
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Controlled alkali etching of MOFs with secondary building units for low-concentration CO2 capture†
Hong
Dong
 ,
Lihua
Li
and
Can
Li
*
,
Lihua
Li
and
Can
Li
*
State Key Laboratory of Catalysis, Dalian National Laboratory for Clean Energy, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, Dalian 116023, China. E-mail: canli@dicp.ac.cn
First published on 15th July 2023
Abstract
Low-concentration CO2 capture is particularly challenging because it requires highly selective adsorbents that can effectively capture CO2 from gas mixtures containing other components such as nitrogen and water vapor. In this study, we have successfully developed a series of controlled alkali-etched MOF-808-X (where X ranges from 0.04 to 0.10), the FT-IR and XPS characterizations revealed the presence of hydroxyl groups (–OH) on the zirconium clusters. Low-concentration CO2 capture experiments demonstrated improved CO2 capture performance of the MOF-808-X series compared to the pristine MOF-808 under dry conditions (400 ppm CO2). Among them, MOF-808-0.07 with abundant Zr–OH sites showed the highest CO2 capture capacity of 0.21 mmol g−1 under dry conditions, which is 70 times higher than that of pristine MOF-808. Additionally, MOF-808-0.07 exhibited fast adsorption kinetics, stable CO2 capture under humid air conditions (with a relative humidity of 30%), and stable regeneration even after 50 cycles of adsorption and desorption. In situ DRIFTS and 13C CP-MAS ssNMR characterizations revealed that the enhanced low-concentration CO2 capture is attributed to the formation of a stable six-membered ring structure through the interaction of intramolecular hydrogen bonds between neighboring Zr–OH sites via a chemisorption mechanism.
Introduction
Direct air capture (DAC) of CO2 has emerged as a promising carbon negative approach to achieving carbon neutrality.1–5 However, the extremely low concentration of CO2 in the air (∼410 ppm) presents a significant challenge. Various solid adsorption materials such as zeolite, activated carbon, porous silicon, coordination polymers, metal–organic frameworks (MOFs), and covalent organic frameworks (COFs) have been explored for CO2 capture.6–20 MOFs, with their diverse structures and post-modification functionalization,21 show potential for CO2 adsorption, but are still challenging for low-concentration CO2 capture, especially for DAC. Currently, only a few ultra microporous MOFs and bioinspired MOFs are capable of capturing ultra-low concentrations of CO2.22–31 One of the most widely studied strategies for achieving low-concentration CO2 adsorption is amine modification, which has ultra-strong affinity for CO2 molecules.32–37 However, amine adsorption suffers from low adsorption kinetics, low amine efficiency and loss of amines, limiting its practical application.38,39 Therefore, the development of non-amine modified low-concentration CO2 adsorption MOFs-based materials is necessary. Zr-based MOFs with secondary building units (SBUs) show promise due to their SBUs and high coordination numbers.40 Controlled etching of these MOFs exposes more M–OH sites, and the microporous environment of MOFs enhances local CO2 enrichment and capture, but exploration in this area is still limited.In this study, we synthesized a series of controlled alkali-etched MOF-808-X (X: 0.04–0.10) materials with enhanced low-concentration CO2 capture capacity under simulated air conditions compared to the pristine MOF-808. Among these materials, MOF-808-0.07 exhibited a CO2 capture capacity of 0.21 mmol g−1 under simulated air conditions, which is 70 times higher than that of the pristine MOF-808. Additionally, MOF-808-0.07 displayed excellent stability over 50 cycles of adsorption and desorption. In situ DRIFTS and 13C CP-MAS ssNMR analysis revealed that the increased low-concentration CO2 capture capacity is attributed to the formation of a stable six-membered ring structure through the interaction of intramolecular hydrogen bonds between neighbouring Zr–OH sites in the micro-mesoporous environment of MOF-808-X.
Results and discussion
MOF-808 was synthesized according to the reported method.40,41 And MOF-808-X (X: 0.04–0.10) series were prepared by various degrees of alkali etching of MOF-808 (Scheme 1). In Fig. 1a, the FT-IR analysis of these samples reveals that the infrared absorption peaks at 1630 and 1400–1600 cm−1, which correspond to the stretching vibration peak of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and the benzene ring, respectively, display varying degrees of weakening. This suggests that the benzene ring in MOF-808 has undergone degradation to different extents. In Fig. 1b, the powder X-ray diffraction (PXRD) patterns of the as-synthesized MOF-808 are shown, which match well with the simulated PXRD pattern obtained from single crystal analysis.40 However, the PXRD peaks of the MOF-808-X (X: 0.04–0.10) series gradually weaken with increasing etching degree, until all XRD diffraction peaks disappear. Scanning electron microscopy (SEM) images of the as-synthesized MOF-808 exhibit octahedral morphology (Fig. S1a†), consistent with previous literature reports. The alkali-etched MOF-808-X (X: 0.04–0.10) series show almost the same morphology as MOF-808 with varying degrees of etching (Fig. S1b–f†).
