
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCombining lipid-mimicking-enabled transition metal and enzyme-mediated catalysis at the cell surface of E. coli†
Tristan
Wegner‡
a,
Alexander
Dombovski‡
 b,
Katrin
Gesing
b,
Alexander
Köhrer
c,
Matthias
Elinkmann
c,
Uwe
Karst
c,
Frank
Glorius
*a and
Joachim
Jose
*b
b,
Katrin
Gesing
b,
Alexander
Köhrer
c,
Matthias
Elinkmann
c,
Uwe
Karst
c,
Frank
Glorius
*a and
Joachim
Jose
*b
aUniversity of Münster, Institute of Organic Chemistry, Münster, Germany. E-mail: glorius@uni-muenster.de
bUniversity of Münster, Institute of Pharmaceutical and Medicinal Chemistry, Münster, Germany. E-mail: joachim.jose@uni-muenster.de
cUniversity of Münster, Institute of Inorganic and Analytical Chemistry, Münster, Germany
First published on 6th October 2023
Abstract
Being an essential multifunctional platform and interface to the extracellular environment, the cell membrane constitutes a valuable target for the modification and manipulation of cells and cellular behavior, as well as for the implementation of artificial, new-to-nature functionality. While bacterial cell surface functionalization via expression and presentation of recombinant proteins has extensively been applied, the corresponding application of functionalizable lipid mimetics has only rarely been reported. Herein, we describe an approach to equip E. coli cells with a lipid-mimicking, readily membrane-integrating imidazolium salt and a corresponding NHC–palladium complex that allows for flexible bacterial membrane surface functionalization and enables E. coli cells to perform cleavage of propargyl ethers present in the surrounding cell medium. We show that this approach can be combined with already established on-surface functionalization, such as bacterial surface display of enzymes, i.e. laccases, leading to a new type of cascade reaction. Overall, we envision the herein presented proof-of-concept studies to lay the foundation for a multifunctional toolbox that allows flexible and broadly applicable functionalization of bacterial membranes.
Introduction
Biological membranes are an essential component of cellular life. They provide basic cellular structure and compartmentalization from the extracellular space and mediate essential cellular processes such as recognition, signaling and transport events.1,2 To do so, membranes are generally composed of numerous different classes of biomolecules including lipids, proteins and decorating carbohydrates.1,2 The exact membrane composition and architecture however largely differ, e.g. depending on species, cell type or organelle, thus indicating its outstanding relevance for constituting a cell's or compartment's functional nature.3Given their huge importance as a multifunctional cellular platform and interface to the extracellular environment, biological membranes have been identified as valuable targets to modify and manipulate cells and cellular behavior, but also to implement artificial, new-to-nature functions into cells. For example, bacterial surface display has been applied for decorating bacterial membrane surfaces with a wide variety of different proteins of interest, thus enabling targeted modification of cell function and interaction.4–6
Another possibility to modify biological membranes is the use of lipid mimetics that readily integrate into lipid bilayers and thus enable manipulation and functionalization of such without the need for genetic modification and expression of recombinant proteins.
While this approach has been applied in various ways in model bilayers and mammalian cell membranes there are only a few examples of the functional integration of lipid mimetics into bacterial cell membranes.7 Recently, we reported a new class of imidazolium-based lipid analogs that exhibit an amphiphilic structure similar to natural lipids. The structure of these analogs consists of a hydrophobic backbone linked to a polar imidazolium headgroup and allows flexible tuning and modification of their function and biological properties.8 In particular, a clickable imidazolium-based cholesterol analog was developed that has been shown to readily integrate into the plasma membrane of mammalian cell lines, where it could be employed for membrane surface functionalization with a fluorescent dye.9,10 However, more commonly imidazolium salts serve as precursors to form N-heterocyclic carbenes (NHCs) that are employed as electron-rich ligands to tune properties and reactivity of transition metal complexes/catalysts in organic synthesis.11 (NHC-)transition metal complexes have, however, also been reported to enable classic synthetic organic reactions in biological environments.12–16
Herein, we sought to equip E. coli cells with new-to-nature functionality by implementing an artificial lipid mimetic NHC–palladium (Pd) complex into the bacterial membrane that subsequently enables the bacteria to mediate cleavage of propargyl ethers present in the surrounding medium. As a proof-of-concept cells equipped with such a Pd-containing lipid were shown to be able to decage a propargyl-protected coumarin or phenol derivative. We show that the derivative, as deprotected, can subsequently serve as a substrate for laccase displayed on the surface of E. coli, thus overall allowing the cells to mediate a new-to-nature cascade reaction on their surface that results from the combination of both the membrane-embedded NHC–Pd lipid analog and the displayed enzyme. We envision this proof-of-concept study to serve as a starting point for a platform that allows flexible and broadly applicable implementation of artificial reactivities into cellular membranes and thus to potentially enable cells to perform new-to-nature reactions that might have useful applications e.g. in biotechnology or medicine.
Results and discussion
Integration of CHIMs into E. coli membranes
We recently reported the design, synthesis, and application of cholesterol-based imidazolium salts (CHIMs). These structurally versatile and readily customizable lipid mimetics could be shown to readily integrate into the plasma membrane of mammals with their unpolar cholesterol part being buried in the unpolar region of the membrane and their polar, readily modifiable imidazolium headgroup being located at the polar surface region of the membrane. In this context, the attachment of a clickable azide linker enabled the functionalization of membrane surfaces either via membrane incorporation of pre-clicked derivatives or via on-membrane surface click chemistry.9,10Herein, we sought to expand CHIM applicability for membrane surface functionalization in bacterial cells using the example of E. coli. E. coli represents a widely used model organism for Gram-negative prokaryotes and is a highly important workhorse in the field of synthetic biology and biotechnology.17,18 We therefore envisioned that an innovative approach for membrane surface functionalization of such organisms could be of particularly high value. In comparison to mammalian plasma membranes, E. coli membranes exhibit a vastly different structure. For example, they do not contain cholesterol but structurally closely related hopanoids instead.19 In addition, the cell envelope of E. coli consists of different layers, with a phospholipid bilayer constituting the inner membrane that is surrounded by a thin peptidoglycan layer and another, outer asymmetric lipid bilayer. In the latter, lipopolysaccharides represent a major component that shields the outer membrane from the surrounding environment, whereas the inner layer is composed of phospholipids.20 Against this backdrop, we first wanted to investigate whether a CHIM analog could nonetheless integrate into the E. coli outer membrane, as it was previously shown for mammalian plasma membranes. For this purpose, the clickable CHIM analog CHIM-L was conjugated with the dibenzocyclooctyne (DBCO)-containing fluorescent dye DBCO-PEG4-Fluor 545 via the strain-promoted azide–alkyne cycloaddition (SPAAC) click reaction and subsequently cells of E. coli BL21 (DE3) were incubated with the clicked molecule (Fig. 1A). After incubation for 90 minutes at 37 °C and subsequent thorough washing with PBS, flow cytometer analysis showed a single homogeneous cell population with increased fluorescence intensity in comparison to cells incubated with natural cholesterol (CHOL) and the CHIM derivative CHIM-H that were equally pre-treated with the DBCO-PEG4-Fluor 545 dye and analysed as non-clickable negative controls (Fig. 1B). This was indicative of homogeneous cell labeling by the conjugated lipid. To rule out potential unspecific, non-membrane integration-related binding modes, e.g. coordination to surface proteins, the experiment was repeated with the addition of 5% bovine serum album to the labeling medium. This resulted in an identical labeling efficiency and confirmed that the labelling procedure was reproducible. The fact that the presence of albumin had no impact on the cell labelling indicates that an unspecific interaction of the conjugated lipid with (surface) proteins as the major mode of cellular binding seems unlikely (Fig. S3†). Fluorescence microscopy of the labelled cells indicated that the fluorophore was – as expected – located at the cell envelope and not in the cytosol (Fig. 1C). Such highly fluorescent cells were absent in the control sample using non-clickable lipid CHIM-H (Fig. S4†). With regard to the high molecular weight of the clicked CHIM-L derivative (∼1.5 kDa) its diffusion through the outer membrane, e.g. via porins (i.e. OmpF/OmpC), β-barrel proteins that allow passive diffusion of molecules with a size exclusion of around 600 Da, seems rather unlikely.21 To further elucidate the exact localization of the CHIM-L conjugate, we additionally performed an osmotic shock experiment with labeled E. coli cells in order to isolate and analyse the outer membrane separately from the rest of the cell. After osmotic shock two different species could be observed: on the one hand small and highly fluorescent particles which appeared to be outer membrane vesicles as reported before22 and on the other hand weakly fluorescent roundish spheroplasts (Fig. S5 and S6†). The presence of spheroplasts clearly indicated the successful stripping of the outer membrane from the bacterial cells. While the substantially higher fluorescence intensity in the outer membrane vesicles indeed indicates a preferential outer membrane localization of the fluorescent conjugate it is not clear whether the observed weak fluorescence in the spheroplasts originates from the fluorescent conjugate that was present in the inner membrane beforehand or integrated due to release of the fluorescent CHIM-L conjugate during osmotic shock. Either way, it is worth noting that also in the spheroplasts the fluorophore appears to be solely located in the membrane compartment rather than in the cytosol, thus demonstrating the distinct membrane affinity of the fluorescent CHIM-L conjugate. Altogether, the obtained results indicate that CHIM-L specifically integrates into the cell envelope of E. coli with a localization of the analogue in the outer membrane being most likely. To the best of our knowledge, this is the first time to show that a eukaryotic membrane lipid mimetic can be integrated and anchor a fluorescent dye into the membrane compartment of E. coli. While a fluorophore was introduced in this case, CHIM-L should principally allow membrane anchoring of any clickable compound. Notably, CHIM-L integration appears to have no visible effect on cell morphology as E. coli's typical rod shape seemed to be preserved (Fig. 1C).
