
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMn(III)-mediated C–P bond activation of diphosphines: toward a highly emissive phosphahelicene cation scaffold and modulated circularly polarized luminescence†
Bo
Yang
a,
Suqiong
Yan
a,
Chengbo
Li
c,
Hui
Ma
a,
Fanda
Feng
a,
Yuan
Zhang
a and
Wei
Huang
 *ab
*ab
aState Key Laboratory of Coordination Chemistry, School of Chemistry and Chemical Engineering, Nanjing University, Nanjing 210093, P. R. China. E-mail: whuang@nju.edu.cn
bShenzhen Research Institute of Nanjing University, Shenzhen 51805, P. R. China
cSchool of Materials and Energy, University of Electronic Science and Technology of China, Chengdu 610000, P. R. China
First published on 24th August 2023
Abstract
Transition metal mediated C–X (X = H, halogen) bond activation provides an impressive protocol for building polyaromatic hydrocarbons (PAHs) in C–C bond coupling and annulation; however, mimicking both the reaction model and Lewis acid mediator simultaneously in a hetero-PAH system for selective C–P bond cleavage faces unsolved challenges. At present, developing the C–P bond activation protocol of the phosphonic backbone using noble-metal complexes is a predominant passway for the construction of phosphine catalysts and P-center redox-dependent photoelectric semiconductors, but non-noble metal triggered methods are still elusive. Herein, we report Mn(III)-mediated C–P bond activation and intramolecular cyclization of diphosphines by a redox-directed radical phosphonium process, generating phosphahelicene cations or phosphoniums with nice regioselectivity and substrate universality under mild conditions. Experiments and theoretical calculations revealed the existence of the unusual radical mechanism and electron-deficient character of novel phosphahelicenes. These rigid quaternary bonding skeletons facilitated versatile fluorescence with good tunability and excellent efficiency. Moreover, the enantiomerically enriched crystals of phosphahelicenes emitted intense circularly polarized luminescence (CPL). Notably, the modulated CPL of racemic phosphahelicenes was induced by chiral transmission in the cholesteric mesophase, showing ultrahigh asymmetry factors of CPL (+0.51, −0.48). Our findings provide a new approach for the design of emissive phosphahelicenes towards chiral emitters and synthesized precursors.
Introduction
Organic diphosphines, as a brilliant ligand family for the construction of catalysts, drug discoveries, and semiconducting materials, determine the regioselectivity and stereoselectivity in catalytic processes,1–4 bioactivities,5,6 and optoelectronic properties7,8 because of their robust chiral skeleton, chelating capacity, and commercial availability. Economically, developing synthesis protocols for C–P bond activation from available diphosphines is a concise route for expanding ligand libraries and functional materials.9–14 Nevertheless, the exploitation of efficient activation and reconstruction between C–P and C–H bonds remains a significant obstruction due to high bonding energy, strong coordinative ability, and intricate selectivity, where noble metal catalysts ([Pd], [Au], and [Rh]) play a critical role.14–19Morandi's group pioneered the C–P bond activation of diphosphines via Pd2(dba)3-catalyzed metathesis (Scheme 1a), and the PPh3 fragment was eliminated for the cyclopalladated intermediate. Finally, novel neutral phospha[5]helicenes (PIII) were generated and further racemic phospha[5]helicene (PV) oxides were isolated.16 Subsequently, Wang's group modified the reaction to break the intramolecular cyclization of pre-activated phosphoniums.15 In addition, Cu(OTf)2-mediated C–H bond activation at 8,8′-positions in binaphthyl produced an achiral bisphosphonium with a low yield of 24%.20 This method spread in the laboratory for the preparation of achiral phosphoniums.21,22 Theoretically, another potential cationic phosphahelicene might be produced after eliminating the PPh2 unit and rebuilding the C–P bond at the 2′-position. However, this valued phosphahelicene cation ([1b]+) and its properties have not been captured and explored (Scheme 3). These above described electron-deficient cationic phosphahelicenes that integrated an active C–P bond and a prochiral axis to enhance the oxidative addition efficacy endowed the enantioselective cross-coupling from C–P to C–C bonds with excellent stereoselectivity and regioselectivity.23 We envisioned that the tunability of metal sources in the pursuit of altering the activation of radical cations and elimination pathways of the intermediates would change the reactivity and selectivity of cyclization.12,16
 | ||
| Scheme 1 (a) Representative C–P bond activation and reconstitution of phosphines. (b) Mn(III)-mediated C–P bond activation and cyclization strategy in this work. (c) Schematic evolution of phospha[5]helicenes. | ||
Recently, heterohelicenes have shown arresting renaissance due to their tunable electronic structures, making them widely used in asymmetric catalysis and optoelectronics.24–28 Notably, the electronic factors are highly dependent on the oxidation state of the phosphorus center in phosphahelicenes. Most neutral phosphahelicenes (PIII) yielded lower quantum efficiency and insufficient CPL because of high nonradiative rates and the fluxional molecular conformation in the excited states (i.e., analogues phospha[5]helicenes 1 and 2 are not luminescent, Scheme 1c).29–33 To overcome this drawback, we speculated that introducing a quaternary bonding environment for the P-atom could increase the steric hindrance to reset the electronic transition nature in phosphahelicenes, thereby inhibiting the nonradiative decay of emitters in excited states.22,34 Furthermore, the cationic phosphahelicene has high stability and electron-defect character in the quaternary C–P bonding environment, providing a new opportunity for the design of chiral donor–acceptor (D–A) modules.35–37 On the other hand, exploring chiral transmission toward increased CPL of racemic [5]helicenes is still rare.38,39 Hence, developing highly emissive cationic phosphahelicene scaffolds and chiral self-assembly tactics is an essential extension of heterohelicenes for photoelectric devices and synthesized precursors.
Unprecedentedly, we reported a novel MnX3 (X = Cl, Br) mediated protocol to construct cationic phosphahelicenes and phosphoniums from diphosphines (Scheme 1b) via intramolecular cyclization with good regioselectivity, substrate scope, and yields (up to 92%). Mechanistically, redox-directed C–P bond cleavage and annulation were confirmed by a radical phosphonium path of the reaction. The intense fluorescence of the products can be regulated from blue to orange (ΦPLQY up to 77%). Importantly, modulated CPL was obtained for this series of emissive phosphahelicenes in a ternary cholesteric mesophase by chiral transmission, showing ultrahigh asymmetry factors (+0.51, −0.48). Mechanistic study demonstrated that the increased CPL critically depended on chiral fluorescence resonance energy transfer (FRET) and helical superstructures rather than selective Bragg reflection.
Results and discussion
Exploration of the C–P bond activation method and substrate scope
Inspired by the redox-related radical cation mechanisms in the annulation of redox-active Lewis acid mediators,40 certain metal halide complexes have been predicted and chosen as targets. The corresponding diphosphines were acquired from commercial sources or prepared via the Monsanto method and characterized by NMR (nuclear magnetic resonance) and SCXRD (single crystal-XRD) (Fig. S2–S7 and Table S2†).41 After metal-mediator screening in the presence of oxygen, a better yield of [1b]+[X]− was achieved with MnCl2 or MnBr2 at room temperature or under modest heating (complex counterions in the products were exchanged into chloride ions in brine), but was ineffective with MnI2 because of oxidative decomposition of iodide ions (Table 1, entries 1–5). Notably, the conversion speed of intramolecular cyclization was rapid under heating, which was faster than that at room temperature. In consideration of the conversion speed and yield, the reaction should be conducted at an elevated temperature (30 °C) for 5–10 minutes (entries 1–3). The presence of O2 could boost the reaction efficiency but inert argon inhibited the conversion (entry 1). Other non-halogenated Mn(II)/Mn(III) complexes led to negative results under the same conditions, generating BINAPO and the corresponding stable complexes (entries 6–9). The other metal halide candidates, i.e., FeCl3, CoCl2, NiCl2, and CuCl2, are noneffective in this reaction (entries 10–13). All control experiments indicated that it was not only the redox potential for metal cations but also the halogen ion that displayed a unique efficacy in the reaction. Considering the polarity gap between substrates, MnCl2, and ionic products, satisfactory conversion rates and isolated yields were obtained in the mixed solvents at 30 °C for [1b]+[Cl]− (entries 1 and 14–18).|
|
||||
|---|---|---|---|---|
| Entry | Mediator | Solvent | T (°C) | Yieldc (%) |
a Reaction conditions: BINAP (0.16 mmol), metal-mediator (3.0 eq.). aMixed-solvent (Vchloroform![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :VEtOH = 2:1, 10 mL), under an O2 atmosphere (1 bar) at corresponding temperature for 5–10 minutes.
b 10 mol%.
c Isolated yields for helicenes and BINAPOs (in parentheses).
d ND represents not detected.
e Ar instead of O2. :VEtOH = 2:1, 10 mL), under an O2 atmosphere (1 bar) at corresponding temperature for 5–10 minutes.
b 10 mol%.
c Isolated yields for helicenes and BINAPOs (in parentheses).
d ND represents not detected.
e Ar instead of O2.
