B NMR spectrum of 4 features two sharp signals corresponding to the B(C6F5)4 (δ −16.2 ppm) and MeB(C6F5)3 (δ −15.0 ppm) anions. The latter chemical shift is comparable to that in previously reported scandium contact ion pairs involving a MeB(C6F5)3 anion.51–53 The 19F NMR spectrum shows the expected set of three signals for a free B(C6F5)4 anion, accompanied by another set of signals for the MeB(C6F5)3 anion {δ −133.27 (o-F), −157.57 (p-F), −162.09 (m-F)}. The large difference in the chemical shift between the meta and para fluorine signals {Δδm,p} of the MeB(C6F5)3 anion (4.52 ppm) is indicative of a contact ion pair where the borate interacts with the metal centre via the methyl group.54 Contact ion pairing is also indicated by the high frequency chemical shift of the BMe methyl group in the 1H NMR spectrum (1.51 ppm), relative to that of the free anion (1.13 ppm).54
Analogous reactivity (leading to a monocation rather than a dication) has previously been reported.51,55,56 Piers et al. described the reaction of [(nacnac)Sc(CH2SiMe3)2] {nacnac = ArNC(tBu)CHC(tBu)NAr; Ar = 2,6-C6H3iPr2} with B(C6F5)3 to afford the contact ion pair [(nacnac)Sc(CH2SiMe2CH2SiMe3)][MeB(C6F5)3],51 and Gordon et al. reported the reaction of [(κ3-L)Lu(CH2SiMe3)2] {L = NC5H3(CMeNAr)(CMe2NAr)-2,6; Ar = 2,6-C6H3iPr2} with B(C6F5)3 in the presence of THF to form the solvent-separated ion pair [(κ3-L)Lu(CH2SiMe2CH2SiMe3)(THF)][MeB(C6F5)3].55 Notably, the 1H NMR chemical shifts for the CH2SiMe2CH2SiMe3 ligand and MeB(C6F5)3− anion in 4 are very similar to those in [(nacnac)Sc(CH2SiMe2CH2SiMe3)][MeB(C6F5)3]: 0.02 (SiMe3), −0.20 (SiMe2), 1.16 (α-CH2), −0.73 (γ-CH2) and 1.51 (BMe) ppm.51
The reaction of 3 with a slight excess of [CPh3][B(C6F5)4] in bromobenzene was also investigated, and with rapid stirring in the presence of 5 equiv. of toluene, the dicationic monoalkyl complex [(XII2)Sc(CH2SiMe3)(ηx-toluene)n][B(C6F5)4]2 (5; Scheme 3) was generated over the course of 2 hours. Compound 5 gave rise to a ScCH2 signal at 0.70 ppm (2H) in the 1H NMR spectrum (Fig. 4), and 63.9 ppm in the 13C NMR spectrum. The expected 3JC,H correlations were observed between the ScCH2 and SiMe3 groups in the 1H–13C HMBC NMR spectrum, and 11B and 19F NMR spectra confirmed that the B(C6F5)4 anions are intact, ruling out C6F5 transfer from boron to scandium. Compound 5 exhibits apparent C2v symmetry in solution (between 25 and −33 °C; measurements at lower temperature were not possible due to the melting point of bromobenzene, insolubility of 5 in toluene, and decomposition of 5 in dichloromethane), indicating that the alkyl group migrates rapidly between positions above and below the plane of the ligand backbone, or less likely, is located within the plane of the ligand backbone.
Dicationic 5 is poorly soluble, so the reaction afforded a bright orange solution accompanied by a dark orange oil which precipitated over the course of the 2 hour reaction period. The formation of 5 was accompanied by previously described byproducts of trimethylsilylmethyl group abstraction by the trityl cation: Ph3CH as well as a product attributed to “Ph3CCH2SiMe3” (one of two typically observed isomers).‡,19,57
Bromobenzene solutions of 5, in the presence of five equivalents of toluene, were stable for hours at room temperature, with a substantial amount of the dication remaining after two days. However, in the absence of toluene, the 1:1 reaction of [CPh3][B(C6F5)4] with 3 in C6D5Br resulted in multiple unidentified products, as well as SiMe4, indicative of decomposition. This strongly suggests that compound 5 is stabilized by toluene π-coordination (i.e. n = 1). Efforts to obtain crystals of 5 were unsuccessful, and 1H NMR spectra at −33 °C did not show separate signals for free and coordinated toluene (consistent with the apparent C2v symmetry of 5 at this temperature). However, arene π-coordination has frequently been observed for sterically similar monoalkyl monocations of the XA2 ligand, such as [(XA2)ZrMe(η6-toluene)]+,20 [(XA2)Th(CH2SiMe3)(η6-benzene)]+, [(XA2)Th(CH2Ph)(η6-toluene)]+,29 [(XA2)U(CH2SiMe3)(η6-benzene)]+, and [(XA2)U(CH2SiMe3)(η3-C6H5R)]+ (R = Me or F).30
Dicationic 4 (a contact ion pair) showed negligible ethylene polymerization activity in fluorobenzene under 1 atm of ethylene at room temperature. By contrast, dicationic 5 proved to be a highly3 active catalyst for ethylene polymerization: exposure of a 0.2 mM solution of 5 in toluene/o-difluorobenzene to 1 atm of ethylene at room temperature (with a water bath around the flask),§ followed by quenching with acidified methanol after 3 minutes, afforded a polymerization activity of 870 kg mol−1 h−1 atm−1 (Table 1). A similar activity of 740 kg mol−1 h−1 was obtained when the reaction was quenched after 2 minutes. However, a lower activity of 570 kg mol−1 h−1 atm−1 was achieved after 5 minutes, perhaps as a result of ineffective stirring due to precipitation of substantial amounts of polymer, catalyst entrapment within the precipitated polymer, and/or catalyst decomposition arising from the exothermic nature of the reaction. Attempted polymerization in neat o-difluorobenzene afforded (with quenching after 3 minutes) a lower polymerization activity of 170 kg mol−1 h−1 atm−1, presumably due to significant catalyst decomposition in the absence of toluene.