 | ||
| Fig. 1 (A) Schematic experimental procedure of E. coli membrane functionalization using fluorophore-clicked CHIM-L. (B) Histogram overlay from flow cytometer analysis of E. coli cells after membrane labeling with a dye-CHIM-L conjugate. The respective lipids were allowed to react (24 h at room temperature) with the fluorescent DBCO dye (DBCO-PEG4-Fluo 545) and subsequently applied for integration into the membrane of E. coli BL21(DE3). Integration proceeded for 90 min at 37 °C and shaking at 800 rpm in 1 mL PBS with a final concentration of the lipids of 10 μM and the cell mass accounting for an OD of 0.4. Afterwards, the cells were washed three times by repeatedly centrifuging (11.000 rcf, 1 min), removing the supernatant and resuspending in 100 μL PBS. Histograms of control samples were recorded using non-clickable lipids CHOL and CHIM-H (black histograms); a histogram of a fluorescently labeled cell sample was recorded using clickable CHIM-L (red histogram). In contrast to CHOL and CHIM-H, CHIM-L has an azide moiety and therefore is the prerequisite for the SPAAC reaction with DBCO. The dotted line approximates the median fluorescence intensity of unlabeled samples. FI = fluorescence intensity [a.u.] (561 nm laser, 582/15 nm bandpass filter). (C) Fluorescence microscopy of E. coli cells labeled with the dye-CHIM-L conjugate as described in B (100× optical magnification, excitation 562/40 nm, emission 624/40 nm). (I–VI) Examples of labeled E. coli cells with sufficient fluorescence intensity in order to recognize the spatial density of the fluorescence signal. Co-localization of the dye-CHIM-L conjugate (red fluorescence, IV–VI) and the membrane compartment (brightfield, I–III) of the cells was evident. Labeled cells showed unaltered coliform morphology and regular length. | ||
Membrane functionalization by metalorganic CHIMs
Having evidence that CHIMs could indeed be used to functionalize the outer membrane surface of E. coli, we next sought to apply CHIMs for implementing new-to-nature functionality to the bacterial surface. For this purpose, the – in comparison to other lipid mimetics – unique feature of the CHIM analogs of being able to form an NHC headgroup was exploited. Such an NHC headgroup can serve as a coordinating ligand for transition metals and therefore be used to load biological membranes with a catalytically active metal complex. Transition metals such as ruthenium, gold, copper and palladium (Pd) have been extensively exploited for mediating synthetic organic reactions in biological environments.13–16,23 Even though there are several reports of applying Pd-promoted chemistry in bacteria24 and Ru-loaded artificial metalloproteins have been developed to enable bacterial surface modification,25 to the best of our knowledge, there is no example for applying a membrane-anchoring catalytically active lipid-mimetic. Such a small molecule transition metal complex would facilitate new-to-nature reactions on the surface of (bacterial) cellular membranes without the need for genetic engineering. To realize this approach, we synthesized CHIM–Pd, a CHIM-based palladium–NHC–allyl complex (Fig. 2, top left). Palladium–NHC– allyl complexes are known to facilitate a variety of reactions in organic synthesis but have also been applied in aqueous media, e.g. for mediating Suzuki couplings, or in biological systems, e.g. as anticancer agents.26,27 They can be readily prepared from the commercially available dimeric allyl–palladium complex and the respective imidazolium salt precursor. Herein, we chose a N-isopropylated CHIM derivative instead of the N-methylated salt as an imidazolium precursor for the synthesis of CHIM–Pd since more bulky N-substituents have been described to be beneficial for complex stability in aqueous medium and to lead preferentially to monocarbene complex formation.27 At the same time, isopropyl substituents were expected to still be small enough to only marginally affect faithful membrane integration.28 The integration of CHIM–Pd was quantified by single-cell inductively coupled plasma mass spectrometry (scICP-MS). In contrast to fluorescence measurements, E. coli cells do not contain any endogenous Pd, which might cause a basal background signal. Thus, this approach was expected to give additional information regarding the extent of bacterial CHIM incorporation. NHC–Pd, the corresponding Pd complex without a membrane-anchoring cholesterol backbone was investigated as presumably a non-integrating negative control (Fig. 2, bottom left). Cells were incubated with 100 μM CHIM–Pd or NHC–Pd and subjected to scICP-MS analysis. NHC–Pd treatment resulted in minor Pd incorporation of 6 fg/cell (Fig. 2, right, Table S1†). In contrast, CHIM–Pd-treated cells contained 276 ± 8 fg per cell and hence a 46-fold higher amount of Pd (median values). These results clearly demonstrated that only CHIM–Pd led to a substantial Pd