
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRhodium-catalyzed atroposelective access to trisubstituted olefins via C–H bond olefination of diverse arenes†
Xiaohan
Zhu
,
Ruijie
Mi
,
Jie
Yin
,
Fen
Wang
* and
Xingwei
Li
 *
*
School of Chemistry and Chemical Engineering, Shaanxi Normal University, Xi'an 710062, China. E-mail: fenwang@snnu.edu.cn; lixw@snnu.edu.cn
First published on 3rd July 2023
Abstract
The atroposelective synthesis of axially chiral acyclic olefins remains a daunting challenge due to their relatively lower racemization barriers, especially for trisubstituted ones. In this work, atroposelective C–H olefination has been realized for synthesis of open-chain trisubstituted olefins via C–H activation of two classes of (hetero)arenes in the coupling with sterically hindered alkynes. The employment of phenyl N-methoxycarbamates as arene reagents afforded phenol-tethered olefins, with the carbamate being a traceless directing group. The olefination of N-methoxy-2-indolylcarboxamides afforded the corresponding chiral olefin by circumventing the redox-neutral [4 + 2] annulation. The reactions proceeded with excellent Z/E selectivity, chemoselectivity, regioselectivity, and enantioselectivity in both hydroarylation systems.
Introduction
As an important phenomenon of chirality, atropisomerism that results from hindered rotation along a single bond has received increasing attention. Axially chiral structures have found tremendous applications in synthetic chemistry, pharmaceutical chemistry, catalysis, and material sciences.1 As a result, catalytic asymmetric construction of axially chiral scaffolds has attracted great attention in the past several decades.2–6 Previous studies have been primarily devoted to synthesis of binaphthalene-based atropisomers and other related 6–6 biaryl systems.3 However, the investigation of axially chiral molecules with a relatively lower rotational energy barrier lags behind, such as five-membered biaryls,4 chiral amides,5 and axially chiral olefins.6 Particularly, atroposelective construction of axially chiral styrenes remains daunting in modern asymmetric organic synthesis. This is ascribed to limited synthetic methods and low configurational stability of the styrenes that are prone to racemization. In addition, a specific E/Z geometry is necessary to maintain the atropo-stability.Given the high reactivity and abundance of unsaturated reagents,7–9 alkenes8 and alkynes9 have been widely applied as substrates in C–H bond asymmetric catalysis. In particular, metal-catalyzed C–H functionalization of arenes with unsaturated reagents has provided tremendous synthetic methods in atroposelective catalysis. Transformations of an existing olefin moiety constitutes a straightforward approach to access axially chiral olefins. In this context, Shi10 realized ortho C–H functionalization of the arene ring assisted by a directing group tethered to a tetrasubstituted olefin, resulting in size-increase along the C–C axis (Scheme 1b). Alternatively, the same group realized C(vinyl)–H functionalization of trisubstituted olefins, leading to tetrasubstituted product.11 Asymmetric functionalization of alkynes provides another straightforward avenue for construction of axially chiral olefins, where sterically hindered alkynes are typically employed to ensure atropo-stability of the olefin product. In 2021, our group reported 1,2-dicarbofunctionalization of sterically hindered alkynes via C–H activation of arenes bearing two classes of migratable directing groups.12 The Song group recently tackled the limitation of sterically hindered alkynes and disclosed copper- and palladium-catalyzed atroposelective arylboration of simple diarylacetylenes using sterically hindered aryl bromides.13 Metal-catalyzed hydrofunctionalization of alkynes offers a convenient approach to prepare trisubstituted olefins. Our group realized palladium-catalyzed atroposelective hydrophosphination of 1-indoylacetylenes, affording vinylphosphines that are potentially chiral ligands.14 Later, Wang reported related hydrophosphinylation of 1-alkynylnaphthol via copper catalysis.15 The Zhu group reported hydroarylation of 1-alkynylnaphthalene via Ni-catalyzed reductive coupling between alkyne and aryl halides.16 Alternatively, organocatalysis delivers powerful protocols to access axially chiral styrenes via addition of a relatively bulky nucleophile (such as sulfinate) to sterically hindered and electronically activated alkyne such as 1-alkynyl-2-naphthol and variants, which reacts via a vinylidene–quinone methide,17 as have been elegantly reported by Yan. In addition, Tan reported the first atroposelective addition of C-based nucleophiles to sterically hindered alkynals via an iminium ion intermediate.18
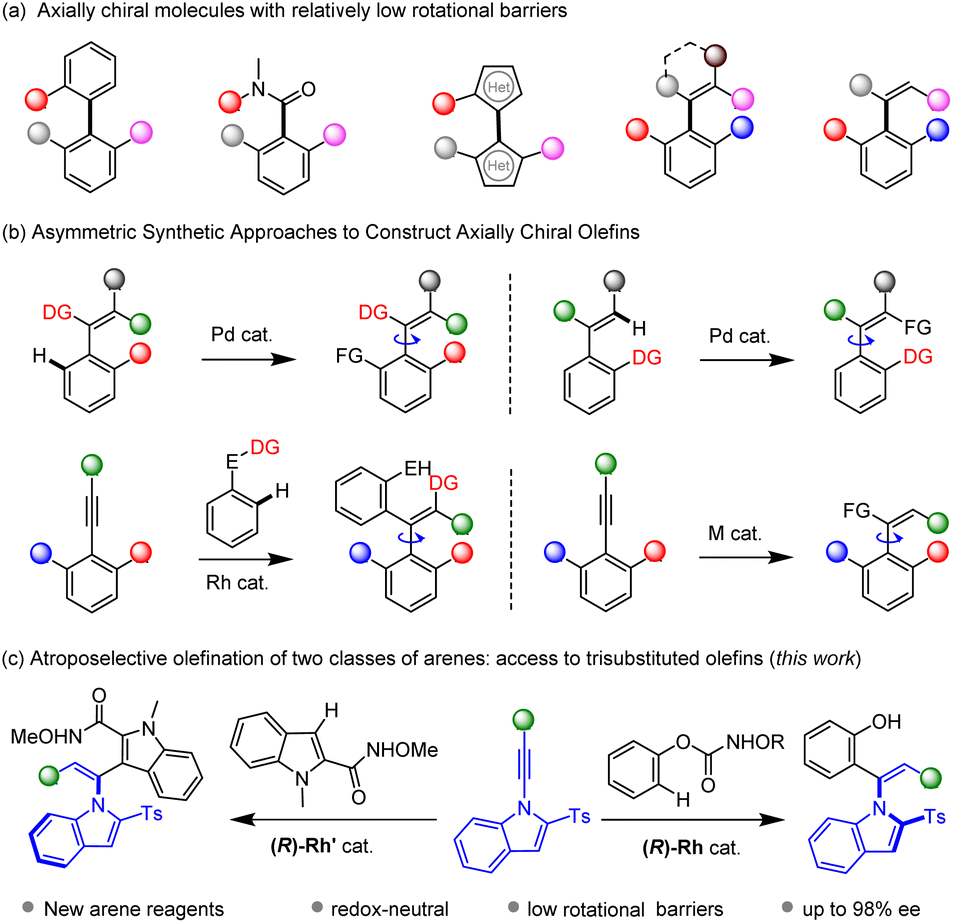 | ||
| Scheme 1 Axially chiral olefins via C–H activation of functionalization of alkynes. | ||
C–H bond activation has emerged as a powerful strategy in modern asymmetric synthesis owing to the abundance of hydrocarbons.19 While increasing reports on asymmetric synthesis of chiral olefins have been documented via this strategy, these systems are mostly restricted to tetrasubstituted ones starting from a pre-existing olefin unit.20 In contrast, trisubstituted olefins generally bear a much lower racemization barrier due to molecular fluxionality and distortion of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond. Consequently, they have been largely underexplored due to the synthetic challenges: the reaction kinetic barrier must be lower than that of the subsequent racemization. Although hydroarylation of alkenes have been extensively studied,21 the atroposelective hydroarylation of alkynes is highly challenging because internal alkynes are necessary, and the alkene product may suffer from low atropo-stability with the introduced of a relatively small aryl group. We focused on hydroarylation of sterically hindered and electronically activated alkynes such as N-alkynylindoles via substrate activation. Meanwhile, (hetero)arenes bearing a functionalizable directing group are employed as suitable reagents, especially by a traceless directing group. We hereby report RhIII-catalyzed atroposelective C–H olefination of two classes of arenes with excellent Z/E selectivity and enantioselectivity under mild conditions (Scheme 1c).
