
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAcyl-caged rhodamines: photo-controlled and self-calibrated generation of acetyl radicals for neural function recovery in early AD mice†
Xiao
Luo‡
 *a,
Zhonghui
Zhang‡
a,
Jie
Wang‡
c,
Xueli
Wang
d,
Yani
Zhang
e,
Jinquan
Chen
d,
Guangbo
Ge
e,
Wen
Yang
*c,
Xuhong
Qian
*ab,
Yang
Tian
*a and
Youjun
Yang
*b
*a,
Zhonghui
Zhang‡
a,
Jie
Wang‡
c,
Xueli
Wang
d,
Yani
Zhang
e,
Jinquan
Chen
d,
Guangbo
Ge
e,
Wen
Yang
*c,
Xuhong
Qian
*ab,
Yang
Tian
*a and
Youjun
Yang
*b
aShanghai Engineering Research Center of Molecular Therapeutics and New Drug Development, Shanghai Key Laboratory of Green Chemistry and Chemical Processes, School of Chemistry and Molecular Engineering, East China Normal University, Dongchuan Road 500, Shanghai 200241, China. E-mail: xluo@chem.ecnu.edu.cn; ytian@chem.ecnu.edu.cn; xhqian@ecnu.edu.cn
bState Key Laboratory of Bioreactor Engineering, Shanghai Key Laboratory of Chemical Biology, School of Pharmacy, East China University of Science and Technology, Meilong Road 130, Shanghai 200237, China. E-mail: youjunyang@ecust.edu.cn
cDepartment of Molecular and Cellular Biochemistry, School of Medicine, Shanghai Jiaotong University, Chongqing South Road 280, Shanghai 200025, China. E-mail: yangwen@shsmu.edu.cn
dState Key Laboratory of Precision Spectroscopy, East China Normal University, Dongchuan Road 500, Shanghai 200241, China
eInstitute of Interdisciplinary Integrative Medicine Research, Shanghai University of Traditional Chinese Medicine, Cailun Road 1200, Shanghai 201203, China
First published on 14th September 2023
Abstract
The biological function of radicals is a broad continuum from signaling to killing. Yet, biomedical exploitation of radicals is largely restricted to the theme of healing-by-killing. To explore their potential in healing-by-signaling, robust radical generation methods are warranted. Acyl radicals are endogenous, exhibit facile chemistry and elicit matrix-dependent biological outcomes. Their implications in health and disease remain untapped, primarily due to the lack of a robust generation method with spatiotemporal specificity. Fusing the Norrish chemistry into the xanthene scaffold, we developed a novel general and modular molecular design strategy for photo-triggered generation of acyl radicals, i.e., acyl-caged rhodamine (ACR). A notable feature of ACR is the simultaneous release of a fluorescent probe for cell redox homeostasis allowing real-time monitoring of the biological outcome of acyl radicals. With a donor of the endogenous acetyl radical (ACR575a), we showcased its capability in precise and continuous modulation of the cell redox homeostasis from signaling to stress, and induction of a local oxidative burst to promote differentiation of neural stem cells (NSCs). Upon intracerebral-injection of ACR575a and subsequent fiber-optical activation, early AD mice exhibited enhanced differentiation of NSCs toward neurons, reduced formation of Aβ plaques, and significantly improved cognitive abilities, including learning and memory.
Introduction
With extraordinary versatility and exquisite specificity, life exploits radicals both constructively and destructively.1 They are intermediates of energy metabolism, mediators of redox signaling, and weapons against foreign pathogens.2 Half a century after the scientific discipline of radical biology was established,3 the theme of biomedical exploitation of radicals is still restricted largely to the destructive domain, i.e., healing by killing malign cells or invading pathogens.4 Superfluous generation of highly oxidative radicals promotes biomacromolecular damage, cell senescence and eventual cell death.5 For example, photo-/chemo-/sono-dynamic therapies were developed as an intervention strategy for cancers or drug-resistant infections.6 In sharp contrast, the signaling use of radicals toward biomedical challenges is rare. With nitric oxide for angina or other hypertensive conditions, the constructive use of radicals has been proved to be viable for a long time.7 Nevertheless, such a healing-by-signaling use of other chemically less-lame radicals is extremely difficult and remains largely unexplored, yet carries hope for neurodegenerative diseases. Neural stem cells (NSCs) reside in a hypoxic niche, i.e., the hippocampal dentate gyrus (DG).8 An oxidative burst could trigger their neural differentiation, and promote neurogenesis for function recovery of early Alzheimer's disease (AD).9 However, the brain tissue of the AD patients is unfortunately prone to further oxidative injuries and deterioration of the pathological condition. Therefore, a delicate radical generation method is warranted, not to disrupt the redox homeostasis beyond the signaling domain for minimal collateral oxidative damage, but to induce a transient local oxidative burst for promoted NSC neurogenesis.Endogenous radicals are diverse in nature and a carbon-centered radical, in particular an acyl radical, is preferred for healing-by-signaling purposes. First, carbon is an element with low electronegativity and hence carbon-centered radicals are less likely to induce oxidative injuries compared to radicals based on oxygen, nitrogen, sulfur, or chlorine.10 Second, the biological outcome of carbon-based radicals, e.g., acyl radicals, is context-dependent. In the biological milieu, acyl radicals are speciated into two different secondary radicals, including acylperoxyl radicals and ketyl radicals (Fig. 1).11 Acylperoxyl radicals, generated through combination with oxygen, are oxidative and therefore can potentially tilt the redox homeostasis toward oxidation for cell signaling.12 Yet, their oxidation potential (1.6 V) is much lower than that of hydroxyl radicals (2.7 V) and less likely to illicit the unwanted indiscriminative oxidation of cellular components.13 Ketyl radicals, generated upon nucleophilic attack of acyl radicals by the ubiquitous NH2/SH, are reductive and therefore buffer the oxidative capacity of acylperoxyl radicals.14 The two secondary radicals from acyl radicals form a redox-buffer to limit oxidative injuries. Third, the therapeutic potential of acyl radicals, e.g., acetyl, is strongly supported by their endogeneity. Acetyl was identified in ex vivo reactions of biological substrates, e.g., radical oxidation of aldehydes (e.g., acetaldehyde),15 peroxynitrite oxidation of dicarbonyl species (e.g., diacetyl16 or methylglyoxal17), and photolysis of aqueous pyruvic acid.18 Finally, acyl radicals have been harnessed in treating diseases. Isoniazid is a frontline drug for tuberculosis. Its pharmacological mechanism involves oxidative metabolism to the corresponding isonicotinic acyl radical, which adds to NAD+ to inhibit mycolic acid biosynthesis and Mycobacterium tuberculosis growth.19
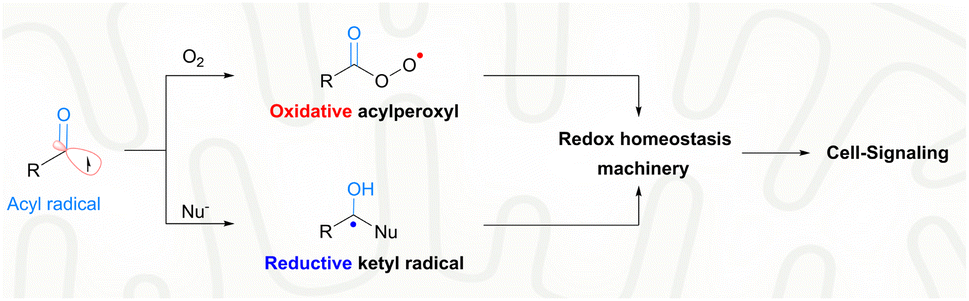 | ||
| Fig. 1 The chemical reactivities of acyl radicals to fine-tune the cellular redox homeostasis. | ||
Acyl radicals can be facilely generated as elegantly showcased by the organic community, i.e., by the homolytic rupture of a RC(O)–X bond, carbonylation of a carbon-centered radical, and fragmentation of the C–C bond in ketones and ketoacids/esters, which typically involve the use of hazardous organic reagents or photocatalysts.20 Yet, for biological use and to render a robust spatiotemporal control of the acyl radical generation, light is the ideal triggering method, due to its biological compatibility, on–off switchability and intensity tunability. A self-calibration mechanism is incorporated into this proposed acyl donor, i.e., the release of a fluorescent probe for mitochondria membrane potential (MMP), which is a robust biochemical index of the cell redox status.21 This allows real-time read-out of the fluctuation of the local redox homeostasis upon the release of the acetyl radical, avoiding oxidative injuries, and facilitating pharmacological studies.
