
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceElectron delocalization of robust high-nuclear bismuth-oxo clusters for promoted CO2 electroreduction†
Baoshan
Hou‡
a,
Haiyan
Zheng‡
a,
Kunhao
Zhang
c,
Qi
Wu
a,
Chao
Qin
 a,
Chunyi
Sun
*a,
Qinhe
Pan
b,
Zhenhui
Kang
d,
Xinlong
Wang
*ab and
Zhongmin
Su
b
a,
Chunyi
Sun
*a,
Qinhe
Pan
b,
Zhenhui
Kang
d,
Xinlong
Wang
*ab and
Zhongmin
Su
b
aKey Lab of Polyoxometalate Science of Ministry of Education, National & Local United Engineering Laboratory for Power Battery Institution, Northeast Normal University, Changchun, Jilin 130024, China. E-mail: wangxl824@nenu.edu.cn; suncy009@nenu.edu.cn
bKey Laboratory of Advanced Materials of Tropical Island Resources, Ministry of Education, Hainan University, Haikou, 570228, China
cShanghai Synchrotron Radiation Facility, 239 Zhangheng Road, Pudong New District, Shanghai, 200120, China
dInstitute of Functional Nano & Soft Materials, Jiangsu Key Laboratory for Carbon-Based Functional Materials & Devices, Soochow University, Suzhou 215123, Jiangsu, China
First published on 7th August 2023
Abstract
The integration of high activity, selectivity and stability in one electrocatalyst is highly desirable for electrochemical CO2 reduction (ECR), yet it is still a knotty issue. The unique electronic properties of high-nuclear clusters may bring about extraordinary catalytic performance; however, construction of a high-nuclear structure for ECR remains a challenging task. In this work, a family of calix[8]arene-protected bismuth-oxo clusters (BiOCs), including Bi4 (BiOC-1/2), Bi8Al (BiOC-3), Bi20 (BiOC-4), Bi24 (BiOC-5) and Bi40Mo2 (BiOC-6), were prepared and used as robust and efficient ECR catalysts. The Bi40Mo2 cluster in BiOC-6 is the largest metal-oxo cluster encapsulated by calix[8]arenes. As an electrocatalyst, BiOC-5 exhibited outstanding electrochemical stability and 97% Faraday efficiency for formate production at a low potential of −0.95 V vs. RHE, together with a high turnover frequency of up to 405.7 h−1. Theoretical calculations reveal that large-scale electron delocalization of BiOCs is achieved, which promotes structural stability and effectively decreases the energy barrier of rate-determining *OCHO generation. This work provides a new perspective for the design of stable high-nuclear clusters for efficient electrocatalytic CO2 conversion.
Introduction
Electrochemical CO2 reduction (ECR) has been considered as a promising technology to achieve a carbon-neutral cycle.1,2 ECR is characterized by multielectron and proton transfer3–6via multiple pathways, which leads to a variety of reduction products;7 therefore, the activity, selectivity and stability of electrocatalysts are crucial to the development of ECR. To comprehensively optimize the catalytic reactivity and selectivity, various strategies have been explored, such as heteroatom doping,8 alloy/dealloying,9 oxidation state adjustment,10 and organic moiety control,11 with the prevailing model locally at individual active sites.12,13 Recently, the effect of electron delocalization on electrocatalytic activity has attracted increasing attention, by which the energetic barriers of key intermediates could be modulated.14,15 Moreover, the additional charge (lost or gained) could uniformly distribute over all atoms, and the stability of catalysts during the electrochemical process may be increased which will help to solve the intractable dynamic reconstruction issue of electrocatalysts.16–18 Clusters are atomic-precision architectures located between atoms and nanomaterials.19 The electronic properties of some clusters have been exhibited to resemble those of atoms,20–23 and thus, large-scale electron delocalization may appear in clusters,24 yet, related research on catalysis is in its infancy. High-nuclear clusters are famous for their high surface-atom ratio, which could supply abundant metal active species for electrocatalysis.25,26 However, the construction of stable high-nuclear clusters with ECR activity is still a huge challenge.27,28Over the past decades, Bi-based materials have been extensively investigated as electrocatalysts because of their activity toward high-valued HCOOH production.29–34 In this work, we chose t-butylcalix[8]arene (TBC[8]) as a ligand to assemble with Bi3+, achieving unprecedented high-nuclear Bi-oxo clusters (BiOCs).35,36 Controlled hydrolysis of Bi3+ ions to promote crystallization is a basic prerequisite for obtaining high-nuclear BiOCs.37,38 Chelating organoamine with weakly coordinated N-donors, such as ethylenediamine (en), can exert a slow release effect on oxophilic Bi3+ ions through coordination (i.e., providing low concentrations of Bi3+ ions), thus avoiding rapid precipitation.39,40 Besides, the hydrolysis-limited ligands with strong coordination ability can delay the hydrolysis of Bi3+ ions.41,42 In addition, the introduction of second metal ions into BiOCs is another effective strategy to achieve a deep self-assembly to form high-nuclear BiOCs.43,44 As a macrocyclic octamer of phenols, TBC[8] can simultaneously grab more Bi3+ ions through multiple pre-organized phenoxy (hydroxyl) sites and delay the hydrolysis of Bi3+. Meanwhile, the continuous carbon backbone of TBC[8] can further provide an additional stabilization effect for BiOCs.
Herein, a family of calix[8]arene-protected bismuth-oxo clusters, including 4-nuclear Bi4 (BiOC-1/2), 9-nuclear Bi8Al (BiOC-3), 20-nuclear Bi20 (BiOC-4), 24-nuclear Bi24 (BiOC-5) and 42-nuclear Bi40Mo2 (BiOC-6), were successfully assembled from TBC[8] and Bi3+ ions under solvothermal conditions. The Bi40Mo2 in BiOC-6 is the largest calix[8]arene-based metal-oxo cluster reported so far. The properties of the solvents had a great influence on the assembly of these Bi–O clusters. The pure methanol environment produced only small-sized Bi4 clusters. Meanwhile the introduction of DMF significantly increased the nuclearity of Bi8Al and Bi20. Increasing the ratio of methanol in methanol/DMF ultimately gave rise to high-nuclear Bi24 and the largest Bi40Mo2. As ECR catalysts, such BiOCs exhibit high structural stability, outstanding turnover frequency (TOF) and extraordinary Faraday efficiency (FE) towards HCOOH generation over a wide potential range. It is noteworthy that the FEHCOOH of BiOC-5 reaches 97% at −0.95 V, which is 16% higher than that of BiOC-4. The analysis of the electron gain and loss process by DFT calculations indicates that a nuclear-related electron-delocalization effect is achieved in these BiOCs. Catalytic mechanism studies verify that *OCHO is a key intermediate for HCOOH formation and electron delocalization plays an essential role in lowering the free energy of the *OCHO production step.