O and the benzene ring, respectively, display varying degrees of weakening. This suggests that the benzene ring in MOF-808 has undergone degradation to different extents. In Fig. 1b, the powder X-ray diffraction (PXRD) patterns of the as-synthesized MOF-808 are shown, which match well with the simulated PXRD pattern obtained from single crystal analysis.40 However, the PXRD peaks of the MOF-808-X (X: 0.04–0.10) series gradually weaken with increasing etching degree, until all XRD diffraction peaks disappear. Scanning electron microscopy (SEM) images of the as-synthesized MOF-808 exhibit octahedral morphology (Fig. S1a†), consistent with previous literature reports. The alkali-etched MOF-808-X (X: 0.04–0.10) series show almost the same morphology as MOF-808 with varying degrees of etching (Fig. S1b–f†).
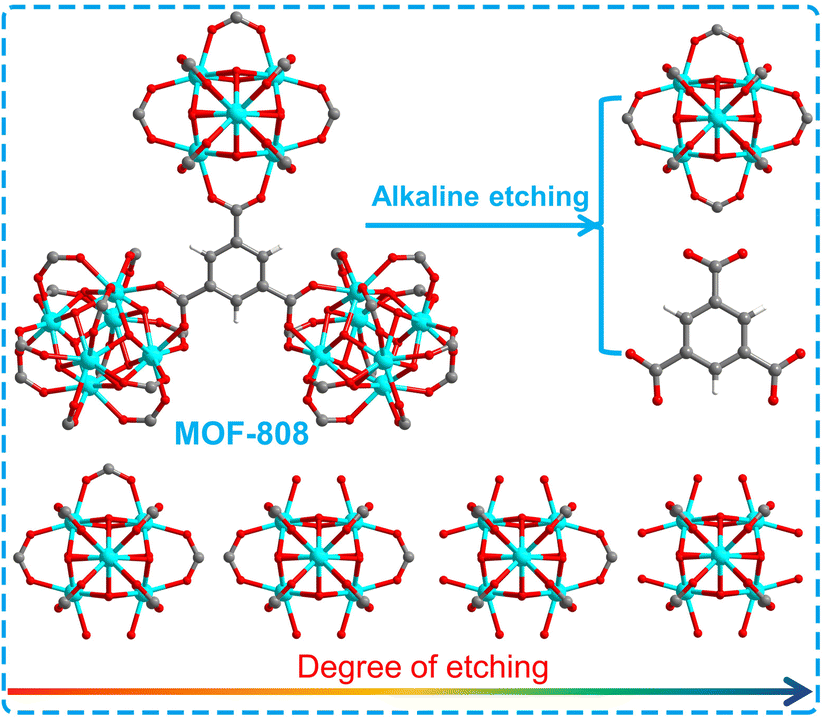 | ||
| Scheme 1 Schematic diagram of degree of etching for MOF-808 and series of MOF-808-X (X: 0.04–0.10). | ||
 | ||
| Fig. 1 (a) FT-IR spectra, (b) PXRD patterns and (c–f) XPS survey, and high-resolution XPS spectra of the C 1s, O 1s, and Zr 3d of MOF-808 and MOF-808-X (X: 0.04–0.1). | ||
X-ray photoelectron spectroscopy (XPS) analyses were conducted to investigate the electronic structure of MOF-808 and MOF-808-X (X: 0.04–0.07) series (Fig. 1c). In Fig. 1d, the C 1s high-resolution spectrum of MOF-808 and MOF-808-X series displays two distinct binding energy peaks at 284.8 and 288.5 eV, corresponding to the binding energy peaks of C–C and C–O. The O 1s high-resolution spectrum of MOF-808 in Fig. 1e shows a binding energy peak of C–O–Zr bond at 532.5 eV. However, a new binding energy peak appeared in the O 1s HR-XPS spectrum at 530.5 eV, which gradually increased with the increase of the alkali etching degree of MOF-808, and the new binding energy peak was attributed to the Zr–OH generated by alkali etching. Moreover, it is obvious from Fig. 1f that the binding energy peak of Zr 3d is shifted towards a lower binding energy in MOF-808-X series compared to the pristine MOF-808. These results suggest that electron-donating groups exist on the Zr site.
N2 adsorption and desorption isotherms were employed to further characterize the pore structure and BET surface area of MOF-808 and MOF-808-X series. The isotherms of these materials exhibit a typical type I adsorption pattern (as shown in Fig. S2†), indicating the presence of micro-mesoporous structure. The BET specific surface area of MOF-808 was found to be 1614 m2 g−1, whereas for MOF-808-X (X: 0.04–0.07) series, the BET specific surface area gradually decreases with the increase in the degree of etching and is found to be 300, 229, 225, 221, and 144 m2 g−1, respectively. This suggests that the BET surface area changes as the degree of etching increases due to the gradual collapse of the MOF-808 framework.