| Solvent | Polymerization time (min) | Yield (g) | Activity (kg mol−1 h−1 atm−1) | M w (g mol−1) | M w/Mn |
|---|---|---|---|---|---|
|
a The catalyst solution was generated in situ by stirring 15 mg of [(XII2)Sc(CH2SiMe3)2][B(C6F5)4] (3) with 1 equiv. of [CPh3][B(C6F5)4] in 3 mL of solvent (either a 1:2 mixture of toluene and o-C6H4F2, or neat o-C6H4F2) for 2 hours, followed by the addition of an additional 40 mL of solvent (either a 3:1 mixture of toluene and o-C6H4F2, or neat o-C6H4F2).
b Values from GPC are relative to polyethylene standards. Additional data (Mp, Mz, Mv and Mp) is provided in the ESI.
|
|||||
| Toluene/o-C6H4F2 | 2 | 0.239 | 741 | 65700 |
1.40 |
| Toluene/o-C6H4F2 | 3 | 0.420 | 868 | 111880 |
1.41 |
| Toluene/o-C6H4F2 | 5 | 0.456 | 565 | 202100 |
2.06 |
| o-C6H4F2 | 3 | 0.135 | 168 | 170550 |
1.29 |
The polydispersity of the polyethylene produced in these experiments was fairly narrow, with values of 1.40 and 1.41 when the reaction was carried out in toluene/o-difluorobenzene and quenched after 2 or 3 minutes, respectively (Table 1), indicative of single-site catalysis. The weight average molecular weight (Mw) of the polyethylene also increased with increasing reaction time; from 66 kg mol−1 when the reaction was quenched after 2 minutes, to 112 kg mol−1 after 3 minutes, and 202 kg mol−1 after 5 minutes, (although after 5 minutes, a higher polydispersity of 2.06 was observed).
The polymerization activity of 5 is comparable to that of [Y(CH2SiMe3)3(THF)2] activated with 5 equiv. of [HNMe2Ph][B(C6F5)4] in toluene to generate a dication in situ {activities between 272 and 1840 kg mol−1 h−1 atm−1 were obtained under 5 atm of ethylene in the presence of 200 equiv. of MAO or AlR3; R = iBu or CH2SiMe3}, although it is notable that under comparable conditions, the analogous reaction with [Sc(CH2SiMe3)3(THF)2] exhibited negligible polymerization activity.4
Summary and conclusions
Palladium-catalysed coupling afforded the neutral XII2 pincer ligand, 2,7-di-tert-butyl-4,5-bis(1,3-diisopropylimidazolin-2-imino)-9,9-dimethylxanthene (XII2), and reaction with YCl3(THF)3.5 provided [(XII2)YCl3] (1). However, compound 1 failed to react cleanly with 3 equiv. of LiCH2SiMe3, and the reaction of XII2 with [Y(CH2SiMe3)3(THF)2] afforded a complex mixture of products. To access group 3 alkyl complexes without the intermediacy of [(XII2)Y(CH2SiMe3)3], the XII2 ligand was protonated to form [H(XII2)][B(C6F5)4], and subsequent reaction with [M(CH2SiMe3)3(THF)2] (M = Y, Sc) directly afforded the cationic dialkyl complexes [(XII2)M(CH2SiMe3)2][B(C6F5)4] {M = Y (2) and Sc (3)}. Cations 2 and 3 feature substantially shorter M–C and M–N bond distances than in the isostructural neutral AII2 complexes, [(AII2)M(CH2SiMe3)2] (M = Y or Sc),18 indicative of a much more electrophilic metal centre in 2 and 3.Compound 2 did not react cleanly with B(C6F5)3 or [CPh3][B(C6F5)4]. By contrast, reaction of 3 with B(C6F5)3 in C6D5Br afforded the dicationic contact ion pair [(XII2)Sc(CH2SiMe2CH2SiMe3)][MeB(C6F5)3][B(C6F5)4] (4), which features a CH2SiMe2CH2SiMe3 ligand, presumably resulting from methyl anion abstraction from silicon with concomitant migration of the neighbouring CH2SiMe3 group from scandium to silicon. This type of reactivity is uncommon, but has previously been observed in the synthesis of a scandium and a lutetium alkyl monocation. Alternatively, reaction of 3 with [CPh3][B(C6F5)3] in C6F5Br/toluene or o-C6H4F2/toluene afforded dicationic [(XII2)Sc(CH2SiMe3)(ηx-toluene)n][B(C6F5)4]2 (5). Compounds 4 and 5 are extremely rare examples of monometallic rare earth alkyl dications, especially those free from ether solvent or crown-ether coordination. The reactivity of 3 with CPh3+ also contrasts that of [(AII2)Y(CH2SiMe3)2] with CPh3+ which generated [(AII2-CH2SiMe3)Y(CH2SiMe3)2][B(C6F5)4] (AII2-CH2SiMe3 is a neutral tridentate ligand with a central amine donor flanked by imidazolin-2-imine groups) in approx. 20% yield, accompanied by HCPh3, an unidentified paramagnetic product, and a colourless precipitate, likely via a pathway involving initial oxidation of the acridanide ligand backbone.18
Compounds 2–4 showed negligible ethylene polymerization activity. By contrast, dicationic 5 is a highly active catalyst (up to 870 kg mol−1 h−1 atm−1 in o-C6H4F2/toluene under 1 atm of ethylene at room temperature). This illustrates the utility of highly electrophilic dicationic alkyl species for olefin polymerization, and highlights the ability of the neutral XII2 pincer ligand to serve as a robust ancillary ligand, capable of stabilizing extremely reactive organometallic species. Ligand rigidity can be anticipated to be particularly important in the case of neutral ligands, to prevent dissociation of one or more neutral donor. The XII2 ligand is sterically analogous to our dianionic XA2 pincer ligand (4,5-bis(2,6-diisopropylanilido)-2,7-di-tert-butyl-9,9-dimethylxanthene), which has been employed by our group and others for the synthesis of highly reactive organometallic complexes of the early transition metals and f-elements, as well as a range of reactive main group species.