Results and discussion
A family of TBC[8]-protected bismuth-oxo clusters with different nuclearities were constructed by varying the type of solvent or introducing second metal ions using Bi(NO3)3 and TBC[8] with the assistance of organic amines. When the solvent was CH3OH, only two 4-nuclear BiOC-1 (Bi4) and BiOC-2 (Bi4) were obtained. Meanwhile BiOC-4 (Bi20) and BiOC-5 (Bi24) with higher nucleation were formed after the introduction of DMF into the reaction system, indicating that the solvent has an important influence on the nucleation process of BiOCs. In addition, the availability of the heterometallic clusters BiOC-3 (Bi8Al) and BiOC-6 (Bi40Mo2) indicates that BiOCs are quite compatible with other metal ions and the introduction of heterometallic clusters is beneficial for structural modulation (Fig. 1, S1 and Table S1†). Meanwhile, the flexible coordination mode of the TBC[8] ligand is another important factor for the structural diversity of BiOCs. A total of 10 different coordination modes of TBC[8]–Bi were found in the structures of BiOC-1 to 6 (Fig. S2†). The eight phenolic hydroxyl groups of the TBC[8] ligands can simultaneously coordinate with 4–8 Bi ions and exhibit flexible and diverse coordination patterns. One of the most typical patterns is TBC[8]–Bi4 (Fig. S2b†), where the four central phenolic hydroxyl groups are coordinated with two Bi ions via the μ2–η1:η1 form, while the other four marginal phenolic hydroxyl groups are coordinated to one Bi ion with Bi–Bi distances of approximately 3.67 Å and 3.69 Å. It is worth mentioning that the coordination pattern of TBC[8]–Bi8 found in BiOC-6 is very rare among TBC[8] complexes (Fig. S2j†) and can be described as the four marginal phenolic hydroxyl groups in TBC[8]–Bi4 also adopt the μ2–η1:η1 form to coordinate with other four Bi ions. These results indicate the promise of enriching the structural diversity of BiOCs by modulating the appropriate reaction conditions and the coordination pattern of TBC[8] ligands.To better understand the structures of this family of BiOCs, we describe them in terms of different degrees of aggregation of TBC[8]. BiOC-1 and BiOC-2 represent the two structurally simplest TBC[8]–Bi4 isomeric monomers, in which each Bi4 cluster is bridged to a TBC[8] ligand on one side, and the rest of the coordination sites are occupied by ethylenediamine or methanol (Fig. 1a, b, and S3†). The different distortion angles of TBC[8] lead to structural variations.45 Meanwhile, the structure of BiOC-3 (Bi8Al) is a TBC[8]–Bi4 dimer interconnected by the central Al3+ ion in which two TBC[8] ligands are located on the pole positions (Fig. 1c). If more TBC[8] ligands are introduced on the BiOCs surface, larger metal-oxo clusters need to be formed to reduce the steric hindrance between the organic ligands. The structure of BiOC-4 (Bi20) confirms that the smallest cluster constructing the TBC[8] trimer is Bi20, and three TBC[8] ligands are uniformly distributed on the surface (Fig. 1d). The tetramer BiOC-5 (Bi24) is composed of four TBC[8]–Bi5 subunits wrapped around the middle Bi4 cluster with a tetrahedral conformation (Fig. 1e). BiOC-6 (Bi40Mo2) is the largest metal-oxo cluster encapsulated by the TBC[8] ligand, which is composed of two Bi20 clusters bridged by two {MoO4} tetrahedra (Fig. 1f and S4†). Structural analysis of BiOC-1–6 confirms that TBC[8] is a compatible macrocyclic ligand for building high-nuclear metal-oxo clusters with strong metal ion trapping ability and flexible coordination modes.