Due to the presence of numerous Zr–OH sites in the MOF-808-X series, we were prompted to investigate the CO2 adsorption characteristics of these materials. As shown in Fig. 2a, the CO2 adsorption isotherms of the MOF-808-X series demonstrate improved CO2 adsorption at low pressures compared to the pristine MOF-808. Particularly, the MOF-808-X series materials with Zr–OH sites exhibit a strong affinity for CO2 at low concentrations, as evidenced by the steepness of the CO2 adsorption isotherms and the attainment of a plateau at very low pressures. Further analysis of the CO2 adsorption behaviour (Fig. 2b) within a low-pressure range of 400 ppm reveals that MOF-808-0.07 exhibits a high CO2 uptake of 0.28 mmol g−1, which is comparable to the values obtained for MOF-808-0.04 (0.01 mmol g−1), MOF-808-0.05 (0.08 mmol g−1), MOF-808-0.06 (0.16 mmol g−1), and the pristine MOF-808 (0.008 mmol g−1). This highlights the significantly enhanced CO2 uptake and the interactions between CO2 and Zr–OH sites in the MOF-808-X series materials compared to the pristine MOF-808. Additionally, the MOF-808-X series exhibits excellent thermal stability up to 200 °C (Fig. S3†).
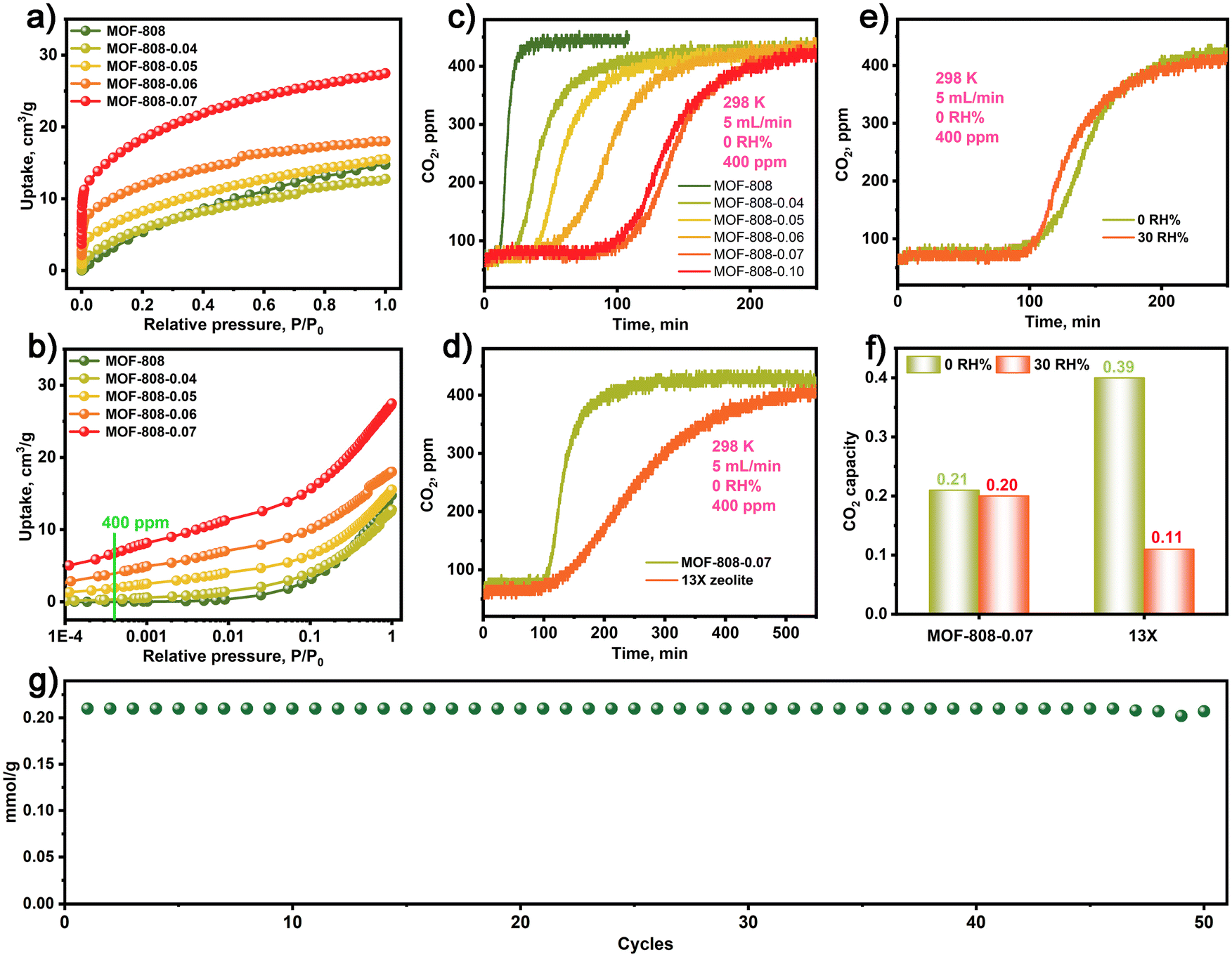 | ||
| Fig. 2 (a) and (b) CO2 adsorption isotherm for MOF-808 and MOF-808-X (X: 0.04–0.07) series. The dynamic CO2 breakthrough curves for MOF-808 and MOF-808-X (X: 0.04–0.07) series (c) and 13X zeolite under dry conditions (d). (e) The dynamic CO2 breakthrough curves for MOF-808-0.07 at 0 and 30 RH%. (f) Comparison of CO2 capture capacity for MOF-808-0.07 and 13X zeolite at 0 and 30 RH%, respectively. (g) The cycling stability of CO2 capture for MOF-808-0.07. | ||
The dynamic CO2 capture performance of MOF-808 and MOF-808-X series were assessed in a fixed-bed reactor packed with a column of simulated ambient air (400 ppm CO2 and argon as balance gas) under flow conditions (5 mL min−1) at 298 K. The detailed experimental procedure is provided in the ESI.†Fig. 2c depicts the short-term CO2 breakthrough process of pristine MOF-808 in simulated dry air conditions (0 RH%), resulting in low CO2 capture capacities of 0.003 mmol g−1. In contrast, MOF-808-X (X: 0.04–0.10) series exhibited long-term dynamic CO2 breakthrough processes with enhanced CO2 capture capacity compared to the pristine MOF-808. The dynamic CO2 capture capacity of MOF-808-X (X: 0.04–0.10) series under simulated dry air conditions were 0.06, 0.09, 0.13, 0.21, and 0.205 mmol g−1, respectively. Notably, the MOF-808-0.07 demonstrated the highest CO2 capture capacity, which is a 70-fold