C bond. Consequently, they have been largely underexplored due to the synthetic challenges: the reaction kinetic barrier must be lower than that of the subsequent racemization. Although hydroarylation of alkenes have been extensively studied,21 the atroposelective hydroarylation of alkynes is highly challenging because internal alkynes are necessary, and the alkene product may suffer from low atropo-stability with the introduced of a relatively small aryl group. We focused on hydroarylation of sterically hindered and electronically activated alkynes such as N-alkynylindoles via substrate activation. Meanwhile, (hetero)arenes bearing a functionalizable directing group are employed as suitable reagents, especially by a traceless directing group. We hereby report RhIII-catalyzed atroposelective C–H olefination of two classes of arenes with excellent Z/E selectivity and enantioselectivity under mild conditions (Scheme 1c).
Results and discussion
Extensive studies were conducted to explore the olefination of diverse arenes using alkyne 2a as a coupling reagent. Various directing group such as pyridine (oxide), isoquinoline, amide and nitrone all failed to give efficient coupling or promising enantioselectivity. To enhance the effectiveness and applicability of the directing group in arenes, O-phenyl carbamate with a traceless directing group was investigated. The N-group was screened in the coupling with alkyne 2a (Table 1). No olefination product was detectable when simple phenyl hydroxycarbamate was employed. A series of N-substituted phenyl carbamate were then extensively investigated. Indeed, some N-alkoxy carbamates exhibited activity, affording an ortho-alkenylated phenol product with the carbamate being a traceless directing group. O-Phenyl-N-methoxy carbamate was identified as a suitable substrate with good yield and promising enantioselectivity (88![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :12 e.r.).
:12 e.r.).
|
|
|||||||
|---|---|---|---|---|---|---|---|
| R | H | Me | i Pr | t Bu | Ph | Boc | Bn |
| a Reaction conditions: 1 (0.12 mmol), 2a (0.1 mmol), (R)-Rh1 cat. (4 mol%) and NaOAc (0.12 mmol) in MeOH (2 mL) at 25 °C for 24 h. The e.r. was determined by HPLC analysis using a chiral stationary phase. | |||||||
| Yield (%) | N.D. | 67 | 46 | N.D. | N.D. | N.D. | 23 |
| e.r. | — | 88:12 |
85:15 |
— | — | — | 70:30 |
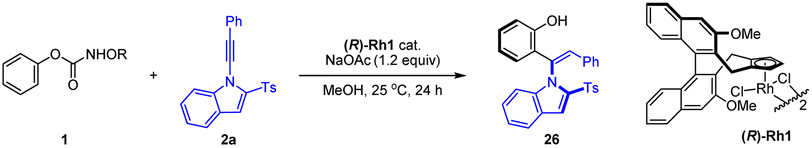
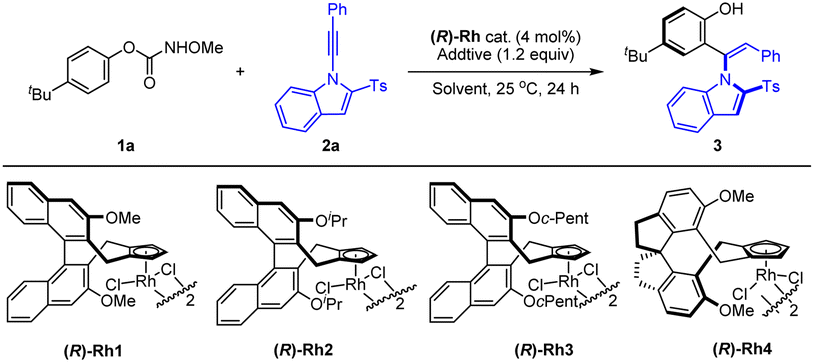
Encouraged by the preliminary results, we next examined the effect of the chiral catalyst on the reaction of a paratBu-substituted carbamate (Table 2), and the reaction proceeded with high yield and enantioselectivity to afford product 3 under the same reaction conditions when catalyzed by the (R)-Rh1 catalyst, indicative of the substrate effect. Switching to other catalysts ((R)-Rh2–4) only led to inferior reactivity and enantioselectivity (entries 2–4). Further investigation of solvent effects revealed that MeOH outperformed other common solvents (entries 5–8). The screening of base additives showed that some common carboxylates of sodium, potassium, and cesium slightly increased the enantioselectivity (entries 9–11). Decreasing the temperature to 10 °C resulted in higher enantioselectivity (96.5:3.5 e.r., entry 12). Further prolonging the reaction time to 48 h led to excellent yield with no change of the enantioselectivity (entry 14, conditions A).