loading of E. coli cells. Consequently, the Pd loading seems to be reliant on the cholesterol backbone, thereby corroborating the assumption that the CHIM backbone leads to bacterial membrane integration. Presuming that CHIM–Pd would indeed be embedded into the cell membrane with its headgroup oriented to the cell surface as previously reported for other CHIMs,9,10 this should enable Pd-mediated reactions in the exterior of CHIM–Pd-treated cells. A commonly applied reaction using palladium salts or complexes in biological systems is the depropargylation of propargyl ethers. Such reactions can enable biorthogonal Pd-mediated prodrug release or enable readout of depropargylation activity by decaging a fluorophore.16,29,30 Herein, a propargylated umbelliferone derivative (pro-umbelliferone) that yields fluorescent umbelliferone after Pd-mediated progargyl cleavage was utilized to evaluate the activity of the synthesized Pd–NHC complexes. The uncaging reaction and resulting increase in fluorescence intensity caused by accumulating depropargylated umbelliferone were tracked over time (Fig. 3A–D). First, both complexes were directly added to the cell-free substrate buffer to test their basic depropargylation ability under the applied experimental conditions. An increase in fluorescence intensity at 460 nm over time was observed for both compounds, thus showing their basic ability to mediate the uncaging of pro-umbelliferone in the substrate buffer (Fig. 3A and C). Notably, the depropargylation activity of CHIM–Pd seemed to be superior in comparison to that of NHC–Pd as 10× higher concentrations of NHC–Pd had to be applied to achieve a similar increase in fluorescence intensity over time. This could be due to the lipid mimetic structure of CHIM–Pd, which may allow the formation of micelles and thus a colocalization with the nonpolar coumarin substrate in an aqueous medium. Also, the comparatively large steric demand of the cholesterol backbone could have positive effects on the activity of the palladium catalyst.30 Subsequently, E. coli cells were incubated with both complexes, in the following referred to as “palladization”. After incubation for over 90 minutes at 37 °C, the cells were thoroughly washed and suspended in a substrate buffer containing pro-umbelliferone to investigate the incubated cells' ability for depropargylation. Notably, the palladization of cells and the uncaging reaction were performed in separate vessels to exclude any potential Pd-sticking to plasticware and thus to ensure that the uncaging reaction was exclusively mediated by Pd that was immobilized on the cell surface. Only the CHIM–Pd incubated cells led to an increase in fluorescence intensity over time reaching a plateau after 16 h. The concentration of uncaged umbelliferone at the plateau phase was quantified to be 8.6 μM (Fig. 3B, Fig. S8†). In contrast, no change in fluorescence intensity could be observed for the NHC–Pd incubated cells (Fig. 3D). Notably, also in this case NHC–Pd was applied at a 10× higher concentration than CHIM–Pd. While the reaction yield could readily be quantified, determining the turnover number would require the exact number of palladized cells per sample. The number of cells could be roughly estimated using the OD of the cell suspension. Assuming that an OD of 1.0 corresponds to 8.6 × 107 cells/mL,31 this would result in a number of 8.6 × 106 cells per sample, not taking into account that cells could have been lost during washing. With 276 fg Pd as measured per single cell, this would result in 2.6 × 10−12 mmol Pd per cell and a total of around 20 nmol Pd per sample. The total amount of Pd as applied to a sample was exactly 20 nmol (200 μL of 100 μM CHIM–Pd solution). This could either indicate that CHIM–Pd was completely incorporated into the cells or could be due to inaccuracies in deriving the cell number from the OD. Being aware of these uncertainties, the turnover number would be determined to be 0.09. This value is based on the assumptions that no cells were lost after washing and that every Pd atom accounts for one catalytically active CHIM–Pd molecule. Thus, it is likely that the actual turnover number is higher than estimated. In a similar fashion as with pro-umbelliferone, we demonstrated that CHIM–Pd loaded cells can also depropargylate other substrates as we exemplarily showed for propargylated fluorescein (Fig. S7,† bottom). With pro-umbelliferone, the product yield was significantly higher in cell-containing samples compared to cell-free samples (Fig. 3A and B). This suggests an enhanced effect of the membrane environment on reactivity. For the fluoresceine-derived substrate, however, an opposite effect was observed (Fig. S7,† bottom). Importantly, a control experiment with non-palladized cells did not reveal any increase in fluorescence intensity, thus demonstrating that the propargyl ether protection group was stable under