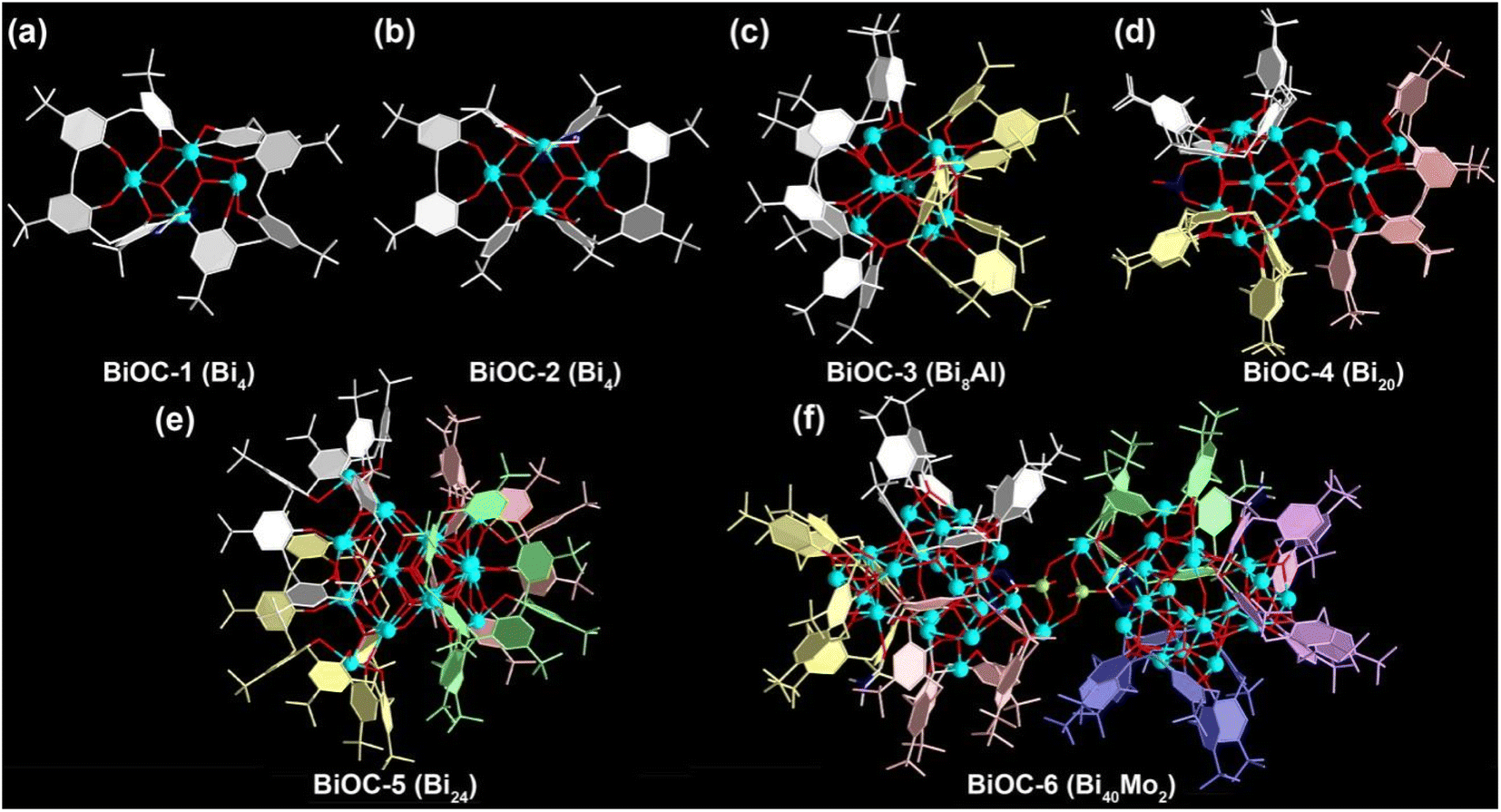 | ||
| Fig. 1 The crystal structures of BiOC-1 (a), BiOC-2 (b), BiOC-3 (c), BiOC-4 (d), BiOC-5 (e), and BiOC-6 (f). Atom color code: sky blue: Bi; red: O; white: C; blue: N; light green: Mo; teal: Al. | ||
To explore the feasibility of high-nuclear BiOCs as CO2 reduction electrocatalysts, BiOC-4 and BiOC-5 were selected as electrocatalysts due to their high yields. Chemical stability studies show that both of them can retain their crystallinity under ambient conditions and in aqueous solutions at pH = 0.3–14, as confirmed by PXRD patterns (Fig. S12–S15†). The ECR electrocatalytic activity and selectivity of BiOC-4 and BiOC-5 are evaluated in a two-compartment gas-tight electrochemical H-type cell. Their linear cyclic voltammetry (LSV) curves are recorded under CO2-saturated (pH = 7.4) and N2-saturated (pH = 8.7) 0.5 M KHCO3 solutions (Fig. 2a) to preliminary characterize their activity. Both of them demonstrate a higher current density and more positive onset potential in the CO2-saturated electrolyte compared with N2-saturated conditions, which verifies that ECR is preferred to the competitive HER at the cathode. The cathodic current density of BiOC-5 abruptly takes off at ∼−0.8 V and reaches a total current density of 28.1 mA cm−2 at −1.1 V. Meanwhile, BiOC-5 demonstrates a larger cathodic current density than BiOC-4 (16.3 mA cm−2), suggesting that BiOC-5 has favorable activity for ECR.
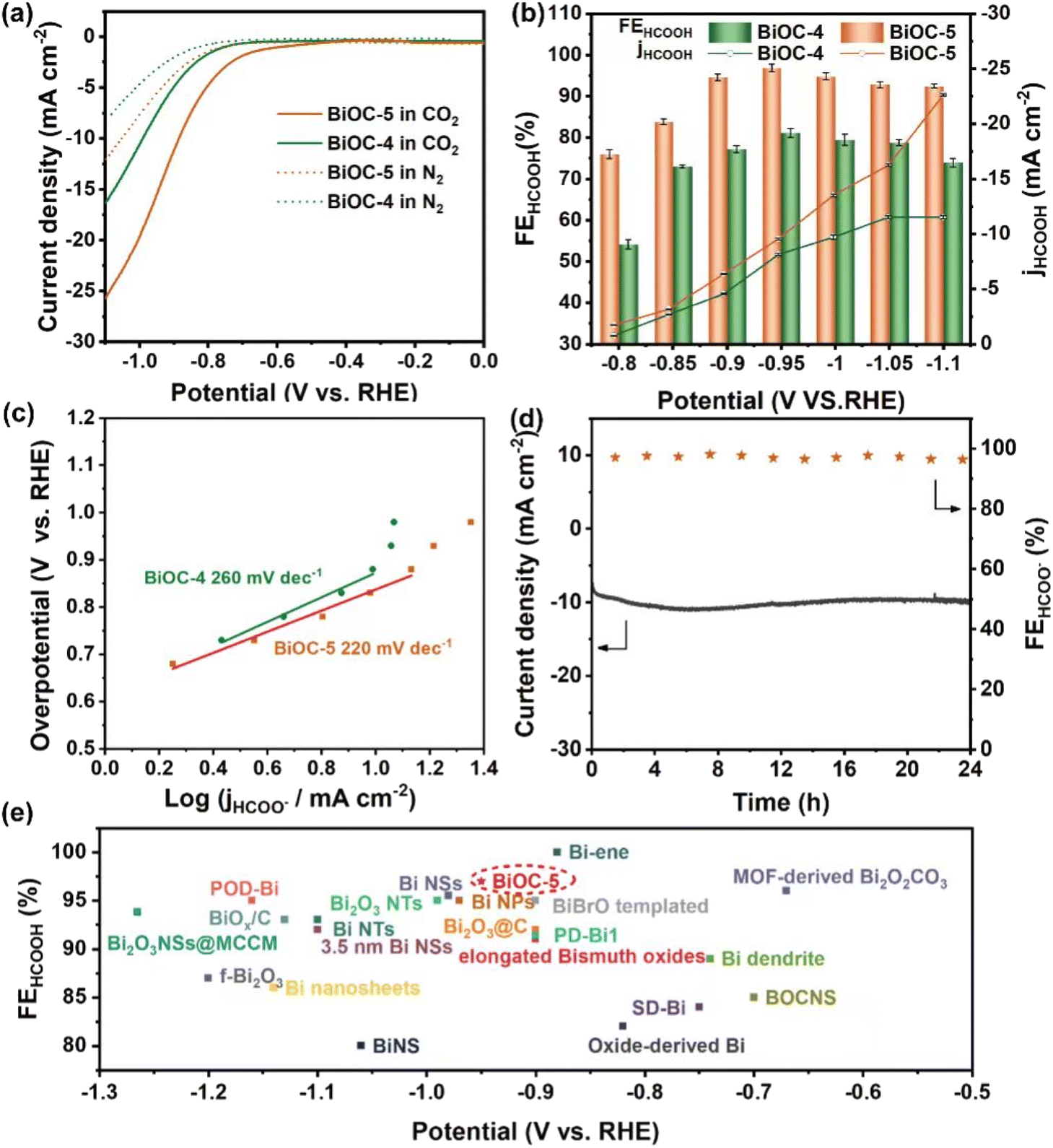 | ||
| Fig. 2 (a) Polarization curves of BiOC-4 and BiOC-5 in N2 or CO2 saturated 0.5 M KHCO3. (b) Faradaic efficiencies of production and the current densities of formate at different working potentials in a CO2-saturated 0.5 M KHCO3 electrolyte for BiOC-4 and BiOC-5. (c) Nyquist plots for BiOC-4 and BiOC-5. (d) Stability test of BiOC-5 at −0.95 V vs. RHE for 24 h. (e) Comparison of FEHCOOH with previously documented reports. | ||
The electrocatalytic ECR selectivity of BiOC-4 and BiOC-5 was biased in the selected potential range (−0.8 to −1.1 V) (Fig. S16 and S17†). The gaseous and liquid products were quantitatively analyzed by gas chromatography (GC) and ion chromatography (IC) after one-hour electrolysis. Fig. 2b illustrates that HCOOH is the major liquid product over the applied potential range from −0.8 to −1.1 V, and CO and H2 were minor gaseous products. To illuminate the origination of the product, control experiments under N2-saturated conditions were performed. No carbon-reduction product (CO and HCOOH) is found by GC and IC, verifying that the carbon-reduction products result from CO2 reduction. To be more credible, the 1H NMR spectrum was employed to check the liquid product and the results are consistent with that of IC analysis (Fig. S18†). Under CO2-saturated conditions, BiOC-5 maintains a high FE of over 92% in a large potential window between −0.9 and −1.1 V and reaches a maximum of 97% at −0.95 V. In contrast, the maximum FEHCOOH over BiOC-4 is less than 82%. The FEHCOOH of BiOC-5 is not only superior to that of BiOC-4 but also ranks among the best activity values of Bi-based ECR catalysts (Fig. 2e and Table S2†). The corresponding FEH2 and FECO are depicted in Fig. S19 and S20.† Then, the partial current density of formate (jHCOOH) for both BiOC-4 and BiOC-5 was calculated. BiOC-5 presents a larger jHCOOH of 22.6 mA cm−2, almost twice that of BiOC-4 (11.7 mA cm−2).