|
|
|||||
|---|---|---|---|---|---|
| Entry | Cat. | Additive | Solvent | Yield (%) | e.r. |
| a Reaction conditions: 1 (0.12 mmol), 2a (0.1 mmol), (R)-Rh cat. (4 mol%) and NaOAc (0.12 mmol) in MeOH (2 mL), 25 °C, 24 h, isolated yield. The e.r. was determined by HPLC analysis using a chiral stationary phase. b 10 °C. c 0 °C. d 10 °C, 48 h. N.D. = not detectable. | |||||
| 1 | (R)-Rh1 | NaOAc | MeOH | 84 | 95:5 |
| 2 | (R)-Rh2 | NaOAc | MeOH | 52 | 90:10 |
| 3 | (R)-Rh3 | NaOAc | MeOH | 31 | 91:9 |
| 4 | (R)-Rh4 | NaOAc | MeOH | 36 | 14:86 |
| 5 | (R)-Rh1 | NaOAc | PhMe | 28 | 93.5:6.5 |
| 6 | (R)-Rh1 | NaOAc | DCE | 46 | 94:6 |
| 7 | (R)-Rh1 | NaOAc | TFE | 20 | 87.5:12.5 |
| 8 | (R)-Rh1 | NaOAc | EA | 19 | 91:9 |
| 9 | (R)-Rh1 | NaOPiv | MeOH | 80 | 95:5 |
| 10 | (R)-Rh1 | KOAc | MeOH | 83 | 95.5:4.5 |
| 11 | (R)-Rh1 | CsOAc | MeOH | 82 | 95.5:4.5 |
| 12b | (R)-Rh1 | NaOAc | MeOH | 81 | 96.5:3.5 |
| 13c | (R)-Rh1 | NaOAc | MeOH | 72 | 96.5:3.5 |
| 14d | (R)-Rh1 | NaOAc | MeOH | 92 | 96.5:3.5 |
With the optimal conditions in hand, we next explored the scope and limitation of this coupling system. Initially, the scope of the 1-indolylacetylenes was investigated (Scheme 2). The coupling of these alkynes with electron-donating (alkyl and alkoxyl), electron-withdrawing (ester), and halogen (Cl and Br) groups at different positions of the indole ring afforded the axially chiral olefins in excellent yields and enantioselectivities (4–12, 91.5:8.5 to 99:1 e.r.). Extension of the alkyne terminus to phenyl groups bearing a large rang of electron-donating, -withdrawing, and halogen substituents at the meta and para positions were fully compatible (13–23). Of note, alkynes bearing an ortho-substituted phenyl was also compatible despite the somewhat enhanced steric effect, giving the desired product in excellent enantioselectivity and efficiency (20 and 21, >96:4 e.r.). The alkyne terminus was also successfully expanded to a 2-thienyl, affording the corresponding product 23 in 86% yield and 95.5:4.5 e.r. The bulky 2-substituent in the indole was not restricted to a sulfonyl group, and N-alkynylindoles bearing a 2-diphenylphosphoryl group also reacted smoothly in high enantioselectivity under the standard reaction conditions (25, 91.5:8.5 e.r.). The absolute configuration of the product 17 was determined by X-ray crystallography (CCDC 2243043†) to be (R), and the rest were assigned by analogy. To obtain the conformational stability of this class of product, racemization studies have been conducted for 12, and a ΔG≠rac = 28.9 kcal mol−1 was obtained in PhMe at 80 °C, indicating the relatively low stability.
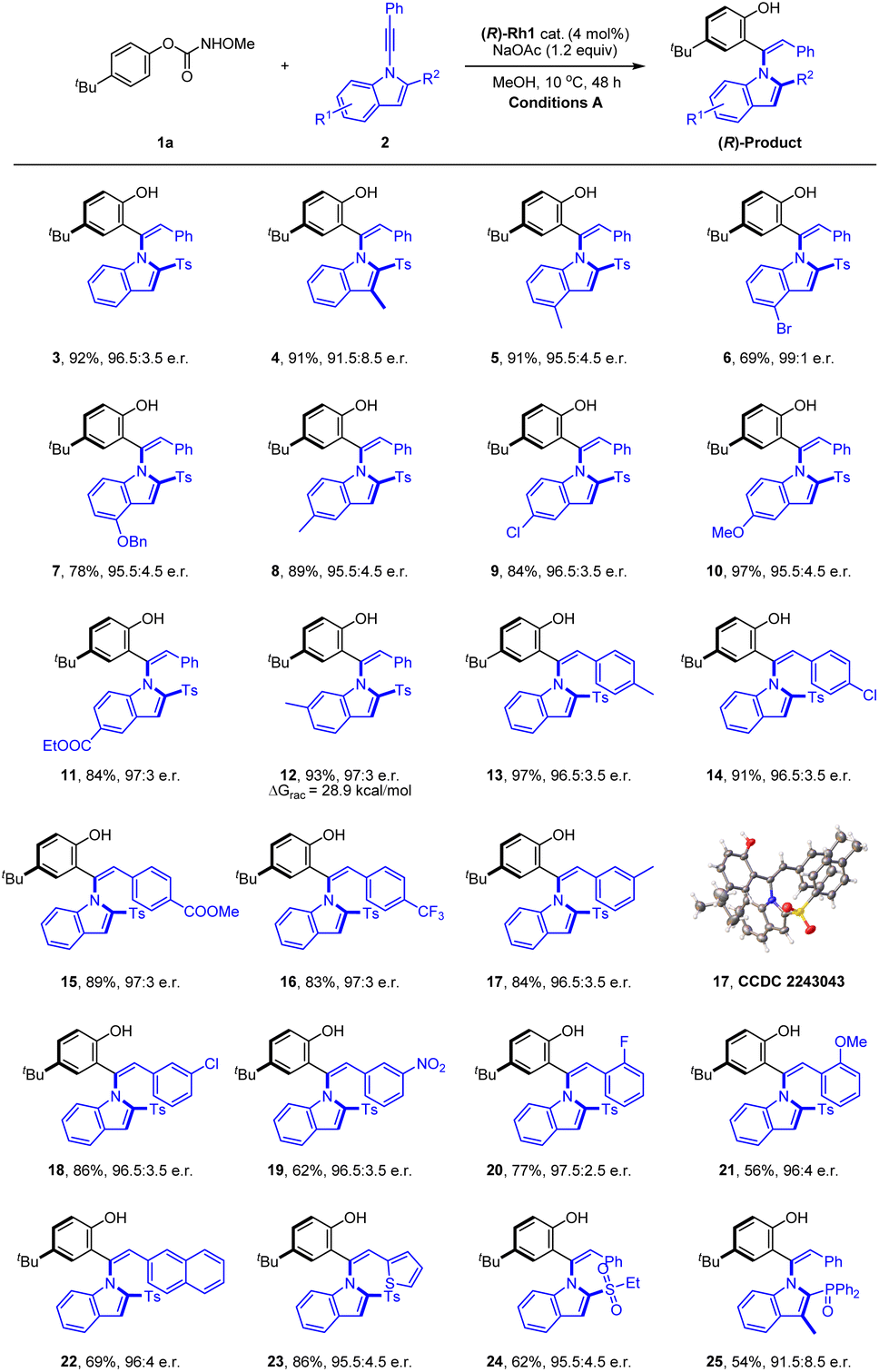 | ||
| Scheme 2 Scope of the alkynes in hydroarylation reactions. [a] Reaction conditions A: 1a (0.12 mmol), 2 (0.1 mmol), (R)-Rh1 cat. (4 mol%) and NaOAc (0.12 mmol) in MeOH (2 mL), 10 °C, 48 h, isolated yield. The e.r. was determined by HPLC analysis using a chiral stationary phase. | ||
We next evaluated the generality of phenyl methoxy-carbamate in the coupling with alkyne 2a under the optimal conditions, where pronounced substituent effects were observed (Scheme 3). Simple phenyl methoxycarbamate was tolerated in this reaction and attenuated enantioselectivity was obtained (26, 90.5:9.5 e.r.). Carbamates bearing an electron-donating group at para position of the benzene ring tend to react with high yield and enantiomeric ratios (27–30). Similarly, carbamates with a halogen, vinyl, or ester group at the para position all tend to react in good yields and slightly lower atroposelectivities (31–34). The presence of meta Me, OMe, and Br groups was also well tolerated, and the corresponding product was obtained with good yield and enantioselective control (35–37). Overall, the reaction enantioselectivity is affected by both the steric and electronic effects of the substituent. Moreover, an estrone-derived carbamate also proved effective in this coupling system with high d.r., albeit with a moderate conversion and a low regioselectivity, and the product (38) could be potentially useful in pharmaceutical development. Unfortunately, no reaction occurred when an ortho-substituted O-phenyl methoxycarbamate was employed as a substrate even under harsh conditions.