increase in CO2 uptake capacity compared to the pristine MOF-808. Although the MOF-808-X series exhibited lower CO2 capture capacity than the 13X zeolite (0.39 mmol g−1) under simulated dry air conditions, they demonstrated faster adsorption kinetics than 13X zeolite, as illustrated by the sharper breakthrough profile for MOF-808-0.07 compared to 13X (Fig. 2e). Additionally, Fig. 2d indicates that MOF-808-0.07 exhibited almost the same CO2 breakthrough curves under simulated dry and humid air conditions (0 and 30 RH%). In contrast, the 13X zeolite in Fig. 2f exhibited significantly reduced CO2 capture capacity under humid air conditions (30 RH%), indicating that MOF-808 has higher moisture resistance. Moreover, Fig. 2g demonstrates that MOF-808-0.07 exhibited stable CO2 capture performance with minimal losses after 50 cycles (Fig. S4†). In addition, the MOF-808-0.07 after CO2 capture was evaluated by FT-IR, PXRD, SEM, XPS and N2 adsorption and desorption isotherms, all results show the structure integrity for MOF-808-0.07 in CO2 capture processing (Fig. S5–S9†). The above results indicate that the MOF-808-0.07 has superior CO2 adsorption–desorption stability.
In order to illustrate the low-concentration CO2 adsorption process of MOFs containing Zr-SBUs at low concentrations, we synthesized a series of MOFs with different M-SBUs, including MIL-101-Fe with Fe3-SBU cluster, MIL-101-Cr with Cr3-SBU cluster, and MIL-125-Ti with Ti4-SBU cluster. Through controlled etching, as confirmed by PXRD analysis (Fig. S10†), we obtained MOFs with varying degrees of etching. The dynamic CO2 capture results revealed that all MOFs with varying degrees of etching exhibited a CO2 capture process, but their capture capacities were not comparable to that of MOF-808-X with controlled etching with Zr6-SBU cluster (Fig. S11†). This suggests that MOFs with higher coordination numbers exhibit superior CO2 capture abilities.
To investigate the desorption kinetics of MOF-808-0.07 in dry air conditions, we employed temperature programmed desorption (TPD) to evaluate its desorption energy. The activation energies of desorption for MOF-808-0.07 were calculated using the method proposed by Cvetanovic and Amenomiya, by measuring the TPD-CO2 signal at different heating rates, as presented in Fig. 3a and b.42 Our results demonstrate that MOF-808-0.07 exhibits a higher desorption energy (56.51 kJ mol−1) than 13X zeolite (48.14 kJ mol−1 (ref. 42)) under simulated dry air conditions (Table S1†), indicating that CO2 adsorption by MOF-808-0.07 occurs through chemical adsorption.