To estimate the electrocatalytic reactivity of BiOC-4 and BiOC-5, the electrochemically active surface area (ECSA) is analyzed, firstly. From the values of electrochemical double-layer capacitance (Cdl) based on cyclic voltammetry (CV) curves, we can find that BiOC-5 has a larger Cdl than BiOC-4, suggesting that BiOC-5 can provide more available active sites for contact with the ECR electrolyte (Fig. S21†). This provides a prerequisite for the high catalytic activity of BiOC-5. Next, the turn-around frequency (TOF) was calculated (for calculation details, please see the ESI†). BiOC-4 presents a TOF of 276.59 h−1 for HCOOH formation at −1.1 V, while BiOC-5 shows an impressive reaction rate with a TOF of 405.7 h−1 at −1.1 V, which outperforms the performance of crystalline Bi-based electrocatalysts.46,47 Catalytic durability is one of the crucial parameters for evaluating the application potential of electrocatalysts. Therefore, a long-time electrolysis experiment was performed at a potential of −0.95 V. The current curve illustrates that BiOC-5 can keep the current density roughly unchanged in a 24 h reaction (Fig. 2d). It worth noting that the FEHCOOH of BiOC-5 still remains at higher than 95% over this reaction period. These results unambiguously demonstrate that BiOC-5 is a robust electrocatalyst for ECR.
The reaction kinetics for HCOOH production are studied. BiOC-5 exhibits a relatively lower Tafel slope of 220 mV dec−1 compared to BiOC-4 (260 mV dec−1), evidencing the favorable kinetics of BiOC-5 for ECR (Fig. 2c). Again, this means that the initial electron transfer for the formation of the *OCHO intermediate on the catalyst surface is the rate-determining step. Furthermore, electrochemical impedance (EIS) was performed for both catalysts at −0.95 V (Fig. S22†). According to the results of the Nyquist plots, BiOC-5 possesses a much smaller semicircle diameter, reflecting faster electron transfer efficiency.
Since Bi-based materials often undergo dynamic reconstruction during ECR, to determine whether the structure of BiOCs is maintained, X-ray absorption near edge structure (XANES) and extended X-ray absorption fine structure (EXAFS) spectroscopy were performed, taking BiOC-5 as an example.
Both the absorption edge and white line peak of the XANES spectrum for BiOC-5 nearly overlap with that of the Bi2O3 reference, suggesting that Bi atoms in BiOC-5 are Bi3+ species (Fig. 3a). After ECR, the absorption edge of BiOC-5 only slightly shifts toward lower energy, indicating that the oxidation state of Bi is mainly retained. By fitting the recorded spectrum with the window from 1 to 6 Å, we obtained the bond lengths of Bi foil, Bi2O3, BiOC-5 before and BiOC-5 after catalysis (Fig. 3b).48 The EXAFS curves of the Bi L3-edge reveal the main peak at 1.6 Å, corresponding to the scattering path of the Bi–O bond. The Bi–Bi oscillation of metallic–Bi at 3.3 Å was not observed during the long-term electrochemical process, which illuminates the robust chemical stability of BiOC-5 during ECR.15
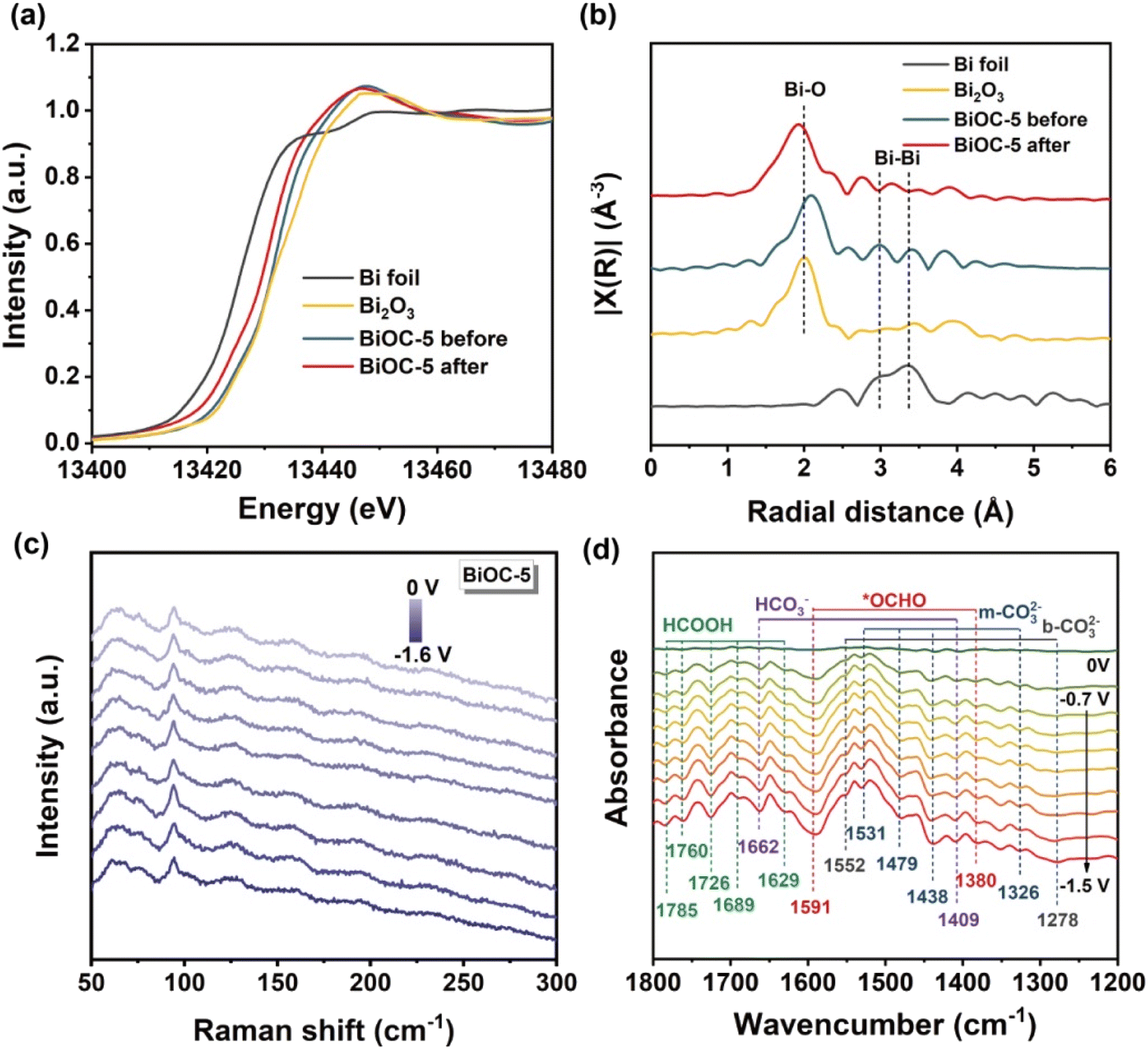 | ||
| Fig. 3 (a) Bi L3-edge XANES spectra of Bi foil, Bi2O3, BiOC-5 before and BiOC-5 after. (b) EXAFS in R-space for Bi foil, Bi2O3, BiOC-5 before and BiOC-5 after. (c). In situ Raman spectra of CO2 reduction on BiOC-5. (d) In situ ATR-FTIR spectra of BiOC-5 during the ECR. | ||
Besides, a series of spectral analyses were performed to further reveal the structural stability during ECR. The PXRD pattern and FT-IR spectrum of the electrocatalysts after stability testing were almost identical to before catalysis, indicating that BiOC-4 and BiOC-5 can maintain their structural integrity during ECR (Fig. S23–S26†). XPS spectral analysis did not detect Bi0 before and after the ECR experiment, and the oxidized state of Bi was well preserved (Fig. S27†). The in situ Raman spectrum provides persuasive evidence that the structure of BiOCs can be preserved as expected (Fig. 3c). Furthermore, the morphology of the BiOC catalyst is largely preserved as shown by SEM images (Fig. S28†). In a word, all the above results demonstrated that BiOCs possess extraordinary stability toward ECR.