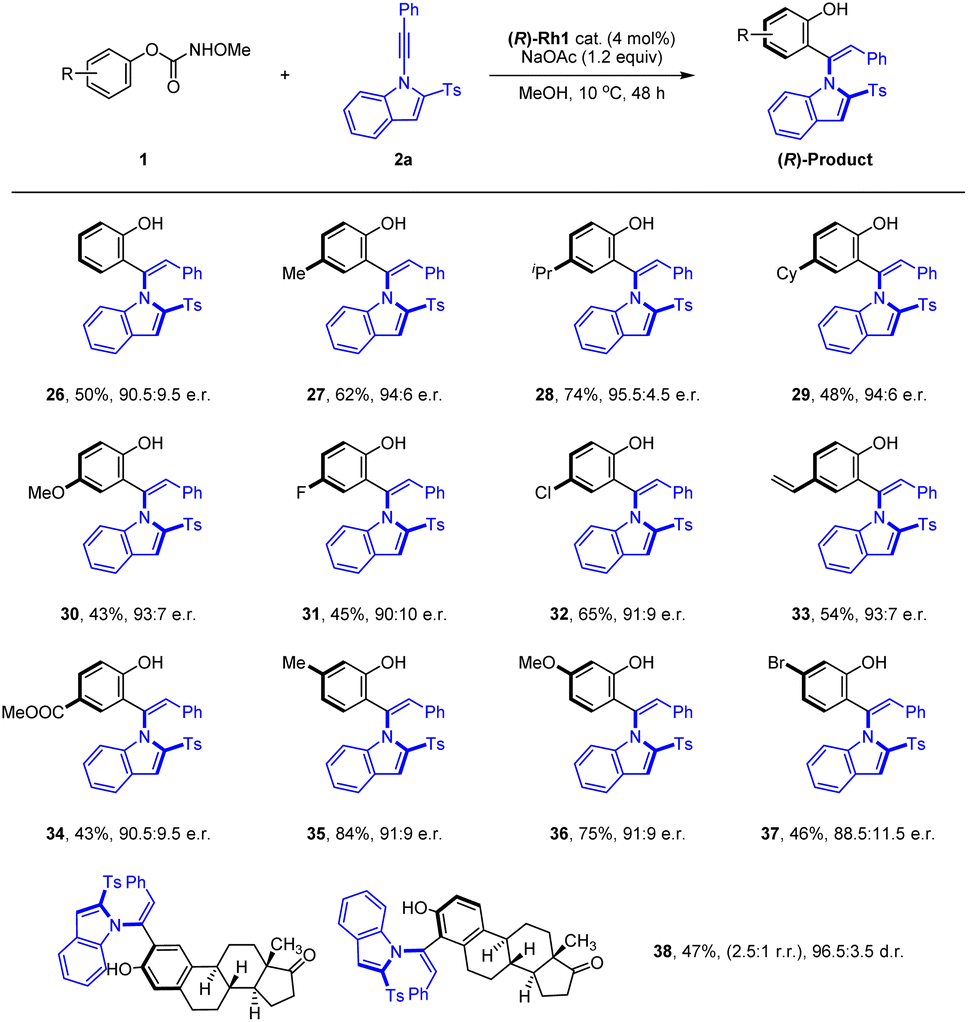 | ||
| Scheme 3 Scope with respect to phenyl methoxycarbamates in C–H Olefination. [a] Reaction conditions A: 1a (0.12 mmol), 2 (0.1 mmol), (R)-Rh1 cat. (4 mol%) and NaOAc (0.12 mmol) in MeOH (2 mL), 10 °C, 48 h, isolated yield. The e.r. was determined by HPLC analysis using a chiral stationary phase. | ||
To better explore the scope of arene reagents in atroposelective C–H olefination, typically reactive heteroarenes such as indoles have been investigated. We have identified N-methoxy-2-indolylcarboxamide (39) as a suitable candidate (Scheme 4). Extensive studies have been conducted to control both the enantioselectivity and the chemical selectivity because it readily underwent redox-neutral [4 + 2] annulation via cleavage of an internal oxidizing N–OMe bond. Optimizations of the catalyst, solvent, and additive revealed that the rhodium spirocyclic Cp catalyst (R)-Rh4 (ref. 22) outperformed the rest, and the choice of iPrOH solvent was essential to suppress the [4 + 2] side reaction (see ESI†). Both excellent enantioselectivity and reactivity were realized under these conditions. The scope of this atroposelective system was briefly explored by using various alkyne reagents. It was found that introduction of alkyl, alkoxyl, halogen, ester, and CF3 group to the different positions of the alkyne has been well-tolerated, and excellent enantioselectivities ranging from 94.5:5.5 to 99:1 e.r. have been realized (40–49). The absolution configuration of product 41 was determined to be (R) by X-ray crystallography (CCDC 2260294†). The conformational stability of product 41 was also investigated (90 °C, toluene), and a racemization barrier of 30.2 kcal mol−1 was obtained, which is only slightly higher than that of the product 12. In contrast to the high reactivity of the indolyl N–Me substrate, no reaction was observed when the corresponding protic NH reagent was used, likely due to the N–N chelation that inhibited subsequent C–H activation (substrate inhibition).