 | ||
| Fig. 3 (a) TPD results for CO2 desorption from MOF-808-0.07 in dry conditions after being saturated with CO2 from a gas stream of 400 ppm CO2. (b) Microkinetic analysis assuming first order desorption. | ||
In order to verify the adsorbed species in CO2 capture for MOF-808-0.07, the in situ diffuse reflectance infrared Fourier transform spectroscopy (in situ DRIFTS) of MOF-808-0.07 with adsorbing CO2 in simulated dry air (MOF-808-0.07-CO2) was carried out. Fig. 4a shows two distinct infrared absorption peaks at 1685 and 3000–3600 cm−1 in the in situ DRIFTS spectra of MOF-808-0.07-CO2 after heat treatment (140 °C), corresponding to the stretching vibration peak of CO (–OCO2H), and –OH (M-OH with broad peak and hydrogen-bonding), respectively. The results display that the CO2 adsorption within the MOF-808-0.07 framework is in the form of bicarbonate species and hydrogen bonding interactions under dry conditions. Furthermore, the heat-treated MOF-808-0.07 is subjected to in situ CO2 adsorption again in dry conditions, the in situ DRIFTS spectra in Fig. 4b show obvious infrared absorption peaks in 1685 and 3000–3500 cm−1 corresponding to the stretching vibration peak of CO (–OCO2H) and hydrogen-bonding. After heat treatment again, the infrared absorption peak of CO and hydrogen-bonding gradually disappeared again (Fig. 4c), demonstration of the breaking of hydrogen bonding and the successful complete desorption of CO2. Further elucidating the adsorption–desorption stability, the second in situ CO2 adsorption also showed that the CO and hydrogen-bonding infrared absorption peak gradually strengthens with various adsorption time (Fig. 4d). As a comparison, the control experiments of pristine MOF-808-CO2 shows no obvious infrared absorption peak for CO2 desorption at 140 °C. And the heat-treated MOF-808 is subjected to in situ CO2 adsorption again in dry conditions along with various time, the in situ DRIFTS spectra show no obvious change in infrared absorption peaks (Fig. S12†). The results show that parent MOF-808 does not have low concentration CO2 adsorption capacity. Based on the above in situ DRIFTS results, showing that the alkali etched MOF-808-0.07 has enhanced low concentration CO2 capture capacity compared to parent MOF-808 under dry air conditions due to the presence of Zr–OH adsorption sites.
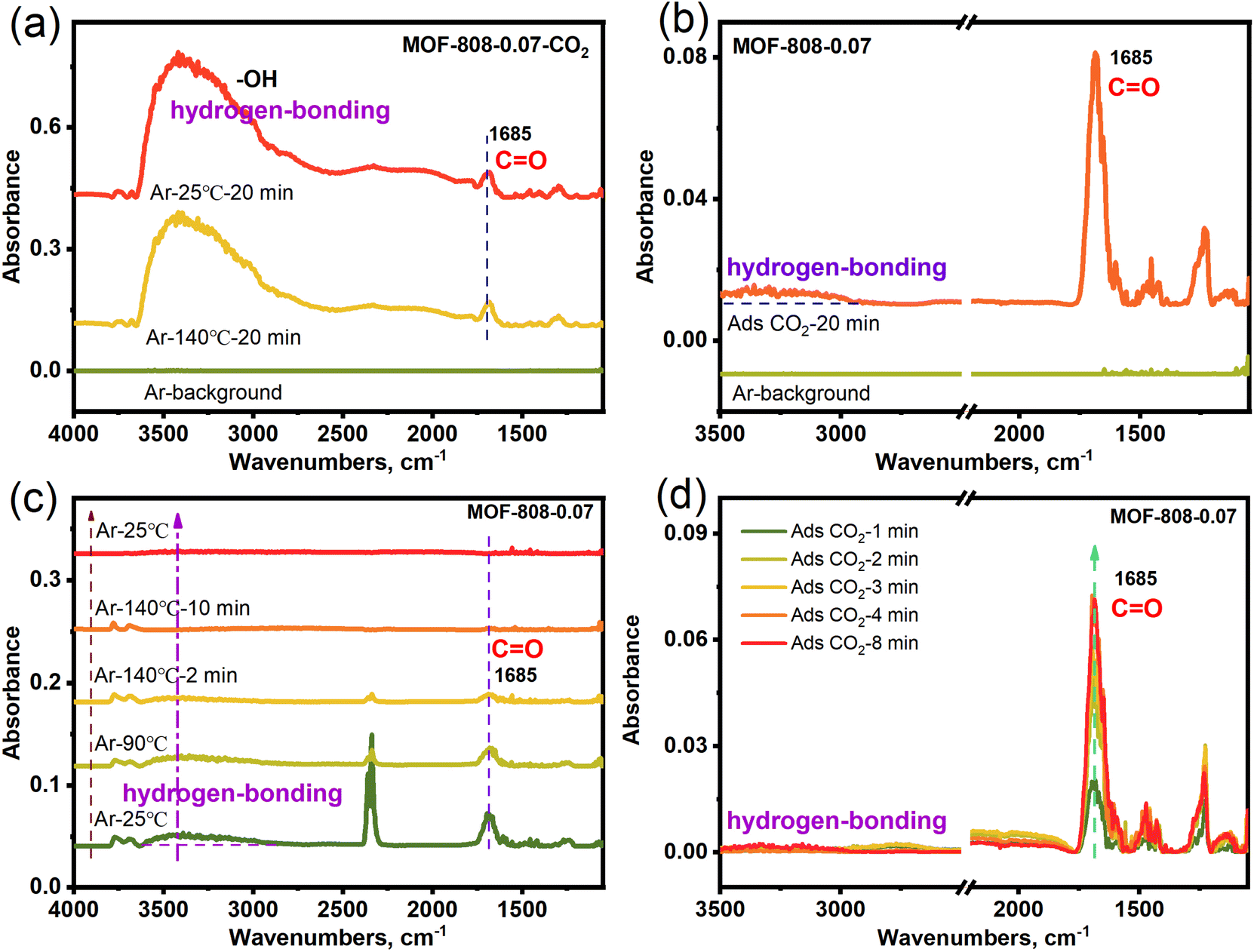 | ||
| Fig. 4 In situ DRIFTS of (a) MOF-808-0.07-CO2 for desorption CO2, (b) MOF-808-0.07 for adsorption CO2, (c) MOF-808-0.07-CO2 for the 2nd desorption CO2, (d) MOF-808-0.07 for the 2nd adsorption CO2. | ||
To elucidate the formation of –OCO2H species under dry conditions, solid-state cross-polarization magic-angle spinning (CP-MAS) 13C NMR experiments were conducted on variant MOF-808-0.07 to investigate the change in chemical species before and after capturing 13CO2 (isotopic gas). Fig. 5 displays the 13C CP-MAS ssNMR spectrum of the pristine MOF-808 without adsorbed CO2, showing no observable chemical shifts, indicating complete etching of the carbon framework in MOF-808. Upon adsorption of 13CO2 under dry conditions, two distinct chemical shifts appeared in the 13C CP-MAS ssNMR spectrum at δ13C = 166.8 and 164.4 ppm. Combining these results with the in situ DRIFTS data, it can be inferred that these shifts are attributed to –OCO2H groups and –OCO2H groups involved in intramolecular hydrogen bonding, respectively.