|
||||
| 1 | MnCl2 | CHCl3:EtOH |
30 | 56(37), NDe |
| 2 | MnCl2 | CHCl3:EtOH |
60 | 51(40) |
| 3 | MnCl2 | CHCl3:EtOH |
80 | 52(40) |
| 4 | MnBr2 | CHCl3:EtOH |
30 | 54(42) |
| 5 | MnI2 | CHCl3:EtOH |
30 | Trace |
| 6 | MnCO3 | CHCl3:EtOH |
80 | NDd |
| 7 | Mn(OTf)2 | CHCl3:EtOH |
80 | NDd |
| 8 | Mn(acac)3 | CHCl3:EtOH |
80 | NDd |
| 9 | Mn(OAc)3 | CHCl3:EtOH |
80 | NDd |
| 10 | FeCl3 | CHCl3:EtOH |
80 | NDd |
| 11 | CoCl2 | CHCl3:EtOH |
80 | NDd |
| 12 | NiCl2 | CHCl3:EtOH |
80 | NDd |
| 13 | CuCl2 | CHCl3:EtOH |
80 | NDd |
| 14 | MnCl2 | Toluene | 80 | Trace |
| 15 | MnCl2 | Chloroform | 80 | 38(35) |
| 16 | MnCl2 | Ethanol | 80 | <5(11) |
| 17 | MnCl2 | THF | 80 | 42(47) |
| 18 | MnCl2 | Acetonitrile | 80 | 29(34) |
| 19 | MnCl2b | CHCl3:EtOH |
30 | Trace |

Based on the optimized conditions in hand, the substrate scope was investigated for BINAPs/BIPHEPs (Scheme 2). Derivatives BINAP-4-Me and BINAP-3,5-Me2 with methyl groups at different positions in phenyls converted into [2b]+[Cl]− and [3b]+[Cl]− with 54% and 56% yields. Partial BINAPs showed slow cyclization at room temperature due to lower activity, which required proper heating (50–80 °C, Scheme 2). BINAPs with electron-donating methyl groups at ortho, meta, and para-positions converted into [11b]+[Cl]−, [12b]+[Cl]−, and [13b]+[Cl]− with depressed yields (37–42%), but the BINAPOs could be reduced and transformed into initial BINAPs by HSiCl3 and utilized next time. Besides, ethyl, methoxyl, tertiary butyl, and mesitylene substituted BINAPs displayed higher yields (61–75%). Encouragingly, electron-deficient BINAPs (fluorine, trifluoromethyl, and ester) showed the highest yields (up to 85%). It was worth noting that the extensional phenyls, biphenyls, and naphthyls had an inconspicuous influence on intramolecular cyclization. Similarly, biphenyl diphosphines (BIPHEPs) cyclized into corresponding phosphoniums with moderate yields (39–67%). Unfortunately, the P,O-embedded phosphonium [8b]+[Cl]− did not generate in this reaction, which was related to the flexible aryl ether motion of the DPEPhos substrate and longer P–P distance (4.88 Å) than that of BINAP (4.20 Å), leading to a different chelation environment for the intermediate (Fig. S7†). Furthermore, 5,5′-modified BINAPs showed satisfactory tolerance (40–73%) equal to those of 6,6′-modified BINAPs. Nevertheless, the 7,7′-modified BINAPs exhibited excellent cyclization, getting the highest yields (68–92%) of phospha[5]helicenes, which could be relevant to variational electronic structures and an elevated competitive rate between cyclization and oxidation. Finally, gram-scale reactions were performed with good yields (Scheme 2). To the best of our knowledge, this method represented the first C–P bond activation discovery in diphosphines using non-noble metal mediators. This family of helicenium products could be transformed into asymmetric monophosphines by Pd-catalyzed stereoselective cleavage of the C–P bond and cross-coupling according to a recent report.23
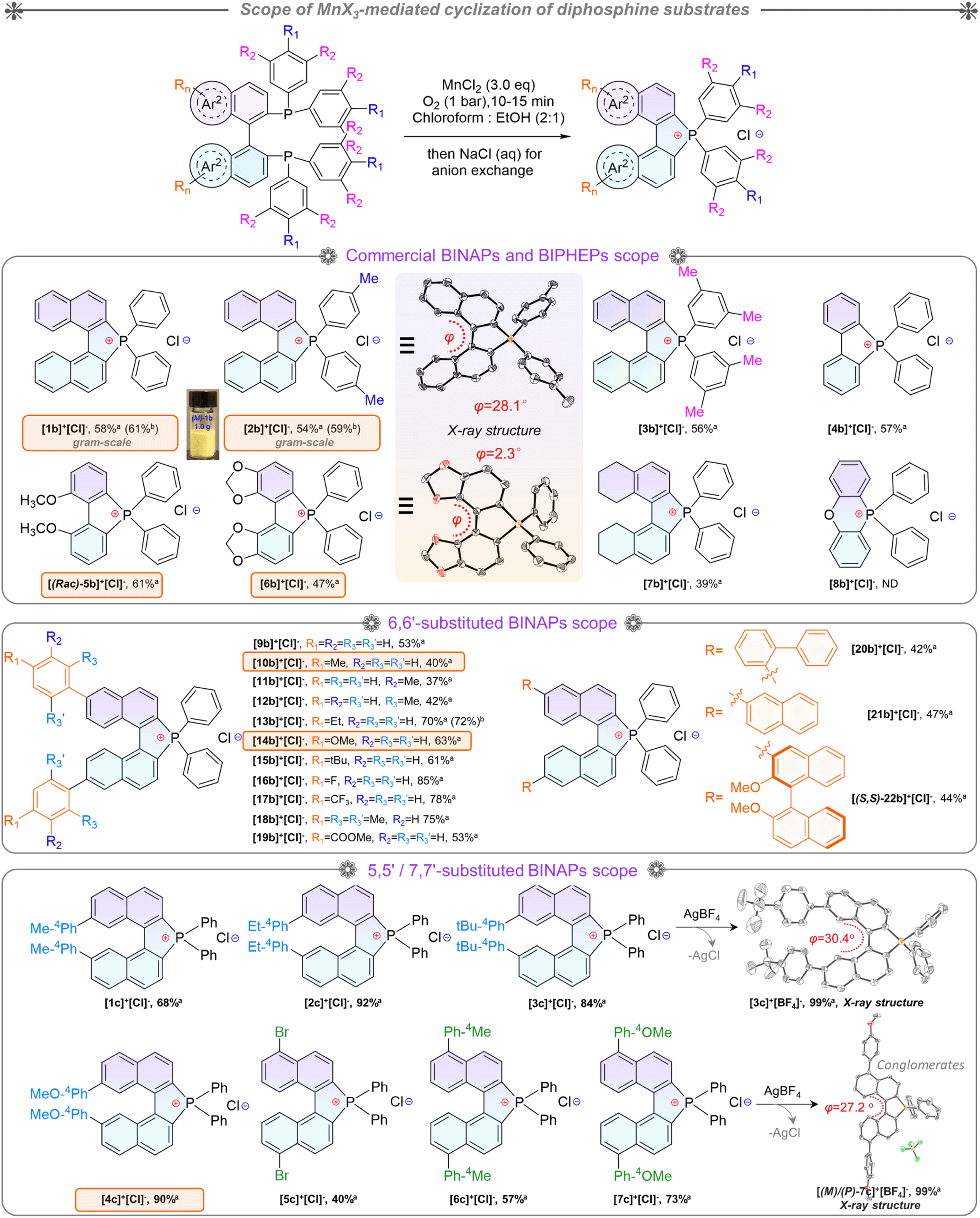 | ||
| Scheme 2 Diphosphine scope of intramolecular cyclization. Reaction conditions: diphosphines (0.2 mmol), MnCl2 (3.0 eq.), mixed-solvent (10 mL), under an O2 atmosphere (1 bar) at corresponding temperature for 10 minutes. Corresponding reaction temperatures: 30 °C: [1b]+[Cl]−–[3b]+[Cl]−, 50 °C: [4b]+[Cl]−–[7b]+[Cl]−, [9b]+[Cl]−–[18b]+[Cl]−, and [1c]+[Cl]−–[7c]+[Cl]−, and 80 °C: [8b]+[Cl]−; [20b]+[Cl]−–[22b]+[Cl]−. aIsolated yields of the 0.2 mmol-scale reaction. bIsolated yields of the gram-scale reaction. | ||
Indeed, this cyclization was highly stereospecific, where the product chirality inherited the BINAPs. However, the enantiomeric purity decreased in solution due to a low interconversion barrier at room temperature.23,42 Enantiomerically enriched [2b]+[Cl]− and [2b]+[BF4]− were acquired via rapid synthesis, chromatographic purification, and their single-crystal growth at low temperature to guarantee adequate enantiomeric purity. Combined with the observations of SCXRD and uniform morphologies (Fig. S8, S9, S16, S17, and S21†), these proofs affirmed the formation of homochiral single crystals. The homochiral crystals were used for the kinetic study of chirality. Initially, we attempted to evaluate the ee values of compounds using chiral HPLC (at 25 °C) but failed. To this end, we then employed the temperature and time-dependence of the rate constant to calculate the ee values and thermodynamic parameters of the racemization process via CD (circular dichroism) spectra (see the ESI for detailed determination, Fig. S1†). According to the CD ellipticities between homochiral crystals and pristine products in solution, the average ee value (for 5 parallel samples) of pristine product [2b]+[Cl]− was about 73%. Finally, as a summary of Eyring plotting and calculations for [2b]+[Cl]−, ΔH‡ = 12.5 kcal mol−1 and ΔS‡ = −25.4 cal mol−1 K−1 were determined, and ΔG‡exp = 20.1 kcal mol−1, k = 1.5 × 10−4 s−1, and t1/2 (half-life) = 77.1 min at 298.15 K were successfully elucidated, whose ΔG‡exp value was smaller than that of carbon[5]helicene (∼25 kcal mol−1 at 298.15 K).43–45 The ΔG‡exp value was slightly below the margin of the activation barrier (∼22.2 kcal mol−1) required for conformational atropo-enantiomers to be resolvable.46 In addition, the interconversion of helical conformation was also considered via density functional theory (DFT) calculations, and the isomerization crossed over the saddle-shaped transition state with a low isomerization barrier (ΔG‡cal = 24.1 kcal mol−1, at the B3LYP/6-31G* level) of [(P)/(M)-2b]+ (Fig. S38†). The degraded ee value observed in our experiments was reasonable, and the experimental ΔG‡cal value was smaller by 4.0 kcal mol−1 than the theoretical one. The reason for the latter may be the problem of the accuracy of the basis set/functionality.47 The lower ΔG‡cal 18.6 kcal mol−1 of [(P)/(M)-1b]+ facilitated the loss of stereochemical purity completely and the heterochiral crystal packing pattern (Fig. 4g). Consequently, (P)-[2b]+ manifested homochiral crystallization, which could inherit (S)-BINAP handedness and higher stability (Fig. S38†). The robust isomerization barriers and well-tailored D–A modules of phospha[5]helicene cations could be realized by introducing strong electron donors at 7,7′/8,8′-positions, and this project was still in progress.