Pd-free experimental conditions (Fig. S7,† top). In order to exclude intracellular activation, cells were palladized as described and incubated with pro-umbelliferone. After running the reaction for standard time, cells and supernatant were separated by centrifugation and fluorescence was determined for the cell fraction and the supernatant. It turned out that the fluorescence (due to the activation of pro-umbelliferone) was exclusively found in the supernatant (30.762 a.u.) and not in the cell fraction (503 a.u.) (Fig. S9†). This clearly indicated that the reaction occurred at the cell surface and ruled out any intracellular activation. Since in previous studies CHIMs have been used in mammalian cells, we additionally investigated whether CHIM–Pd-mediated depropargylation could also be applied in HEK-293 cells (Fig. S10†). However, the standard incubation conditions (basic Tris buffer, pH 8.0) as established here resulted in rapid lysis of the cells. Using a less basic buffer led to improved cell viability and a measurable increase in fluorescence intensity for palladized vs. untreated HEK-293 cells. Nevertheless, the observed increase in fluorescence intensity for palladized HEK-293 cells was barely two-fold when compared to the control. This low depropargylation activity could have been due to the low activation of the Pd(II)-allyl catalyst which is known to require base-mediated activation to be converted into the catalytically active Pd(0) species.29,30,32 Thus, although the experiment showcases the general potential of CHIM–Pd or related catalyst systems to be applied in mammalian cells, further investigations are necessary to optimize catalyst and reaction compatibility.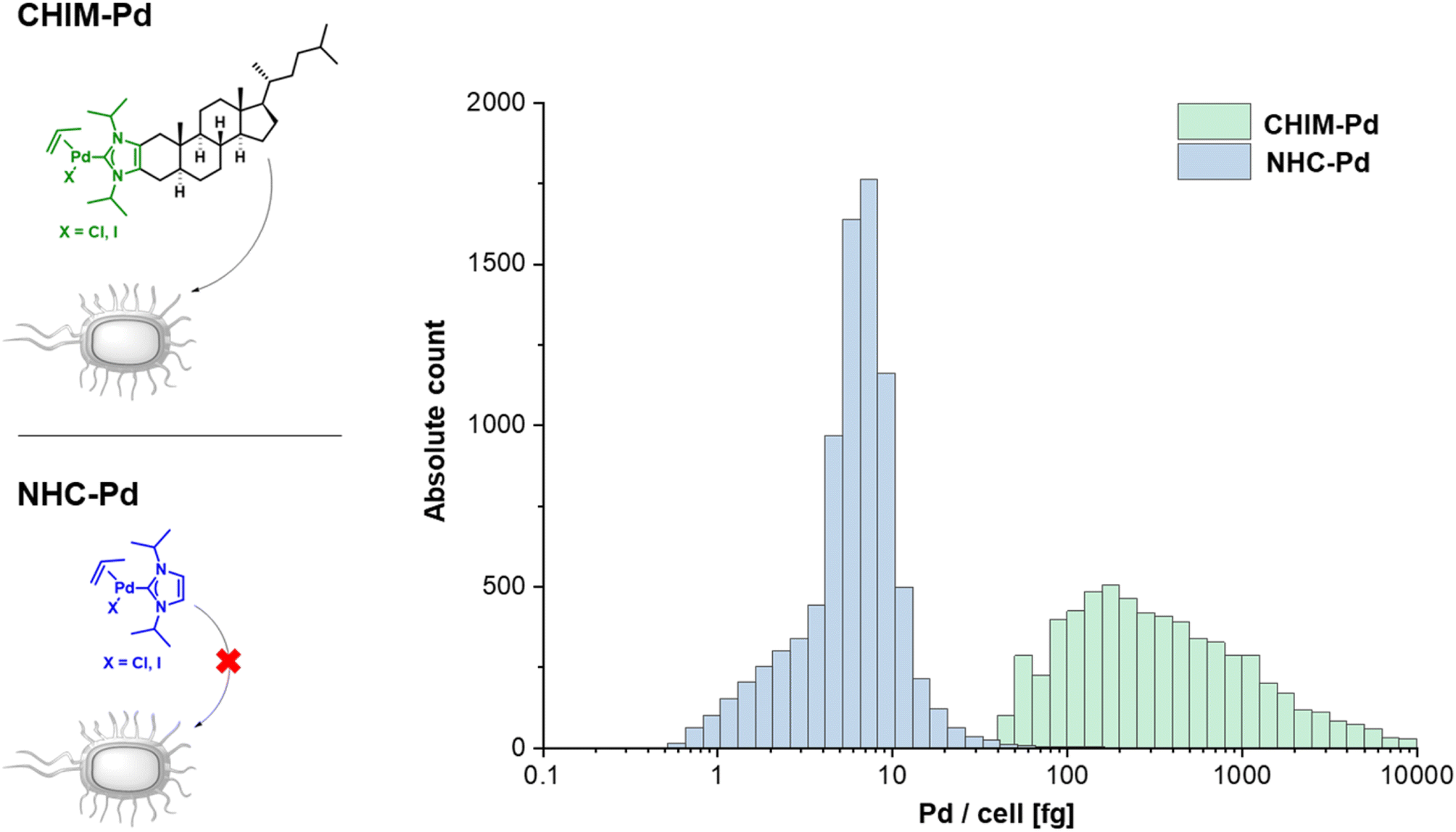 | ||
| Fig. 2 scICP-MS quantification of Pd on E. coli cells treated with CHIM–Pd (green) or NHC–Pd (blue). Integration proceeded for 90 min at 37 °C and shaking at 1.200 rpm in 200 μL Tris buffer pH 8.0 with a final concentration of CHIM–Pd and NHC–Pd of 100 μM and the cell mass accounting for an OD of 0.5. Afterwards, the cells were washed three times by repeatedly centrifuging (20.000 rcf, 2 min), removing the supernatant and resuspending in 100 μL Tris buffer pH 8.0. An overlay of mass distribution histograms of both samples is shown. To show the two data sets in one graph, x-values are displayed on a logarithmic scale. An equal-width binning was chosen with ten bins per logarithmic interval. | ||
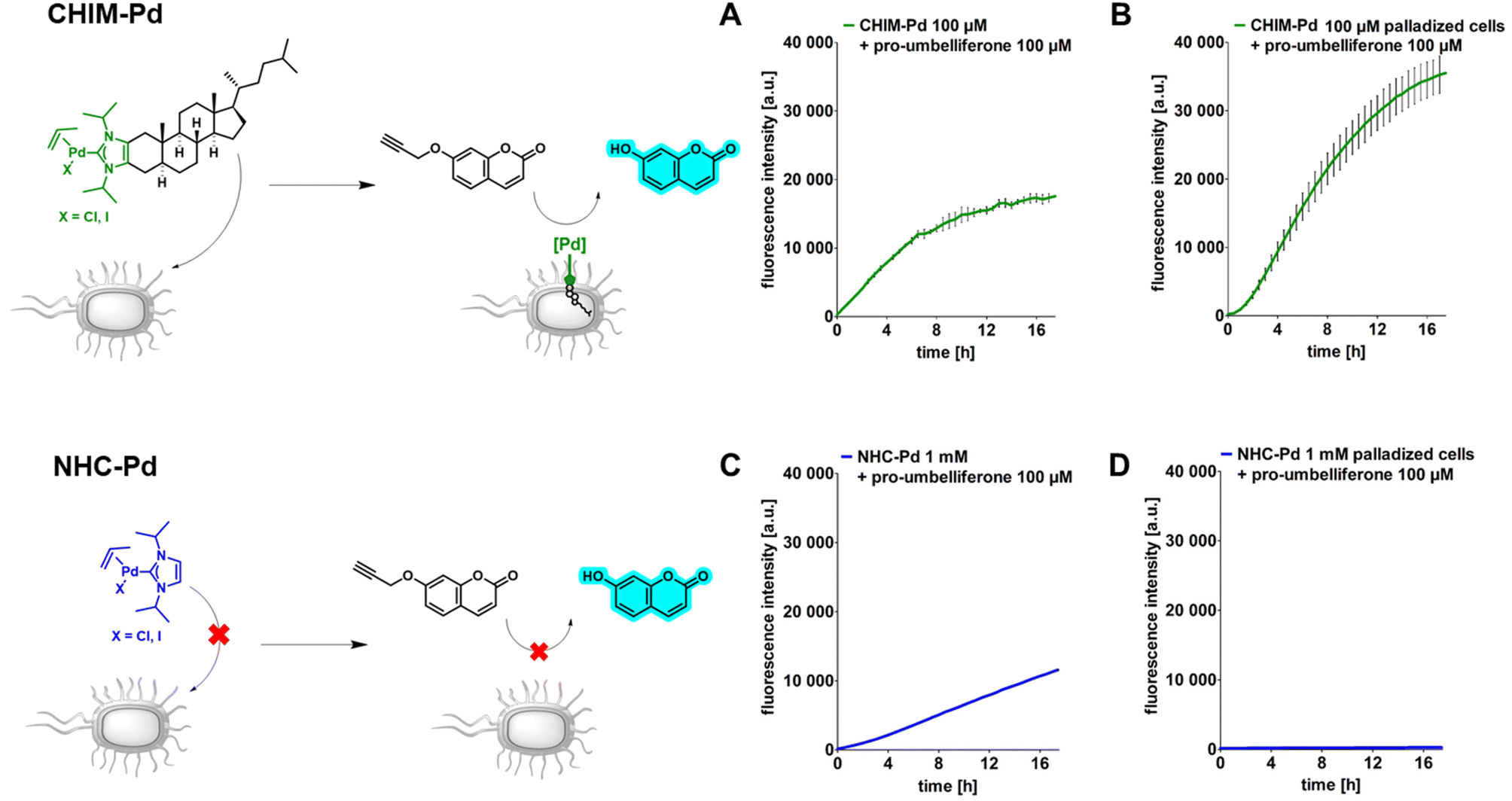 | ||
| Fig. 3 Spectrofluorometric measurement of the Pd-mediated uncaging reaction of pro-umbelliferone. CHIM–Pd-mediated uncaging is shown with blue lines T(A and B) and NHC–Pd-mediated uncaging with red lines (C and D). Data points represent the mean value of three independent samples and the SD is represented as black bars. (A) CHIM–Pd was directly added into the substrate buffer and enabled the Pd-mediated uncaging reaction of the substrate. (B) CHIM–Pd was used to palladize E. coli cells. Palladization proceeded for 90 min at 37 °C and shaking at 1.200 rpm in 200 μL Tris buffer pH 8.0 with a final concentration of 100 μM CHIM–Pd and the cell mass accounting for an OD of 0.5. Afterwards, the cells were washed three times by repeatedly centrifuging (20.000 rcf, 2 min), removing the supernatant and resuspending in 100 μL Tris buffer pH 8.0. CHIM–Pd-treated cells were subsequently resuspended in substrate buffer (200 μL of a 100 μM pro-umbelliferone solution in Tris buffer pH 8.0) to enable the Pd-mediated uncaging reaction by membrane-integrated CHIM–Pd, thus confirming successful palladization of the cells. (C) NHC–Pd was directly added into the substrate buffer and enabled the Pd-mediated uncaging reaction of the substrate. (D) 1 mM NHC–Pd was used to palladize E. coli cells under the same conditions as described in B. NHC–Pd-treated cells were washed and resuspended in the substrate buffer, but due to the lack of incorporated Pd pro-umbelliferone remained caged. | ||
In summary, these results indicated that CHIM–Pd is applicable to decorate the surface of E. coli cells with Pd and subsequently enable these cells to perform a new-to-nature depropargylation reaction. In accordance with the scICP-MS measurements the control experiment using NHC–Pd suggested once more that the integration of the CHIM–Pd complex indeed relies on its cholesterol backbone since NHC–Pd-incubated cells, even at high concentrations of 1 mM, did not show any depropargylation activity. Most probably, this was due to the fact that the complex is not able to anchor in the bacterial membrane and therefore is being removed during the washing step. The dependency on its cholesterol anchor for enabling bacterial incorporation as well as its observed capability to react with substrates in the surrounding cell medium further corroborates the assumption that CHIM–Pd indeed integrates into E. coli's outer membrane compartment.