These considerations led to our rational design of acyl-caged rhodamines (ACRs), a general and modular approach for photo-triggered and self-calibrated acyl radical generation. Both the nature of the radical-of-interest and the spectral properties of the fluorochromic template could be judiciously selected. We thoroughly investigated their photolysis and acyl radical generation with robust spectroscopic methods. With an acetyl radical donor (ACR575a) as an example, we further showcase its application in the precise modulation of cell redox homeostasis and in in vivo stimulation of NSC differentiation in AD mice. Notably, early AD mice that received ACR575a stimulation exhibited improved cognition and memory, substantiating the strategy of AD treatment via stimulated neurogenesis.
Results and discussion
Design rationale and synthesis of ACRs
Integration of the classic Norrish type I reaction22 to the rhodamine scaffold led to the design of acyl-caged rhodamines (ACRs). ACR575a/p were prepared via a Lewis acid-catalyzed condensation of a bis-N,N-diethylamino substituted diphenyl ether (1) with a 1-phenylpropane-1,2-dione (3) to produce 4 in a 48% yield. Subsequent reaction of 4 and MeLi or nBuLi generated ACR575a or ACR575p, respectively, in a ca. 90% yield (Fig. 2a). The crystal structures of ACR575a/p indicate that the oxygen atom of the carbonyl group points away from the bridging oxygen of the xanthene core, presumably to minimize repulsion (Fig. 2b). Acyl-caged Si–rhodamines ACR670a and ACR670p were prepared analogously. Mechanistically, an n–π* transition furnishes a triplet biradical intermediate (6) upon photo-excitation, which further decomposes via β-fragmentation to the desired acetyl radical (9) and a tertiary benzylic radical (7). The acyl radical potentiates the cell redox homeostasis for cell-signaling purposes, and compound 8 (a rhodamine fluorophore) from 1 − e− oxidation of a tertiary benzylic radical (7) is a MMP probe for real time monitoring of the cell redox homeostasis (Fig. 3a).23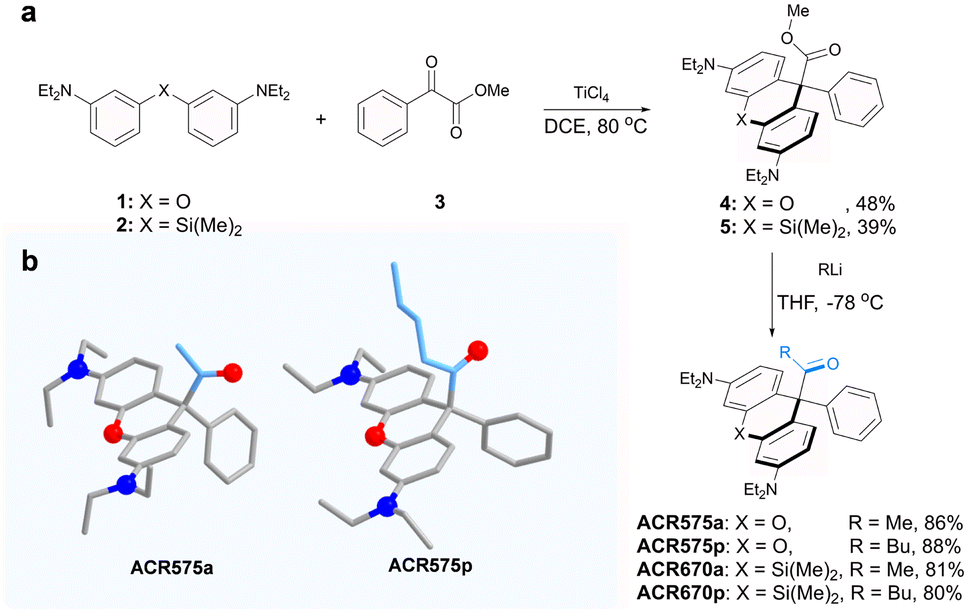 | ||
| Fig. 2 (a) The synthetic procedures for acyl-caged rhodamines (ACRs). (b) The crystal structures of ACR575a and ACR575p. | ||
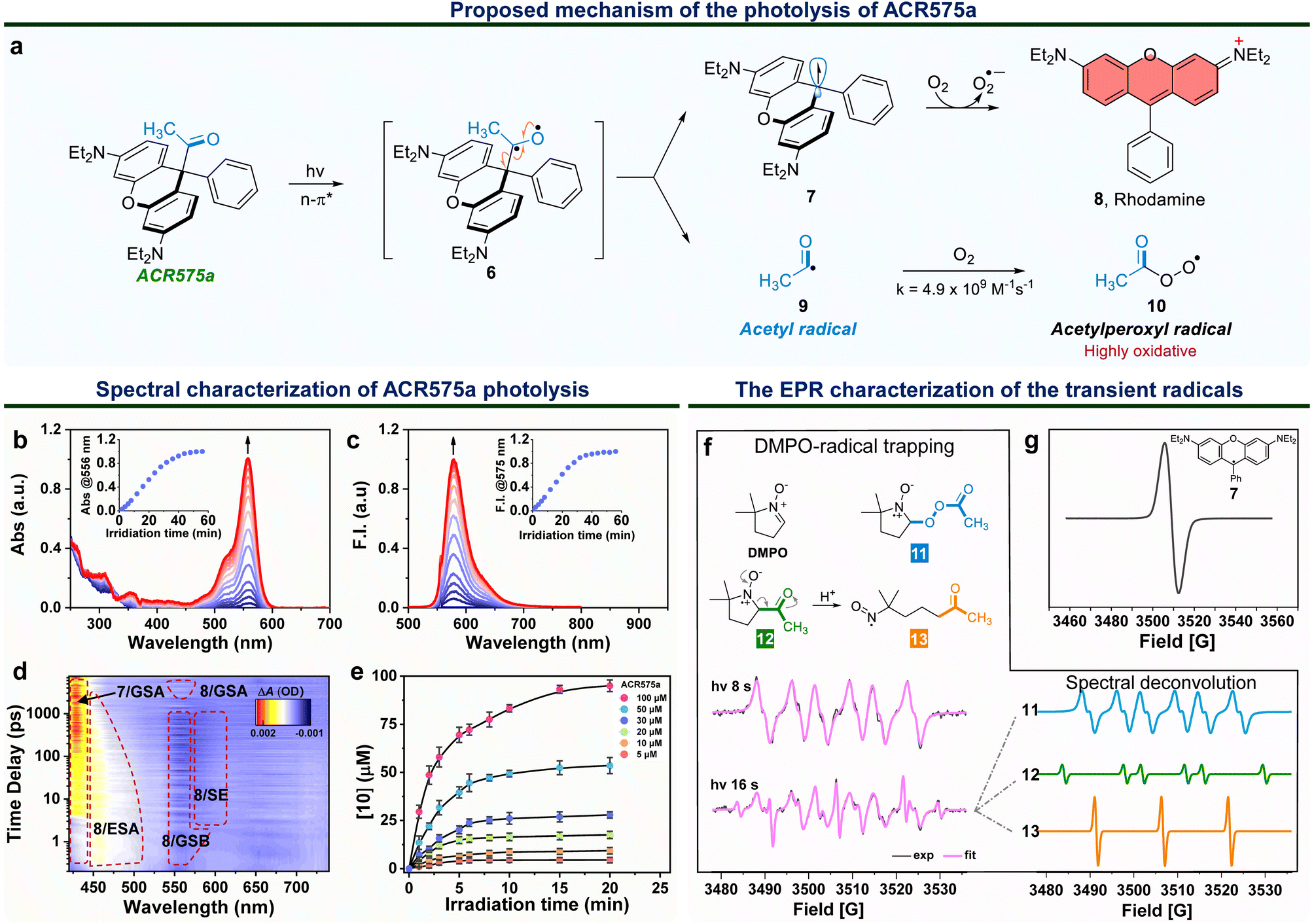 | ||
| Fig. 3 The molecular design of a photo-triggered acetyl radical donor (ACR575a). (a) The proposed photolysis mechanism of ACR575a. (b) The changes of the UV-vis absorption and (c) fluorescence emission spectra of the ACR575a solution (2 μM) in phosphate buffer (50 mM, pH = 7.4, with 10% MeCN) upon irradiation by a 375 nm UV light. Inset: absorbance @ 556 nm or fluorescence intensity @ 575 nm along with the irradiation time (min). (d) The femtosecond transient absorption spectra of ACR575a (0–7.48 ns) following a 330 nm laser pump pulse in CH3CN. (e) Fluorescence-based detection of peroxyl radicals generated upon photolysis of ACR575a with a ratiometric probe (light source: a 405 nm laser). (f) The radical trapping chemistry by DMPO upon photolysis of ACR575a. The EPR spectra of ACR575a (1 mM) in CH3CN with DMPO (10 mM) recorded after irradiation with a UV LED light for 8 s or 16 s. (g) The EPR spectrum to confirm the generation of a tertiary benzylic radical (7) without the use of DMPO. | ||
Spectral studies on the formation of rhodamine and acetylperoxyl radicals as the photolytic products of ACR575a
The photolysis of ACR575a was studied in phosphate buffer (50 mM, pH = 7.4, with 10% CH3CN) with UV-vis absorption and fluorescence spectroscopies. ACR575a did not exhibit visible absorption or fluorescence due to the sp3 hybridization of the central carbon and hence the lack of a push–pull scaffold in its structure. A solution of ACR575a was irradiated with a UV LED light (375 ± 25 nm, 20 mW cm−2, Fig. S3†). An absorption band at ca. 556 nm emerged along with an emission band at 575 nm upon photo-excitation, presumably from the rhodamine (8) (Fig. 3b and c). The absorbance and emission enhancements were linearly proportional to the irradiation time before reaching a plateau. The final stable photolysis products of ACR575a were confirmed by NMR and MS to be rhodamine 8 and acetic acid (Fig. S7†). The relative uncaging quantum yield of ACR575a in phosphate buffer was determined to be 0.037 with BisCMNB-FL (0.13)24 as a reference (Fig. S5†). The photo-decomposition of ACR575a to rhodamine 8 was also feasible in other solvents, e.g., CH3CN, CH2Cl2, DMSO and EtOH (Fig. S4 and S6†). A general trend is that the photolysis was faster in less-polar solvents and slower in polar protic solvents, due to H-bonding of the non-bonding lone pair of the carbonyl oxygen. The valuable mechanistic insights from this phenomenon helped to identify