Experimental
General experimental
An argon-filled M-Braun UNIlab glovebox equipped with a −29 °C freezer was employed for the manipulation and storage of all air sensitive compounds, and reactions were performed on a double manifold vacuum line (with all glass–glass connections, rather than connections via hose tubing) using standard techniques. A Fisher Scientific Ultrasonic FS-30 bath was used to sonicate reaction mixtures where indicated. The scandium and yttrium compounds reported in this article are very air and moisture sensitive, and the vacuum line operated at <5 mTorr. Argon (99.999%) and ethylene (99.999%) were purchased from Praxair, and both gases were further purified by passage through an Oxisorb-W scrubber from Matheson Gas Products.Hexanes and THF were initially dried and distilled at atmospheric pressure from sodium/benzophenone, while toluene was dried and distilled at atmospheric pressure from sodium. These solvents were then stored over an appropriate drying agent (toluene, THF = Na/Ph2CO; hexanes = Na/Ph2CO/tetraglyme). Fluorobenzene, 1,2-difluorobenzene, C6D5Br, and CD2Cl2 were dried over 4 Å molecular sieves, and C6D6 was dried over sodium/benzophenone; these solvents were degassed via three freeze–pump–thaw cycles. All solvents were introduced to reactions or solvent storage flasks via room temperature vacuum transfer with condensation at −78 °C (except for C6D5Br which was distilled at elevated temperature in vacuo).
4,5-Dibromo-2,7-di-tert-butyl-9,9-dimethylxanthene,19 [Y(CH2SiMe3)3(THF)2],58 [Sc(CH2SiMe3)3(THF)2],59 B(C6F5)3,60,61 and 1,3-diisopropylimidazolin-2-imine62 were synthesized using literature procedures. [H(Et2O)2][B(C6F5)4] was prepared using a modification of the literature synthesis:63 a 2.0 M solution of HCl in OEt2 (100 mL) was added via canula to a −78 °C solution of K[B(C6F5)4] (10.0 g) in OEt2 (30 mL); the reaction was stirred for 4 h at −30 °C before removal of the cold bath, filtration to remove KCl, and evaporation of the filtrate to dryness; the resulting white solid was washed with 10 mL of OEt2 and then dried in vacuo to afford 6.5–8.0 g of the product.
LiCH2SiMe3 (1.0 M in pentane), 1,3-diisopropylimidazolium chloride, HCl (2.0 M in OEt2), anhydrous YCl3, anhydrous ScCl3, Pd(OAc)2, NaOtBu, and DPEPhos were purchased from Sigma Aldrich. [CPh3][B(C6F5)4] was purchased from Alfa Aesar. K[B(C6F5)4] was purchased from Boulder Scientific. YCl3(THF)3.5 and ScCl3(THF)3 were obtained by refluxing anhydrous MCl3 (M = Y, Sc) in dry THF for 24 hours, followed by removal of solvent under vacuum. Deuterated solvents were purchased from Cambridge Isotope Laboratories.
Combustion elemental analyses were performed by Midwest Microlab, LLC in Indianapolis, Indiana. NMR spectroscopy [1H, 13C{1H}, DEPT-Q, COSY, HSQC, 1H–13C and 1H–29Si HMBC, 19F, 11B, 29Si] was performed on a Bruker AV-600 or AV-500 Spectrometer. All 1H NMR and 13C NMR spectra were referenced relative to SiMe4 using the resonance of the deuterated solvent (13C NMR) or protio impurity in the deuterated solvent (1H NMR); in 1H NMR, 7.16 ppm for C6D6, 5.32 ppm for CD2Cl2, and 7.30, 7.02 and 6.94 ppm for C6D5Br; in 13C NMR, 128.06 ppm for C6D6, 54.00 ppm for CD2Cl2, and 130.90, 129.41, 126.24 and 122.17 ppm for C6D5Br. All NMR spectra were obtained at 298 K unless otherwise specified.
X-ray crystallographic analyses were performed on crystals coated in Paratone oil and mounted on a Bruker SMART APEX II diffractometer with a 3 kW sealed-tube Mo generator and APEX II CCD detector in the McMaster Analytical X-Ray (MAX) Diffraction Facility. A semi-empirical absorption correction was applied using redundant and symmetry related data. Raw data was processed using XPREP (as part of the APEX v2.2.0 software), and solved by intrinsic (SHELXT)64 methods. Structures were completed by difference Fourier synthesis and refined with full-matrix least-squares procedures based on F2. In all cases, non-hydrogen atoms were refined anisotropically and hydrogen atoms were generated in ideal positions and then updated with each cycle of refinement. Refinement was performed with SHELXL65 in Olex2.66 Two equivalents of disordered solvent (C6H5F or C6H14) were masked in the structures of 2 and 3 using the BYPASS method within Olex2.67 Definitive identification of the masked solvent as either C6H5F or C6H14 was not possible given the identical number of electrons in both solvents. For 2, two equivalents of C6H14 were masked, while for 3, two equivalents of C6H5F were masked, because the difference map for the un-masked data contained maxima (in the void between molecular units) that roughly corresponded to a 6-membered ring.
Polyethylene molecular weights and molecular weight distributions were obtained using a Polymer Labs (now an Agilent company) PL-220 gel permeation chromatograph with 1,2,4-trichlorobenzene as the solvent at a flow rate of 1 mL min−1 and at 145 °C. 2,6-Di-tert-butyl-4-methylphenol (BHT) at a concentration of 0.5 g L−1 was used as a stabilizer in the solvent. An injection volume of 400 μL was used with a nominal polymer concentration of 1 g L−1. Dissolution of the stabilized sample was carried out by heating at 150 °C for 5 h with occasional agitation. Three Waters HT-6E columns (7.8 × 300 mm) were used and calibrated with a broad linear polyethylene standard (Phillips Marlex® BHB 5003) whose molecular weight had previously been determined.