Combining CHIM–Pd with a surface-displayed enzyme
Based on the results obtained from fluorescence microscopy (Fig. 1) we concluded that integration of CHIM-L into the E. coli outer membrane did not lead to detectable cell lysis and that bacterial cells were able to maintain their native morphology and membrane integrity. Against this backdrop, we sought to explore whether CHIM–Pd complexes could be combined with surface-displayed proteins to combine these two different approaches of bacterial membrane functionalization. In the past, heterologous proteins were displayed on the surface of E. coli by replacing the DNA sequence of the natural passenger of autotransporter proteins with the DNA sequence of a protein of interest, an approach which was termed “autodisplay”.6,33 Among drug target proteins and inhibitor peptide libraries, the technique was extensively used to display various catalytically active enzymes such as esterases, nitrilases, truncated variants of cytochrome P450 enzymes and many more on the surface of bacterial cells.5,34–37 Recently, we reported successful combinations of different surface-displayed enzymes on E. coli. Combining alcohol dehydrogenase displaying cells with cyclohexanone monooxygenase displaying cells facilitated a cascade reaction of cyclohexanol to cyclohexanone and sequentially to ε-caprolactone in a two-cell approach.37 We also reported that two functional enzymes were successfully combined by co-display in a single-cell approach. Here, truncated variants of human cytochrome P450 1A2 (CYP1A2) and human cytochrome P450 reductase (CPR) were co-displayed on E. coli. The surface-displayed CYP1A2 was shown to enable enzymatic oxidation of several structurally different substrates while the CPR supplied NADPH-derived electrons for the regeneration of FeII within the active center of CYP1A2.36 However, to the best of our knowledge, there are no reports of combining artificial transition metal complex- and enzyme-mediated reactions on the surface of live cells. Herein, we combined the reactivity of the outer membrane integrated CHIM–Pd with a surface-displayed laccase. Laccases are multicopper oxidases and were chosen here because of the plethora of established model substrates available for performing readily accessible activity assays utilizing chromogenic substrates such as 2,6-dimethoxyphenol (2,6-DMP).38 2,6-DMP is a monophenolic compound that is commonly used to quantify the laccase or peroxidase activity of enzymes. Even though not all steps of the underlying reaction mechanism are fully understood yet, it is assumed that the interacting enzyme abstracts a proton and an electron from the phenolic hydroxyl group of 2,6-DMP and transfers them onto elemental oxygen (laccases) or hydrogen peroxide (peroxidases). 2,6-DMP is thereby converted into its respective phenoxy radical. Two radical species form a covalent C–C bond and after additional oxidation, the orange-colored compound coerulignone is formed.39,40 Based on literature proposals about the reaction mechanism of laccases, we concluded that caging the phenolic hydroxyl group with any substituent should fully halt the formation of coerulignone. Thus, propargylated 2,6-DMP (pro-2,6-DMP) should no longer be an accepted laccase substrate (Fig. 4A). However, it should be readily decageable by CHIM–Pd to subsequently yield laccase-sensitive 2,6-DMP. Herein, we therefore synthesized the Pd-cleavable propargylated 2,6-DMP derivative as a potential probe to assay both the activity of CHIM–Pd and laccase simultaneously. As a laccase CotA from Bacillus coagulans was chosen. CotA laccase was expressed as a maximized autotransporter mediated expression (MATE-CotA) fusion protein on the surface of E. coli BL21 ΔcueO.34 The strain E. coli BL21 ΔcueO lacks E. coli's chromosomal encoded laccase CueO, so the only laccase activity of the strain results from the surface-displayed CotA. Cultures of E. coli BL21 ΔcueO pMATE-CotA were grown in an LB medium. For incorporation of copper into the CotA laccase, the medium was supplemented with 400 μM CuCl2. Cells were incubated at 37 °C at 200 rpm until they reached the exponential growth phase. The cultures were then supplemented with 0.2% L-arabinose (w/v) to induce gene expression of the gene coding for the CotA laccase autotransporter fusion protein, stationary incubated at 37 °C for 21 h and washed with Tris buffer pH 8.0. CotA laccase displaying cells were then transferred into the substrate buffer with a concentration of 10 mM of either 2,6-DMP or pro-2,6-DMP. The formation of orange-colored coerulignone was tracked spectrometrically at 468 nm (Fig. 4B). For 2,6-DMP an increase in absorption could be observed in Pd-free and palladized cells, whereas pro-2,6-DMP was not converted under Pd-free conditions. Therefore, the obtained results indicate that caging 2,6-DMP indeed prevented the formation of coerulignone, confirming our initial hypothesis. On the other hand, 2,6-DMP was reliably converted to coerulignone to almost the same extent in both Pd-free and palladized cells with surface-displayed laccase, thus indicating that additional palladization of the laccase-displaying cells did not lead to substantial loss of enzymatic activity. Thus, pro-2,6-DMP was assumed to be a suitable substrate to probe whether both the uncaging reaction and following oxidation, would occur through the simultaneous presence of CHIM–Pd and CotA laccase on the surface of E. coli.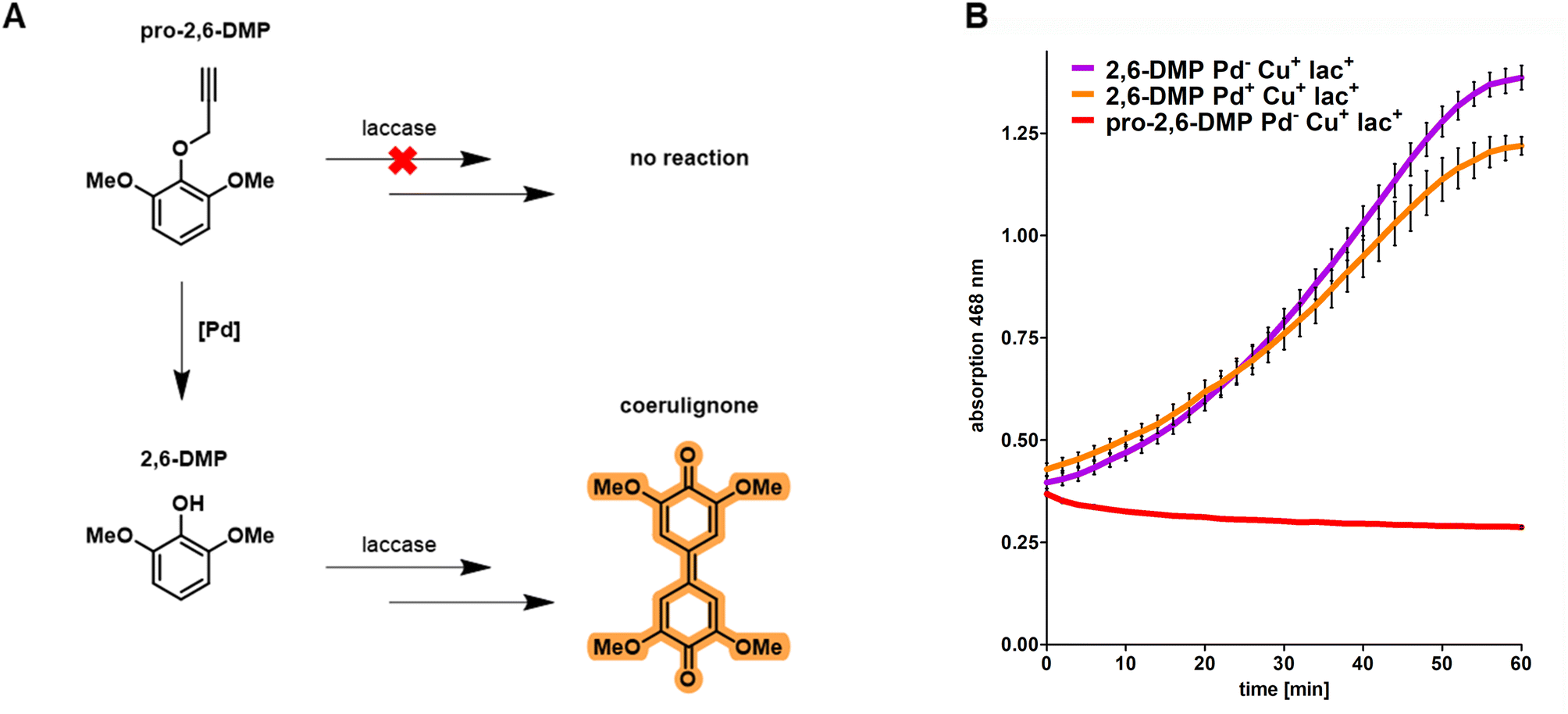 | ||
| Fig. 4 (A) Proposed concept of the herein-performed chromogenic laccase activity assay. While laccases are generally known to convert 2,6-DMP to orange-colored coerulignone by a multi-step reaction involving oxyradical formation, dimerization and oxidation, it was hypothesized that propargylated pro-2,6-DMP would not be an accepted laccase substrate due to blockage of the hydroxy group. However, pro-2,6-DMP should be readily decageable by CHIM–Pd to yield laccase-sensitive 2,6-DMP. Thus, pro-2,6-DMP was hypothesized to serve as a potentially suitable substrate to probe the ability of laccase and CHIM–Pd to act in a combined, cascade-like fashion on the surface of E. coli. (B) Spectrometric measurement of laccase-mediated coerulignone formation. Laccase activity was measured by the formation of orange-colored coerulignone and detected by an increased absorption at 468 nm. The caged substrate pro-2,6-DMP is not converted to coerulignon by surface-displayed CotA laccase under Pd-free conditions (red). In contrast, 2,6-DMP is readily converted to coerulignone in both Pd-free (purple) and palladized (orange) cells with surface-displayed CotA laccase. Palladization proceeded for 90 min at 37 °C and shaking at 1.200 rpm in 200 μL Tris buffer pH 8.0 with a final concentration of 100 μM CHIM–Pd and the cell mass accounting for an OD of 0.5. Afterwards, the cells were washed three times by repeatedly centrifuging (20.000 rcf, 2 min), removing the supernatant and resuspending in 100 μL Tris buffer pH 8.0. Notably, the palladization only leads to a minor impairment of the catalytic activity (purple and orange). The presence of cells in the substrate buffer contributed to a basal level of absorption that is not linked to the formation of coerulignone. | ||