the lone pair of the carbonyl pointing away from the xanthene core to be the one involved in the n–π* excitation.The kinetics of ACR575a photolysis were studied with transient absorption measurement in CH3CN with 330 nm excitation (Fig. 3d). An intense positive absorption band peaked at ∼430 nm appeared instantaneously within the initial 4 ps upon photo-excitation and was persistent up to the upper limit (7.48 ns) of the time window of our system. This transient signal was attributed to the ground state absorption (GSA) of the triarylmethyl radical 7.25 A laser line at 405 nm also readily triggered the photolysis of ACR575a. Though the acetyl radical is not oxidative, the acetylperoxyl radical is. Indeed, the oxidative capacity of 10 was readily detected with a ratiometric fluorescent probe26 in a dose-dependent fashion (Fig. 3e), to delineate its potential to modulate cell redox homeostasis.
The EPR studies of ACR575a verified the proposed photolysis mechanism
The short-lived radical intermediates generated upon photolysis of ACR575a were identified with EPR (Fig. 3f and g). A solution of ACR575a in CH3CN was irradiated with UV light. The hallmark EPR peak of the stable tertiary benzylic radical (7)27 was observed (Fig. 3g). Identification of other transient radical species necessitated a spin trap, i.e., 5,5-dimethylpyrroline-N-oxide (DMPO) (Fig. 3f). A solution of ACR575a (1 mM) with DMPO (10 mM) was irradiated with UV light for varying durations of time and the EPR spectra of the resulting solutions were then acquired and analyzed. When a solution of ACR575a was briefly irradiated for 8 s, a 6-line EPR signal (g = 2.0055, αN = 13.198 G, αβH = 8.00 G) in agreement with the adduct of a DMPO/CH3(CO)OO radical (11) was observed. With a longer irradiation time of 16 s, the EPR signal of the adduct of DMPO/acetyl radical (12, g = 2.0054, αN = 13.95 G, αβH = 17.90 G) was identified through deconvolution, along with a nitroxide radical (13, g = 2.0056, αN = 15.25 G) generated in situ through an E2-elimination of 12 (Fig. 3f). This experiment suggested that the formation of 13 was not significant until O2 was consumed.Taken together, the mechanism of the photolysis of ACR575a was fully explained and the potential of ACR575a as a photo-triggered and photo-calibrated acetyl radical donor was unambiguously demonstrated.
The design strategy of ACR575a is general and modular
The molecular design of the acyl-caged rhodamine (ACR) scaffold as a photo-triggered acyl radical donor allows further structural modifications. We confirmed that the acetyl group within ACR575a could be altered to other acyls of interest, such as the pentanoyl of ACR575p (Fig. 2a). Furthermore, the rhodamine scaffold could also be replaced by silicon–rhodamine to construct an acetyl (ACR670a) or a pentanoyl radical donor (ACR670p) (Fig. 2a), which would produce a bright red-emitting Si–rhodamine for self-calibration upon photolysis. The photochemistry of the three new acyl donors, i.e., ACR575p, ACR670a, and ACR670p, was spectrally examined in phosphate buffer (Fig. 4a–c) and organic solvents (Fig. S4 and S6†). Analogous to ACR575a, irradiation by 375 nm light induced their photolysis, as evidenced by the increase of their absorption and emission bands. A general trend is that pentanoyl-caged rhodamines (ACR575p or ACR670p) were photolyzed faster than the acetyl-caged analogues (ACR575a or ACR670a). This phenomenon was interesting and reasonable. The butyl group of ACR575p is sterically bulkier than the methyl group of ACR575a, which disrupts the H-bonding of H2O to the carbonyl group, and hence renders the microenvironment of the carbonyl group in ACR575p less polar than that of ACR575a. The valuable mechanistic insights from this phenomenon helped to identify the lone pair of the carbonyl pointing away from the xanthene core to be the one involved in the n–π* excitation. The relative uncaging quantum yields of ACR575p, ACR670a and ACR670b in phosphate buffer were determined to be 0.047, 0.041 and 0.029, respectively (Fig. S5†). The ground state absorption signal originated from the triarylmethyl radical was also observed with transient absorption spectroscopy in less than 10 ps, to confirm the quick homolysis of the acyl–trityl bond within ACRs initialed upon photo-excitation (Fig. 4d–f). Finally, acyl radicals along with their secondary radicals were trapped by DMPO and identified with EPR (Fig. 4h–j).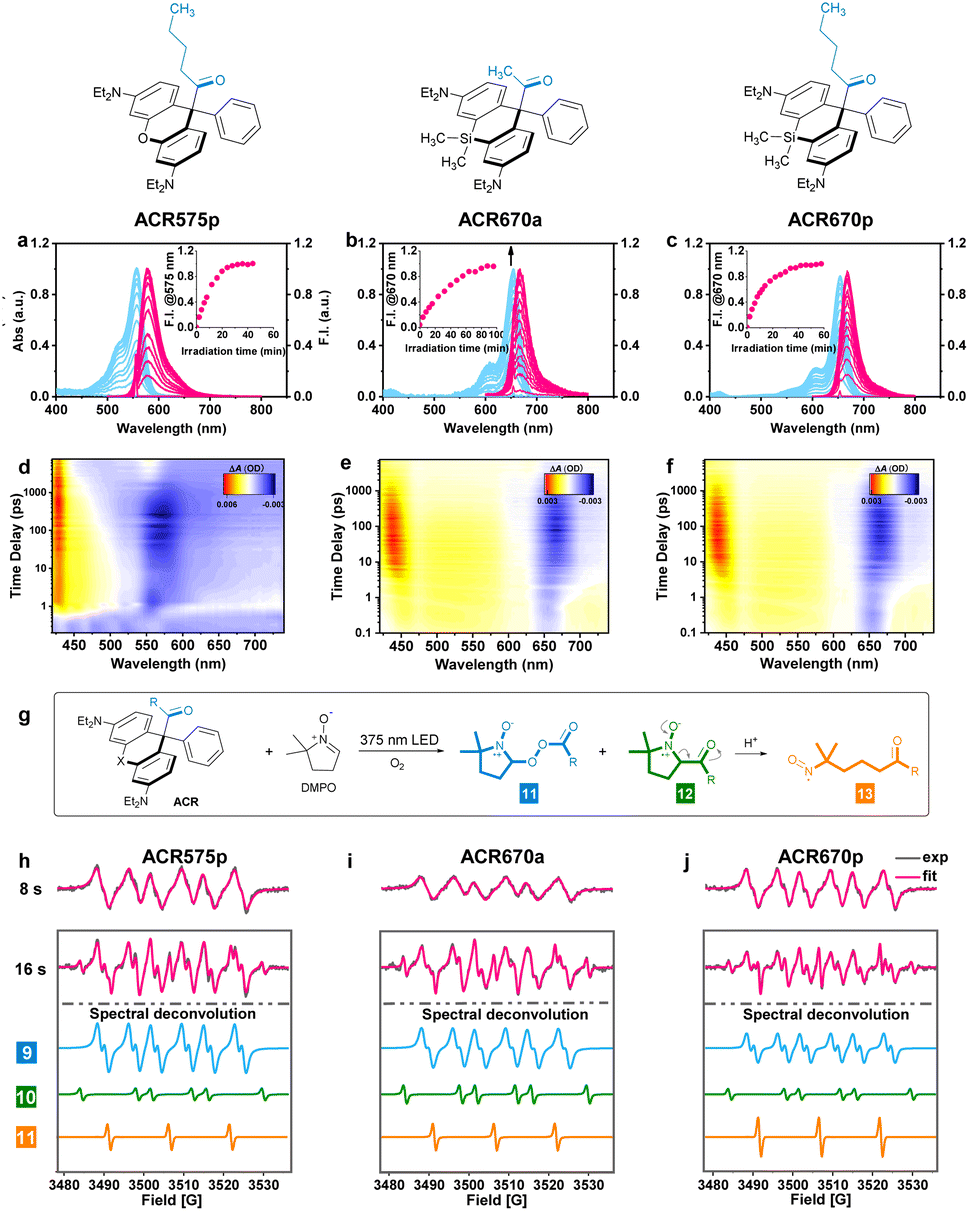 | ||