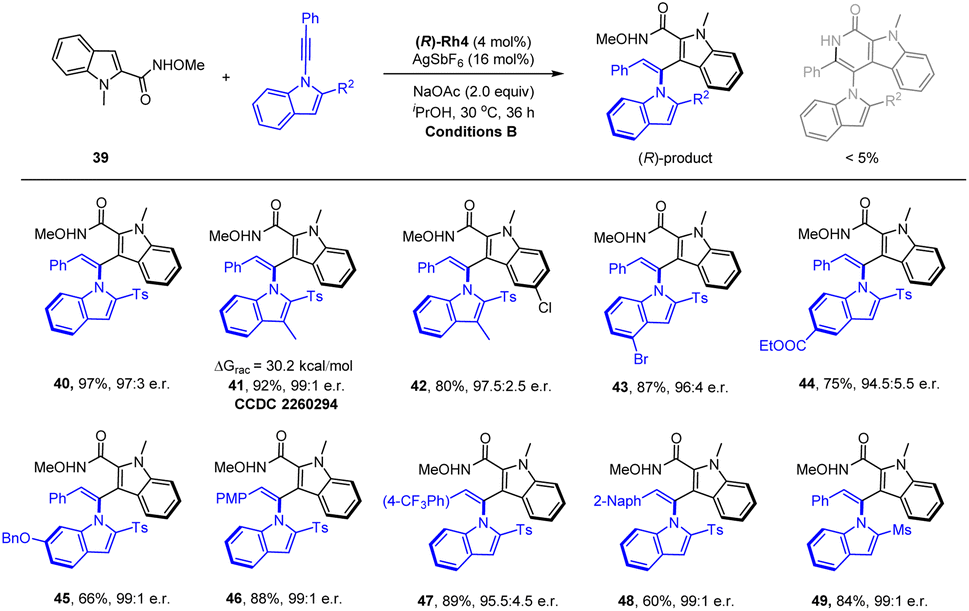 | ||
| Scheme 4 Atroposelective C–H olefination of 2-Indolylcarboxamides. [a] Reaction conditions B: indolylcarboxamide (0.1 mmol), alkyne 2 (0.12 mmol), (R)-Rh4 (4 mol%), AgSbF6 (16 mol%), and NaOAc (0.2 mmol) in iPrOH (2 mL), 30 °C, 36 h, isolated yield. The e.r. was determined by HPLC analysis using a chiral stationary phase. | ||
Synthetic applications of representative products were next demonstrated (Scheme 5). To explore the scalability of this protocol, the reaction of carbamate 1a was performed at a mmol scale under a reduced catalyst leading, affording (R)-3 in 90% yield with 96.5:3.5 e.r. Oxidative C–O cyclization has been realized when the (R)-3 was treated with I2 under mild conditions, affording a C–N axially chiral biaryl 50 in a moderate yield (47%) and excellent enantioselectivity (95.5:4.5 e.r.). Electrophilic bromination of 3 with NBS was accomplished in 68% yield with 92.5:7.5 e.r. of the product 51. O-Triflation (52) of the axially chiral indole–phenol with Tf2O followed by the Sonogashira coupling with phenylacetylene afforded alkyne 53 in good yield. In all cases, only slight erosion of enantiopurity was detected.
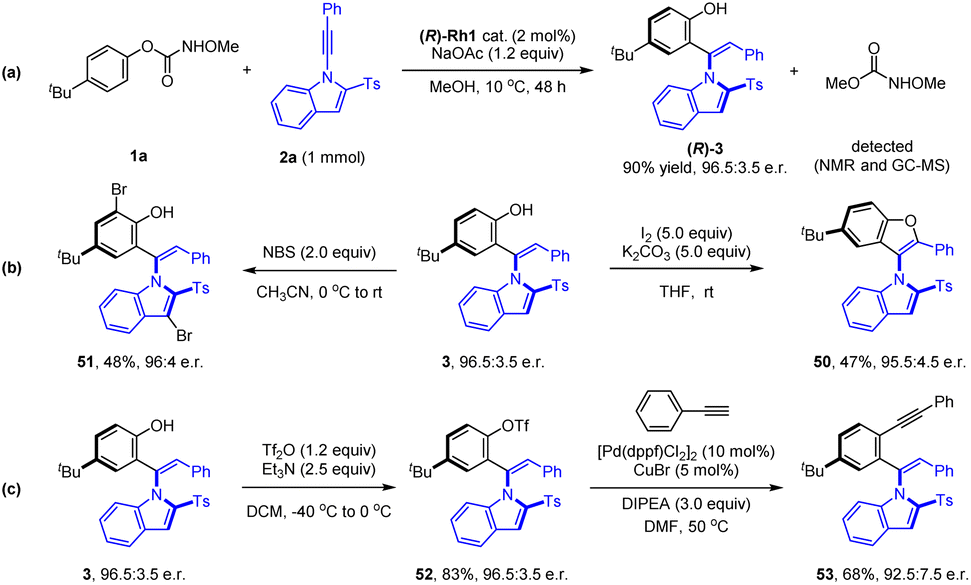 | ||
| Scheme 5 Scale-up synthesis and transformations of selected products. | ||
Experimental studies have been briefly conducted to gain some insight into the mechanism of this reaction (Scheme 6). A control experiment using simple phenol as a substrate has been conducted under the standard conditions, but no desired product was detected (Scheme 6a). This observation indicated necessity of the carbamate directing group, and the phenol moiety was derived from decay of this traceless directing group. Indeed, an OMe carbamate was detected as a co-product (see Scheme 5a). The reaction likely follows a C–H activation pathway based on previously related reports of phenols bearing a heterocyclic directing group, and kinetic isotope effect (KIE) was then determined from two parallel reactions using 1 and 1-d5. A rather large value of KIE = 3.6 was obtained based 1H NMR analysis of the coupled products, indicating that the cleavage of the C–H bond is involved in the rate-limiting step (Scheme 6b). A Hammett plot of log(kR/kH) for various para-substituted phenyl methoxycarbamates revealed a decent linear correlation with a slight negative slope (Scheme 6c), suggesting stabilization of positive charge in the transition state of the turnover-limiting step. This is consistent with a C–H activation process where a more electron-rich arene is more susceptible to C–H activation. To explore the mechanism of the indolylcarboxamide system, a stoichiometric C–H activation reaction between the amide 39 and the Rh-4 complex was conducted in the presence of a base followed by saturation with PPh3,9b affording the 18-electron complex 54 in high yield as a single stereoisomer (Scheme 6d) that has been characterized by X-ray crystallography (CCDC 2265864†). Complex 54 was indeed catalytically active when designated as a catalyst for the coupling of amide 39 and 2a, affording coupled product 40 in high yield with 98.5:1.5 e.r. In complex 54, the bulky amide directing group is pointed inward for minimized repulsions with the chiral ligand, which will then dictate the orientation of an incoming alkyne 2a in the catalytic cycle (replacement of the PPh3 by the alkyne). A stereochemical control model9b,12,23 is then proposed to account for the observed enantioselectivity of the product 40 when catalyzed by the (R)-Rh4 catalyst on the basis of our previous report (Scheme 6e). The 2-Ts group of the alkyne is aligned upward, as dictated by the chiral environment of the rhodacycle for minimized steric repulsions with the arene ring. Enantio-determining migratory insertion of the Rh–C bond is followed by protonolysis of the resulting rhodium vinyl bond, affording the observed (R) product.