 | ||
| Fig. 5 Stacked plots of solid-state 13C CP-MAS NMR spectra of MOF-808-0.07 before and after adsorption of 13CO2. | ||
Based on the above in situ DRIFTS and 13C CP-MAS ssNMR characterizations, we proposed a possible mechanistic of low-concentration CO2 capture process in MOF-808 series. (1) When the two Zr–OH sites within the MOF-808-X framework are distanced apart, each Zr–OH site can adsorb one CO2 molecule, forming Zr–O2COH species (Fig. 6a). (2) When the neighbouring Zr–OH sites within the MOF-808-X framework are in close proximity. As shown in Fig. 6b, first, a Zr–OH site adsorbs a CO2 molecule to form a Zr–O2COH species, and the Zr–O2COH species forms intramolecular hydrogen bonding with the neighbouring Zr–OH site. Subsequently, the neighbouring Zr–OH re-adsorbs a CO2 molecule with it to form two opposing Zr–O2COH species, which interact to form a stable six-membered ring structure through the interaction of intramolecular hydrogen bonding to complete an adsorption process.
 | ||
| Fig. 6 Proposed low-concentration CO2 capture mechanism for MOF-808-X series. | ||
Conclusions
In conclusion, we have demonstrated that controlled alkali etching of MOF-808 leads to the formation of MOF-808-X series, which exhibit significantly enhanced low-concentration CO2 capture compared to the pristine MOF-808 under dry air conditions. Among the MOF-808-X series, MOF-808-0.07 displays the highest CO2 capture capacity of 0.21 mmol g−1 in simulated dry air conditions, which is 70 times higher than the pristine MOF-808. The desorption kinetics of the MOF-808-0.07 also show higher desorption energy compared to the commonly used 13X zeolite. Our control experiments suggest that MOFs with high coordination numbers show higher CO2 capture performance under dry air conditions. Furthermore, in situ DRIFTS and 13C CP-MAS ssNMR results indicate that the enhanced low-concentration CO2 capture is due to the formation of a stable six-membered ring structure through intramolecular hydrogen bonds between Zr–OH sites of neighbouring micro-mesoporous environments of MOF-808-X. Overall, these findings suggest the potential of MOF-808-X series as promising materials for low-concentration CO2 capture.Data availability
The authors declare that all data supporting the findings of this study are available from the corresponding author upon reasonable request.Author contributions
Hong Dong: conceptualization, data curation, formal analysis, investigation, funding acquisition, writing – original draft, writing – review & editing. Lihua Li: investigation and formal analysis, Can Li: supervision, funding acquisition, project administration and writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Youth Program of the National Natural Science Foundation of China (No. 22102178), Project funded by China Postdoctoral Science Foundation (No. 2021M693119), and the Special Research Assistant Funding Project of Chinese Academy of Sciences (No. 2021000135). This work was also conducted by the Fundamental Research Center of Artificial Photosynthesis (FReCAP), financially supported by the National Natural Science Foundation of China (NSFC) under Grant No. 22088102. H. Dong would like to thank Shiyanjia Lab for the XPS and 13C CP-MAS ssNMR analysis.Notes and references
- A. Kumar, D. G. Madden, M. Lusi, K. J. Chen, E. A. Daniels, T. Curtin, J. J. Perry and M. J. Zaworotko, Angew. Chem., Int. Ed., 2015, 54, 14372–14377 CrossRef CAS PubMed.
- X. Y. Shi, H. Xiao, H. Azarabadi, J. Z. Song, X. L. Wu, X. Chen and K. S. Lackner, Angew. Chem., Int. Ed., 2020, 59, 6984–7006 CrossRef CAS PubMed.