| Fig. 4 The spectral studies of the ACR photolysis processes. (a–c) The changes of UV-vis absorption and fluorescence emission spectra of the ACR solution (2 μM) in phosphate buffer (50 mM, pH = 7.4, with 10% MeCN) upon irradiation by a 375 nm UV light. Inset: fluorescence intensity at 575 or 670 nm along with the irradiation time (min). (d–f) The femtosecond transient absorption spectra of the ACRs (0–7.48 ns) following a 330 nm laser pump pulse in CH3CN. (g) The radical trapping chemistry by DMPO upon photolysis of the ACRs. (h–j) The EPR spectra of the ACRs (1 mM) in CH3CN with DMPO (10 mM) recorded after irradiation with a UV LED light for 8 s or 16 s. | ||
Precise modulation of the cellular redox status by photoactivation of ACR575a
ACR575a exhibited good membrane permeability and low cytotoxicity, as tested in HeLa cells (Fig. S10g†). The judicious site-specific photoactivation of ACR575a in a subset of HeLa cells in the entire field-of-view was achieved by confocal fluorescence microscopy with a 405 nm laser (Fig. 5a and b), confirming the photoactivation feasibility of ACR575a in intracellular milieu. The rise of cell oxidative status upon photo-triggered decomposition of ACR575a was detected with CellROX Deep Red28 (Fig. S10f†). As previously noted, a highlight of the molecular design of ACR575a is that rhodamine 8 is a probe for MMP, which is a sensitive index of the mitochondrial oxidative status.29 Such a feedback mechanism allows the redox-modulatory effect of the acetyl radical generated upon photolysis of ACR575a to be conveniently assessed. Upon irradiation of ACR575a-incubated HeLa cells, the fluorescence of rhodamine 8 appeared and gradually enhanced (Fig. 5c). Within the first ca. 40 s, the average cellular fluorescence intensity enhanced linearly with respect to time, due to the accumulation of 8 in the mitochondria (Fig. 5b). Apparently, the production of low doses of acetyl was tolerated by the cells and MMP was not noticeably affected. From ca. 40–80 s, the cellular fluorescence intensity continued to enhance, yet the linearity was gradually lost (Fig. 5d), suggesting that the oxidative insult was strong enough to dial the redox homeostasis toward oxidative eustress/stress to start to impact the MMP. After the fluorescence intensity reached a plateau after ca. 80 s, a rapid drop of the fluorescence intensity was observed, reminiscent of the collapse caused by the MMP decoupling chemical FCCP30 (Fig. S10b†). Apparently, the sustained release of acetyl in normoxic cells induced an abrupt rise of mitochondrial redox status, which subsequently activated uncoupling proteins (UCPs)31 to lower the MMP, as an innate defense against oxidative stress. Beyond ca. 150 s, the fluorescence intensity gradually leveled off and ceased to decrease further. The cellular Ca2+ signaling pathways are closely linked to redox-signaling.32 The photolysis of ACR575a-incubated cells was also accompanied by a rise of cytosolic [Ca2+] (Fig. 5d), probably through oxidation and activation of IP3R or RyRs on the ER.33 With this study, we successfully showcased the capability of ACR575a in precisely modulating the cell redox status through the entire continuum from homeostasis to a mild imbalance, and eventually to oxidative eustress/stress.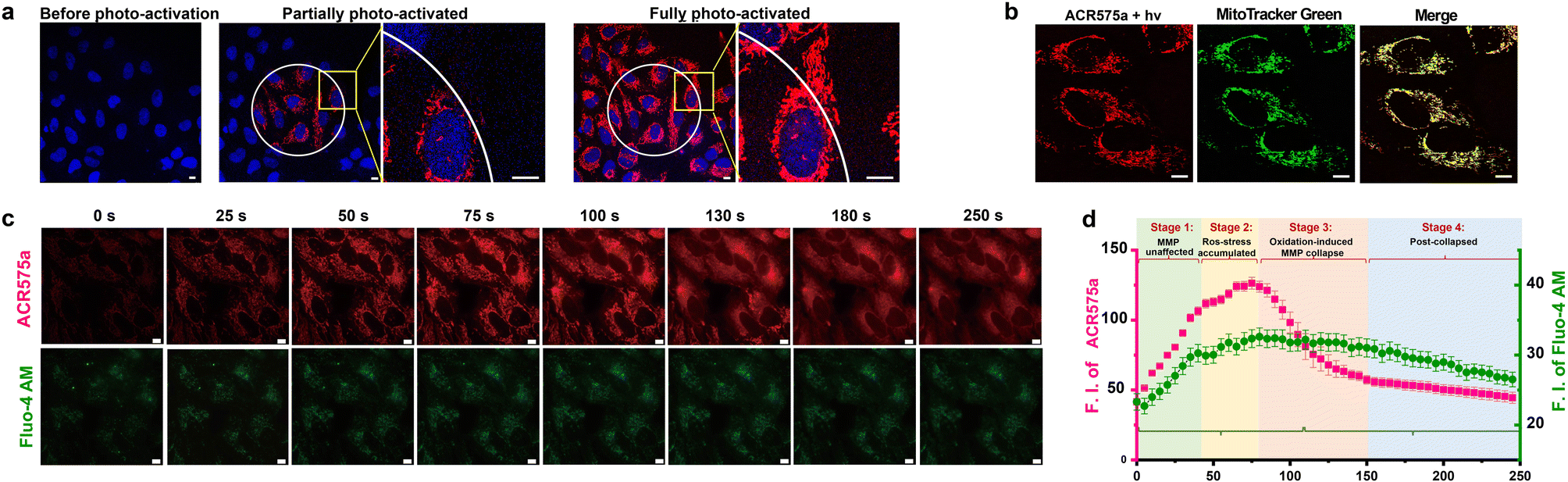 | ||
| Fig. 5 Photoactivation of ACR575a in live HeLa cells for redox modulation. (a) The confocal images of HeLa cells incubated with ACR575a (red) upon laser-irradiation (405 nm) and the nuclei were counterstained by Hoechst 33342 (blue). (b) A co-localization study of the fluorophore (rhodamine 8, red) generated upon ACR575a photolysis with MitoTracker Green (green). Pearson's coefficient = 0.87. (c) The epi-fluorescence images of the photo-irradiated HeLa cells incubated with ACR575a (red) and a calcium probe Fluo-4 AM (green). (d) The average cellular fluorescence intensity of the two channels in Fig. 5c with respect to the irradiation time. Error bars: SD, n = 3. Scale bar: 10 μm. | ||
The cellular photoactivation behavior of ACR575p was similar to that of ACR575a (Fig. S10†). The potential of Si–rhodamine as a MMP probe was also evaluated. Si–rhodamine mainly localized to mitochondria exemplified by a high Pearson's correlation coefficient of 0.84 with the mitochondria-specific dye MitoTracker Green (Fig. S10c†). The administration of FCCP also induced a decrease of its fluorescent intensity although to a less extent compared with that of rhodamine 8 (Fig. S10c†). The photolysis of ACR670a/p-incubated cells was accompanied by a rise-then-fall signal change pattern for Si–rhodamine (Fig. S10d†), demonstrating their potential in modulating the cell redox homeostasis. Considering that acetyl radicals are endogenous and rhodamine 8 is a more sensitive MMP probe than Si–rhodamine, ACR575a was chosen for the further exploration of the potential of acyl radicals in NSC differentiation manipulation.