2,7-Di-tert-butyl-4,5-bis(1,3-diisopropylimidazolin-2-imino)-9,9-dimethylxanthene (XII2)
A mixture of Pd(OAc)2 (46.7 mg, 0.208 mmol), DPEPhos (156 mg, 0.29 mmol), NaOtBu (479 mg, 2.49 mmol) was dissolved in approximately 20 mL of toluene in a 100 mL bomb, and stirred at room temperature for 10 minutes in the glovebox. A solution of 1,3-diisopropylimidazolin-2-imine (720 mg, 4.3 mmol) and 4,5-dibromo-2,7-di-tert-butyl-9,9-dimethylxanthene (1.00 g; 2.08 mmol) in approximately 30 mL of toluene was added to the reaction mixture. The reaction mixture was sealed in the 100 mL bomb and placed in a room temperature oil bath which was heated slowly (over several hours) to 95 °C, and maintained at this temperature for 48 h {note: placing the flask directly into a 95 °C oil bath, or cooling and re-heating before the 48 h at 95 °C had elapsed, generated undesired byproducts}. The reaction solution was passed through a pad of Celite and volatiles were removed under reduced pressure to afford a dark brown oil. Under air, the product was extracted using approximately 60 mL of CH2Cl2 and was washed with 60 mL of water. The resulting aqueous layer was additionally extracted with 30 mL of CH2Cl2 and the organic layers were combined. The organic layer was dried over MgSO4 and gravity filtered. Volatiles were removed from the filtrate under reduced pressure to afford a sticky dark brown solid. This solid was dissolved in approximately 50 mL of hexanes and was passed through a pad of Celite, yielding a dark orange filtrate. The filtrate was concentrated to approximately 10 mL and was placed at −18 °C overnight. The resulting beige powder was dried for 12 h at 80 °C under reduced pressure to yield 609 mg of XII2 (45% yield). 1H NMR (C6D6, 600 MHz, 298 K): δ 7.13 (s. 4H, CH1,8 & CH3,6), 5.92 (s, 4H, N-CH), 4.45 (sept, 4H, 3JH,H 7 Hz, CHMe2), 1.82 (s, 6H, CMe2), 1.38 (s, 18H, CMe3) 1.00 (d, 24H, 3JH,H 7 Hz) ppm. 13C{1H} NMR (C6D6, 151 MHz, 298 K): δ 144.64 (s, NCN), 144.47 (s, C2,7), 142.77 (s, C4,5), 140.09 (s, C11,12) 130.46 (s, C10,13), 118.00 (s, CH1,8), 112.02 (CH3,6), 107.73 (s, N-CH), 45.26 (s, CHMe2), 35.57 (s, C9). 34.58 (s, CMe3), 32.03 (s, CMe2 & CMe3), 21.78 (s, CHMe2) ppm. C41H60N6O1 (652.95 g mol−1): calcd C 75.42, H 9.26, N 12.87%; found. C 75.63, H 9.47, N 12.51%.[(XII2)YCl3] (1)
A solution of XII2 (100 mg, 0.153 mmol) in 5 mL of THF was added dropwise to a solution of [YCl3(THF)3.5] (69 mg, 0.153 mmol) in THF (5 mL) at room temperature and the reaction was stirred for 1 hour. Approximately 40 mL of hexanes was added to the reaction, resulting in the precipitation of a white solid. The solution was decanted, and the white solid was further dried under vacuum for 1 hour. The solid was then washed with hexanes (3 × ∼5 mL) and dried under vacuum for 2 hours, yielding 1 as an air-sensitive white solid (106 mg, 82% yield). 1H NMR (CD2Cl2, 600 MHz, 298 K): δ 7.07 (s, 4H, N-CH), 6.81 (s, 2H, CH1,8), 5.65 (s, 2H, CH3,6), 5.08 (sept, 4H, 3JH,H 7 Hz, CHMe2), 1.73 (s, 6H, CMe2), 1.54 (d, 12H, 3JH,H 7 Hz, CHMe2), 1.29 (d, 12H, 3JH,H 7 Hz, CHMe2), 1.20 (s, 18H, CMe3) ppm. 13C{1H} NMR (CD2Cl2, 151 MHz, 298 K): δ 148.39 (s, NCN), 147.12 (s, C2,7), 141.66 (s, C4,5), 137.89 (C11,12), 129.23(s, C10,13), 114.79 (s, N-CH), 112.94 (s, CH1,8), 109.72 (s, CH3,6), 48.81 (s, CHMe2), 34.75 (s, CMe3), 34.38 (s, C9), 34.04 (s, CMe2), 31.50 (s, CMe3), 23.63, 23.33 (2 × s, CHMe2) ppm. C41H60N6O1Cl3Y (848.22 g mol−1): calcd C 58.06, H 7.13, N 9.91%; found. C 58.33, H 7.16, N 9.43%.[H(XII2)][B(C6F5)4]·0.5hexane
A solution of [H(Et2O)2][B(C6F5)4] (127 mg, 0.153 mmol) in approximately 10 mL of C6H5F was added dropwise to a solution of XII2 (1) (100 mg, 0.153 mmol) in C6H5F (5 mL) at room temperature. The reaction was stirred for 1 hour and 40 mL of hexanes was added, which resulted in precipitation of a white solid. The solution was decanted, and the solid was dried under vacuum. The solid was further washed with hexanes (3 × ∼5 mL) and then dried under vacuum for 4 hours, yielding [H(XII2)][B(C6F5)4]·0.5 hexane as a white solid (182 mg, 86% yield). 1H NMR (CD2Cl2, 600 MHz, 298 K): δ 7.03 (s, 2H, CH1,8), 6.83 (br s, 4H, N-CH), 6.40 (br s, 2H, CH3,6), 5.85 (br s, 1H, NH), 4.38 (sept, 4H, 3JH,H 7 Hz, CHMe2), 1.67 (s, 6H, CMe2), 1.29 (d, 3JH,H 7 Hz, 24H, CHMe2), 1.26 (s, 18H, CMe3) ppm. 13C{1H} NMR (CD2Cl2, 151 MHz, 298 K): δ 146.76 (s, C2,7), 142.3 (br s, NCN), 138.82 (s, C11,12), 130.71 (s, C10,13), 124.5 (br s, C4,5), 115.62 (s, CH1,8), 113.38 (br s, CH3,6 & NCH), 35.52 (s, C9), 35.02 (s, CMe3), 32.61 (s, CMe2), 31.73 (s, CMe3), 22.40 (s, CHMe2) ppm. C68H68N6O1F20B1 (1376.08 g mol−1): calcd C 59.35, H 4.98, N 6.11%; found. C 59.75, H 5.48, N 5.55%.[(XII2)Y(CH2SiMe3)2][B(C6F5)4] (2)