Mechanistic studies by control experiments and DFT calculations
Control experiments and calculations were performed to understand the reaction mechanism (Scheme 3a). First, the cyclization showed no conversion using the catalytic amount of MnCl2 (10 mol%, entry 19), indicating that excess counterions from MnCl2 were essential for the charge balance in the ionic product. The cyclization was suppressed in argon, which could be related to the redox process in the reaction. To gain more insight into this speculation, we conducted Mn(III)-prompted studies in the presence of necessary counterionic chloridions (HCl or pyridine hydrochloride). The Mn(III) ion possessed appropriate oxidizing power (Ered = 1.51 V, vs. H+/H2) and can oxidize the BINAP (Eox(onset) = 1.46 V vs. H+/H2, Fig. S22†) to give radical phosphonium and cyclization products. The reduction potential of the metal complexes was crucial to this cyclization, as demonstrated by the high reactivity of activated Mn(III)Cl3 in contrast to the lack of reactivity of Fe3+ (0.77 V), Co2+ (0.28 V), Ni2+ (−0.23 V), and Cu2+ (0.16 V) in the reaction. Indeed, when Mn(OAc)3 was injected into a mixture of BINAP and MnCl2 under inert Ar conditions, the mixture also underwent rapid oxidation and cyclization to form [1b]+, indicating that in situ Mn(III)Cl3 triggered the single electron transfer from the P-atom to Mn(III) (Scheme 3a). Unexpectedly, photocyclization took place in the absence of MnCl2 under UV radiation in the air with a meager yield but increased moderately with the addition of counter chloridions (HCl or pyridine hydrochloride, yields from 3% to 14%), suggesting the homolysis of the C–P bond in the photoinduced excited state (Scheme 3a, Fig. S24†).48,49 Other low-energy light sources (405 nm or indoor light) cannot activate cyclization owing to the unmatched absorption energy of BINAP (Fig. S25†). Furthermore, both standard MnCl2-mediated and photo-induced annulations were suppressed in radical scavenger TEMPO. These results concluded that the intramolecular cyclization proceeded via a radical pathway.20 Furthermore, the dark experiments had a similar cyclization yield to the MnCl2-mediated system (Scheme 3a). | ||
| Scheme 3 (a) Control experiments of the reactions. (b) Plausible mechanism. | ||
The DFT calculated HOMO of BINAP revealed that electrons were dominated on the P-moieties (Fig. 1a), giving the preferential oxidation of losing one electron in the P-atom and generating the corresponding radical cation 3-[BINAP]+˙ after single electron transfer (SET) oxidation by Mn(III)Cl3. Spin densities analysis demonstrated that P-atoms had the highest spin densities in radical cation 3-[BINAP]+˙ and were preferred sites for the radical attack (Fig. 1a). The calculated Gibbs free energies of the critical transition states also supported the observation that phosphonium attack at the C2′-position was favorable with a low barrier (ΔG‡ = 19.8 kcal mol−1, Fig. 1b), which was reasonable for the observed mild cyclization conditions at near room temperature. With the above results and previous studies on metal-triggered cleavage and annulation of C–P bonds in hand, a plausibly radical phosphonium pathway is depicted in Scheme 3b.16,50–53 The preferential coordination of BINAP with MnCl2 formed an Mn(BINAP)Cl2 intermediate (1a), after which the presence of oxygen caused the oxidation of Mn(II) to Mn(III) complexes (2a). The matched redox potential in the Mn(III) complex led to the formation of radical phosphonium 3-[BINAP]+˙via the SET oxidation between the P-atom and Mn(III)-center. The subsequent attacks with the C2′-position produced the cyclized radical cation 4-[BINAP]+˙, which eliminated the PPh2 residue to form thermodynamically stable [1b]+.54,55
 | ||
| Fig. 1 (a) DFT calculated HOMO diagrams and Mulliken charge of the substrate BINAP (left), and spin densities of radical phosphonium of 3-[BINAP]+˙ (right). (b) Calculated Gibbs free-energy diagrams of the formation of a five-membered ring via the radical phosphonium pathway. | ||
Crystallographic structure determination
Single crystals of enantiomeric and racemic [2b]+[X]− were grown from the purified compounds by slow volatilization in mixed solvents (details are listed in the ESI†). [(6b)+]2[MnCl4]2− crystals were obtained from the reaction mixtures in Schlenk tubes after slow cooling and volatilization. [(M)-2b]+[Cl]−, [(P)-2b]+[Cl]−, and [(P)-2b]+[Br]− crystallized in the chiral P212121 space group with regular cuboid morphology (Fig. 2j and S8–S10†). For [(M)-2b]+, the quaternary P-atom occupied the center and coordinated with chiral binaphthyl and 4-tolyl wings to form a distorted tetrahedron topology, where the C–P bond lengths were determined to be 1.76–1.79 Å, which were close to DFT optimized values (1.79–1.80 Å) and shorter than those of precursor BINAPs (1.83–1.85 Å, Fig. 2a). The bond angle of φ(C1P1C12) was 94.6° in the five-membered ring, which was smaller than that of the opposite unclosed wing φ(C21P1C28) (114.0°). The selected dihedral angle (θ) for the binaphthyl unit and torsion angle (φ) for the binaphthyl axis were determined at 37.1° and 28.1° due to steric hindrance at the 8,8′-positions of hydrogen atoms (Fig. 2a). This structural constraint should cause a compression of left-handedness helicity (absolute configuration was confirmed using a reliable flack parameter of 0.03(5), Table S3†), where the dihedral angle (θ = 37.1°) of [(M)-2b]+ was reduced to about 44.9° compared with that of BINAPs (θ = 82.0°). [(M)-2b]+[Cl]− crystals were acquired from the (R)-BINAPs precursor, while [(P)-2b]+[Cl]− was derived from (S)-BINAPs, which demonstrated that stereochemistry might be preserved throughout the preparation and crystallization. The nonplanar tetrahedron backbone interrupted π–π interactions, but multiple H⋯Cl bondings (2.53–2.87 Å) existed between adjacent