Photo-irradiation of ACR575a-treated NSCs exhibited enhanced neural differentiation
The commitment of NSC differentiation toward neurons is the fundamental basis of neurogenesis, which is believed to persist into adulthood.34 Effective strategies to activate neural differentiation and suppress NSC self-renewal are sought after to enhanced neurogenesis, which could eventually lead to improved cognition. Kempermann recently articulated that a pulsed rise of cell ROS preceded NSC differentiation.9,35 Such a pulsed rise of ROS is not readily achieved via conventional redox modulation methods, including disruption of the cell antioxidant machinery, or superfluous generation of highly oxidative oxygen(/nitrogen) based reactive species. ACR575a is an optimal choice to address this challenge. First, continuous irradiation of NSCs with a 405 nm laser (10 mW cm−2) for 14 min did not induce a significant decrease of cell viability and was used in further triggering the photolysis of ACR575a (Fig. 6a, gray line). Then, NSCs were incubated with varying doses of ACR575a (5, 10, 20, 30 and 50 μM, respectively) and then irradiated by a 405 nm laser. Cell viability was negatively correlated with both the dose of ACR575a and irradiation time. Four conditions resulting in a cell viability of 85% were chosen for NSC stimulation, i.e., 5 μM/8 min, 10 μM/6 min, 20 μM/4 min, and 30 μM/1 min, respectively (Fig. 6a). NSCs that underwent these treatments were further cultured for 3 days before being immunofluorescently stained with a neuron-specific antibody (Tujl) and an astrocyte-specific antibody (GFAP). The flow cytometry data indicated that the differentiation of NSCs was greatly enhanced compared to a control group (Fig. 6c). | ||
| Fig. 6 Photoactivation of ACR575a induces the differentiation of NSCs in vitro. (a) The viability of NSCs with various concentrations of ACR575a with respect to the irradiation time. (b) The immunofluorescence analysis, (c) the flow cytometry analysis, and (d) statistics of NSC differentiation to neurons (Tuj1, red) and astrocytes (GFAP, green) upon irradiation of ACR575a incubated cells. n = 4. Data are shown as mean ± SEM. Statistical significance was determined by two-sided Welch's ANOVA. *p < 0.05, **p < 0.01, ***p < 0.001. Scale bar: 10 μm. | ||
An optimal neuronal differentiation efficiency of in total 44.6 ± 2.6% was found with the stimulation condition of 20 μM/4 min, in which the neuron differentiation efficiency was 28.9 ± 2.3%, and the astrocyte differentiation efficiency was 15.7 ± 1.7% (Fig. 6d). These results were further verified by confocal fluorescence imaging (Fig. 6b).
In vivo photoactivation of stereotactically injected ACR575a led to a cognitive recovery of early AD mice
Having verified the capability of ACR575a to promote NSC differentiation to neurons and astrocytes, the potential of ACR575a for in situ stimulation of NSC differentiation and neurofunction recovery was further evaluated. NSCs are in the subgranular zone (SGZ) of the dentate gyrus, a hypoxic niche in the hippocampus.36ACR575a was stereotactically injected into the SGZ of APPswe/PS1dE9 double transgenic mice (2× Tg-AD, 14 or 26 weeks old), a mouse model of Alzheimer's disease. Upon injection, the mice were raised for a specified period before being sacrificed. The brain slices and the major vital organs were collected and irradiated with a 405 nm laser line to activate ACR575a, the fluorescence of the photolysis product of which was a reliable indicator of the biological distribution of ACR575a (Fig. S13†). ACR575a was readily membrane-permeable and quickly taken up by the NSCs within 20 min (Fig. S13a†). Over the next few hours, fluorescence was also found in the extracellular region. The signal intensity peaked at 6 hours and gradually diminished until 34 hours (Fig. S13b†). After 4 days, ACR575a was hepatically and renally metabolized before being excreted (Fig. S13f†). No obvious damage to the vital organs of the mice was identified through H&E staining to confirm its good biocompatibility (Fig. S13g†).An optical fiber was implanted into the SGZ region for site-specific photo-activation of ACR575a. A group of five 14 w AD mice was intracerebrally injected with ACR575a (5 μL, 20 μM) and irradiated for 4 min by a 405 nm laser. Over the next 28 days, the mice were treated by this procedure for a total of six times before the mice were first evaluated for cognitive functions, i.e., learning and memory, with the Morris water maze (MWM), and then sacrificed for pathology studies via immunological staining and western blot analyses (Fig. 7a). Against the spatial acquisition tests on the 1st day, the treated AD mice averaged a latency of 81.2 ± 5.6 s before reaching the platform, not far behind that of 63.0 ± 5.8 s of the wild-type (WT) mice (Fig. 7c). In comparison, the untreated mice (Fig. 7b, E3), the mice treated with PBS (Fig. 7b, E4), and the ACR575a-injected and yet unirradiated mice (Fig. 7b, E5) all averaged a latency of 107.4 ± 2.7 s. The treated group, now 18 w old at the time, remarkably outperformed the 14 w early AD mice (Fig. 7b, E2). During the next seven days of training, the latency of the healthy WT mice and treated mice was significantly reduced to 5.6 ± 3.5 s and 22.6 ± 8.4 s, respectively (Fig. 7d). In comparison, the improvement was minimal for all three control groups (E3–E5) and the 14 week early AD mouse (E2). The platform of the MWM was then removed to evaluate the memory of these groups of mice (Fig. 7e–g). During the fixed internal of 150 s, the healthy wild type spent an average of 78.9 ± 12.4 s in the correct quadrant and crossed the original position of the platform an average of 9 ± 2 times. The treated group spent 63.9 ± 10.6 s in the correct quadrant and crossed the platform 4 ± 2 times, while the untreated group scored an average of 34.8 ± 12.5 and 2 ± 1 times. Taking these results together, the in vivo photo-irradiation of ACR575a-injected exhibited a notable effect in early AD mice of cognitive improvement, including learning and memory. Then, the brain slices were immunologically stained to find that the treated early AD mice exhibited higher signal intensities for both GFAP and Tuj1 (Fig. 7h), suggesting that the capability of ACR575a to promote NSC differentiation in vitro was successfully translated to in situ in early AD mice. These results were further corroborated with western blot studies (Fig. 7i). Through an in vitro experiment, the effect of acetyl radicals to inhibit the formation of Aβ-aggregates was also observed (Fig. S13c–e†). Indeed, the Aβ-plaques of the ACR575a-treated mice were noticeably reduced compared to untreated early AD mice (Fig. 7j and k). We also attempted to treat late AD mice (26 w) with ACR575a. However, no noticeable beneficial effect was discovered through the MWM experiment (Fig. 7l–q) and the Aβ-plaques of the treated mice did not reduce (Fig. 7r and s). The Tuj1/GFAP staining of the brain slices of 26 week late-stage AD mice was not carried out considering that the viability and neuro-differentiation capability of NSCs deteriorate with the progression of AD.37 All of these results indicated that the in vivo photogeneration of acetyl radicals from ACR575a can promote in situ NSC differentiation and attenuate Aβ-aggregation, leading to cognition recovery of early AD mice.