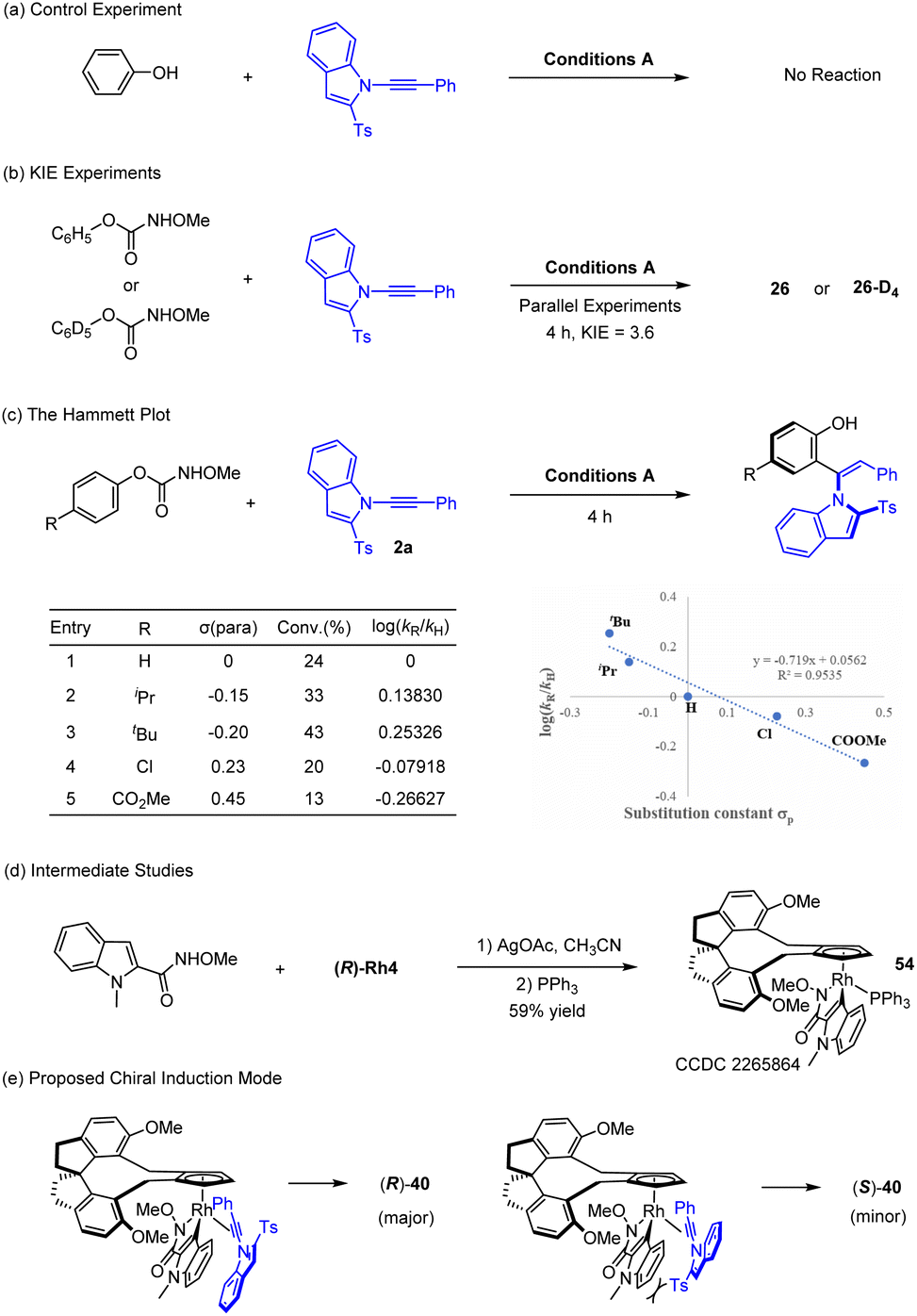 | ||
| Scheme 6 Mechanistic studies. | ||
Conclusions
In conclusion, we have successfully developed an efficient and redox-neutral approach for atroposelective synthesis of axially chiral trisubstituted styrenes using two classes of arenes. The coupling systems proceeded via C–H activation–olefination of the arene with a sterically hindered alkynes as a result of dynamic kinetic transformation of the alkyne. In the case of O-phenyl carbamate substrates, the carbamate servers as a traceless directing group, affording synthetically useful phenols. One the other hand, efficient C–H olefination of 2-indolylcarboxamide was realized by successful suppression of the [4 + 2] redox-neutral annulation reaction. Relatively low atropo-stability of these chiral olefins have been evoked. In both systems, the hydroarylation reactions proceeded with excellent Z/E-selectivity, chemoselectivity, regioselectivity, and enantioselectivity. This coupling system provides a potentially useful protocol to access chiral olefins that may find applications in asymmetric synthesis.Data availability
Further details of the experimental procedure, 1H, and 13C NMR, HPLC spectra, and X-ray crystallographic data for products 17, 41 and 54 are available in the ESI.†Author contributions
X. L. and F. W. conceived the idea and directed the project. X. Zhu, R. Mi and J. Yin performed the experiments. X. L. and F. W. wrote the manuscript.Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
Financial support from the NSFC (22101167) and the SNNU is gratefully acknowledged.Notes and references
- For recent reviews, see: (a) E. Kumarasamy, R. Raghunathan, M. P. Sibi and J. Sivaguru, Chem. Rev., 2015, 115, 11239–11300 CrossRef CAS; (b) J. E. Smyth, N. M. Butler and P. A. Keller, Nat. Prod. Rep., 2015, 32, 1562–1583 RSC; (c) G. Bringmann, T. Gulder, T. A. M. Gulder and M. Breuning, Chem. Rev., 2011, 111, 563–639 CrossRef CAS PubMed; (d) Y.-M. Li, F.-Y. Kwong, W.-Y. Yu and A. Chan, Coord. Chem. Rev., 2007, 251, 2119–2144 CrossRef CAS; (e) S. Erbas-Cakmak, D. A. Leigh, C. T. McTernan and A. L. Nussbaumer, Chem. Rev., 2015, 115, 10081–10206 CrossRef CAS PubMed; (f) S. T. Toenjes and J. L. Gustafson, Future Med. Chem., 2018, 10, 409–422 CrossRef CAS PubMed; (g) X.-Z. Bao, J. Rodriguez and D. Bonne, Angew. Chem., Int. Ed., 2020, 59, 12623–12634 CrossRef CAS; (h) J. Clayden, W. J. Moran, P. J. Edwards and S. R. Laplante, Angew. Chem., Int. Ed., 2009, 48, 6398–6401 CrossRef CAS PubMed.