Cascade reaction on the E. coli surface by palladization of CotA laccase displaying cells
Having confirmed that the surface-displayed CotA laccase does indeed not accept caged pro-2,6-DMP as a substrate, we sought to simultaneously exploit the combined reactivities of CHIM–Pd and surface-displayed CotA laccase on a single cell sample. For this purpose, CotA laccase displaying cells were additionally palladized with CHIM–Pd and subjected to the previously described chromogenic assay using pro-2,6-DMP as a substrate (Fig. 5A). As controls, we selectively inhibited the Pd-mediated decaging of pro-2,6-DMP or enzyme-mediated oxidation of 2,6-DMP. To inhibit pro-2,6-DMP decaging, we omitted the palladization with CHIM–Pd. To inhibit the enzyme-mediated oxidation of 2,6-DMP we either omitted the surface display of CotA laccase by using non-transformed E. coli BL21 ΔcueO cells or omitted copper supplementation to prevent the surface-displayed CotA laccase from loading copper which is essential for enzymatic activity. Our previous results already suggested that conversion of caged pro-2,6-DMP to coerulignone cannot occur without prior palladization (Fig. 4B). Indeed, coerulignone formation was fully absent in samples containing non-palladized, CotA displaying cells supplemented with copper (Fig. 5B, red), thus demonstrating that decaging of pro-2,6-DMP does not occur without palladization and that neither copper supplementation, nor CotA laccase affects the stability of the caged substrate. Combining palladization with either CotA laccase surface display without copper supplementation (Fig. 5B, green) or non-displaying cells with copper supplementation (Fig. 5B, blue) resulted in a mediocre formation of coerulignone. This observation can likely be linked to autoxidation of decaged 2,6-DMP rather than biocatalytic activity since the samples were either lacking the enzyme (Fig. 5B, blue) or copper (Fig. 5B, green). The autoxidation of 2,6-DMP in an alkaline environment (the herein discussed assay was performed in basic Tris buffer at a pH of 8.0) is well reported in the literature39 and we therefore concluded that the mediocre formation of coerulignone is related to successful decaging of pro-2,6-DMP and subsequent autoxidation without biocatalytic contribution. This was experimentally confirmed by investigating coerulignone formation in cell-free 2,6-DMP-containing assay buffer. Here indeed an increase in absorption over time was observed under the applied conditions (Fig. S12†). Only when palladization was combined with CotA displaying cells that were additionally supplemented with copper throughout the growth- and protein expression phases, could a substantial formation and an increased formation rate of coerulignone clearly exceeding the beforehand observed autoxidative coerulignone formation be observed (Fig. 5B, orange, Fig. S13†). Overall, the obtained results demonstrate the ability of the surface-displayed CotA laccase and membrane-embedded CHIM–Pd to act in a combined, cascade-like fashion to convert pro-2,6-DMP to coerulignone within uniform cells of E. coli. Since the active site of the surface-displayed CotA laccase was shown to be located at the cell surface (Fig. S11†) and the observed cascade reaction indicates co-localization of both the laccase and CHIM–Pd, the obtained results additionally support our hypothesis that the CHIM–Pd complex is indeed residing in the outer membrane, with its NHC–Pd-allyl headgroup mediating the depropargylation of the surrounding substrate at the cell surface.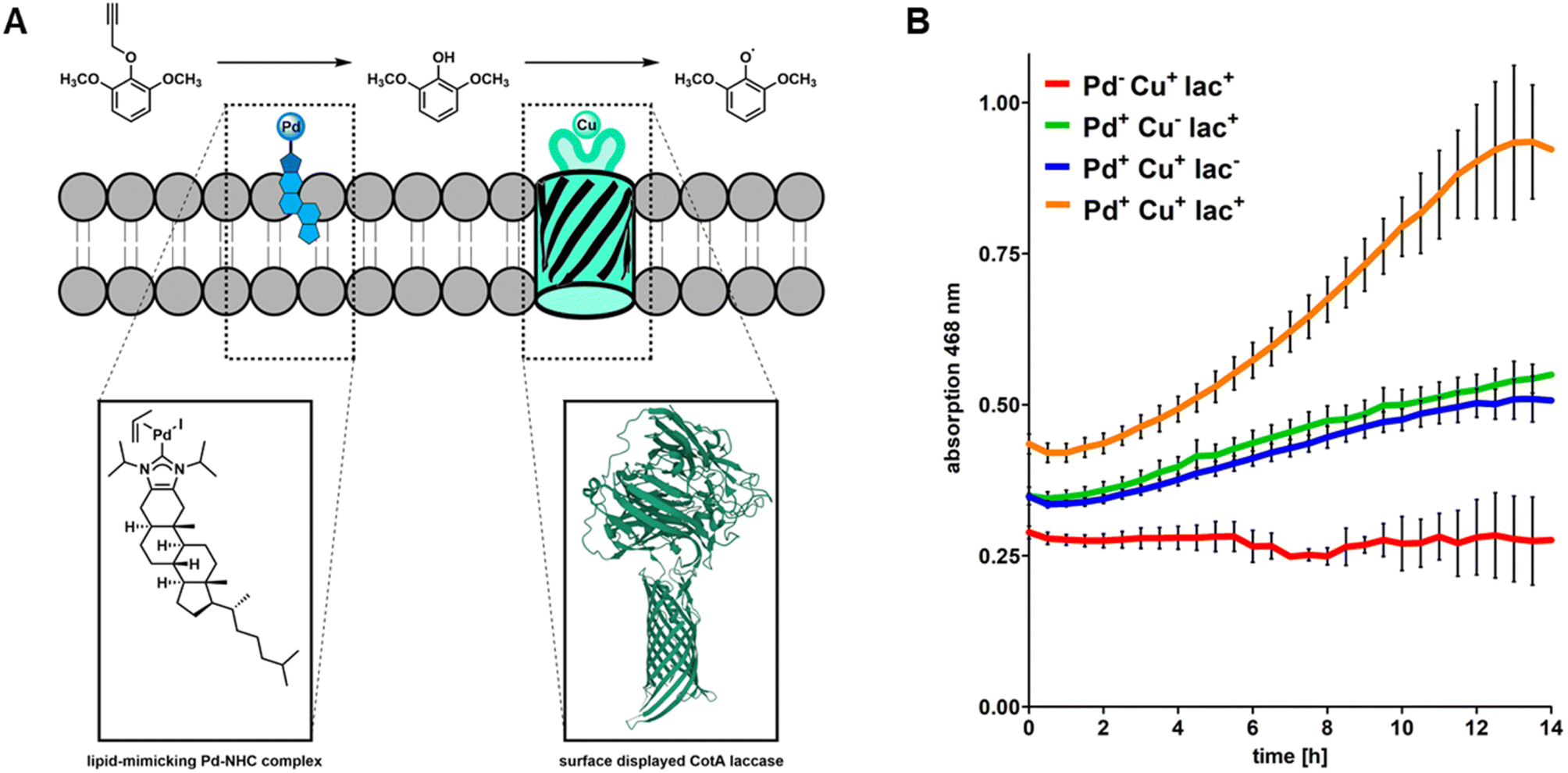 | ||
| Fig. 5 A) Schematic concept of the herein performed CHIM–Pd-/CotA laccase-mediated cascade reaction assay on the surface of E. coli. (B) Spectrometric measurement of Pd- and enzyme-mediated coerulignone formation by membrane-embedded CHIM–Pd (Pd) and CotA laccase (lac) in E. coli cells. Palladization proceeded for 90 min at 37 °C and shaking at 1.200 rpm in 200 μL Tris buffer pH 8.0 with a final concentration of 100 μM CHIM–Pd and the cell mass accounting for an OD of 0.5. Afterwards, the cells were washed three times by repeatedly centrifuging (20.000 rcf, 2 min), removing the supernatant and resuspending in 100 μL Tris buffer pH 8.0. The presence of cells in the substrate buffer contributed to a basal level of absorption that is not linked to coerulignone. Data points represent the mean value of three independent samples and the SD is represented by black bars. The most extensive coerulignone formation was observed in CHIM–Pd palladized samples with both surface-displayed CotA laccase and copper supplementation which is essential for laccase activity (orange). Palladized samples lacking either copper supplementation (green) or CotA laccase (blue) show mediocre coerulignone formation due to the autoxidation of decaged 2,6-DMP. Coerulignon formation was fully absent in non-palladized samples (red). | ||
Conclusions