ionic pairs as well as CH2Cl2 solvent, and the chloridions were restricted in the voids by cationic helicenes (Fig. 2c and g). The homochiral blocks were linked by the strongly interlaced H-bonds (Fig. 2d). However, the racemic [(Rac)-2b]+[Br]− crystallized in the centrosymmetric C2/c space group with a long rhomboid shape and exhibited obvious H⋯Br interactions (2.46–2.97 Å) between neighboring ionic pairs. Therefore, this affirmed that the installation of a quaternary P-atom embedded at the helical tetrahedron can result in large steric resistance that destroys the π-stacking of phosphahelicenes (Fig. S21†). Notably, all ionic crystals emitted intense fluorescence, distinct from weakly emissive trivalent phosphahelicenes and their oxides (1 and 2 are nonemissive, Scheme 1c).16 Importantly, 0-D hybrid [(6b)+]2[MnCl4]2− crystals were captured in the reaction mixture, and the organic cation [6b]+ exhibited a distinguishing construction compared to [(P)-2b]+ (Fig. 2b, i, and l). The flat 4,4′-bibenzo[d][1,3]dioxole unit (θ = 0°, φ = 2.2°) adopted horizontal conformation due to inexistent steric hindrance of dioxole units, where flat rings favored the formation of strong H-bonding (2.55–2.60 Å) and π–π (3.65 Å) interactions in antiparallel dimers (Fig. 2i and S12b†), demonstrating that chirality was annihilated in [6b]+. The computationally reduced density gradient (RDG) maps using DFT calculations revealed direct intermolecular noncovalent interactions (NCIs) between dimers as well (Fig. S12d†).56 Interestingly, the crystals of [(6b)+]2[MnCl4]2− emitted intense green phosphorescence at 511 nm owing to the 4T1(G) → 6A1 radiative decay in [MnCl4]2−, but [6b]+[Cl]− allowed deep blue fluorescence (Fig. 2l, 5d, and S46†). This fact suggested that the excited energy transfer between [MnCl4]2− and donor [6b]+ in the rigid crystal could be beneficial for generating sensitized phosphorescence because of their appropriate energy levels.57,58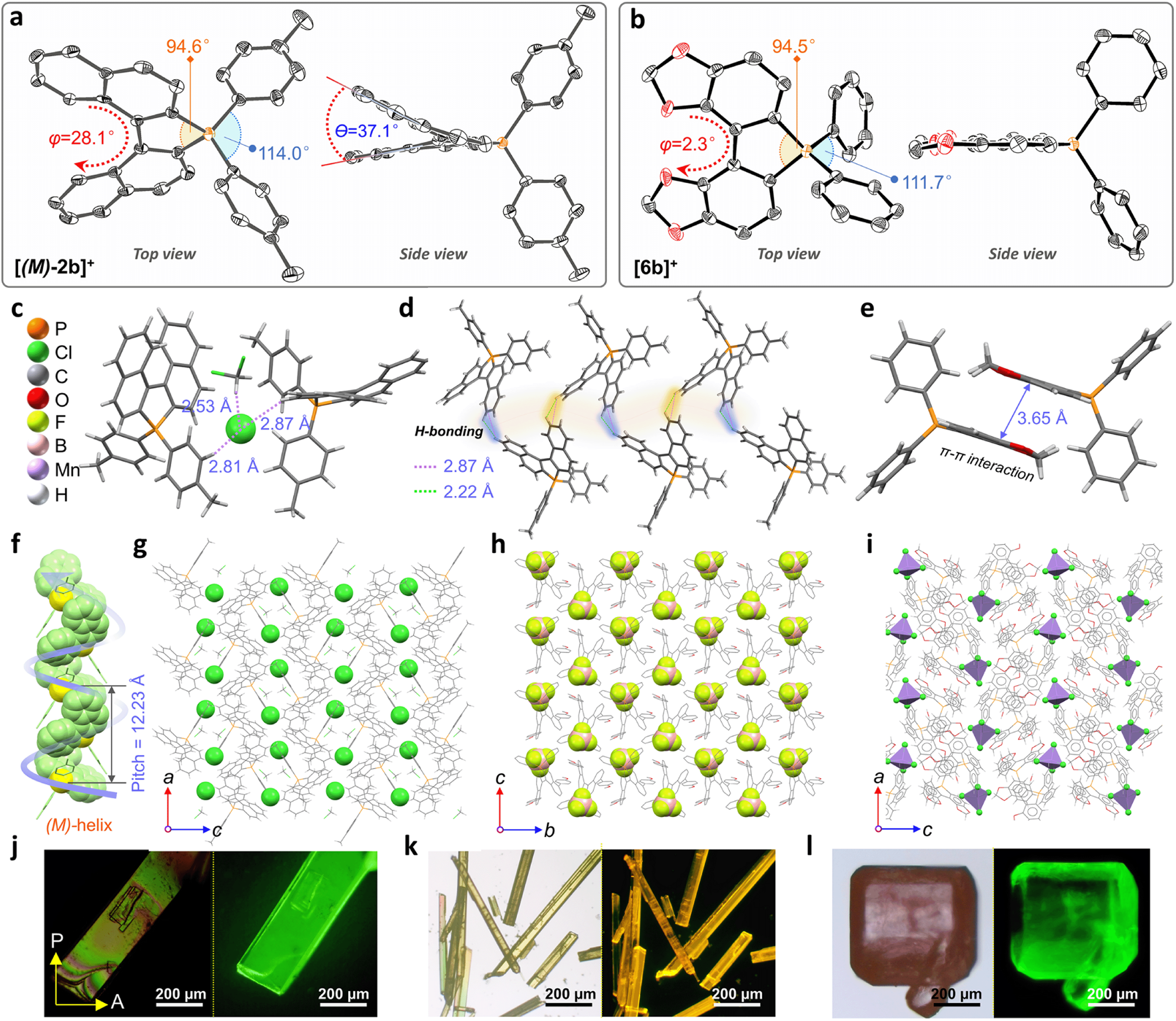 | ||
| Fig. 2 (a) ORTEP drawing of [(M)-2b]+[Cl]− and (b) complex [6b]2+[MnCl4]2− with thermal ellipsoids set at the 50% probability level. The hydrogen atoms, solvents, and anions are omitted for clarity. (c) Multiple H⋯Cl bonding interactions between two molecules and CH2Cl2 of [(M)-2b]+[Cl]−. (d) Interlaced hydrogen bonding interactions in adjacent molecules of [(M)-2b]+[Cl]−. (e) Dimeric packing of [6b]+ units in [(6b)+]2[MnCl4]2−. (f and g) Crystal packing of [(M)-2b]+[Cl]− and (h) [(M)-7c]+[BF4]− and (i) [(6b)+]2[MnCl4]2− complexes. (j) Polarized optical microscope (left) and fluorescence microscope images (right, excited at 365 nm) of [(M)-2b]+[Cl]− and (k) [(M)-7c]+[BF4]− crystals. (l) Bright-field optical microscope (left) and fluorescence microscope images (right, excited at 365 nm) of [(6b)+]2[MnCl4]2− crystals. | ||
As mentioned above, the installation of the diverse anions at the ionic crystals of phosphahelicens can bring out robust NCI in the packing of those molecules. This might also explain well why our devotion to growing single crystals of other phosphahelicenes (the small halide ion acted as the counterion) ultimately failed, since a more torsional aromatic substituent at the edge-position might significantly result in loose stacking and an unfitting cavity of molecules during the crystallization process. Therefore, the large anion exchange strategy was tried to obtain high-quality single crystals to gain insight into structural information on crystallizing-induced assembly and chiral character. As expected, a series of single crystals were obtained by anion exchange in equivalent halide scavenger (AgBF4 or AgPF6) solutions. [Cl]− anions in enantiomerically enriched [(P)-2b]+[Cl]− and [(M)-2b]+[Cl]− could be exchanged with [BF4]− or [PF6]− anions smoothly, while the crystallographic and stereochemical information was maintained (Fig. S16 and S17†), indicating that the initial chiral bias of cations could be inherited and racemization was hindered during the recrystallization.59,60 In contrast, the prepared [1b]+[BF4]− and [1b]+[PF6]− crystals bearing the PPh2 moieties exhibited expected heterochiral arrangements regardless of stereochemical conformation for precursors (Fig. S14 and S15†). This result demonstrated the racemic nature of the prepared [1b]+[Cl]− under the same conditions, where the isomerization barrier was lower than that of [2b]+[Cl]− by about 5.5 kcal mol−1 (Fig. 4g and S38†). In [(Rac)-3c]+[BF4]−, two edge aromatic rings suffered from folded orientation, huge repulsion, and distortion, thus severely enlarging their dihedral angle for the binaphthyls (θ = 43.6°) and edge phenyls (θ' = 72.9°) as well as the persistence of chiral configuration. In addition, the (M)-isomer or (P)-isomer accumulated into 1-D columnar superhelices along the b-axis with opposite handedness via tight π–π stacking (3.42–3.87 Å) and alternate rotation about 81° and 99° (Fig. 3a and S18g†), where the helical pitch was 23.96 Å. Unexpectedly, homochiral [(M)-7c]+[BF4]− and [(P)-7c]+[BF4]− conglomerates were crystalized in racemic solutions (Fig. 3b), but this chiral self-sorting tendency has not been observed in [(Rac)-3c]+[BF4]−, [(Rac)-1b]+[BF4]−, and [(Rac)-1b]+[PF6]−, manifesting that the chiral self-discrimination was dependent on the subtle variation of cations (Fig. S14, S15, S18, and S19†).60
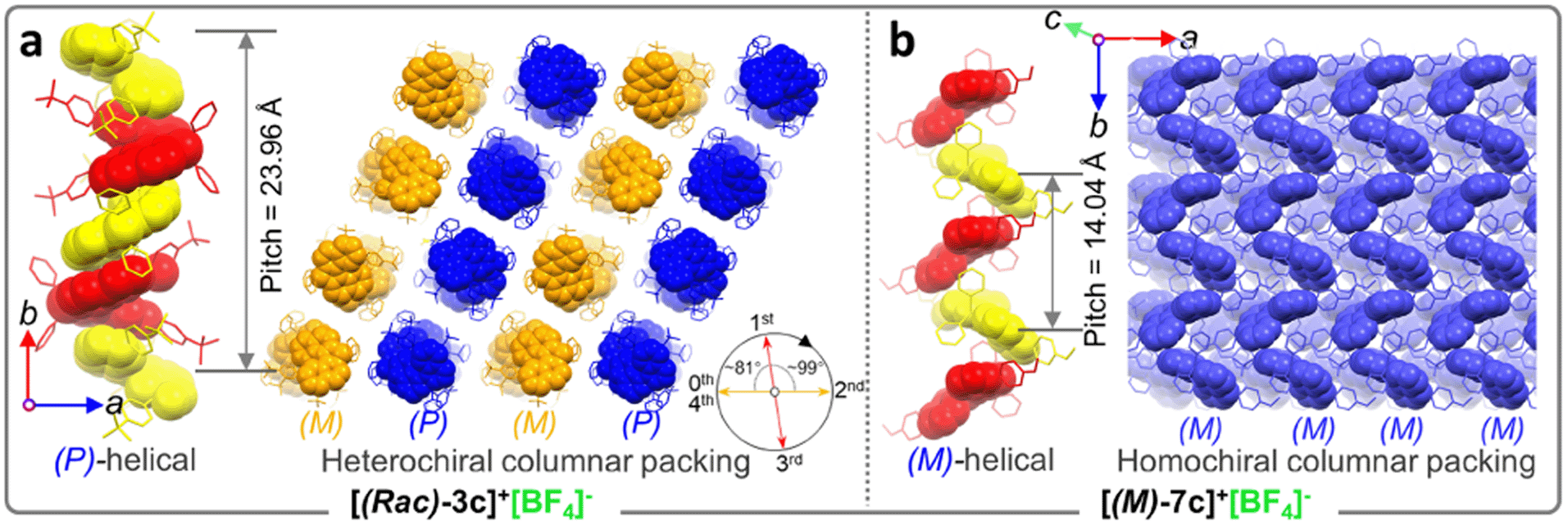 | ||
| Fig. 3 (a) Crystal structure arrangements of [(Rac)-3c]+[BF4]− and (b) [(M)-7c]+[BF4]−. The hydrogen atoms and anions are omitted for clarity. | ||
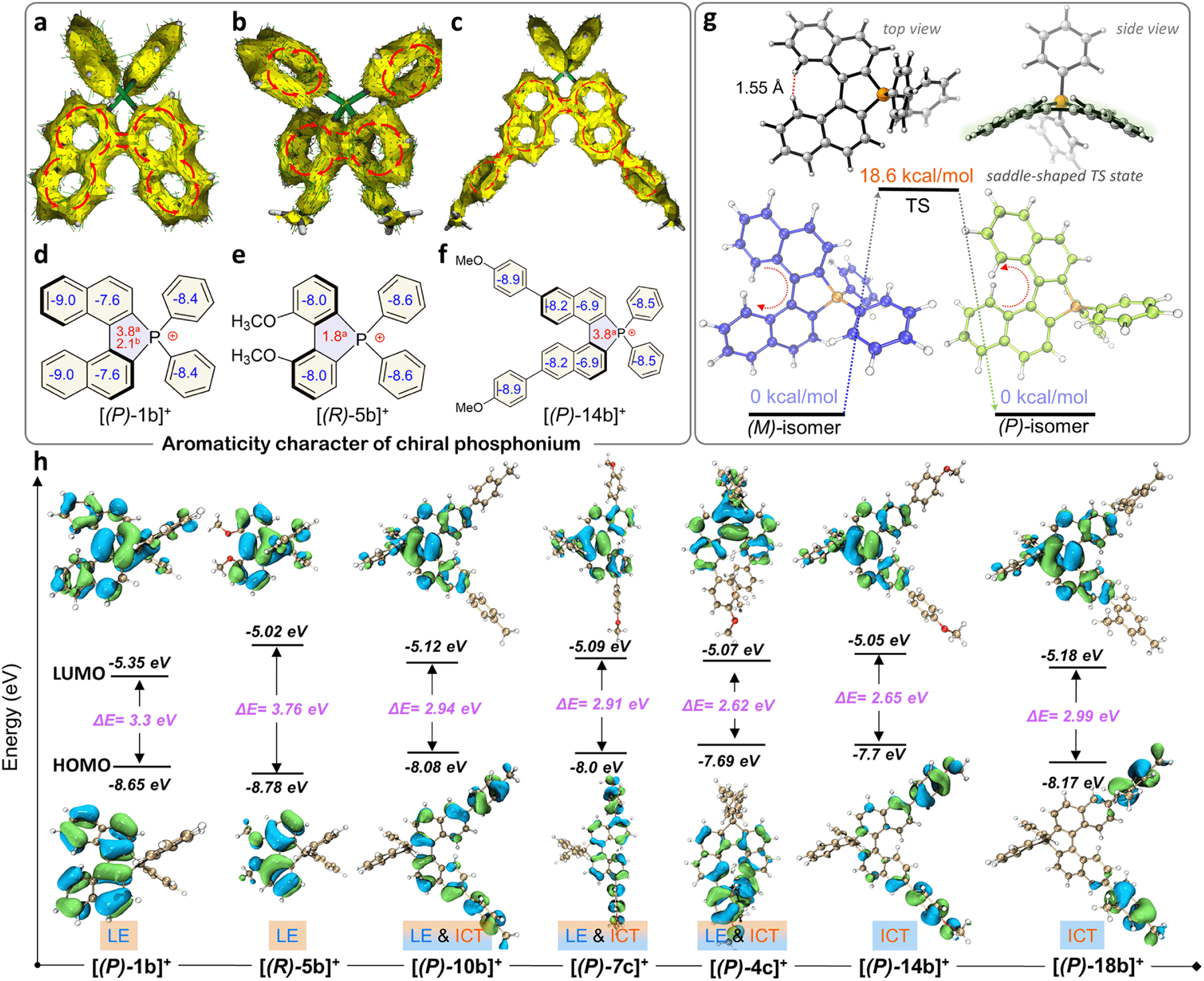 | ||