- For some recent reviews: (a) D. Bonne and J. Rodriguez, Chem. Commun., 2017, 53, 12385–12393 RSC; (b) D. Bonne and J. Rodriguez, Eur. J. Org. Chem., 2018, 2018, 2417–2431 CrossRef CAS; (c) S. Zhang, G. Liao and B.-F. Shi, Chin. J. Org. Chem., 2019, 39, 1522–1528 CrossRef CAS; (d) G. Liao, T. Zhou, Q.-J. Yao and B.-F. Shi, Chem. Commun., 2019, 55, 8514–8523 RSC; (e) T.-Z. Li, S.-J. Liu, W. Tan and F. Shi, Chem.–Eur. J., 2020, 26, 15779–15792 CrossRef CAS PubMed; (f) B.-C. Da, S.-H. Xiang, S. Li and B. Tan, Chin. J. Chem., 2021, 39, 1787–1796 CrossRef CAS; (g) J. K. Cheng, S.-H. Xiang, S.-Y. Li, L. Ye and B. Tan, Chem. Rev., 2021, 121, 4805–4902 CrossRef CAS; (h) J. K. Cheng, S.-H. Xiang and B. Tan, Acc. Chem. Res., 2022, 55, 2920–2937 CrossRef CAS; (i) H.-H. Zhang and F. Shi, Acc. Chem. Res., 2022, 55, 2562–2580 CrossRef CAS PubMed.
- For some representative reviews: (a) J. Wencel-Delord, A. Panossian, F. R. Leroux and F. Colobert, Chem. Soc. Rev., 2015, 44, 3418–3430 RSC; (b) A. Link and C. Sparr, Chem. Soc. Rev., 2018, 47, 3804–3815 RSC; (c) P. Loxq, E. Manoury, R. Poli, E. Deydier and A. Labande, Coord. Chem. Rev., 2016, 308, 131–190 CrossRef CAS.
- G. Centonze, C. Portolani, P. Righi and G. Bencivenni, Angew. Chem., Int. Ed., 2023, e202303966 Search PubMed.
- (a) V. C. Fäseke and C. Sparr, Angew. Chem., Int. Ed., 2016, 55, 7261–7264 CrossRef PubMed; (b) A. J. Fugard, A. S. K. Lahdenperä, J. S. J. Tan, A. Mekareeya, R. S. Paton and M. D. Smith, Angew. Chem., Int. Ed., 2019, 58, 2795–2798 CrossRef CAS PubMed; (c) Q.-J. Yao, P.-P. Xie, Y.-J. Wu, Y.-L. Feng, M.-Y. Teng, X. Hong and B.-F. Shi, J. Am. Chem. Soc., 2020, 142, 18266–18276 CrossRef CAS PubMed.
- For some representative reviews on axially chiral olefins: (a) J. Feng and Z.-H. Gu, SynOpen, 2021, 05, 68–85 CrossRef CAS; (b) S. Wu, S.-H. Xiang, J. K. Cheng and B. Tan, Tetrahedron Chem, 2022, 1, 100009 CrossRef.
- (a) Y.-S. Jang, Ł. Woźniak, J. Pedroni and N. Cramer, Angew. Chem., Int. Ed., 2018, 57, 12901–12905 CrossRef CAS PubMed; (b) S.-G. Wang, Y. Liu and N. Cramer, Angew. Chem., Int. Ed., 2019, 58, 18136–18140 CrossRef CAS PubMed; (c) L. Liu, H. Song, Y.-H. Liu, L.-S. Wu and B.-F. Shi, ACS Catal., 2020, 10, 7117–7122 CrossRef CAS; (d) P.-J. Hu, B.-X. Liu, F. Wang, R.-J. Mi, X.-X. Li and X.-W. Li, ACS Catal., 2022, 12, 13884–13896 CrossRef CAS; (e) J.-Y. Li, P.-P. Xie, T. Zhou, P.-F. Qian, Y.-B. Zhou, H.-C. Li, X. Hong and B.-F. Shi, ACS Catal., 2022, 12, 9083–9091 CrossRef CAS.
- (a) Z.-J. Jia, C. Merten, R. Gontla, C. G. Daniliuc, A. P. Antonchick and H. Waldmann, Angew. Chem., Int. Ed., 2017, 56, 2429–2434 CrossRef CAS PubMed; (b) E. A. Trifonova, N. M. Ankudinov, A. A. Mikhaylov, D. A. Chusov, Y. V. Nelyubina and D. S. Perekalin, Angew. Chem., Int. Ed., 2018, 57, 7714–7718 CrossRef CAS PubMed; (c) X.-F. Yang, G.-F. Zheng and X.-W. Li, Angew. Chem., Int. Ed., 2019, 58, 322–326 CrossRef CAS PubMed; (d) W.-J. Cui, Z.-J. Wu, Q. Gu and S.-L. You, J. Am. Chem. Soc., 2020, 142, 7379–7385 CrossRef CAS PubMed; (e) G.-Z. Li, X.-Q. Yan, J.-J. Jiang, H. Liang, C. Zhou and J. Wang, Angew. Chem., Int. Ed., 2020, 59, 22436–22440 CrossRef CAS PubMed; (f) R.-J. Mi, Z.-Y. Ding, S.-J. Yu, R. H. Crabtree and X. Li, J. Am. Chem. Soc., 2023, 145, 8150–8162 CrossRef CAS PubMed.
- (a) M.-M. Tian, D.-C. Bai, G.-F. Zheng, J.-B. Chang and X.-W. Li, J. Am. Chem. Soc., 2019, 141, 9527–9532 CrossRef CAS PubMed; (b) F. Wang, Z.-S. Qi, Y.-X. Zhao, S.-L. Zhai, G.-F. Zheng, R.-J. Mi, Z.-Y. Huang, X.-L. Zhu, X.-M. He and X.-W. Li, Angew. Chem., Int. Ed., 2020, 59, 13288–13294 CrossRef CAS PubMed; (c) P.-J. Hu, L.-H. Kong, F. Wang, X.-L. Zhu and X.-W. Li, Angew. Chem., Int. Ed., 2021, 60, 20424–20429 CrossRef CAS PubMed; (d) W.-W. Zhang, Q. Wang, S.-Z. Zhang, C. Zheng and S.-L. You, Angew. Chem., Int. Ed., 2023, 62, e202214460 CAS.
- H. Song, Y. Li, Q.-J. Yao, L. Jin, L. Liu, Y.-H. Liu and B.-F. Shi, Angew. Chem., Int. Ed., 2020, 59, 6576–6580 CrossRef CAS PubMed.
- L. Jin, Q.-J. Yao, P.-P. Xie, Y. Li, B.-B. Zhan, Y.-Q. Han, X. Hong and B.-F. Shi, Chem, 2020, 6, 497–511 CAS.
- R.-J. Mi, H.-H. Chen, X.-K. Zhou, N. Li, D.-Q. Ji, F. Wang, Y. Lan and X.-W. Li, Angew. Chem., Int. Ed., 2022, 61, e202111860 CrossRef CAS PubMed.
- W.-Y. Li, S.-L. Chen, J.-H. Xie, Z.-W. Fan, K. Yang and Q.-L. Song, Nat. Synth., 2023, 2, 140–151 CrossRef.
- D.-Q. Ji, J.-R. Jing, Y. Wang, Z.-S. Qi, F. Wang, X.-P. Zhang, Y. Wang and X.-W. Li, Chem, 2022, 8, 3346–3362 CAS.