The ability of an imidazolium-based cholesterol analog (CHIM) to integrate into the outer membrane of E. coli and to functionalize its surface was investigated. We showed that a clickable CHIM derivative (CHIM-L) can be applied to fluorescently label the cell envelope of E. coli while preserving the cell’s natural rod-shaped morphology, thus demonstrating its basic ability to anchor into the bacterial membrane without fundamentally affecting cell integrity. Based on this, we designed and synthesized a lipid-mimicking CHIM–palladium allyl complex (CHIM–Pd) and showed that this complex can be applied to enable E. coli cells to perform a new-to-nature ether cleavage of a propargylated coumarin derivative present in the surrounding medium. In this context, we showed that the cholesterol backbone of CHIM–Pd is essential for bacterial incorporation and for enabling propargyl ether cleavage. This indicates that CHIM–Pd is integrated into the bacterial outer membrane, thereby functionalizing its surface with a catalytically active palladium–NHC complex. Finally, we demonstrated that this approach can be combined with established bacterial surface display methods by simultaneously modifying E. coli cells with CHIM–Pd as well as with a surface-displayed laccase which was shown to allow the cells to perform a transition metal complex-enzyme-mediated cascade reaction converting pro-2,6-DMP to coerulignone. Overall, we envision the herein presented proof-of-concept studies to lay the foundation for a multifunctional toolbox that allows flexible and broadly applicable functionalization of bacterial membranes using a variety of CHIM-based metal catalysts and/or combinations of such with surface-displayed proteins of choice. As CHIM–Pd did not show a substantial impact on the catalytic activity of the displayed laccase, the tertiary structure and function of the displayed protein seem to be nearly unaffected by CHIM implementation. This might be exploited in the future, e.g. to display proteins with an affinity towards a certain epitope and facilitate the targeting of palladized cells towards a cell or tissue of interest. This combination could allow for the spatial release of a labeling compound or prodrug in the vicinity of the addressed site. Additionally, given the already reported successful application of CHIM derivatives in membrane vesicles as well as in mammalian cell membranes,9,10 the use of CHIM-based metal catalysts might not be limited to bacterial membrane functionalization but can also potentially be extended to other types of (cellular) membranes. However, here initial experiments in mammalian cells showed that careful catalyst tuning and reaction optimization depending on the investigated cell type might be crucial to obtain compatible and efficient catalyst systems.Author contributions
Tristan Wegner: investigation, formal analysis, validation, writing – original draft, visualization. Alexander Dombovski: investigation, formal analysis, validation, writing – original draft, visualization. Katrin Gesing: investigation, formal analysis. Alexander Köhrer: investigation, formal analysis, visualization. Matthias Elinkmann: software. Uwe Karst: methodology, resources. Frank Glorius: conceptualization, methodology, resources, writing – review & editing, supervision, funding acquisition. Joachim Jose: conceptualization, methodology, resources, writing – review & editing, supervision, funding acquisition.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) under SFB 858 and Chembion (Chemical Biology of Ion Channels, GRK2515) for generous financial support.References
- H. Watson, Essays Biochem., 2015, 59, 43–69 CrossRef PubMed.
- P. L. Yeagle, The Membranes of Cells, Elsevier, Amsterdam, 2016 Search PubMed.
- T. Harayama and H. Riezman, Nat. Rev. Mol. Cell Biol., 2018, 19, 281–296 CrossRef CAS PubMed.
- (a) J. Jose, Appl. Microbiol. Biotechnol., 2006, 69, 607–614 CrossRef CAS PubMed; (b) E. van Bloois, R. T. Winter, H. Kolmar and M. W. Fraaije, Trends Biotechnol., 2011, 29, 79–86 CrossRef CAS PubMed; (c) L. Han, Y. Zhao, S. Cui and B. Liang, Appl. Biochem. Biotechnol., 2018, 185, 396–418 CrossRef CAS PubMed.
- J. Schüürmann, P. Quehl, G. Festel and J. Jose, Appl. Microbiol. Biotechnol., 2014, 98, 8031–8046 CrossRef PubMed.
- J. Jose and T. F. Meyer, Microbiol. Mol. Biol. Rev., 2007, 71, 600–619 CrossRef CAS PubMed.
- (a) T. Wegner, R. Laskar and F. Glorius, Curr. Opin. Chem. Biol., 2022, 71, 102209 CrossRef CAS PubMed; (b) D. Artner, A. Oblak, S. Ittig, J. A. Garate, S. Horvat, C. Arrieumerlou, A. Hofinger, C. Oostenbrink, R. Jerala, P. Kosma and A. Zamyatina, ACS Chem. Biol., 2013, 8, 2423–2432 CrossRef CAS PubMed; (c) I. Nilsson, S. Y. Lee, W. S. Sawyer, C. M. Baxter Rath, G. Lapointe and D. A. Six, J. Lipid Res., 2020, 61, 870–883 CrossRef CAS PubMed.
- (a) D. Wang, C. Richter, A. Rühling, P. Drücker, D. Siegmund, N. Metzler-Nolte, F. Glorius and H.-J. Galla, Chem. - Eur. J., 2015, 21, 15123–15126 CrossRef CAS PubMed; (b) Da Wang, D. H. de Jong, A. Rühling, V. Lesch, K. Shimizu, S. Wulff, A. Heuer, F. Glorius and H.-J. Galla, Langmuir, 2016, 32, 12579–12592 CrossRef CAS PubMed; (c) P. Drücker, A. Rühling, D. Grill, D. Wang, A. Draeger, V. Gerke, F. Glorius and H.-J. Galla, Langmuir, 2017, 33, 1333–1342 CrossRef PubMed; (d) L. Rakers and F. Glorius, Biophys. Rev., 2018, 10, 747–750 CrossRef CAS PubMed.
- A. L. L. Matos, F. Keller, T. Wegner, C. E. C. Del Castillo, D. Grill, S. Kudruk, A. Spang, F. Glorius, A. Heuer and V. Gerke, Commun. Biol., 2021, 4, 720 CrossRef CAS PubMed.
- (a) L. Rakers, D. Grill, A. L. L. Matos, S. Wulff, D. Wang, J. Börgel, M. Körsgen, H. F. Arlinghaus, H.-J. Galla, V. Gerke and F. Glorius, Cell Chem. Biol., 2018, 25, 952–961 CrossRef CAS PubMed; (b) T. Wegner, A. L. L. Matos, K. Porte, K. Mehring, M. Pierau, H. Horstmeier, V. Gerke and F. Glorius, Org. Biomol. Chem., 2023, 21, 4817–4822 RSC.
- M. N. Hopkinson, C. Richter, M. Schedler and F. Glorius, Nature, 2014, 510, 485–496 CrossRef CAS PubMed.
- M. Jalal, B. Hammouti, R. Touzani, A. Aouniti and I. Ozdemir, Mater. Today: Proc., 2020, 31, 122–129 CrossRef PubMed.
- Y. Liu and Y. Bai, ACS Appl. Bio Mater., 2020, 3, 4717–4746 CrossRef CAS PubMed.
- P. K. Sasmal, C. N. Streu and E. Meggers, Chem. Commun., 2013, 49, 1581–1587 RSC.
- A. Seoane and J. L. Mascareñas, Eur. J. Org Chem., 2022, e202200118 CrossRef CAS PubMed.
- (a) P. Destito, C. Vidal, F. López and J. L. Mascareñas, Chem. - Eur. J., 2021, 27, 4789–4816 CrossRef CAS PubMed; (b) J. J. Soldevila-Barreda and N. Metzler-Nolte, Chem. Rev., 2019, 119, 829–869 CrossRef CAS PubMed.
- (a) V. Singh, Microbial Cell Factories Engineering for Production of Biomolecules, Elsevier, Amsterdam, 2021 Search PubMed; (b) A. Valle and J. Bolívar, in Microbial Cell Factories Engineering for Production of Biomolecules, ed. V. Singh, Elsevier, Amsterdam, 2021, Chapter 8, pp. 115–137 Search PubMed.
- Z. D. Blount, eLife, 2015, 4, e05826 CrossRef PubMed.
- J. P. Sáenz, D. Grosser, A. S. Bradley, T. J. Lagny, O. Lavrynenko, M. Broda and K. Simons, Proc. Natl. Acad. Sci. U.S.A., 2015, 112, 11971–11976 CrossRef PubMed.