In situ FTIR measurements were conducted to identify the intermediates during the ECR process. As illustrated in Fig. 3d, the peaks appear at 1326, 1438, 1479 and 1531 cm−1 attributed to the stretching vibration of m-CO32−, and at 1278 and 1552 cm−1 belonging to the stretching vibration of b-CO32−.49 The bands at 1409 and 1662 cm−1 in the spectra are attributed to HCO3−.49 Most importantly, the *OCHO intermediate signals detected at 1380 and 1591 cm−1 are generally regarded as key intermediates in the electrochemical CO2 → HCOOH conversion and increase gradually with the applied potentials varying between −0.7 and −1.5 V.50 Besides, the characteristic peaks of HCOO− (1629, 1689, 1727, 1760, and 1785 cm−1) start to appear with increasing voltage.51
To gain a deep understanding of the high stability and the outstanding catalytic activity and selectivity of BiOC catalysts, theoretical calculations were performed. The charge distribution on the LUMO orbital and the localized orbital locator (LOL) maps suggest that significant electron delocalization occurs throughout the BiOC clusters (Fig. S29 and S30†). The orbital delocalization index (ODI) of LUMO orbitals is a significant factor to evaluate the extent of spatial delocalization. The small ODI value of BiOC-5 in contrast to BiOC-4 (1.56 vs. 5.58) indicates that BiOC-5 possesses a much wider range of electron delocalization. These results are further corroborated by the projected density of states (PDOS) of all Bi atoms in BiOC-4 and BiOC-5 (Fig. 4a and b). The roughly equivalent contributions of each Bi atom on the non-occupied orbitals demonstrate that each Bi atom could accept additional electrons. Compared to BiOC-4, the PDOS of BiOC-5 is closer to the Fermi energy level, which indicates a stronger electron-donating capability for BiOC-5, while the narrower band gap provides a better electron transport capability. Furthermore, the vertical ionization energy was calculated for BiOC-4 and BiOC-5, the charge of which is neutralized by protons (Table S3†).24 The results present that BiOC-4 needs 156.112 kcal mol−1 to ionize the first electron, which is larger than 79.759 kcal mol−1 required by BiOC-5. Therefore, we speculate that electron delocalization is achieved in such metal-oxo clusters, which enables the entire cluster to act as an active catalytic site and may facilitate the catalytic process. The electron delocalization on the metal-oxo cluster also contributes to the structural stability in the electrocatalytic CO2RR process.52,53
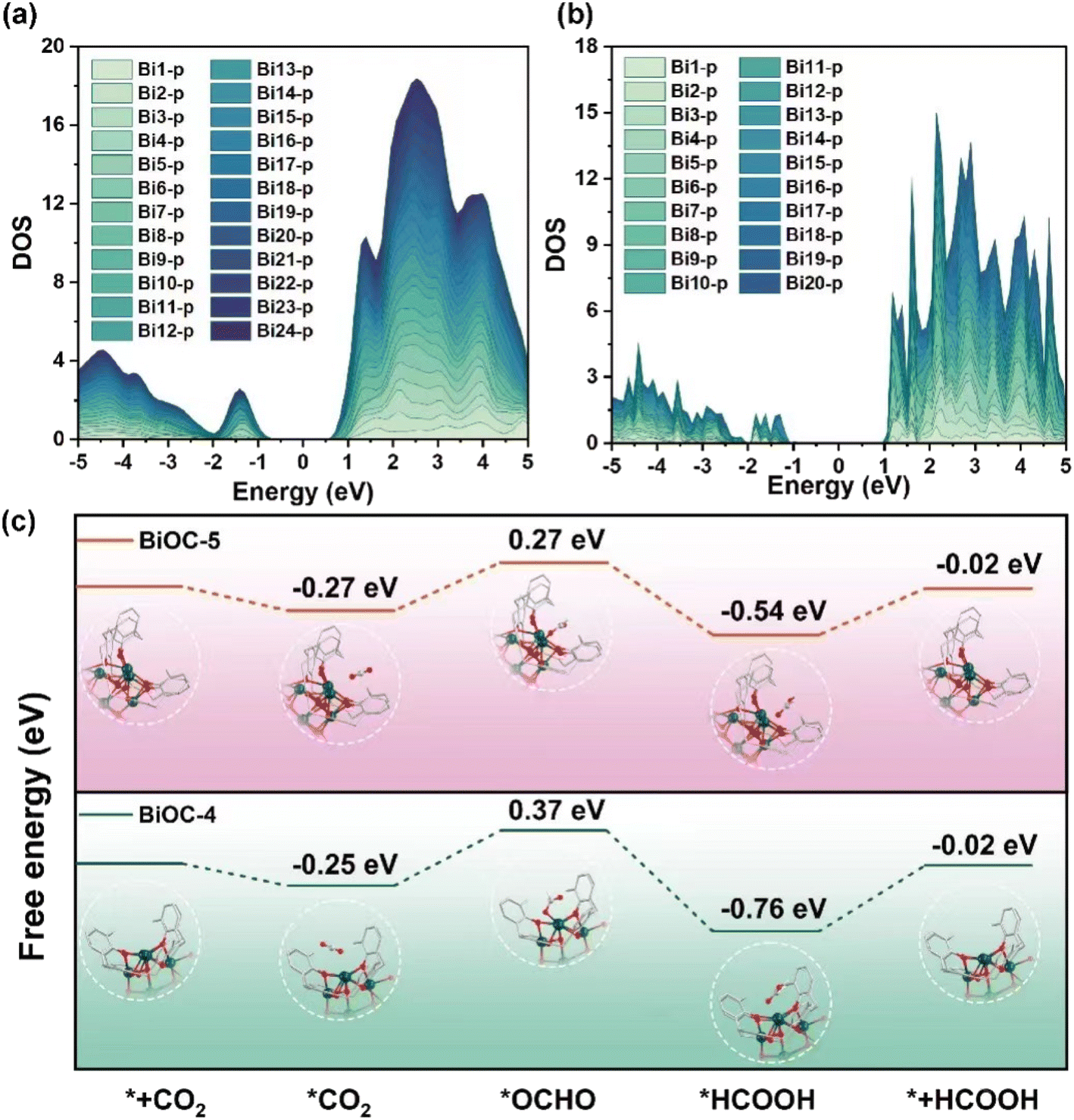 | ||
| Fig. 4 (a and b) PDOS of all Bi atoms for BiOC-4 and BiOC-5. (c) Free-energy diagrams for HCOOH on BiOC-4 and BiOC-5. | ||
The reaction energy pathways for CO2 reduction to HCOOH and the competing hydrogen evolution reaction were explored by density functional theory (DFT) calculations. Both BiOC-4 and BiOC-5 have lower energy barriers for *OCHO formation than for H* formation (Fig. S31†). Therefore, HCOOH production is preferential on BiOC-4 and BiOC-5. Moreover, the hydrophobic skeleton of BiOC-4 and BiOC-5 (contact angle in Fig. S32†) also helps to suppress hydrogen production. Fig. 4c illustrates the optimized structures and energies of the CO2RR pathway involved in each dominant intermediate. For BiOC-5, the Gibbs free energy change (ΔG) of *OCHO formation is calculated to be +0.54 eV, whereas the ΔG for BiOC-4 is +0.62 eV. It implies