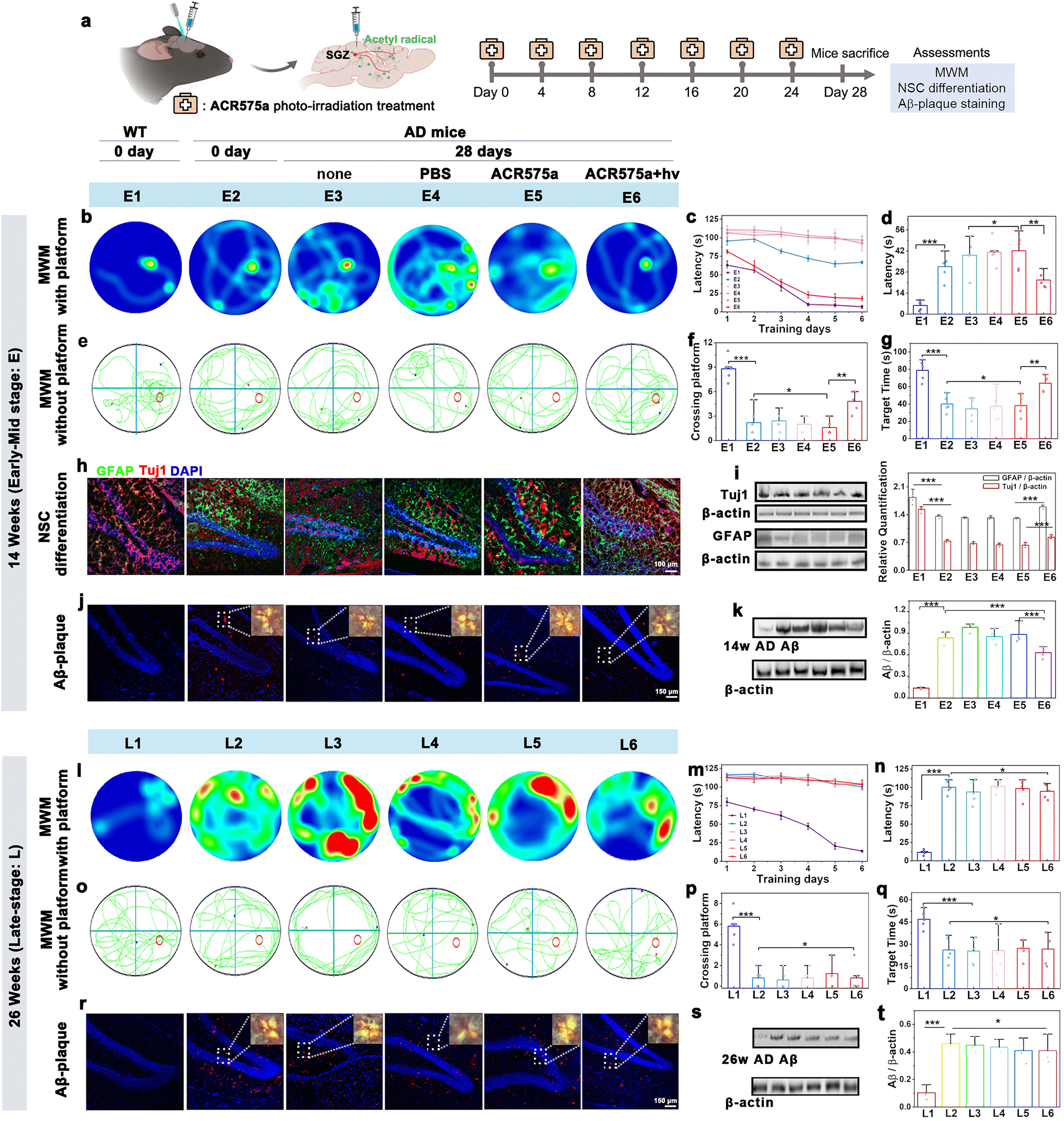 | ||
| Fig. 7 Evaluation of the therapeutic potential of ACR575a for AD mice. (a) ACR575a-based treatment of AD mice involves intracerebral injection of ACR575a in SGZ, and subsequent fiber-optical irradiation. Each AD mouse received 6 treatments prior to cognition-function evaluation by the Morris water maze test, immunofluorescence staining, and Aβ-plaque staining. The escape track plots in the MWM with a platform of (b) the 14 week AD mice (E1–E6) and (l) the 26 week AD mice (L1–L6) on the 7th day after different treatment plans. (c) Escape latency to reach the safety platform for E1–E6 from day 1 to day 6, and (d) on day 7. The escape track plots in MWM without a platform on the 7th day for (e) E1–E6 and (o) L1–L6. (f) Number of times crossing the hidden platform located area and (g) time spent in the target quadrant for E1–E6. (h) Representative hippocampal section images of E1–E6 immunostained for neurons (β3-Tuj1, red), astrocytes (GFPA, green) and nuclei (DAPI, blue). (i) Immunoblotting analysis and quantification of the relative levels of GFAP and Tuj1 in (h). (j) Immunofluorescence of Aβ in the hippocampus of 14 week (E1–E6) and (r) 26 week (L1–L6) AD mice (inset: the polarizing microscopic images of Aβ). (k) Immunoblotting analysis and quantification of relative levels of Aβ in the hippocampus of E1–E6 and (s) L1–L6. (m) Escape latency to reach the safety platform for L1–L6 from day 1 to day 6 and (n) on day 7. (p) Number of times crossing the hidden platform located area and (q) time spent in the target quadrant for L1–L6. n = 5 mice for E1–E6 and L1–L6. The means ± SD are shown (t-test, * < 0.05, ** < 0.01, *** < 0.001). | ||
Proteomics analysis of NSCs with the stimulation of acetyl radicals
The redox signaling is mediated by the dynamic modulation of the cell-redox buffering network comprising numerous chemical and biochemical entities. Redox signaling mandates the alteration of the cell protein expression profile. Therefore, proteomic analyses of NSCs with and without acetyl radical generation are a valuable approach to probe the mechanism of the acetyl radicals behind the observed phenotypic neural function recovery of early AD mice (Fig. 8). Analyses revealed two-fold upregulation of 640 proteins and downregulation of 357 proteins (Fig. 8a and b). Among them, the enrichment of mitochondrial inner membrane proteins was identified by GO enrichment analysis (cell component) (Fig. 8b). This is consistent with the fact that ACR575a was found to specifically localize in mitochondria and mitochondria are the central hub of cell redox-signaling. Also, many of the proteins that were up/down-regulated are related to neural degenerative diseases (Fig. 8d). This strongly suggests that acetyl radicals could impact neuronal function-related gene expression through modulating the redox homeostasis. Further elucidation of the molecular biological mechanism of ACR575a in NSCs is our ongoing endeavor.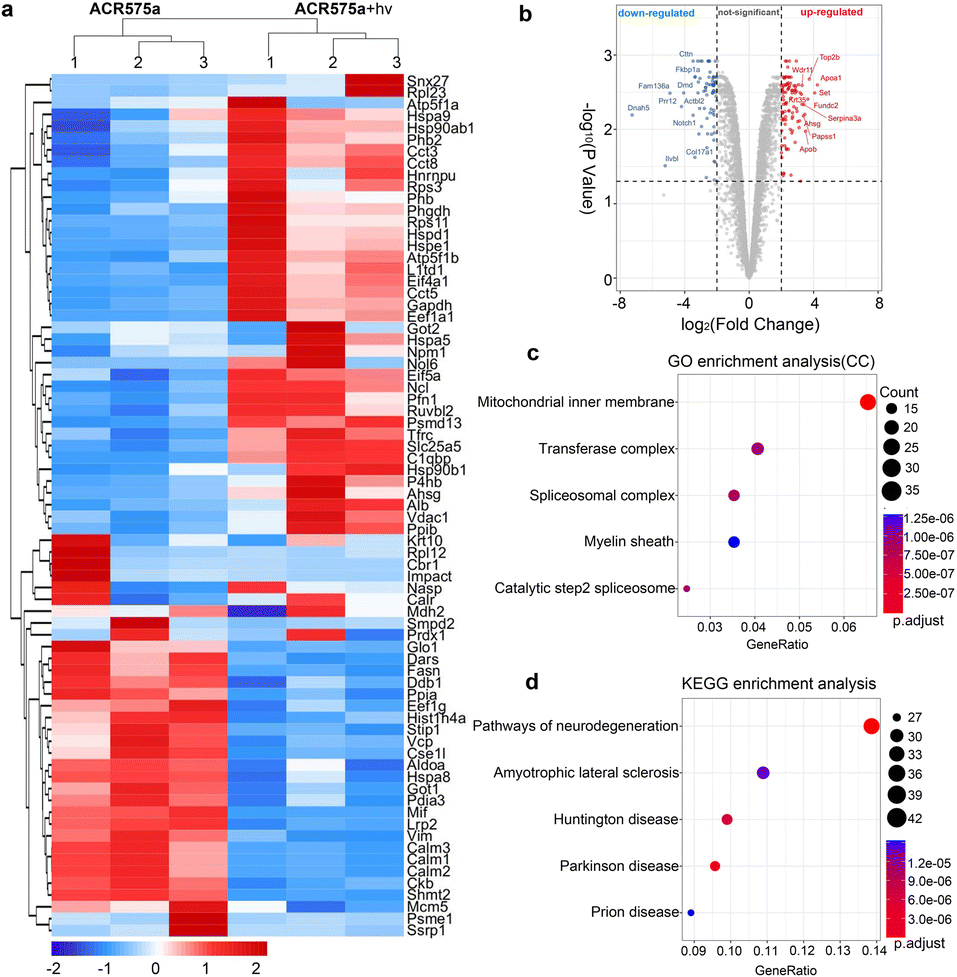 | ||
| Fig. 8 Proteomics analysis of NSCs with and without the stimulation of acetyl radicals. (a) Heatmap of gene expression of NSCs upon treatment with ACR575a or ACR575a + hv. (b) Volcano map of the genes quantified with TMT-labeling in ACR575a or ACR575a + hv treated NSCs. (c) GO enrichment analysis (cell component) of genes with fold-change ≥ 2. (d) KEGG pathway analysis with the same genes as in (c). | ||
Conclusions
Radicals are biological. Yet, radical biology has been a challenging field due to their diversity, high reactivity and short-term existence. The biological outcome of radicals is an elusive function of their chemical nature, flux and matrix, and a broad continuum from signaling to stress to damaging. The progress of radical biology has been a source of inspiration for novel therapeutic strategies. The destructive nature of oxidative radicals has been harnessed in treating cancers or resistant bacterial infections by killing malign cells or invading pathogens. In contrast, the signaling use of radicals for disease treatment is rare and challenging and yet may trigger a paradigm change in disease intervention. A notable example is the identification of nitric oxide as the endothelial-derived relaxing factor. This led to the mechanistic understanding of nitroglycerin for angina and the development of pharmaceuticals for hypertensive conditions.Acetyl radicals are known to be endogenous and yet their biological implications remain untapped. This work offers a powerful molecular tool for acyl biology, chemical biology and potentially medicine. We report that acyl-caged rhodamine is a modular and general molecular mechanism for photo-triggered and feedback-controlled generation of acyl radicals of interest. ACR575a, an acetyl radical donor, was showcased to precisely modulate the cell redox homeostasis, promote NSC differentiation, and have therapeutic effects in early AD mice. Amid the increasingly heavy society burden and the poor results of the intensive drug-development endeavors against Alzheimer's disease, the success of ACR575a offers an alternative drug-development strategy, besides the heavily exploited targeted-therapies. This work will also stimulate further exploration of the profound implications of acyl radicals in health and disease.