- (a) A. Kutschera and S. Ranf, Biochimie, 2019, 159, 93–98 CrossRef CAS PubMed; (b) P. van der Ley, P. de Graaff and J. Tommassen, J. Bacteriol., 1986, 168, 449–451 CrossRef CAS PubMed; (c) T. J. Silhavy, D. Kahne and S. Walker, Cold Spring Harbor Perspect. Biol., 2010, 2, a000414 Search PubMed.
- (a) T. Nakae, Crit. Rev. Microbiol., 1986, 13, 1–62 CrossRef CAS PubMed; (b) H. Nikaido and M. Vaara, Microbiol. Rev., 1985, 49, 1–32 CrossRef CAS PubMed.
- M. Park, G. Yoo, J.-H. Bong, J. Jose, M.-J. Kang and J.-C. Pyun, Biochim. Biophys. Acta, 2015, 1848, 842–847 CrossRef CAS PubMed.
- (a) B. Lozhkin and T. R. Ward, Bioorg. Med. Chem., 2021, 45, 116310 CrossRef CAS PubMed; (b) A. Sathyan, L. Deng, T. Loman and A. R. A. Palmans, Catal. Today, 2023, 418, 114116 CrossRef CAS; (c) H. Madec, F. Figueiredo, K. Cariou, S. Roland, M. Sollogoub and G. Gasser, Chem. Sci., 2023, 14, 409–442 RSC; (d) C. C. James, B. de Bruin and J. N. H. Reek, Angew. Chem., Int. Ed., 2023, e202306645 CAS.
- (a) J. Li, S. Lin, J. Wang, S. Jia, M. Yang, Z. Hao, X. Zhang and P. R. Chen, J. Am. Chem. Soc., 2013, 135, 7330–7338 CrossRef CAS PubMed; (b) C. D. Spicer, T. Triemer and B. G. Davis, J. Am. Chem. Soc., 2012, 134, 800–803 CrossRef CAS PubMed; (c) N. Li, C. P. Ramil, R. K. V. Lim and Q. Lin, ACS Chem. Biol., 2015, 10, 379–384 CrossRef CAS PubMed; (d) N. Li, R. K. V. Lim, S. Edwardraja and Q. Lin, J. Am. Chem. Soc., 2011, 133, 15316–15319 CrossRef CAS PubMed; (e) Y. Era, J. A. Dennis, L. E. Horsfall and S. Wallace, JACS Au, 2022, 2, 2446–2452 CrossRef CAS PubMed.
- T. Heinisch, F. Schwizer, B. Garabedian, E. Csibra, M. Jeschek, J. Vallapurackal, V. B. Pinheiro, P. Marlière, S. Pankec and T. R. Ward, Chem. Sci., 2018, 9, 5383–5388 RSC.
- (a) M. S. Viciu, O. Navarro, R. F. Germaneau, R. A. Kelly, W. Sommer, N. Marion, E. D. Stevens, L. Cavallo and S. P. Nolan, Organometallics, 2004, 23, 1629–1635 CrossRef CAS; (b) M. S. Viciu, R. F. Germaneau, O. Navarro-Fernandez, E. D. Stevens and S. P. Nolan, Organometallics, 2002, 21, 5470–5472 CrossRef CAS; (c) N. Marion, O. Navarro, J. Mei, E. D. Stevens, N. M. Scott and S. P. Nolan, J. Am. Chem. Soc., 2006, 128, 4101–4111 CrossRef CAS PubMed; (d) T. Scattolin, E. Bortolamiol, F. Rizzolio, N. Demitri and F. Visentin, Appl. Organomet. Chem., 2020, 34, e5876 CrossRef CAS; (e) T. Scattolin, E. Bortolamiol, F. Visentin, S. Palazzolo, I. Caligiuri, T. Perin, V. Canzonieri, N. Demitri, F. Rizzolio and A. Togni, Chem. - Eur. J., 2020, 26, 11868–11876 CrossRef CAS PubMed; (f) T. Scattolin, I. Caligiuri, L. Canovese, N. Demitri, R. Gambari, I. Lampronti, F. Rizzolio, C. Santo and F. Visentin, Dalton Trans., 2018, 47, 13616–13630 RSC.
- J. M. Asensio, R. Andrés, P. Gómez-Sal and E. de Jesús, Organometallics, 2017, 36, 4191–4201 CrossRef CAS.
- A. Rühling, D. Wang, J. B. Ernst, S. Wulff, R. Honeker, C. Richter, A. Ferry, H.-J. Galla and F. Glorius, Chem. - Eur. J., 2017, 23, 5920–5924 CrossRef PubMed.
- (a) M. Pal, K. Parasuraman and K. R. Yeleswarapu, Org. Lett., 2003, 5, 349–352 CrossRef CAS PubMed; (b) J. Li, J. Yu, J. Zhao, J. Wang, S. Zheng, S. Lin, L. Chen, M. Yang, S. Jia, X. Zhang and P. R. Chen, Nat. Chem., 2014, 6, 352–361 CrossRef CAS PubMed; (c) M. Martínez-Calvo, J. R. Couceiro, P. Destito, J. Rodríguez, J. Mosquera and J. L. Mascareñas, ACS Catal., 2018, 8, 6055–6061 CrossRef PubMed; (d) S. Learte-Aymamí, C. Vidal, A. Gutiérrez-González and J. L. Mascareñas, Angew. Chem., Int. Ed., 2020, 59, 9149–9154 CrossRef PubMed; (e) M. Miller, B. Askevold, H. Mikula, R. H. Kohler, D. Pirovich and R. Weissleder, Nat. Commun., 2017, 8, 15906 CrossRef CAS PubMed.
- S. E. Coelho, F. S. S. Schneider, D. C. de Oliveira, G. L. Tripodi, M. N. Eberlin, G. F. Caramori, B. de Souza and J. B. Domingos, ACS Catal., 2019, 9, 3792–3799 CrossRef CAS.
- A. Gratz, A. Bollacke, S. Stephan, C. Nienberg, M. Le Borgne, C. Götz and J. Jose, Microb. Cell Factories, 2015, 14, 74 CrossRef PubMed.
- N. Marion and S. P. Nolan, Acc. Chem. Res., 2008, 41, 1440–1449 CrossRef CAS PubMed.
- J. Maurer, J. Jose and T. F. Meyer, J. Bacteriol., 1997, 179, 794–904 CrossRef CAS PubMed.
- (a) C. Detzel, R. Maas, A. Tubeleviciute and J. Jose, Appl. Microbiol. Biotechnol., 2013, 97, 4887–4896 CrossRef CAS PubMed; (b) E. Kranen, C. Detzel, T. Weber and J. Jose, Microb. Cell Fact., 2014, 13, 19 CrossRef PubMed; (c) J. Schüürmann, P. Quehl, F. Lindhorst, K. Lang and J. Jose, Biotechnol. Bioeng., 2017, 114, 1658–1669 CrossRef PubMed.
- S. Sichwart, I. E. P. Tozakidis, M. Teese and J. Jose, Food Technol. Biotechnol., 2015, 53, 251–260 CAS.
- P. Quehl, J. Hollender, J. Schüürmann, T. Brossette, R. Maas and J. Jose, Microb. Cell Fact., 2016, 15, 26 CrossRef PubMed.
- H. Tian, C. Furtmann, F. Lenz, V. Srinivasamurthy, U. T. Bornscheuer and J. Jose, Microb. Biotechnol., 2022, 15, 2235–2249 CrossRef CAS PubMed.
- W. A. Edens, T. Q. Goins, D. Dooley and J. M. Henson, Appl. Environ. Microbiol., 1999, 65, 3071–3074 CrossRef CAS PubMed.
- (a) F. Solano, P. Lucas-Elío, D. López-Serrano, E. Fernández and A. Sanchez-Amat, FEMS Microbiol. Lett., 2001, 204, 175–181 CrossRef CAS PubMed; (b) H. Wariishi, K. Valli and M. H. Gold, J. Biol. Chem., 1992, 267, 23688–23695 CrossRef CAS PubMed.
- E. Breslmayr, M. Hanžek, A. Hanrahan, C. Leitner, R. Kittl, B. Šantek, C. Oostenbrink and R. Ludwig, Biotechnol. Biofuels, 2018, 11, 79 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sc02960c |
| ‡ These authors have contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2023 |