- C. Yang, T.-R. Wu, Y. Li, B.-B. Wu, R.-X. Jin, D.-D. Hu, Y.-B. Li, K.-J. Bian and X.-S. Wang, Chem. Sci., 2021, 12, 3726–3732 RSC.
- F.-T. Sheng, S.-C. Wang, J.-Q. Zhou, C.-P. Cheng, Y. Wang and S.-L. Zhu, ACS Catal., 2023, 13, 3841–3846 CrossRef CAS.
- (a) S.-Q. Jia, Z.-L. Chen, N. Zhang, Y. Tan, Y.-D. Liu, J. Deng and H.-L. Yan, J. Am. Chem. Soc., 2018, 140, 7056–7060 CrossRef CAS PubMed; (b) Y. Tan, S.-Q. Jia, F.-L. Hu, Y.-D. Liu, L. Peng, D.-M. Li and H.-L. Yan, J. Am. Chem. Soc., 2018, 140, 16893–16898 CrossRef CAS PubMed; (c) S. Li, D. Xu, F.-L. Hu, D.-M. Li, W.-L. Qin and H.-L. Yan, Org. Lett., 2018, 20, 7665–7669 CrossRef CAS; (d) Y.-B. Wang, P.-Y. Yu, Z.-P. Zhou, J. Zhang, J. Wang, S.-H. Luo, Q.-S. Gu, K. N. Houk and B. Tan, Nat. Catal., 2019, 2, 504–513 CrossRef CAS; (e) A.-Q. Huang, L.-L. Zhang, D.-M. Li, Y.-D. Liu, H.-L. Yan and W.-J. Li, Org. Lett., 2019, 21, 95–99 CrossRef CAS PubMed.
- S.-C. Zheng, S. Wu, Q.-H. Zhou, L. W. Chung, L. Ye and B. Tan, Nat. Commun., 2017, 8, 15238–15245 CrossRef.
- For reviews on asymmetric C–H functionalization: (a) R. Giri, B.-F. Shi, K. M. Engle, N. Maugel and J.-Q. Yu, Chem. Soc. Rev., 2009, 38, 3242–3272 RSC; (b) C. Zheng and S.-L. You, RSC Adv., 2014, 4, 6173–6214 RSC; (c) C. G. Newton, S.-G. Wang, C. C. Oliveira and N. Cramer, Chem. Rev., 2017, 117, 8908–8976 CrossRef CAS PubMed; (d) T. G. Saint-Denis, R.-Y. Zhu, G. Chen, Q.-F. Wu and J.-Q. Yu, Science, 2018, 359, eaao4798 CrossRef PubMed; (e) Q. Zhang and B.-F. Shi, Chin. J. Chem., 2019, 37, 647–656 CrossRef CAS; (f) J. Loup, U. Dhawa, F. Pesciaioli, J. Wencel-Delord and L. Ackermann, Angew. Chem., Int. Ed., 2019, 58, 12803–12818 CrossRef CAS PubMed; (g) Ł. Woźniak and N. Cramer, Trends Chem., 2019, 1, 471–484 CrossRef; (h) T. Yoshino, S. Satake and S. Matsunaga, Chem.–Eur. J., 2020, 26, 7346–7357 CrossRef CAS PubMed; (i) G. Liao, T. Zhang, Z.-K. Lin and B.-F. Shi, Angew. Chem., Int. Ed., 2020, 59, 19773–19786 CrossRef CAS PubMed; (j) P.-S. Wang and L.-Z. Gong, Acc. Chem. Res., 2020, 53, 2841–2854 CrossRef CAS; (k) T. K. Achar, S. Maiti, S. Jana and D. Maiti, ACS Catal., 2020, 10, 13748–13793 CrossRef CAS; (l) C.-X. Liu, W.-W. Zhang, S.-Y. Yin, Q. Gu and S.-L. You, J. Am. Chem. Soc., 2021, 143, 14025–14040 CrossRef CAS PubMed.
- (a) J. Feng, B. Li, J.-L. Jiang, M.-K. Zhang, W.-B. Ouyang, C.-Y. Li, Y. Fu and Z.-H. Gu, Chin. J. Chem., 2018, 36, 11–14 CrossRef CAS; (b) Y.-B. Wang, Q.-H. Wu, Z.-P. Zhou, S.-H. Xiang, Y. Cui, P.-Y. Yu and B. Tan, Angew. Chem., Int. Ed., 2019, 58, 13443–13447 CrossRef CAS; (c) Y.-Y. Liang, J.-Y. Ji, X.-Y. Zhang, Q.-B. Jiang, J. Luo and X.-D. Zhao, Angew. Chem., Int. Ed., 2020, 59, 4959–4964 CrossRef CAS PubMed; (d) C. Ma, F.-T. Sheng, H.-Q. Wang, S. Deng, Y.-C. Zhang, Y.-C. Jiao, W. Tan and F. Shi, J. Am. Chem. Soc., 2020, 142, 15686–15696 CrossRef CAS; (e) J. D. Jolliffe, R. J. Armstrong and M. D. Smith, Nat. Chem., 2017, 9, 558–562 CrossRef CAS.
- (a) B.-H. Ye, P. A. Donets and N. Cramer, Angew. Chem., Int. Ed., 2014, 53, 507–511 CrossRef CAS PubMed; (b) C. M. Filloux and T. Rovis, J. Am. Chem. Soc., 2015, 137, 508–517 CrossRef CAS PubMed; (c) S. Satake, T. Kurihara, K. Nishikawa, T. Mochizuki, M. Hatano, K. Ishihara, T. Yoshino and S. Matsunaga, Nat. Catal., 2018, 1, 585–591 CrossRef CAS; (d) V. S. Shinde, M. V. Mane, L. Cavallo and M. Rueping, Chem.–Eur. J., 2020, 26, 8308–8313 CrossRef CAS PubMed; (e) S.-J. Lou, Z.-B. Mo, M. Nishiura and Z.-M. Hou, J. Am. Chem. Soc., 2020, 142, 1200–1205 CrossRef CAS; (f) A. Carral-Menoyo, N. Sotomayor and E. Lete, ACS Omega, 2020, 5, 24974–24993 CrossRef CAS PubMed; (g) Y.-H. Liu, P.-P. Xie, L. Liu, J. Fan, Z.-Z. Zhang, X. Hong and B.-F. Shi, J. Am. Chem. Soc., 2021, 143, 19112–19120 CrossRef CAS PubMed.
- J. Zheng, W.-J. Cui, C. Zheng and S.-L. You, J. Am. Chem. Soc., 2016, 138, 5242–5245 CrossRef CAS PubMed.
- (a) B.-H. Ye and N. Cramer, Science, 2012, 338, 504–506 CrossRef CAS PubMed; (b) B.-H. Ye and N. Cramer, J. Am. Chem. Soc., 2013, 135, 636–639 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2243043, 2260294 and 2265864. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3sc02714g |
| This journal is © The Royal Society of Chemistry 2023 |