| Fig. 4 (a–c) DFT calculated AICD plots of [(P)-1b]+, [(R)-5b]+, and [(P)-14b]+ at the CSGT-B3LYP/6-31G(d) level. The red arrows indicate ring current delocalized along the aromatic rings. (d–f) DFT calculated aNICS(0) and bNICS(1) values of [(P)-1b]+, [(R)-5b]+, and [(P)-14b]+ at the GIAO-B3LYP/6-31G(d) level. (g) Calculated racemization barrier (ΔE‡) and conformation for cation [1b]+ between the (M)-isomer and (P)-isomer at the B3LYP/6-31G(d) level. (h) Frontier molecular orbital diagrams and energy gaps (ΔE) for representative phosphonium (B3LYP/6-311G(d) level). | ||
Aromaticity analysis
The quaternary C–P bonding environment and electron-deficient nature of the P-center might bring out unique electronic structures. First, the aromaticity of the skeleton was investigated by anisotropy of the induced current density (AICD) and nucleus independent chemical shifts (NICS) simulations (Fig. 4a–f).61,62 [(P)-2b]+ manifested a diatropic ring current originating from a binaphthyl fragment along the periphery of the helical backbone (Fig. 4a), but the current was broken between the binaphthyl unit and phenyl wings at the penta-heterocycle due to the tetrahedral P(III)-node, confirming the local aromaticity in [(P)-2b]+. [(R)-5b]+ and [(P)-14b]+ had similar aromaticity as well (Fig. 4b and c). NICS analysis was adopted to gain a degree of theoretical aromaticity for [(P)-1b]+ and others (Fig. 4d–f). The negative NICS values revealed typical aromaticity in binaphthyl (NICS(0): −7.6 ppm and −9.0 ppm) and phenyl rings (−8.4 ppm) but antiaromaticity in the P(III) penta-heterocycle (NICS(0): +3.8 ppm, NICS(1): +2.1 ppm), which was consistent with the broken AICD at the P(III)-node. The local aromaticity was similar to that of other azahelicenes (antiaromaticity for heteroatomic linking rings) but different from that of carbohelicenes and O/S-atom embedded ones (global aromaticity).63–65Electronic structures and photophysical properties
Electronic absorption spectra of synthetic compounds are shown in Fig. 5a, S40, and S42.† Representative systems were discussed (structures highlighted in Scheme 2) in this section and others are analyzed in the ESI.† [1b]+[Cl]− gave a strong absorption band from 225 to 310 nm and two weak shoulder peaks at 330 and 412 nm (Fig. 5a). TD-DFT calculations showed that the absorption band of [1b]+ in the longest wavelength region was attributed to the HOMO → LUMO transition (98.8%, f = 0.118, Fig. 5b, S29, and Table S9†). The electronic cloud was mainly distributed on binaphthyl for the HOMO, while the LUMO electronic cloud was primarily located on the binaphthyl and electron-deficient P(III)-pentagon, indicating locally excited (LE) characteristics in [1b]+ (Fig. 4h). [5b]+[Cl]− and [6b]+[Cl]− experienced identifiable hypsochromic shifted absorption with similar LE characteristics due to the reduced conjugated backbone, but extended phospha[5]helicenes ([9b]+[Cl]−–[21b]+[Cl]−) at 6,6′-positions displayed a similar absorption curve and enhanced absorbance (Fig. 5a and S40†). The variation tendency of the optical bandgap agreed well with the computational orbital gaps (ΔE = ELUMO − EHOMO, Fig. 4h). Interestingly, the LE transition was gradually transformed into the intramolecular charge transfer (ICT) state for [(P)-10b]+, [(P)-7c]+, [(P)-14b]+, [(P)-4c]+, and [(P)-18b]+ owing to the torsional electron donor, which could be confirmed by the separated electronic cloud in the HOMO and LUMO.36,37 The HOMO-electrons resided on the extended phenyls, and the LUMO-electrons were dominated by the P(III)-pentagon with high contributions up to 46.8–48.2% (8.3–8.5% on the P-atom, Fig. S35†), suggesting a typical electron-deficient character of the quaternary bonding scaffold, which was distinct from the typical LE nature of trivalent and pentavalent phosphahelicenes.29,31,66 Solvent-dependent UV-vis absorption and fluorescence emission reaffirmed the mutation of LE to ICT of [(P)-14b]+[Cl]− due to the torsional D–A structure and elevated transition dipole moments (Fig. S45†).67 | ||
| Fig. 5 (a) Normalized UV-vis spectra of compounds [1b]+[Cl]−, [5b]+[Cl]−, [6b]+[Cl]−, [1b]+[Cl]−, and [14b]+[Cl]− in chloroform with 2 × 10−5 M at room temperature. (b) Experimental and TD-DFT simulated UV-vis and PL spectra of [1b]+[Cl]−. (c) PL spectra of the representative compounds in chloroform with 1 × 10−4 M at room temperature (λex = 370 nm for [1b]+[Cl]−, [2b]+[Cl]−, [10b]+[Cl]−, and [14b]+[Cl]− and 360 nm for [5b]+[Cl]− and [6b]+[Cl]−). (d) Transient fluorescence decay curves for selective compounds in chloroform (1 × 10−4 M) and their photographs under UV-light. | ||
Remarkably, all compounds were emissive in chloroform and solid states. [1b]+[Cl]− exhibited structureless green emission at 498 nm with moderate PLQY (Φ = 21%) in chloroform (Fig. 5b). TD-DFT calculations suggested that the fluorescence showed the S1 → S0 transition (Fig. 5b). Moreover, the emission of [1b]+[Cl]− powder displayed a slight bathochromic shift together with a retentive full width at half maximum (FWHM = 76 nm) and an enhanced PLQY (Φ = 34%) owing to the rigidity-enhanced environment in the solid and restriction of intramolecular rotation (RIR) of the tetrahedral C–P scaffolds.68 Furthermore, the fluorescence could be adjusted from deep blue to orange for these serial emitters, including deep blue ([6b]+[Cl]−, 405 nm, Φ = 12%), sky blue ([5b]+[Cl]−, 457 nm, Φ = 77%), yellow ([2c]+[Cl]−, 540 nm, Φ = 39%), orange-yellow ([14b]+[Cl]−, 560 nm, Φ = 35%), and orange ([4c]+[Cl]−, 568 nm in the solid state, Φ = 47%) (Fig. 5c, S41, S43, and Table S12†). Their fluorescence lifetimes (τ) varied from 1.7 to 12.9 ns in chloroform (Fig. 5d). The longer decay of [14b]+[Cl]− and [4c]+[Cl]− than [1b]+[Cl]− could be interpreted as slower relaxation kinetics in the ICT state.67 Notably, all methoxy-substituted compounds at different positions exhibited significant Stokes shifts and higher quantum efficiency, which can be attributed to the enhanced ICT feature of the D–A structures, as evidenced by electronic donation and communication of the bridge methoxy group (Tables S10, S12, and Fig. S32a†).