- C. J. J. Haertel, M. McNutt, M. Ozkan, E. S. P. Aradottir, K. T. Valsaraj, P. R. Sanberg, S. Talati and J. Wilcox, Chem, 2021, 7, 2831–2834 CAS.
- X. C. Zhu, W. W. Xie, J. Y. Wu, Y. H. Miao, C. J. Xiang, C. P. Chen, B. Y. Ge, Z. Z. Gan, F. Yang, M. Zhang, D. O'Hare, J. Li, T. S. Ge and R. Z. Wang, Chem. Soc. Rev., 2022, 51, 6574–6651 RSC.
- J. L. Sun, M. Zhao, L. Huang, T. Y. Zhang and Q. Wang, Curr. Opin. Green Sustainable Chem., 2023, 40, 100752 CrossRef CAS.
- M. Oschatz and M. Antonietti, Energy Environ. Sci., 2018, 11, 57–70 RSC.
- S. Choi, J. H. Drese, P. M. Eisenberger and C. W. Jones, Environ. Sci. Technol., 2011, 45, 2420–2427 CrossRef CAS PubMed.
- A. Ghosh, G. N. Reddy, P. K. M. Siddhique, S. Chatterjee, S. Bhattacharjee, R. Maitra, S. E. Lyubimov, A. V. Arzumanyan, A. Naumkin, A. Bhaumik and B. Chowdhury, Green Chem., 2022, 24, 1673–1692 RSC.
- D. E. F. Oliveira, J. A. O. Chagas, A. L. De Lima and C. J. A. Mota, Ind. Eng. Chem. Res., 2022, 61, 10522–10530 CrossRef CAS.
- S. M. W. Wilson and F. H. Tezel, Ind. Eng. Chem. Res., 2020, 59, 8783–8794 CrossRef CAS.
- P. Hu, H. Liu, H. Wang, J. Zhou, Y. Q. Wang and H. B. Ji, J. Mater. Chem. A, 2022, 10, 881–890 RSC.
- W. J. Wang, M. Zhouab and D. Q. Yuan, J. Mater. Chem. A, 2017, 5, 1334–1347 RSC.
- X. Q. Kong, E. Scott, W. Ding, J. A. Mason, J. R. Long and J. A. Reimer, J. Am. Chem. Soc., 2012, 134, 14341–14344 CrossRef CAS PubMed.
- H. Lyu, O. I. F. Chen, N. Hanikel, M. I. Hossain, R. W. Flaig, X. K. Pei, A. Amin, M. D. Doherty, R. K. Impastato, T. G. Glover, D. R. Moore and O. M. Yaghi, J. Am. Chem. Soc., 2022, 144, 2387–2396 CrossRef CAS.
- H. Lyu, H. Z. Li, N. Hanikel, K. Y. Wang and O. M. Yaghi, J. Am. Chem. Soc., 2022, 144, 12989–12995 CrossRef CAS.
- T. M. McDonald, J. A. Mason, X. Q. Kong, E. D. Bloch, D. Gygi, A. Dani, V. Crocella, F. Giordanino, S. O. Odoh, W. S. Drisdell, B. Vlaisavljevich, A. L. Dzubak, R. Poloni, S. K. Schnell, N. Planas, K. Lee, T. Pascal, L. W. F. Wan, D. Prendergast, J. B. Neaton, B. Smit, J. B. Kortright, L. Gagliardi, S. Bordiga, J. A. Reimer and J. R. Long, Nature, 2015, 519, 303–308 CrossRef CAS.
- J. B. Lin, T. T. T. Nguyen, R. Vaidhyanathan, J. Burner, J. M. Taylor, H. Durekova, F. Akhtar, R. K. Mah, O. Ghaffari-Nik, S. Marx, N. Fylstra, S. S. Iremonger, K. W. Dawson, P. Sarkar, P. Hovington, A. Rajendran, T. K. Woo and G. K. H. Shimizu, Science, 2021, 374, 1464–1469 CrossRef CAS.
- Y. Zhou, J. L. Zhang, L. Wang, X. L. Cui, X. L. Liu, S. S. Wong, H. An, N. Yan, J. Y. Xie, C. Yu, P. X. Zhang, Y. H. Du, S. B. Xi, L. R. Zheng, X. Z. Cao, Y. J. Wu, Y. X. Wang, C. Q. Wang, H. M. Wen, L. Chen, H. B. Xing and J. Wang, Science, 2021, 373, 315–320 CrossRef CAS PubMed.
- C. Jia, R. R. Liang, S. X. Gan, S. Y. Jiang, Q. Y. Qi and X. Zhao, Chem. - Eur. J., 2023, 29, e202300186 CrossRef CAS PubMed.
- Y. F. Zeng, R. Q. Zou and Y. L. Zhao, Adv. Mater., 2016, 28, 2855–2873 CrossRef CAS PubMed.
- X. Zhao, Y. X. Wang, D. S. Li, X. H. Bu and P. Y. Feng, Adv. Mater., 2018, 30, 1705189 CrossRef PubMed.