A solution of [H(XII2)][B(C6F5)4]·0.5 hexane (300 mg, 0.218 mmol) in 10 mL of C6H5F was added dropwise to a solution of [Y(CH2SiMe3)3(THF)2] (108 mg, 0.218 mmol) in C6H5F (5 mL) at room temperature and the reaction was stirred for 1 hour. Approximately 40 mL of hexanes was added to the reaction resulting in the precipitation of a white solid. The solution was decanted, and the white solid was dried under vacuum for 1 hour. The solid was washed with hexanes (3 × ∼5 mL) and dried under vacuum for 2 hours, yielding 2 as a highly air-sensitive white solid (323 mg, 93% yield). X-ray quality crystals of 2 were grown by layering hexanes on top of a solution of 2 in C6H5F and cooling to −29 °C. 1H NMR (C6D5Br, 600 MHz, 298 K): δ 7.01 (s, 2H, CH1,8), 6.72 (s, 4H, N-CH), 5.64 (s, 2H, CH3,6), 4.63 (sept, 3JH,H 7 Hz, 4H, CHMe2), 1.68 (s, 6H, CMe2), 1.32 (d, 3JH,H 7 Hz, 12H, CHMe2), 1.16 (s, 18H, CMe3), 1.10 (d, 3JH,H 7 Hz, 12H, CHMe2), −0.19 (s, 18H, SiMe3), −0.77 (appt s, 2H, YCH2) ppm. 13C{1H} NMR (C6D5Br, 151 MHz, 298 K): δ 148.49 (s, C2,7), 147.47 (s, NCN), 140.49 (s, C4,5), 138.01 (C11,12), 130.23 (s, C10,13), 114.68 (s, N-CH), 112.90 (s, CH1,8), 109.28 (s, CH3,6), 48.49 (s, CHMe2), 37.69 (d, 3JH,H 42 Hz, YCH2), 34.57 (s, C9), 34.49 (s, CMe3), 32.14 (s, CMe2), 31.19 (s, CMe3), 23.06, 21.92 (2 × s, CHMe2), 3.46 (s, SiMe3) ppm. C73H82N6O1F20B1Si2Y (1595.32 g mol−1): calcd C 54.96, H 5.18, N 5.27%; found. C 54.63, H 5.23, N 4.91%.[(XII2)Sc(CH2SiMe3)2][B(C6F5)4] (3)
A solution of [H(XII2)][B(C6F5)4]·0.5 hexane (300 mg, 0.218 mmol) in 10 mL of C6H5F was added dropwise to a solution of [Sc(CH2SiMe3)3(THF)2] (98 mg, 0.218 mmol) in C6H5F (5 mL) at room temperature and the reaction was stirred for 1 hour. Approximately 40 mL of hexanes was added to the reaction resulting in the precipitation of a white solid. The solution was decanted, and the white solid was dried under vacuum for 1 hour. The solid was further washed with hexanes (3 × ∼5 mL) and dried under vacuum for 2 hours, yielding 3 as a highly air-sensitive white solid (308 mg, 91% yield). X-ray quality crystals of 3 were grown by layering hexanes on top of a solution of 3 in C6H5F and cooling to −29 °C. 1H NMR (C6D5Br, 600 MHz, 298 K): 6.94 (s, 2H, CH1,8), 6.76 (s, 4H, N-CH), 5.62 (s, 2H, CH3,6), 4.69 (br s, 4H, CHMe2), 1.66 (s, 6H, CMe2), 1.32 (d, 12H, 3JH,H 6 Hz, CHMe2), 1.16 (s, 18H, CMe3), 1.10 (d, 3JH,H 6 Hz, 12H, CHMe2), −0.25 (s, 18H, SiMe3), −0.35 (br s, 4H, ScCH2) ppm. 13C{1H} NMR (C6D5Br, 151 MHz, 298 K): 148.44 (s, NCN), 147.90 (s, C2,7), 140.74 (s, C4,5), 137.72 (C11,12), 129.69 (s, C10,13), 114.66 (s, N-CH), 112.14 (s, CH1,8), 108.32 (s, CH3,6), 48.34 (s, CHMe2), 34.57 (s, C9), 34.46 (s, CMe3), 31.10 (s, CMe3 & CMe2), 23.21, 21.54 (2 × s, CHMe2), 2.87 (s, CH2SiMe3) ppm. Note: the 13C NMR ScCH2 signal was not located from the 1H–13C HSQC NMR spectrum, but may be a very broad peak at 43.9 ppm. C73H82N6O1F20B1Si2Sc (1551.37 g mol−1): calcd C 56.52, H 5.33, N 5.42%; found. C 56.21, H 5.46, N 5.08%.In situ synthesis of [(XII2)Sc(CH2SiMe2CH2SiMe3)][MeB(C6F5)3][B(C6F5)4] (4)
A solution of B(C6F5)3 (9 mg, 0.0176 mmol) in 0.4 mL of C6D5Br was added dropwise to a solution of [(XII2)Sc(CH2SiMe3)2][B(C6F5)4] (3) (25 mg, 0.0161 mmol) in C6D5Br (0.4 mL) at room temperature and the reaction was stirred for 2 hours. 