Chiroptical properties and chiral induced LC self-assembly
Solid chiroptical properties of enantiomerically enriched [2b]+[Cl]− and [2b]+[BF4]− crystals were evaluated at room temperature. The enantiomerically enriched [2b]+[BF4]− crystals afforded mirrored CD signals from 280 to 450 nm (Fig. 6a). However, the solution CD intensity deteriorated quickly at the room temperature of kinetic experiments (Fig. S1 and S48a†). The CPL activity of the solution was observed with moderate dissymmetry factors (±0.94 × 10−3) and decreased with increased radiation time and temperature, indicating loss of enantiomeric purity (Fig. S48c and S48d†).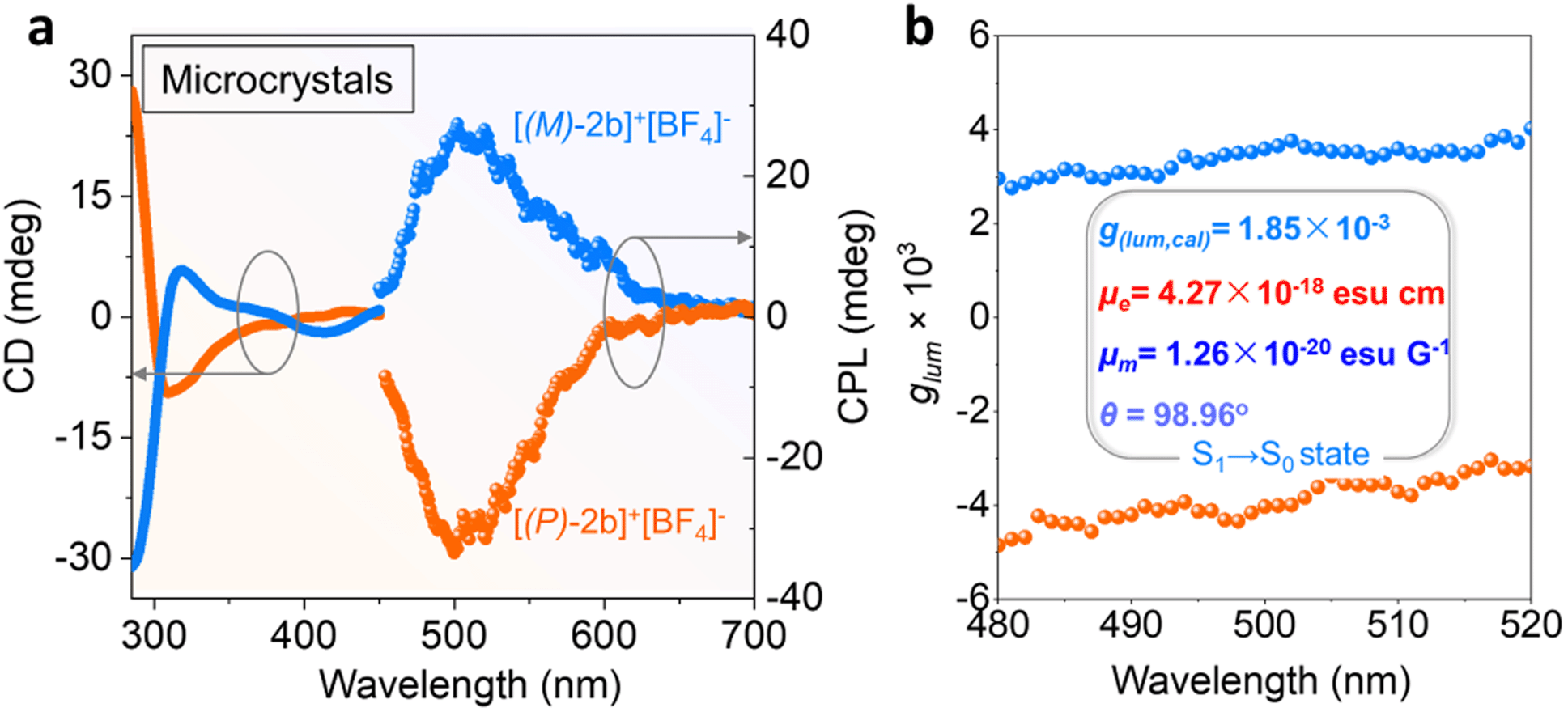 | ||
| Fig. 6 (a) CD and CPL spectra of enantiomerically enriched [2b]+[BF4]− microcrystals at room temperature (λex = 360 nm). (b) Corresponding experimental glum factors of the CPL spectra and TD-DFT simulative electron (μe), magnetic transition dipole moments (μm), and their angle (θe–m), g(lum,cal). | ||
CPL spectra were also collected for crystalline [(M)/(P)-2b]+[BF4]− powder (Fig. 6 and S44†). Angle-dependent CD/CPL (enantiomerically enriched crystals) and extra CPL (racemic crystals) measurements were performed. CD/CPL signs were unchanged when altering the rotation angles, confirming that the contributions of birefringence and linear dichroism to the CD signals were negligible and the resultant chiral spectra were reliable (Fig. S49†). Enantiomerically enriched [2b]+[BF4]− microcrystals exhibited mirrored CPL emission at 496 nm with moderate luminescence dissymmetry factors (glum) of 3.3 × 10−3/−3.5 × 10−3, which originated from S1 → S0 emission. The experimentally negative CPL signal of [(P)-2b]+[BF4]− was consistent with the TD-DFT simulative results (Fig. S45b, see the ESI† for detailed determination). The |glum(exp)| values and PLQY (41%) for [(P)/(M)-2b]+[BF4]− were comparable to those of reported carbo[n]helicenes and heterohelicenes,69 and the order of magnitude was also equivalent to those of pentavalent phosphahelicene oxides.29,31,66 These results reaffirmed that the synergistic construction of a quaternary C–P bonding environment and helical skeleton was an efficient way to prepare CPL active phosphahelicenes.
Controlling dynamic chirality expression is a formidable challenge and an attractive project for prochiral emitters (their chiral conformation only remained at the condensed phase), for instance, propeller-like TPE derivatives, C3-symmetric dendrimers, axial atropisomers, and macrocycles.70,71 The dynamic chiral conformation in [5]helicenes may be further controlled and induced,38,39 but this conjecture has not been illustrated at present in phosphahelicenes. Herein, an attempt has been made to address this issue through chiral induced LC self-assembly for phospha[5]helicenes. First, we designed and synthesized a pair of axial chiral guests-A ((R)/(S)-BINOL-CN) and guests-B (phospha[5]helicenes) and screened for compatibility and helical twist power (HTP) between the LC host (5CB) and guests (Fig. 7a and b). A ternary cholesteric LC system has been prepared by annealing of the polar 5CB host, racemic phosphorus[5]helicenes (2.0 wt%, [6b]+[Cl]− or [2c]+[Cl]− or [4c]+[Cl]−), and chiral BINOL-CN (0.5–4.0 wt%) mixtures (details are listed in the ESI†). Pure 5CB exhibited a Schlieren texture in the traditional nematic phase at room temperature under a polarized optical microscope (POM), while a uniform fingerprint texture was observed in ternary cholesteric LCs with periodic helicity (Fig. 7c and d). As the concentration of (S)-BINOL-CN increased, the average helical pitch of the fingerprint gradually decreased (Fig. S60 and S61†). Finally, the fingerprint texture transformed into an oily streak texture (4.0 wt%), where the XRD diffraction peak was absent in the small-angle region (Fig. S61e†), confirming that the orientational helical superstructures were induced.72,73
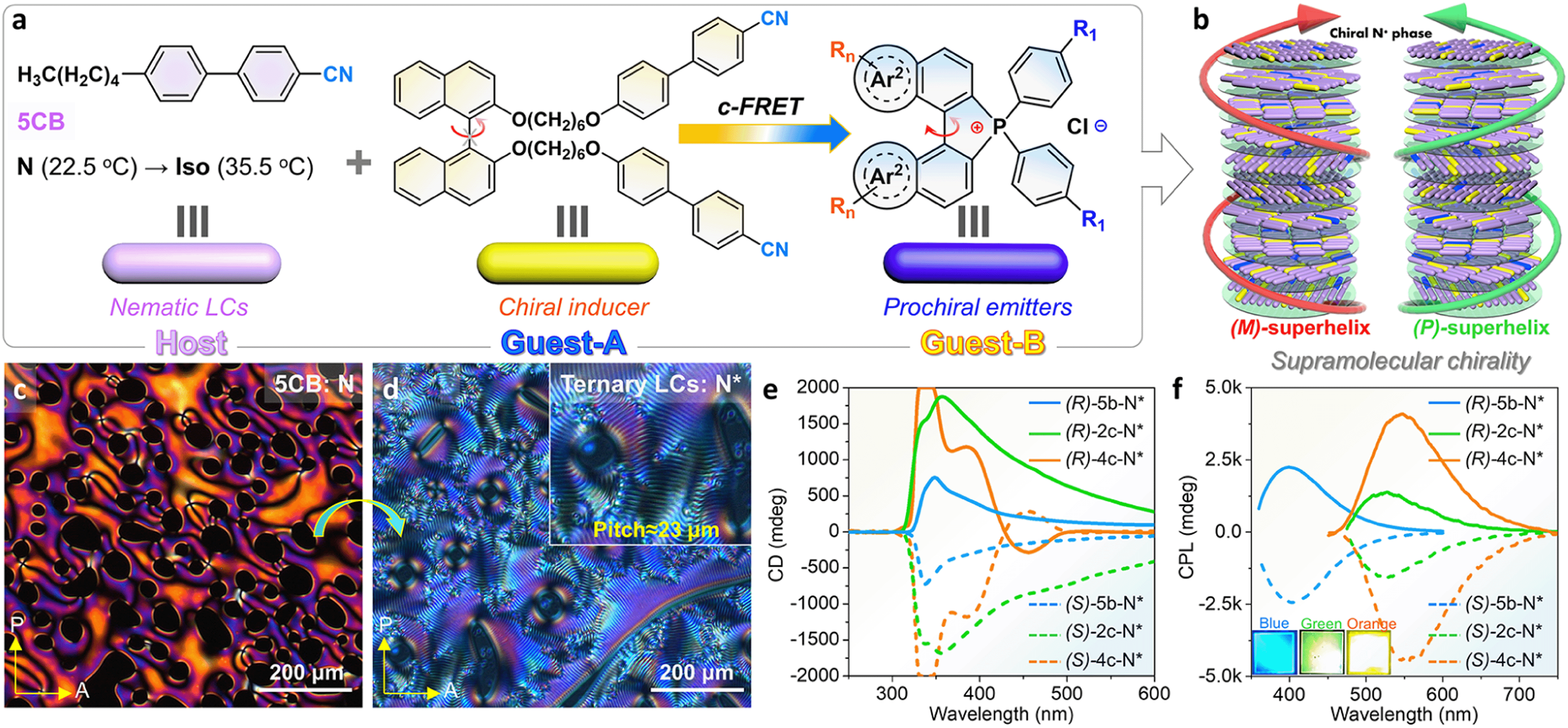 | ||
| Fig. 7 (a) Schematic illustration of the ternary chiral LC system. (b) Superchiral assembly model of the N* mesophase. (c) POM texture of pure 5-CB at 25 °C. (d) POM texture of (S)-5b-N* (1.5 wt% for (S)-guest-B and 2.0 wt% for [5c]+[Cl]−). (e) CD spectra of enantiomeric ternary chiral LC films (guest-A/4.0 wt% and (S)/(R)-guest-B/2.0 wt%) at 25 °C. (f) CPL spectra of enantiomeric ternary chiral LC films (guest-A/4.0 wt% and (S)/(R)-guest-B/2.0 wt%) at 25 °C (λex = 350 nm). | ||
CD spectra of the cholesteric LC film doped with fixed contents of emitters (2.0 wt%) and (S)/(R)-BINOL-CN (4.0 wt%) are shown in Fig. 7e. All prepared (S)-N* and (R)-N* films produced strong negative and positive CD signals at 300–500 nm with large gabs (−0.24 to −0.37, +0.21 to +0.36), respectively. The corresponding CPL spectra of enantiomeric LC films (4c-N*) revealed high glum at 549 nm, up to (+0.51, −0.48), while the enantiomeric 2c-N* and 6b-N* LC films exhibited slightly lower glum at 526 nm (+0.10, −0.14) and 400 nm (+0.22, −0.23), respectively (Fig. 7f). These values were comparable to those of the reported supramolecular helicene polymers and other LC systems.72–75 Furthermore, the CPL emission of phosphahelicenes was blue-shifted about 14–19 nm in LC films compared to their solid fluorescence. The blue-shifted emission was also confirmed in doped PMMA matrices (Fig. S47†), which assuredly revealed a uniform dilution and suppressed π–π stacking of emitters in LCs. Nevertheless, 6b-N* exhibited an integrated deep blue emission due to collective CPL contribution from the host and guests (Fig. S55a and S57a†). Interestingly, the CPL contribution of BINOL-CN has not been observed in 2c-N* and 4c-N* LCs. Although the emission of helicenes can be directly excited at 350 nm, the fluorescence output might be accompanied by partial FRET in this unique system. Both the 5CB host and BINOL-CN donor produced deep blue fluorescence at 382 nm which was predominated by the ICT transition between BINOL and cyanobiphenyls (Fig. S55†), which overlapped with the ICT absorption of the phosphahelicene acceptor (Fig. S56 and S57a†). Hence, we further explored the fluorescence of various molar proportions of [4c]+[Cl]− in host LCs. As molar ratios of helicenes gradually increased, the fluorescence intensity and decay of 5-CB@BINOL-CN appeared to decrease (Fig. S58†). These results provided further evidence for energy transfer from the donor to the acceptor.66,72 In contrast, the guest [6b]+[Cl]− absorption onset peak (370 nm) was out of tune with the host emission region (382 nm), leading to an interrupted energy transfer between the donor and acceptor (Fig. 5a).