Ethical statement
All animal experiments were performed according to the guidelines of the Care and Use of Laboratory Animals formulated by the Ministry of Science and Technology of China and were approved by the Animal Care and Use Committee of East China Normal University (approval no., m+ R20190304. Shanghai. China).Data availability
The synthesis and characterization of all compounds, and crystallographic analysis of all relevant compounds are available in the ESI.†Author contributions
X. L., Y. Y., Y. T., X. Q. and W. Y. devised the project and are the corresponding authors. X. L. designed and performed the chemical synthesis, characterization and spectral recording with oversight from Y. Y. and X. Q., along with support from X. W. and J. C. for the TCSPC. Z. Z. performed all NSC-associated cell experiments and animal tests under the supervision of Y. T., J. W. performed all HeLa cell-associated experiments with oversight from W. Y. Principal manuscript writing was by Y. Y., Y. T., X. L. and Z. Z. with critical feedback from all authors. Y. Z. performed the protein mass spectrometry with oversight from B. G.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 21822805, 21908065, 22078098, 31871430, 21635003, 21811540027, 21827814), China Post-doctoral Science Foundation (No. 2019M651427, 2020T130197), the Research Funds of Happiness Flower ECNU (2020JK2103), the Commission of Science and Technology of Shanghai Municipality (18430711000) and the Innovation Program of Shanghai Municipal Education Commission (201701070005E00020). W. Yang acknowledges the financial support of the Innovative Research Team of High-level Local Universities in Shanghai. We thank Dr Yanxia Zhang at Shanghai Institute of Organic Chemistry (Chinese Academy of Sciences) for her help in EPR data analysis.References
- (a) K. H. Cheeseman and T. F. Slater, Br. Med. Bull., 1993, 49, 481–493 CrossRef CAS; (b) K. B. Beckman and B. N. Ames, Physiol. Rev., 1998, 78, 547–581 CrossRef CAS PubMed; (c) M. Valko, D. Leibfritz, J. Moncol, M. T. Cronin, M. Mazur and J. Telser, Int. J. Biochem. Cell Biol., 2007, 39, 44–84 CrossRef CAS; (d) H. Sies, V. V. Belousov, N. S. Chandel, M. J. Davies, D. P. Jones, G. E. Mann, M. P. Murphy, M. Yamamoto and C. Winterbourn, Nat. Rev. Mol. Cell Biol., 2022, 23, 499–515 CrossRef CAS.
- (a) J. M. McCord and I. Fridovich, J. Biol. Chem., 1969, 244, 6049–6055 CrossRef CAS PubMed; (b) D. C. Wallace, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 8947–8953 CrossRef CAS; (c) A. M. James, Y. Collins, A. Logan and M. P. Murphy, Trends Endocrinol. Metab., 2012, 23, 429–434 CrossRef CAS PubMed; (d) H. Sies and D. P. Jones, Nat. Rev. Mol. Cell Biol., 2020, 21, 363–383 CrossRef CAS.
- (a) J. M. McCord and I. Fridovich, J. Biol. Chem., 1969, 244, 6049–6055 CrossRef CAS; (b) I. Fridovich, Arch. Biochem. Biophys., 1986, 247, 1–11 CrossRef CAS.
- (a) B. Halliwell and J. M. C. Gutteridge, Free Radicals in Biology and Medicine.Oxford University Press, USA, 2015 CrossRef; (b) B. Yang, Y. Chen and J. Shi, Chem. Rev., 2019, 119, 4881–4985 CrossRef CAS.
- (a) M. Valko, C. J. Rhodes, J. Moncol, M. Izakovic and M. Mazur, Chem.-Biol. Interact., 2006, 160, 1–40 CrossRef CAS; (b) H. Sies, Redox Biol., 2015, 4, 180–183 CrossRef CAS PubMed; (c) Q. Lan, P. Yu, K. Yan, X. Li, F. Zhang and Z. Lei, J. Am. Chem. Soc., 2022, 144, 21010–21015 CrossRef CAS.
- (a) I. Rosenthal, J. Z. Sostaric and P. Riesz, Ultrason. Sonochem., 2004, 11, 349–363 CrossRef CAS; (b) P. Agostinis, K. Berg, K. A. Cengel, T. H. Foster, A. W. Girotti, S. O. Gollnick, S. M. Hahn, M. R. Hamblin, A. Juzeniene, D. Kessel, M. Korbelik, J. Moan, P. Mroz, D. Nowis, J. Piette, B. C. Wilson and J. Golab, Ca-Cancer J. Clin., 2011, 61, 250–281 CrossRef PubMed; (c) Z. Tang, Y. Liu, M. He and W. Bu, Angew. Chem., Int. Ed., 2019, 58, 946–956 CrossRef CAS PubMed; (d) X. Wu, Y. Jiang, N. J. Rommelfanger, F. Yang, Q. Zhou, R. Yin, J. Liu, S. Cai, W. Ren, A. Shin, K. S. Ong, K. Pu and G. Hong, Nat. Biomed. Eng., 2022, 6, 754–770 CrossRef CAS PubMed; (e) X. Wei, C. Zhang, S. He, J. Huang, J. Huang, S. Liew, Z. Zeng and K. Pu, Angew. Chem., Int. Ed., 2022, 61, e202202966 CrossRef CAS; (f) C. Zhang and K. Pu, Adv. Mater., 2023, 35, 2303059 CrossRef PubMed.
- (a) J. D. Parker and J. O. Parker, N. Engl. J. Med., 1998, 338, 520–531 CrossRef CAS PubMed; (b) S. Moncada and E. A. Higgs, Br. J. Pharmacol., 2006, 147, S193–S201 CrossRef CAS; (c) D. Tousoulis, A. M. Kampoli, C. Tentolouris, N. Papageorgiou and C. Stefanadis, Curr. Vasc. Pharmacol., 2012, 10, 4–18 CrossRef CAS.
- (a) H. A. Cameron and E. Gould, Neuroscience, 1994, 61, 203–209 CrossRef CAS; (b) T. Duncan and M. Valenzuela, Stem Cell Res. Ther., 2017, 8, 1–9 CrossRef; (c) M. Boldrini, C. A. Fulmore, A. N. Tartt, L. R. Simeon, I. Pavlova, V. Poposka, G. B. Rosoklija, A. Stankov, V. Arango, A. J. Dwork, R. Hen and J. J. Mann, Cell Stem Cell, 2018, 22, 589–599 CrossRef CAS PubMed.
- (a) H. V. Praag, G. Kempermann and F. H. Gage, Nat. Neurosci., 1999, 2, 266–270 CrossRef PubMed; (b) J. Smith, E. Ladi, M. Mayer-Pröschel and M. Nobel, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 10032–10037 CrossRef CAS; (c) K. Wang, T. Zhang, Q. Dong, E. C. Nice, C. Huang and Y. Wei, Cell Death Dis., 2013, 4, e537 CrossRef CAS; (d) C. L. Bigarella, R. Liang and S. Ghaffari, Development, 2014, 141, 4206–4218 CrossRef CAS PubMed; (e) M. Khacho, R. Harris and R. S. Slack, Nat. Rev. Neurosci., 2019, 20, 34–48 CrossRef CAS PubMed; (f) E. P. Moreno-Jiménez, M. Flor-García, J. Terreros-Roncal, A. Rábano, F. Cafini, N. Pallas-Bazarra, J. Ávila and M. Llorens-Martín, Nat. Med., 2019, 25, 554–560 CrossRef.