- C. E. Bien, K. K. Chen, S. C. Chien, B. R. Reiner, L. C. Lin, C. R. Wade and W. S. W. Ho, J. Am. Chem. Soc., 2018, 140, 12662–12666 CrossRef CAS PubMed.
- C. E. Bien, Q. Liu and C. R. Wade, Chem. Mater., 2020, 32, 489–497 CrossRef CAS.
- A. Kumar, C. Hua, D. G. Madden, D. O'Nolan, K. J. Chen, L. A. J. Keane, J. J. Perry and M. J. Zaworotko, Chem. Commun., 2017, 53, 5946–5949 RSC.
- Z. Q. Zhang, Q. Ding, J. Y. Cui, X. L. Cui and H. B. Xing, Sci. China Mater., 2021, 64, 691–697 CrossRef.
- M. D. Jiang, B. Li, X. L. Cui, Q. W. Yang, Z. B. Bao, Y. W. Yang, H. Wu, W. Zhou, B. L. Chen and H. B. Xing, ACS Appl. Mater. Interfaces, 2018, 10, 16628–16635 CrossRef CAS PubMed.
- P. Nugent, Y. Belmabkhout, S. D. Burd, A. J. Cairns, R. Luebke, K. Forrest, T. Pham, S. Q. Ma, B. Space, L. Wojtas, M. Eddaoudi and M. J. Zaworotko, Nature, 2013, 495, 80–84 CrossRef CAS.
- W. Liang, P. M. Bhatt, A. Shkurenko, K. Adil, G. Mouchaham, H. Aggarwal, A. Mallick, A. Jamal, Y. Belmabkhout and M. Eddaoudi, Chem, 2019, 5, 950–963 CAS.
- P. M. Bhatt, Y. Belmabkhout, A. Cadiau, K. Adil, O. Shekhah, A. Shkurenko, L. J. Barbour and M. Eddaoudi, J. Am. Chem. Soc., 2016, 138, 9301–9307 CrossRef CAS PubMed.
- O. Shekhah, Y. Belmabkhout, Z. J. Chen, V. Guillerm, A. Cairns, K. Adil and M. Eddaoudi, Nat. Commun., 2014, 5, 4228 CrossRef CAS PubMed.
- Z. Q. Zhang, Q. Ding, S. B. Peh, D. Zhao, J. Y. Cui, X. L. Cui and H. B. Xing, Chem. Commun., 2020, 56, 7726–7729 RSC.
- L. A. Darunte, A. D. Oetomo, K. S. Walton, D. S. Sholl and C. W. Jones, ACS Sustainable Chem. Eng., 2016, 4, 5761–5768 CrossRef CAS.
- S. Choi, T. Watanabe, T. H. Bae, D. S. Sholl and C. W. Jones, J. Phys. Chem. Lett., 2012, 3, 1136–1141 CrossRef CAS PubMed.
- A. J. Emerson, A. Chahine, S. R. Batten and D. R. Turner, Coord. Chem. Rev., 2018, 365, 1–22 CrossRef CAS.
- R. L. Siegelman, P. J. Milner, A. C. Forse, J. H. Lee, K. A. Colwell, J. B. Neaton, J. A. Reimer, S. C. Weston and J. R. Long, J. Am. Chem. Soc., 2019, 141, 13171–13186 CrossRef CAS PubMed.
- P. Q. Liao, X. W. Chen, S. Y. Liu, X. Y. Li, Y. T. Xu, M. N. Tang, Z. B. Rui, H. B. Ji, J. P. Zhang and X. M. Chen, Chem. Sci., 2016, 7, 6528–6533 RSC.
- R. L. Siegelman, T. M. McDonald, M. I. Gonzalez, J. D. Martell, P. J. Milner, J. A. Mason, A. H. Berger, A. S. Bhovvn and J. R. Long, J. Am. Chem. Soc., 2017, 139, 10526–10538 CrossRef CAS PubMed.
- J. S. A. Carneiro, G. Innocenti, H. J. Moon, Y. Guta, L. Proano, C. Sievers, M. A. Sakwa-Novak, E. W. Ping and C. W. Jones, Angew. Chem., Int. Ed., 2023, 62, e202302887 CrossRef CAS PubMed.
- S. C. Li, M. R. Ceron, H. V. Eshelman, A. J. Varni, A. Maiti, S. Akhade and S. H. Pang, ChemSusChem, 2023, 16, e202201908 CAS.
- H. Furukawa, F. Gandara, Y. B. Zhang, J. C. Jiang, W. L. Queen, M. R. Hudson and O. M. Yaghi, J. Am. Chem. Soc., 2014, 136, 4369–4381 CrossRef CAS PubMed.
- H. Dong, G. Yang, X. Zhang, X. Meng, J. Sheng, X. Sun, Y. Feng and F. Zhang, Chem. - Eur. J., 2018, 24, 17148–17154 CrossRef CAS PubMed.
- D. L. Fu, Y. Park and M. E. Davis, Angew. Chem., Int. Ed., 2022, 61, e202112916 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sc03213b |
| This journal is © The Royal Society of Chemistry 2023 |