To reveal the mechanism of increased CPL emission in N*-LCs, excitation wavelength-dependent CPL spectra of N*-4c were also measured in quartzose cells (Fig. S59†). The CPL spectra of 4c-N* displayed larger glum values in the excitation region of FRET (λem = 350 nm) than direct excitation at 400 nm, demonstrating that chiral FRET could successfully promote the enlargement effect of the induced CPL signal in an intrinsically helical assembly environment.62,76 CPL emissions of emitters were interrelated to chiral BINOL-CN, indicating the formation of chirality transmission and coupling through the π−π, dipole–dipole interactions of the polar aromatic units in the helical host–guest system.76 In addition, no Bragg reflection and structural color were observed in the visible light region due to the sizeable helical pitches, and chirality transmission was interrupted in the phase-separated devices (the N*-LC layer and heliceneslayer were isolated, Fig. S59†), which firmly ruled out the physical reflection of CPL and verified the CPL transmission process.72,77 This suggested that increased CPL of phosphahelicenes benefited from a spiral arrangement and chiral energy transmission.
Conclusions
In summary, we have demonstrated the first case of Mn(III)-mediated C–P bond activation and intramolecular annulation of diphosphines by a radical phosphonium process, which gives access to highly emissive phospha[5]helicene cations and phosphoniums in satisfactory yields and with substrate universality and economic applicability. The electron-deficient structure and stereochemical information were revealed through experiments, calculations, and the anion exchange strategy. Further utilization of this skeleton was illustrated for constructing polychromatic fluorophores, CPL, and a supramolecular chiral LC mesophase, which displayed good tunability, high emission efficiency, and excellent CPL performance with ultrahigh glum values. Our findings provide an innovative synthesis protocol and insight for designing cationic phosphahelicenes via C–P bond activation toward desired bright emission, chiral amplification, and potential catalyst precursors.Data availability
Detailed experimental details and characterizations are listed in ESI.† The data that support the findings of this study are available from the corresponding author on reasonable request.Author contributions
W. H. directed the project and revised the manuscript; B. Y. carried out synthesis and property studies, wrote, and revised the work. S. Y. performed the SC-XRD collections. The other authors provided assistance for optical measurements in the work. All authors have given approval to the final version of the manuscript.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
We gratefully thank the National Natural Science Foundation of China (No. 21871133) and the Natural Science Foundation of Jiangsu Province (No. BK20211146) and the Science, Technology, and Innovation Commission of Shenzhen Municipality (No. JCYJ20180307153251975) for financial support.Notes and references
- A. Miyashita, A. Yasuda, H. Takaya, K. Toriumi, T. Ito, T. Souchi and R. Noyori, J. Am. Chem. Soc., 1980, 102, 7932–7934 CrossRef CAS.
- M. Berthod, G. Mignani, G. Woodward and M. Lemaire, Chem. Rev., 2005, 105, 1801–1836 CrossRef CAS PubMed.
- J.-P. Genet, T. Ayad and V. Ratovelomanana-Vidal, Chem. Rev., 2014, 114, 2824–2880 CrossRef CAS PubMed.
- W. Tang and X. Zhang, Chem. Rev., 2003, 103, 3029–3070 CrossRef CAS PubMed.
- Y. Wang, M. Liu, R. Cao, W. Zhang, M. Yin, X. Xiao, Q. Liu and N. Huang, J. Med. Chem., 2013, 56, 1455–1466 CrossRef CAS PubMed.
- W. Wang, Y. Ling, Y. Zhong, Z. Li, C. Tan and Z. Mao, Angew. Chem., 2022, 134, e202115247 CrossRef.
- L.-Y. Yao, T. K.-M. Lee and V. W.-W. Yam, J. Am. Chem. Soc., 2016, 138, 7260–7263 CrossRef CAS PubMed.
- Y. J. Kong, Z. P. Yan, S. Li, H. F. Su, K. Li, Y. X. Zheng and S. Q. Zang, Angew. Chem., Int. Ed., 2020, 59, 5336–5340 CrossRef CAS PubMed.
- S. A. Macgregor, Chem. Soc. Rev., 2007, 36, 67–76 RSC.
- L. Wang, H. Chen and Z. Duan, Chem.–Asian J., 2018, 13, 2164–2173 CrossRef CAS PubMed.
- Y. H. Lee and B. Morandi, Coord. Chem. Rev., 2019, 386, 96–118 CrossRef CAS.
- T. Jiang, H. Zhang, Y. Ding, S. Zou, R. Chang and H. Huang, Chem. Soc. Rev., 2020, 49, 1487–1516 RSC.
- K. M. Crawford, T. R. Ramseyer, C. J. A. Daley and T. B. Clark, Angew. Chem., 2014, 126, 7719–7723 CrossRef.
- X. Qiu, M. Wang, Y. Zhao and Z. Shi, Angew. Chem., Int. Ed., 2017, 56, 7233–7237 CrossRef CAS PubMed.
- M. Lei, X. Chen, Y. Wang, L. Zhang, H. Zhu and Z. Wang, Org. Lett., 2022, 24, 2868–2872 CrossRef CAS PubMed.
- Z. Lian, B. N. Bhawal, P. Yu and B. Morandi, Science, 2017, 356, 1059–1063 CrossRef CAS PubMed.
- H. Kawai, W. J. Wolf, A. G. Dipasquale, M. S. Winston and F. D. Toste, J. Am. Chem. Soc., 2016, 138, 587–593 CrossRef CAS PubMed.
- K. Baba, M. Tobisu and N. Chatani, Org. Lett., 2015, 17, 70–73 CrossRef CAS PubMed.
- K. Baba, M. Tobisu and N. Chatani, Angew. Chem., Int. Ed., 2013, 52, 11892–11895 CrossRef CAS PubMed.
- S. Nieto, P. Metola, V. M. Lynch and E. V. Anslyn, Organometallics, 2008, 27, 3608–3610 CrossRef CAS.
- X. Zhang, Y. Jiang, Q. Ma, S. Hu and S. Liao, J. Am. Chem. Soc., 2021, 143, 6357–6362 CrossRef CAS PubMed.
- A. Belyaev, Y.-T. Chen, Z.-Y. Liu, P. Hindenberg, C.-H. Wu, P.-T. Chou, C. Romero-Nieto and I. O. Koshevoy, Chem.–Asian J., 2019, 25, 6332–6341 CAS.
- L. Pang, Q. Sun, Z. Huang, G. Li, J. Liu, J. Guo, C. Yao, J. Yu and Q. Li, Angew. Chem., Int. Ed., 2022, 61, e202211710 CrossRef CAS PubMed.
- M. Gicquel, Y. Zhang, P. Aillard, P. Retailleau, A. Voituriez and A. Marinetti, Angew. Chem., Int. Ed., 2015, 54, 5470–5473 CrossRef CAS PubMed.
- Y. Shen and C.-F. Chen, Chem. Rev., 2012, 112, 1463–1535 CrossRef CAS PubMed.
- K. Yavari, P. Aillard, Y. Zhang, F. Nuter, P. Retailleau, A. Voituriez and A. Marinetti, Angew. Chem., Int. Ed., 2014, 53, 861–865 CrossRef CAS PubMed.