1H NMR (C6D5Br, 500 MHz, 252 K): δ 7.09 (s, 2H, C1,8), 6.79, 6.77 (2 × br s, 2H, N-CH), 5.46 (s, 2H, C3,6), 4.91 (sept, 2H, 3JH,H 7 Hz, CHMe2), 4.27 (sept, 2H, 3JH,H 7 Hz, CHMe2), 1.72 (s, 6H, CMe2), 1.51 (br s, 3H, MeB(C6F5)3), 1.26 (d, 6H, 3JH,H 7 Hz, CHMe2), 1.17 (d, 6H, 3JH,H 7 Hz, CHMe2), 1.15 (s, 18H, CMe3), 0.99 (d, 6H, 3JH,H 7 Hz, CHMe2), 0.85 (s, 2H, ScCH2), 0.82 (d, 6H, 3JH,H 7 Hz, CHMe2), −0.12 (s, 9H, SiMe3), −0.31 (s, 6H, SiMe2), −0.66 (s, 2H, CH2SiMe3) ppm. 13C{1H} NMR (C6D5Br, 126 MHz, 252 K): δ 149.89 (s, C2,7), 144.73 (s, NCN), 139.42 (s, C4,5), 136.61 (s, C11,12), 129.2 (s, C10,13), 115.98, 115.38 (2 × s, N-CH), 114.87 (s, CH1,8), 109.05 (s, CH3,6), 65.7 (s, ScCH2), 49.31, 48.47 (2 × s, CHMe2), 34.59 (s, CMe3 & CMe2), 34.30 (s, C9), 30.85 (s, CMe3), 31.4 (s, BMe), 30.02 (s, CMe2), 23.32, 22.61, 21.59, 21.12 (4 × s, CHMe2), 6.60 (s, CH2SiMe3), 2.46 (s, CH2SiMe2), 1.22 (s, CH2SiMe3) ppm. Note: the C,10,13 ScCH2 and BMe13C NMR signals were located via1H–13C HSQC or HMBC NMR. 11B NMR (C6D5Br, 161 MHz, 252 K): −15.02 (s, MeB(C6F5)3), −16.20 (s, B(C6F5)4) ppm. 19F NMR (C6D5Br, 471 MHz, 252 K): −131.76 (br s, o-B(C6F5)4), −133.27 (d, 3JF,F 22 Hz, o-MeB(C6F5)3), −157.57 (br s, p-MeB(C6F5)3), −161.76 (br s, p-B(C6F5)4), −162.09 (br s, m-MeB(C6F5)3), −165.58 (br s, m-B(C6F5)4) ppm.In situ synthesis of [(XII2)Sc(CH2SiMe3){ηx-toluene}][B(C6F5)4]2 (5)
A solution of [CPh3][B(C6F5)4] (15.5 mg, 0.0168 mmol) in 0.4 mL of C6D5Br was added dropwise to a solution of [(XII2)Sc(CH2SiMe3)2][B(C6F5)4] (3) (20 mg, 0.0129 mmol) and 5 equivalents of toluene (11.9 mg) in C6D5Br (0.4 mL) at room temperature. The reaction was stirred vigorously for 2 hours. 1H NMR (C6D5Br, 600 MHz, 298 K): δ 7.11 (s, 2H, CH1,8), 6.87 (s, 4H, N-CH), 5.63 (s, 2H, CH3,6), 4.38 (br s, 4H, CHMe2), 1.65 (s, 6H, CMe2), 1.16 (s, 18H, CMe3), 1.08 (appt t, 3JH,H 6 Hz, 24 H, CHMe2), 0.70 (s, 2H, ScCH2), −0.22 (s, 9H, SiMe3) ppm. 13C{1H} NMR (C6D5Br, 151 MHz, 298 K): δ 151.03 (s, C2,7), 143.93 (s, NCN), 137.90 (s, C4,5), 130.9 (s, C10,13), 116.08 (s, N-CH), 115.10 (s, CH1,8), 108.92 (s, CH3,6), 63.9 (s, CH2SiMe3), 49.27 (s, CHMe2), 35.05 (s, C9), 34.73 (s, CMe3), 30.86 (s, CMe3), 30.4 (s, CMe2), 22.72, 21.39 (2 × s, CHMe2), 1.68 (s, SiMe3) ppm. Note: the C,10,13 ScCH2 and CMe213C NMR signals were located by 1H–13C HSQC or HMBC NMR. 11B NMR (C6D5Br, 161 MHz, 298 K): δ −16.21 (s, B(C6F5)4) ppm. 19F NMR (C6D5Br, 471 MHz, 298 K): δ −131.50 (br s, o-B(C6F5)4), −161.67 (br s, p-B(C6F5)4), −165.44 (br s, m-B(C6F5)4) ppm.Ethylene polymerization
In the glovebox, 15 mg (9.7 μmol) of [(XII2)Sc(CH2SiMe3)2][B(C6F5)4] (3) was dissolved in approximately 1 mL of o-C6H4F2 and 1 mL of toluene. To this was added a solution of 9 mg (9.8 μmol) of [CPh3][B(C6F5)4] in 1 mL of o-C6H4F2, and the solution was stirred vigorously for 2 hours at room temperature. The solution was diluted with 10 mL of o-C6H4F2 and 30 mL of toluene, and the flask was affixed to a vacuum line. A room temperature water bath was placed around the flask, and with rapid stirring, the solution was briefly evacuated before placing the flask under dynamic ethylene (1 atm), causing an exothermic reaction, with the onset of polyethylene precipitation after approx. 1 minute. After 2, 3 or 5 minutes, the solution was opened to air and approximately 5 mL of acidified methanol (10 vol% 12 M HCl(aq.); 90 vol% MeOH) was added. The resulting solid was filtered, washed with methanol and acetone, and then dried at 50 °C under vacuum to afford dry polyethylene. This method was also carried out using the same masses and approximate solution volumes, but replacing toluene with o-C6H4F2.Data availability
All relevant data is in the ESI† or the CCDC.Author contributions
DJHE conceptualized the project. AV performed the synthesis, characterization and polymerization experiments. JSP carried out the solution and refinement of X-ray data. All authors contributed to writing the manuscript and have approved the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
D. J. H. E. thanks NSERC of Canada for a Discovery Grant. We are also grateful to Dr Carlos A. Cruz and Chevron Phillips Chemical Company for GPC analysis of polymer samples, and to Dr Jim Britten of the McMaster Analytical X-ray Diffraction Facility for advice and support with X-ray diffraction.Notes and references
- A. A. Trifonov and D. M. Lyubov, Coord. Chem. Rev., 2017, 340, 10 CrossRef CAS.
- D. Peng, X. Yan, C. Yu, S. Zhang and X. Li, Polym. Chem., 2016, 7, 2601 RSC.
- V. C. Gibson and S. K. Spitzmesser, Chem. Rev., 2003, 103, 283 CrossRef CAS PubMed.
- S. Arndt, T. P. Spaniol and J. Okuda, Angew. Chem., Int. Ed., 2003, 42, 5075 CrossRef CAS PubMed.
- M. U. Kramer, D. Robert, S. Arndt, P. M. Zeimentz, T. P. Spaniol, A. Yahia, L. Maron, O. Eisenstein and J. Okuda, Inorg. Chem., 2008, 47, 9265 CrossRef CAS PubMed.