that this step is thermodynamically more favorable on BiOC-5. Subsequently, these intermediates accept electrons and protons to form adsorbed HCOOH and desorb from the catalyst surface. Furthermore, the maximum energy barriers of BiOC-5 are observed for *OCHO formation, indicating that the first electron/proton transfer was the chemical rate-determining step. Moreover, BiOC-5 shows a great ΔG for *H formation, which could inhibit the generation of H2 compared with BiOC-4. It's conjectured that the electron delocalization effect possibly has an essential contribution in lowering the energy barrier to the formation of *OCHO intermediates in the CO2RR. Besides, 4-core BiOC-1 was further selected to be analyzed and evaluated for ECR to confirm the results and conclusions (Fig. S33†). The results are as predicted where less core-numbered BiOC-1 exhibits the smallest FEHCOOH of 75% and jHCOOH of −9.63 mA cm−2 compared to BiOC-4 and BiCO-5, implying the rationality of the conclusions.
Conclusions
In summary, a family of calix[8]arene-protected bismuth-oxo clusters with robust stability were synthesized and explored as catalysts for the electrochemical reduction of CO2 to HCOOH. Calix[8]arene is proved to be an excellent macrocyclic ligand for the construction of high-nuclear metal-oxo clusters with its strong ion-trapping ability and flexible coordination modes. The Bi40Mo2 cluster in BiOC-6 is the first metal-oxo cluster with more than 40 metal centers constructed from calix[8]arenes, illuminating significant progress in the synthesis of calix[8]arene-based high-nuclear clusters. Notably, BiOCs present excellent stability during an electrocatalytic process, and BiOC-5 achieves high selectivity for formate production over a wide potential range with an outstanding maximum Faraday efficiency of 97% at −0.95 V vs. RHE. Besides, BiOC-5 achieves an impressive TOF of 405.7 h−1, ranking among the highest values of crystalline Bi-based materials. Overall, BiOC-5 exhibits high catalytic selectivity and activity, while maintaining high structural stability for ECR. DFT calculations suggest that electron delocalization occurs in BiOC-5, resulting in the uniform distribution of additional charge on all Bi atoms and low energy for electron departure. This not only guarantees structural stability, but also reliably improves the conversion efficiency of CO2 to HCOOH by decreasing the ΔG of decisive *OCHO formation. This study not only paves the way for the construction of high-nuclear metal-oxo clusters but also illustrates the superiority of clusters as catalysts for CO2 electroreduction.Data availability
All experimental supporting data and procedures are available in the ESI.†Author contributions
C. Y. S. and X. L. W. conceived the idea and designed the experiments. B. S. H. performed the SCXRD measurement and crystal structure analysis. H. Y. Z. performed the electrochemical studies. Q. W. performed the DFT calculations. K. H. Z. performed the single-crystal synchrotron X-ray diffraction measurements. Z. H. K., Q. H. P and Z. M. S. contributed to the general methodology and assisted with data interpretation. C. Q., C. Y. S. and X. L. W. co-drafted the manuscript and reviewed/revised the manuscript. All authors contributed to the analysis of the manuscript.Conflicts of interest
The authors declare no competing interests.Acknowledgements
This work was financially supported by the NSFC of China (No. 21971032), Foundation of Jilin Educational Committee (No. JJKH20221153KJ), and Jilin Provincial Department of Science and Technology (No. 20230508108RC).References
- L. L. Zhuo, P. Chen, K. Zheng, X. W. Zhang, J. X. Wu, D. Y. Lin, S. Y. Liu, Z. S. Wang, J. Y. Liu, D. D. Zhou and J. P. Zhang, Angew. Chem., Int. Ed., 2022, 61, e202204967 CrossRef CAS PubMed.