- (a) D. Griller and K. U. Ingold, Persistent carbon-centered radicals, Acc. Chem. Res., 1976, 9, 13–19 CrossRef CAS; (b) K. Kato and A. Osuka, Angew. Chem., Int. Ed., 2019, 58, 8978–8986 CrossRef CAS.
- (a) C. Chatgilialoglu, D. Crich, M. Komatsu and I. Ryu, Chem. Rev., 1999, 99, 1991–2070 CrossRef CAS; (b) G. Bergonzini, C. Cassani and C. J. Wallentin, Angew. Chem., Int. Ed., 2015, 54, 14066–14069 CrossRef CAS PubMed.
- C. E. Brown, A. G. Neville and D. M. Rayner, Aust. J. Chem., 1995, 48, 363–379 CrossRef CAS.
- G. Merényi, J. Lind and L. Engman, J. Chem. Soc., Perkin Trans. 2, 1994, 12, 2551–2553 RSC.
- Á. Péter, S. Agasti, O. Knowles, E. Pye and D. J. Procter, Chem. Soc. Rev., 2021, 50, 5349–5365 RSC.
- L. S. Nakao, D. Ouchi and O. Augusto, Toxicol, 1999, 12, 1010–1018 CAS.
- J. Massari, D. E. Fujiy, F. Dutra, S. M. Vaz, A. C. O. Costa, G. A. Micke, M. F. M. Tavares, R. Tokikawa, N. A. Assunção and E. J. H. Bechara, Chem. Res. Toxicol., 2008, 21, 879–887 Search PubMed.
- J. Massari, R. Tokikawa, L. Zanolli, M. F. M. Tavares, N. A. As-sunção and E. J. H. Bechara, Chem. Res. Toxicol., 2010, 23, 1762–1770 Search PubMed.
- A. J. Eugene and M. I. Guzman, J. Phys. Chem. A, 2017, 121, 2924–2935 CrossRef CAS.
- (a) D. A. Rozwarski, G. A. Grant, D. H. R. Barton, W. R. Jacobs and J. C. Sacchettini, Science, 1998, 279, 98–102 CrossRef CAS PubMed; (b) L. Qin, C.-H. Huang, L. Mao, B. Shao and B.-Z. Zhu, Free Radical Biol. Med., 2020, 154, 1–8 CrossRef CAS.
- (a) G. Bergonzini, C. Cassani and C. J. Wallentin, Angew. Chem., Int. Ed., 2015, 54, 14066–14069 CrossRef CAS; (b) S. Sultan, M. A. Rizvi, J. Kumar and B. A. Shah, Chem.–Eur. J., 2018, 24, 10617–10620 CrossRef CAS PubMed; (c) A. Banerjee, Z. Lei and M. Y. Ngai, Synthesis, 2019, 51, 303–333 CrossRef CAS PubMed.
- (a) R. S. Balaban, S. Nemoto and T. Finkel, Cell, 2005, 120, 483–495 CrossRef CAS PubMed; (b) L. D. Zorova, V. A. Popkov, E. Y. Plotnikov, D. N. Silachev, I. B. Pevzner, S. S. Jankauskas, V. A. Babenko, S. D. Zorov, A. V. Balakireva, M. Juhaszova, S. J. Sollott and D. B. Zorov, Anal. Biochem., 2018, 552, 50–59 CrossRef CAS PubMed.
- (a) R. G. W. Norrish and C. H. Bamford, Nature, 1936, 138, 1016 CrossRef CAS; (b) S. Majhi, Photochem. Photobiol. Sci., 2021, 20, 1357–1378 CrossRef CAS.
- R. C. Scaduto and L. W. Grotyohann, Biophys. J., 1999, 76, 469–477 CrossRef CAS PubMed.
- T. Kobayashi, Y. Urano, M. Kamiya, T. Ueno, H. Kojima and T. Nagano, J. Am. Chem. Soc., 2007, 129, 6696–6697 CrossRef CAS PubMed.
- G. N. Lewis, D. Lipkin and T. T. Magel, J. Am. Chem. Soc., 1944, 66, 1579–1583 CrossRef CAS.
- H. Huang, F. Dong and Y. Tian, Anal. Chem., 2016, 88, 12294–12302 CrossRef CAS PubMed.
- H. Guo, Q. Peng, X.-K. Chen, Q. Gu, S. Dong, E. W. Evans, A. J. Gillett, X. Ai, M. Zhang, D. Gredgington, V. Coropceanu, F. H. Friend, J. Brédas and F. Li, Nat. Mater., 2019, 18, 977–984 CrossRef CAS PubMed.
- H. Y. Ahn, K. E. Fairfull-Smith, B. J. Morrow, V. Lussini, B. Kim, M. V. Bondar, S. E. Bottle and K. D. Belfield, J. Am. Chem. Soc., 2012, 134, 4721–4730 CrossRef CAS PubMed.
- A. Baracca, G. Sgarbi, G. Solaini and G. Lenaz, Biochim. Biophys. Acta, Bioenerg., 2003, 1606, 137–146 CrossRef CAS PubMed.
- J. P. Brennan, R. Southworth, R. A. Medina, S. M. Davidson, M. R. Duchen and M. J. Shattock, Cardiovasc. Res., 2006, 72, 313–321 CrossRef CAS PubMed.
- K. S. Echtay, D. Roussel, J. St-Pierre, M. B. Jekabsons, S. Cadenas, J. A. Stuart, J. A. Harper, S. J. Roebuck, A. Morrison, S. Pickering, J. C. Clapham and M. D. Brand, Nature, 2002, 415, 96–99 CrossRef CAS.
- (a) L. C. Hool and B. Corry, Antioxid. Redox Signaling, 2007, 9, 409–435 CrossRef CAS PubMed; (b) R. F. Feissner, J. Skalska, W. E. Gaum and S. S. Sheu, Front. Biosci., 2009, 14, 1197 CrossRef CAS PubMed.
- P. S. Brookes, Y. Yoon, J. L. Robotham, M. W. Anders and S. S. Sheu, Am. J. Physiol.: Cell Physiol., 2004, 287, C817–C833 CrossRef CAS.
- (a) S. F. Sorrells, M. F. Paredes, A. Cebrian-Silla, K. Sandoval, D. Qi, K. W. Kelley, D. James, S. Mayer, J. Chang, K. I. Auguste, E. F. Chang, A. J. Gutierrez, A. R. Kriegstein, G. W. Mathern, M. C. Oldham, E. J. Huang, J. M. Garcia-Verdugo, Z. Yang and A. Alvarez-Buylla, Nature, 2018, 555, 377–381 CrossRef CAS; (b) Y. Mu and F. H. Gage, Mol. Neurodegener., 2011, 6, 85 CrossRef PubMed; (c) M. K. Tobin, K. Musaraca, A. Disouky, A. Shetti, A. Bheri, W. G. Honer, N. Kim, R. J. Dave and D. A. Bennett, Cell Stem Cell, 2019, 24, 974–982 CrossRef CAS PubMed.
- V. S. Adusumilli, T. L. Walker, R. W. Overall, G. M. Klatt, S. A. Zeidan, S. Zocher, D. G. Kirova, K. Ntitsias, T. J. Fischer, A. M. Sykes, S. Reinhardt, A. Dahl, J. Mansfeld, A. E. Runker and G. Kempermann, Cell Stem Cell, 2021, 28, 300–314 CrossRef CAS.
- J. Mazumdar, W. T. O'brien, R. S. Johnson, J. C. LaManna, J. C. Chavez, P. S. Klein and M. C. Simon, Nat. Cell Biol., 2010, 12, 1007–1013 CrossRef CAS.
- (a) S. Martín-Suárez, J. Valero, T. Muro-García and J. M. Encinas, Aging Cell, 2019, 18, e12958 CrossRef PubMed; (b) E. P. Moreno-Jiménez, M. Flor-García, J. Terreros-Roncal, A. Rábano, F. Cafini, N. Pallas-Bazarra, J. Ávila and M. Llorens-Martín, Nat. Med., 2019, 25, 554–560 CrossRef PubMed; (c) S. Lewis, Nat. Rev. Neurosci., 2023, 24, 394 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2024836 and 2024838. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3sc03035k |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2023 |