- S. Bambirra, A. Meetsma and B. Hessen, Organometallics, 2006, 25, 3454 CrossRef CAS.
- B. R. Elvidge, S. Arndt, P. M. Zeimentz, T. P. Spaniol and J. Okuda, Inorg. Chem., 2005, 44, 6777 CrossRef CAS PubMed.
- A. Nieland, A. Mix, B. Neumann, H. G. Stammler and N. W. Mitzel, Dalton Trans., 2010, 39, 6753 RSC.
- A. Nieland, A. Mix, B. Neumann, H. G. Stammler and N. W. Mitzel, Z. Naturforsch. B, 2014, 69, 327 CrossRef CAS.
- Pyridine coordinated [YMe(py)6]2+ has also been reported: S. Arndt, B. R. Elvidge, P. M. Zeimentz, T. P. Spaniol and J. Okuda, Organometallics, 2006, 25, 793 CrossRef CAS.
- Dicationic rare earth aryl complexes, [MAr(THF)5]2+, have also been reported: P. M. Zeimentz and J. Okuda, Organometallics, 2007, 26, 6388 CrossRef CAS.
- Dicationic rare earth allyl complexes, [M(η3-C3H5)(THF)6]2+ (M = La, Nd), have also been reported: D. Robert, E. Abinet, T. P. Spaniol and J. Okuda, Chem.–Eur. J., 2009, 15, 11937 CrossRef CAS PubMed.
- B. D. Ward, S. Bellemin-Laponnaz and L. H. Gade, Angew. Chem., Int. Ed., 2005, 44, 1668 CrossRef CAS PubMed.
- B. D. Ward, L. Lukesova, H. Wadepohl, S. Bellemin-Laponnaz and L. H. Gade, Eur. J. Inorg. Chem., 2009, 866 CrossRef CAS.
- Y. Pan, A. J. Zhao, Y. Li, W. Q. Li, Y. M. So, X. M. Yan and G. H. He, Dalton Trans., 2018, 47, 13815 RSC.
- Thorium ispoprene polymerization pre-catalysts, [LTh(CH2SiMe3)3], which require activation with 2 equiv. of [CPh3][B(C6F5)4] have been reported: G. Qin and J. Cheng, Dalton Trans., 2019, 48, 11706 RSC.
- B. M. Schmidt, A. Pindwal, A. Venkatesh, A. Ellern, A. J. Rossini and A. D. Sadow, ACS Catal., 2019, 9, 827 CrossRef CAS.
- A. Vasanthakumar, N. A. G. Gray, C. J. Franko, M. C. Murphy and D. J. H. Emslie, Dalton Trans., 2023, 52, 5642 RSC.
- C. A. Cruz, D. J. H. Emslie, L. E. Harrington, J. F. Britten and C. M. Robertson, Organometallics, 2007, 26, 692 CrossRef CAS.
- K. S. A. Motolko, J. S. Price, D. J. H. Emslie, H. A. Jenkins and J. F. Britten, Organometallics, 2017, 36, 3084 CrossRef CAS.
- K. S. A. Motolko, D. J. H. Emslie and H. A. Jenkins, Organometallics, 2017, 36, 1601 CrossRef CAS.
- K. S. A. Motolko, D. J. H. Emslie and J. F. Britten, RSC Adv., 2017, 7, 27938 RSC.
- C. A. Cruz, T. Chu, D. J. H. Emslie, H. A. Jenkins, L. E. Harrington and J. F. Britten, J. Organomet. Chem., 2010, 695, 2798 CrossRef CAS.
- C. A. Cruz, D. J. H. Emslie, H. A. Jenkins and J. F. Britten, Dalton Trans., 2010, 39, 6626 RSC.
- B. Vidjayacoumar, S. Ilango, M. J. Ray, T. Chu, K. B. Kolpin, N. R. Andreychuk, C. A. Cruz, D. J. H. Emslie, H. A. Jenkins and J. F. Britten, Dalton Trans., 2012, 41, 8175 RSC.
- N. R. Andreychuk, S. Ilango, B. Vidjayacoumar, D. J. H. Emslie and H. A. Jenkins, Organometallics, 2013, 32, 1466 CrossRef CAS.
- N. R. Andreychuk, D. J. H. Emslie, H. A. Jenkins and J. F. Britten, J. Organomet. Chem., 2018, 857, 16 CrossRef CAS.
- C. A. Cruz, D. J. H. Emslie, L. E. Harrington and J. F. Britten, Organometallics, 2008, 27, 15 CrossRef CAS.
- C. A. Cruz, D. J. H. Emslie, C. M. Robertson, L. E. Harrington, H. A. Jenkins and J. F. Britten, Organometallics, 2009, 28, 1891 CrossRef CAS.
- N. R. Andreychuk, B. Vidjayacoumar, J. S. Price, S. Kervazo, C. A. Peeples, D. J. H. Emslie, V. Vallet, A. S. P. Gomes, F. Real, G. Schreckenbach, P. W. Ayers, I. Vargas-Baca, H. A. Jenkins and J. F. Britten, Chem. Sci., 2022, 13, 13748 RSC.
- J. Hicks, P. Vasko, J. M. Goicoechea and S. Aldridge, Nature, 2018, 557, 92 CrossRef CAS PubMed.
- J. Hicks, P. Vasko, J. M. Goicoechea and S. Aldridge, J. Am. Chem. Soc., 2019, 141, 11000 CrossRef CAS PubMed.
- J. Hicks, P. Vasko, A. Heilmann, J. M. Goicoechea and S. Aldridge, Angew. Chem., Int. Ed., 2020, 59, 20376 CrossRef CAS.
- M. M. D. Roy, A. Heilmann, M. A. Ellwanger and S. Aldridge, Angew. Chem., Int. Ed., 2021, 60, 26550 CrossRef CAS PubMed.
- A. Heilmann, M. M. D. Roy, A. E. Crumpton, L. P. Griffin, J. Hicks, J. M. Goicoechea and S. Aldridge, J. Am. Chem. Soc., 2022, 144, 12942 CrossRef CAS PubMed.
- M. M. D. Roy, J. Hicks, P. Vasko, A. Heilmann, A. M. Baston, J. M. Goicoechea and S. Aldridge, Angew. Chem., Int. Ed., 2021, 60, 22301 CrossRef CAS PubMed.