- S. Navarro-Jaén, M. Virginie, J. Bonin, M. Robert, R. Wojcieszak and A. Y. Khodakov, Nat. Rev. Chem., 2021, 5, 564–579 CrossRef PubMed.
- G. B. Wen, B. H. Ren, Y. Zheng, M. Li, C. Silva, S. Q. Song, Z. Zhang, H. Z. Dou, L. Zhao, D. Luo, A. P. Yu and Z. W. Chen, Adv. Energy Mater., 2022, 12, 2103289 CrossRef CAS.
- W. Zhang, C. Q. Huang, J. X. Zhu, Q. C. Zhou, R. H. Yu, Y. L. Wang, P. F. An, J. Zhang, M. Qiu, L. Zhou, L. Q. Mai, Z. G. Yi and Y. Yu, Angew. Chem., Int. Ed., 2022, 61, e202112116 CAS.
- S. Nitopi, E. Bertheussen, S. B. Scott, X. Y. Liu, A. K. Engstfeld, S. Horch, B. Seger, I. E. L. Stephens, K. Chan, C. Hahn, J. K. Nørskov, T. F. Jaramillo and I. Chorkendorff, Chem. Rev., 2019, 119, 7610–7672 CrossRef CAS PubMed.
- Q. Zhao, J. M. P. Martirez and E. A. Carter, J. Am. Chem. Soc., 2021, 143, 6152–6164 CrossRef CAS PubMed.
- Y. H. Wang, Z. Y. Wang, C. T. Dinh, J. Li, A. Ozden, M. G. Kibria, A. Seifitokaldani, C. S. Tan, C. M. Gabardo, M. C. Luo, H. Zhou, F. W. Li, Y. W. Lum, C. McCallum, Y. Xu, M. X. Liu, A. Proppe, A. Johnston, P. Todorovic, T. T. Zhuang, D. Sinton, S. O. Kelley and E. H. Sargent, Nat. Catal., 2019, 3, 98 CrossRef.
- Y. S. Zhou, F. L. Che, M. Liu, C. Q. Zou, Z. Q. Liang, P. D. Luna, H. F. Yuan, J. Li, Z. Q. Wang, H. P. Xie, H. M. Li, P. N. Chen, E. Bladt, R. Quintero-Bermudez, T. K. Sham, S. Bals, J. Hofkens, D. Sinton, G. Chen and E. H. Sargent, Nat. Chem., 2018, 10, 974–980 CrossRef CAS PubMed.
- E. L. Clark, C. Hahn, T. F. Jaramillo and A. T. Bell, J. Am. Chem. Soc., 2017, 139, 15848–15857 CrossRef CAS PubMed.
- P. D. Luna, R. Quintero-Bermudez, C. T. Dinh, M. B. Ross, O. S. Bushuyev, P. Todorović, T. Regier, S. O. Kelley, P. D. Yang and E. H. Sargent, Nat. Catal., 2018, 1, 103–110 CrossRef.
- M. S. Xie, B. Y. Xia, Y. W. Li, Y. Yan, Y. H. Yang, Q. Sun, S. H. Chan, A. Fishere and X. Wang, Energy Environ. Sci., 2016, 9, 1687–1695 RSC.
- Z. W. Huang, J. X. Liang, D. M. Tang, Y. X. Chen, W. Y. Qu, X. L. Hu, J. X. Chen, Y. Y. Dong, D. R. Xu, D. Golberg, J. Li and X. F. Tang, Chem, 2022, 8, 3008–3017 CAS.
- S. Nitopi, E. Bertheussen, S. B. Scott, X. Y. Liu, A. K. Engstfeld, S. Horch, B. Seger, I. E. L. Stephens, K. Chan, C. Hahn, J. K. Nørskov, T. F. Jaramillo and I. Chorkendorff, Chem. Rev., 2019, 119, 7610–7672 CrossRef CAS PubMed.
- H. Sun, L. Chen, L. K. Xiong, K. Feng, Y. F. Chen, X. Zhang, X. Z. Yuan, B. Y. Yang, Z. Deng, Y. Liu, M. H. Rümmeli, J. Zhong, Y. Jiao and Y. Peng, Nat. Commun., 2021, 12, 6823 CrossRef CAS PubMed.
- S. S. He, F. L. Ni, Y. J. Ji, L. Wang, Y. Z. Wen, H. P. Bai, G. J. Liu, Y. Zhang, Y. Y. Li, B. Zhang and H. S. Peng, Angew. Chem., Int. Ed., 2018, 57, 16114–16119 CrossRef CAS PubMed.
- C. S. Cao, D. D. Ma, J. F. Gu, X. Y. Xie, G. Zeng, X. F. Li, S. G. Han, Q. L. Zhu, X. T. Wu and Q. Xu, Angew. Chem., Int. Ed., 2020, 59, 15014–15020 CrossRef CAS PubMed.
- D. Z. Yao, C. Tang, A. Vasileff, X. Zhi, Y. Jiao and S. Z. Qiao, Angew. Chem., Int. Ed., 2021, 60, 18178–18184 CrossRef CAS PubMed.
- J. Y. Duan, T. Y. Liu, Y. H. Zhao, R. O. Yang, Y. Zhao, W. B. Wang, Y. W. Liu, H. Q. Li, Y. F. Li and T. Y. Zhai, Nat. Commun., 2022, 13, 2039–2049 CrossRef CAS PubMed.
- Q. D. Liu and X. Wang, Chem Catal., 2022, 2, 1257–1266 CrossRef CAS.
- S. N. Khanna and P. Jena, Phys. Rev. B: Condens. Matter Mater. Phys., 1995, 51, 13705–13716 CrossRef CAS PubMed.
- C. Bo and J. M. Poblet, Isr. J. Chem., 2011, 51, 228–237 CrossRef CAS.
- Q. Tang, G. X. Hu, V. Fung and D. E. Jiang, Acc. Chem. Res., 2018, 51, 2793–2802 CrossRef CAS PubMed.
- L. R. Liu, P. Li, L. F. Yuan, L. J. Cheng and J. L. Yang, Nanoscale, 2016, 8, 12787–12792 RSC.