- N. Saleh, C. Shen and J. Crassous, Chem. Sci., 2014, 5, 3680–3694 RSC.
- K. Dhbaibi, L. Favereau and J. Crassous, Chem. Rev., 2019, 119, 8846–8953 CrossRef CAS PubMed.
- S. Nishigaki, K. Murayama, Y. Shibata and K. Tanaka, Mater. Chem. Front., 2018, 2, 585–590 RSC.
- R. Mokrai, A. Mocanu, M. P. Duffy, T. Vives, E. Caytan, V. Dorcet, T. Roisnel, L. Nyulászi, Z. Benkő, P.-A. Bouit and M. Hissler, Chem. Commun., 2021, 57, 7256–7259 RSC.
- K. Yavari, W. Delaunay, N. De Rycke, T. Reynaldo, P. Aillard, M. Srebro-Hooper, V. Y. Chang, G. Muller, D. Tondelier, B. Geffroy, A. Voituriez, A. Marinetti, M. Hissler and J. Crassous, Chem.–Asian J., 2019, 25, 5303–5310 CAS.
- Y. Sawada, S. Furumi, A. Takai, M. Takeuchi, K. Noguchi and K. Tanaka, J. Am. Chem. Soc., 2012, 134, 4080–4083 CrossRef CAS PubMed.
- K. Nakano, H. Oyama, Y. Nishimura, S. Nakasako and K. Nozaki, Angew. Chem., 2012, 124, 719–723 CrossRef.
- M. Widhalm, C. Aichinger and K. Mereiter, Tetrahedron Lett., 2009, 50, 2425–2429 CrossRef CAS.
- E. Rémond, J. Fehrentz, L. Liénart, S. Clement, J. Baneres and F. Cavelier, Chem.–Eur. J., 2022, 28, e202201526 CrossRef PubMed.
- L. Frederic, A. Desmarchelier, L. Favereau and G. Pieters, Adv. Funct. Mater., 2021, 31, 2010281 CrossRef CAS.
- Y. Im, M. Kim, Y. J. Cho, J.-A. Seo, K. S. Yook and J. Y. Lee, Chem. Mater., 2017, 29, 1946–1963 CrossRef CAS.
- K. Kawara, G. Tsuji, Y. Taniguchi and S. Sasaki, Chem.–Asian J., 2017, 23, 1763–1769 CAS.
- X. Xiao, Q. Cheng, S. T. Bao, Z. Jin, S. Sun, H. Jiang, M. L. Steigerwald and C. Nuckolls, J. Am. Chem. Soc., 2022, 144, 20214–20220 CrossRef CAS PubMed.
- M. Grzybowski, K. Skonieczny, H. Butenschön and D. T. Gryko, Angew. Chem., Int. Ed., 2013, 52, 9900–9930 CrossRef CAS PubMed.
- D. J. Ager, M. B. East, A. Eisenstadt and S. A. Laneman, Chem. Commun., 1997, 2359–2360 RSC.
- S. Gladiali, A. Dore, D. Fabbri, O. De Lucchi and G. Valle, J. Org. Chem., 1994, 59, 6363–6371 CrossRef CAS.
- A. kumar Swain, K. Kolanji, C. Stapper and P. Ravat, Org. Lett., 2021, 23, 1339–1343 CrossRef PubMed.
- P. Ravat, R. Hinkelmann, D. Steinebrunner, A. Prescimone, I. Bodoky and M. Juríček, Org. Lett., 2017, 19, 3707–3710 CrossRef CAS PubMed.
- R. H. Janke, G. Haufe, E. U. Würthwein and J. H. Borkent, J. Am. Chem. Soc., 1996, 118, 6031–6035 CrossRef CAS.
- M. Oki, Top. Stereochem., 1983, 14, 1–81 CAS.
- M. Toya, T. Omine, F. Ishiwari, A. Saeki, H. Ito and K. Itami, J. Am. Chem. Soc., 2023, 145, 11553–11565 CrossRef CAS PubMed.
- Y. Okamoto, N. Iwamoto, S. Toki and S. Takamuku, Bull. Chem. Soc. Jpn., 1987, 60, 277–282 CrossRef CAS.
- K. Wei, K. Luo, F. Liu, L. Wu and L.-Z. Wu, Org. Lett., 2019, 21, 1994–1998 CrossRef CAS PubMed.
- Chaolumen, M. Murata, A. Wakamiya and Y. Murata, Angew. Chem., Int. Ed., 2017, 56, 5082–5086 CrossRef CAS PubMed.
- M. Zhao, S. H. Pun, Q. Gong and Q. Miao, Angew. Chem., Int. Ed., 2021, 60, 24124–24130 CrossRef CAS PubMed.
- J. Liu, A. Narita, S. Osella, W. Zhang, D. Schollmeyer, D. Beljonne, X. Feng and K. Müllen, J. Am. Chem. Soc., 2016, 138, 2602–2608 CrossRef CAS PubMed.
- Y. Zou, Y. Han, S. Wu, X. Hou, C. H. E. Chow and J. Wu, Angew. Chem., Int. Ed., 2021, 60, 17654–17663 CrossRef CAS PubMed.
- N. Sharma, H.-B. Zou, Y.-M. Lee, S. Fukuzumi and W. Nam, J. Am. Chem. Soc., 2021, 143, 1521–1528 CrossRef CAS PubMed.
- L. Liu, L. Li, X. Wang, R. Sun, M. D. Zhou and H. Wang, Org. Lett., 2021, 23, 5826–5830 CrossRef CAS PubMed.
- E. R. Johnson, S. Keinan, P. Mori-Sánchez, J. Contreras-García, A. J. Cohen and W. Yang, J. Am. Chem. Soc., 2010, 132, 6498–6506 CrossRef CAS PubMed.
- L.-J. Xu, C.-Z. Sun, H. Xiao, Y. Wu and Z.-N. Chen, Adv. Mater., 2017, 29, 1605739 CrossRef PubMed.
- P. Tao, S. J. Liu and W. Y. Wong, Adv. Opt. Mater., 2020, 8, 1–21 Search PubMed.
- T. Buhse, J.-M. Cruz, M. E. Noble-Terán, D. Hochberg, J. M. Ribó, J. Crusats and J.-C. Micheau, Chem. Rev., 2021, 121, 2147–2229 CrossRef CAS PubMed.
- F. Wang, F. Gan, C. Shen and H. Qiu, J. Am. Chem. Soc., 2020, 142, 16167–16172 CrossRef CAS PubMed.
- A. I. Boldyrev and L.-S. Wang, Chem. Rev., 2005, 105, 3716–3757 CrossRef CAS PubMed.
- H. Fallah-Bagher-Shaidaei, C. S. Wannere, C. Corminboeuf, R. Puchta and P. v. R. Schleyer, Org. Lett., 2006, 8, 863–866 CrossRef CAS PubMed.
- E. Cherni, B. Champagne, S. Ayadi and V. Liégeois, Phys. Chem. Chem. Phys., 2019, 21, 14678–14691 RSC.
- G. Naulet, L. Sturm, A. Robert, P. Dechambenoit, F. Röhricht, R. Herges, H. Bock and F. Durola, Chem. Sci., 2018, 9, 8930–8936 RSC.
- T. Luo, Y. Wang, J. Hao, P. A. Chen, Y. Hu, B. Chen, J. Zhang, K. Yang and Z. Zeng, Angew. Chem., Int. Ed., 2023, 62, e202214653 CrossRef CAS PubMed.
- K. Usui, N. Narita, R. Eto, S. Suzuki, A. Yokoo, K. Yamamoto, K. Igawa, N. Iizuka, Y. Mimura, T. Umeno, S. Matsumoto, M. Hasegawa, K. Tomooka, Y. Imai and S. Karasawa, Chem.–Asian J., 2022, 28, e202202922 CAS.
- X. Cao, D. Zhang, S. Zhang, Y. Tao and W. Huang, J. Mater. Chem. C, 2017, 5, 7699–7714 RSC.
- J. Luo, Z. Xie, J. W. Y. Lam, L. Cheng, H. Chen, C. Qiu, H. S. Kwok, X. Zhan, Y. Liu, D. Zhu and B. Z. Tang, Chem. Commun., 2001, 1740–1741 RSC.
- T. Mori, Chem. Rev., 2021, 121, 2373–2412 CrossRef CAS PubMed.
- P. Xing and Y. Zhao, Acc. Chem. Res., 2018, 51, 2324–2334 CrossRef CAS PubMed.
- M. Liu, L. Zhang and T. Wang, Chem. Rev., 2015, 115, 7304–7397 CrossRef CAS PubMed.
- Y. Wu, C. Yan, X.-S. Li, L. H. You, Z.-Q. Yu, X. Wu, Z. Zheng, G. Liu, Z. Guo, H. Tian and W.-H. Zhu, Angew. Chem., Int. Ed., 2021, 60, 24549–24557 CrossRef CAS PubMed.
- B. A. San Jose, S. Matsushita and K. Akagi, J. Am. Chem. Soc., 2012, 134, 19795–19807 CrossRef CAS PubMed.
- X. Yang, X. Jin, T. Zhao and P. Duan, Mater. Chem. Front., 2021, 5, 4821–4832 RSC.
- J. Wade, J. R. Brandt, D. Reger, F. Zinna, K. Y. Amsharov, N. Jux, D. L. Andrews and M. J. Fuchter, Angew. Chem., 2021, 133, 224–229 CrossRef.
- K. Yao, Y. Shen, Y. Li, X. Li, Y. Quan and Y. Cheng, J. Phys. Chem. Lett., 2021, 12, 598–603 CrossRef CAS PubMed.
- J. Yan, F. Ota, B. A. San Jose and K. Akagi, Adv. Funct. Mater., 2017, 27, 1604529 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2130476, 2130479, 2130454, 2130459, 2225461, 2225466–2225474, 2225477–2225478, and 2225482. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3sc03201a |
| This journal is © The Royal Society of Chemistry 2023 |