- A. Heilmann, J. Hicks, P. Vasko, J. M. Goicoechea and S. Aldridge, Angew. Chem., Int. Ed., 2020, 59, 4897 CrossRef CAS.
- J. Hicks, A. Heilmann, P. Vasko, J. M. Goicoechea and S. Aldridge, Angew. Chem., Int. Ed., 2019, 58, 17265 CrossRef CAS PubMed.
- J. Hicks, A. Mansikkamaki, P. Vasko, J. M. Goicoechea and S. Aldridge, Nat. Chem., 2019, 11, 237 CrossRef CAS.
- C. McManus, J. Hicks, X. L. Cui, L. L. Zhao, G. Frenking, J. M. Goicoechea and S. Aldridge, Chem. Sci., 2021, 12, 13458 RSC.
- C. McManus, A. E. Crumpton and S. Aldridge, Chem. Commun., 2022, 58, 8274 RSC.
- J. S. McMullen, A. J. Edwards and J. Hicks, Dalton Trans., 2021, 50, 8685 RSC.
- F. Kramer, M. S. Luff, U. Radius, F. Weigend and F. Breher, Eur. J. Inorg. Chem., 2021, 2021, 3591 CrossRef CAS.
- L. F. Lim, M. Judd, P. Vasko, M. G. Gardiner, D. A. Pantazis, N. Cox and J. Hicks, Angew. Chem., Int. Ed., 2022, 61, e202201248 CrossRef CAS PubMed.
- N. R. Andreychuk and D. J. H. Emslie, Angew. Chem., Int. Ed., 2013, 52, 1696 CrossRef CAS PubMed.
- In XA2 complexes, large xanthene backbone bend angles (e.g. 41–47°) have typically been observed for complexes of smaller metal ions such as magnesium(II) and aluminum(III). See ref. 21 and 23.
- R. A. Kunetskiy, S. M. Polyakova, J. Vavrik, I. Cisarova, J. Saame, E. R. Nerut, I. Koppel, I. A. Koppel, A. Kutt, I. Leito and I. M. Lyapkalo, Chem.–Eur. J., 2012, 18, 3621 CrossRef CAS PubMed.
- S. A. Filimon, D. Petrovic, J. Volbeda, T. Bannenberg, P. G. Jones, C. G. F. von Richthofen, T. Glaser and M. Tamm, Eur. J. Inorg. Chem., 2014, 5997 CrossRef CAS.
- The Y–Nimidazolin-2-imine distances in 2 are also shorter than the corresponding distances of 2.381(2) and 2.358(2) Å in Tamm's [{2,6-C5H3N(CH2N{NHC})2}YCl3] (NHC = 1,3-di-tert-butylimidazol-2-ylidene): T. K. Panda, D. Petrovic, T. Bannenberg, C. G. Hrib, P. G. Jones and M. Tamm, Inorg. Chim. Acta, 2008, 361, 2236 CrossRef CAS.
- R. D. Shannon, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 1976, 32, 751 CrossRef.
- P. G. Hayes, W. E. Piers and M. Parvez, Organometallics, 2005, 24, 1173 CrossRef CAS.
- P. G. Hayes, W. E. Piers and R. McDonald, J. Am. Chem. Soc., 2002, 124, 2132 CrossRef CAS PubMed.
- L. K. Knight, W. E. Piers and R. McDonald, Organometallics, 2006, 25, 3289 CrossRef CAS.
- A. D. Horton, J. de With, A. J. van der Linden and H. van de Weg, Organometallics, 1996, 15, 2672 CrossRef CAS.
- T. M. Cameron, J. C. Gordon, R. Michalczyk and B. L. Scott, Chem. Commun., 2003, 2282 RSC.
- For a related reaction involving methyl abstraction from a CH2SiMe3 ligand on titanium, see: E. Gielens, J. Y. Tiesnitsch, B. Hessen and J. H. Teuben, Organometallics, 1998, 17, 1652 CrossRef CAS.
- P. D. Bolton, E. Clot, N. Adams, S. R. Dubberley, A. R. Cowley and P. Mountford, Organometallics, 2006, 25, 2806 CrossRef CAS.
- F. Estler, G. Eickerling, E. Herdtweck and R. Anwander, Organometallics, 2003, 22, 1212 CrossRef CAS.
- G. L. Cai, Y. D. Huang, T. T. Du, S. W. Zhang, B. Yao and X. F. Li, Chem. Commun., 2016, 52, 5425 RSC.
- J. L. W. Pohlmann and F. E. Brinckmann, Z. Naturforsch. B, 1965, 20, 5 CrossRef.
- S. Lancaster, ChemSpider SyntheticPages, 2001, http://cssp.chemspider.com/215 Search PubMed.
- M. Tamm, D. Petrovic, S. Randoll, S. Beer, T. Bannenberg, P. G. Jones and O. Grunenberg, Org. Biomol. Chem., 2007, 5, 523 RSC.
- P. Jutzi, C. Müller, A. Stammler and H.-G. Stammler, Organometallics, 2000, 19, 1442 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2015, 71, 3 CrossRef.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3 Search PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339 CrossRef CAS.
- P. V. D. Sluis and A. L. Spek, Acta Crystallogr., Sect. A: Found. Crystallogr., 1990, 46, 194 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available: NMR spectra, the X-ray structure of compound 3, and polyethylene GPC data. CCDC 2247081 (2) and 2247082 (3). For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3sc01830j |
| ‡ Two isomers of “Ph3CCH2SiMe3” are frequently observed, giving rise to 1H NMR signals at 2.36 and 2.06 ppm (CH2), and −0.01 and −0.26 ppm (SiMe3) in C6D5Br; see ref. 19 and 57. In the reaction to form 5, only one of these isomers was formed (2.36, −0.02 ppm; relative integration 2:9). By contrast, in the reaction to form B in Fig. 1 (see ref. 15), only the other isomer (2.05, −0.26 ppm) was formed. |
| § The polymerization reactions were exothermic, causing an increase in the solution temperature, despite the room temp. water bath around the flask. |
| This journal is © The Royal Society of Chemistry 2023 |