- Q. D. Liu, Q. H. Zhang, W. X. Shi, H. S. Hu, J. Zhuang and X. Wang, Nat. Chem., 2022, 14, 433–440 CrossRef CAS PubMed.
- B. W. J. Chen, L. Xu and M. Mavrikakis, Chem. Rev., 2021, 121, 1007–1048 CrossRef CAS PubMed.
- Y. Liu, N. O. Weiss, X. D. Duan, H. C. Cheng, Y. Huang and X. F. Duan, Nat. Rev. Mater., 2016, 1, 16042 CrossRef CAS.
- W. H. Fang, L. Zhang and J. Zhang, J. Am. Chem. Soc., 2016, 138, 7480–7483 CrossRef CAS PubMed.
- M. X. Ma, X. L. Ma, G. M. Liang, X. T. Shen, Q. L. Ni, L. C. Gui, X. J. Wang, S. Y. Huang and S. M. Li, J. Am. Chem. Soc., 2021, 143, 13731–13737 CrossRef CAS PubMed.
- N. Han, Y. Wang, H. Yang, J. Deng, J. H. Wu, Y. F. Li and Y. G. Li, Nat. Commun., 2018, 9, 1320–1327 CrossRef PubMed.
- F. P. G. D. Arquer, O. S. Bushuyev, P. D. Luna, C. T. Dinh, A. Seifitokaldani, M. I. Saidaminov, C. S. Tan, L. N. Quan, A. Proppe, M. G. Kibria, S. O. Kelley, D. Sinton and E. H. Sargent, Adv. Mater., 2018, 30, 1802858 CrossRef PubMed.
- H. Yang, N. Han, J. Deng, J. H. Wu, Y. Wang, Y. P. Hu, P. Ding, Y. F. Li, Y. G. Li and J. Lu, Adv. Energy Mater., 2018, 8, 1801536 CrossRef.
- Y. Zhang, X. L. Zhang, Y. Z. Ling, F. W. Li, A. M. Bond and J. Zhang, Angew. Chem., Int. Ed., 2018, 57, 13283–13287 CrossRef CAS PubMed.
- Z. P. Chen, K. W. Mou, X. H. Wang and L. C. Liu, Angew. Chem., Int. Ed., 2018, 57, 12790–12794 CrossRef CAS PubMed.
- P. L. Deng, H. M. Wang, R. J. Qi, J. X. Zhu, S. H. Chen, F. Yang, L. Zhou, K. Qi, H. F. Liu and B. Y. Xia, ACS Catal., 2020, 10, 743–750 CrossRef CAS.
- Z. Wang, H. F. Su, Y. W. Gong, Q. P Qu, Y. F. Bi, C. H. Tung, D. Sun and L. S. Zheng, Nat. Commun., 2020, 11, 308–315 CrossRef CAS PubMed.
- D. M. Espinosa, A. L. Rheinggold and T. A. Hanna, Dalton Trans., 2009, 26, 5226–5238 RSC.
- D. Mansfeld, M. Mehring and M. Schurmann, Angew. Chem., Int. Ed., 2005, 44, 245–249 CrossRef CAS PubMed.
- E. V. Dikarev, H. T. Zhang and B. Li, Angew. Chem., Int. Ed., 2006, 45, 5448–5451 CrossRef CAS PubMed.
- Z. Li, Z. H. Lv, H. Yu, Y. Q. Sun, X. X. Li and S. T. Zheng, CCS Chem., 2022, 4, 2938–2945 CrossRef CAS.
- M. Mehring, Coord. Chem. Rev., 2007, 251, 974–1006 CrossRef CAS.
- L. H. Liu, L. N. Zakharov, A. L. Rheingold and T. A. Hanna, Chem. Commun., 2004, 13, 1472–1473 RSC.
- D. M. Espinosa and T. A. Hanna, Dalton Trans., 2009, 26, 5211–5225 RSC.
- L. Jin, X. X. Li, Y. J. Qi, P. P. Niu and S. T. Zheng, Angew. Chem., Int. Ed., 2016, 55, 13793–13797 CrossRef CAS PubMed.
- J. C. Liu, Q. Han, L. J. Chen, J. W. Zhao, C. Streb and Y. F. Song, Angew. Chem., Int. Ed., 2018, 57, 8416–8420 CrossRef CAS PubMed.
- S. M. Taylor, S. Sanz, R. D. Mclntosh, C. M. Beavers, S. J. Teat, E. K. Brrchin and S. J. Dalgarno, Chem.–Eur. J., 2012, 18, 16014–16022 CrossRef CAS PubMed.
- F. Li, G. H. Gu, C. Choi, P. Kolla, S. Hong, T. S. Wu, Y. L. Soo, J. Masa, S. Mukerjee, Y. Jung, J. S. Qiu and Z. Y. Sun, Appl. Catal., B, 2020, 277, 119241–119250 CrossRef CAS.
- J. W. Shi, S. N. Sun, J. Liu, Q. Niu, L. Z. Dong, Q. Huang, J. J. Liu, R. Wang, Z. F. Xin, D. D. Zhang, J. Y. Niu and Y. Q. Lan, ACS Catal., 2022, 12, 14436–14444 CrossRef CAS.
- C. W. Lee, J. S. Hong, K. D. Yang, K. Jin, J. H. Lee, H. Y. Ahn, H. M. Seo, N. E. Sung and K. T. Nam, ACS Catal., 2018, 8, 931–937 CrossRef CAS.
- S. S. He, F. L. Ni, Y. J. Ji, L. Wang, Y. Z. Wen, H. P. Bai, G. J. Liu, Y. Zhang, Y. Y. Li, B. Zhang and H. S. Peng, Angew. Chem., Int. Ed., 2018, 130, 16346–16351 CrossRef.
- J. J. Liu, N. Li, J. W. Sun, J. Liu, L. Z. Dong, S. J. Yao, L. Zhang, Z. F. Xin, J. W. Shi, J. X. Wang, S. L. Li and Y. Q. Lan, ACS Catal., 2021, 11, 4510–4519 CrossRef CAS.
- X. L. Zu, X. D. Li, W. Liu, Y. F. Sun, J. Q. Xu, T. Yao, W. S. Yan, S. Gao, C. M. Wang, S. Q. Wei and Y. Xie, Adv. Mater., 2019, 31, 1808135 CrossRef PubMed.
- H. C. Hu, H. S. Hu, B. Zhao, P. Cui, P. Cheng and J. Li, Angew. Chem., Int. Ed., 2015, 54, 11681–11685 CrossRef CAS PubMed.
- P. Cui, H. S. Hu, B. Zhao, J. T. Miller, P. Cheng and J. Li, Nat. Commun., 2015, 6, 6331–6335 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2236082–2236087. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3sc02924g |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2023 |