
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIsomerism tunes the diradical character of difluorenopyrroles at constant Hückel-level anti-aromaticity†
Ryotaro
Moriyasu‡
a,
Sergio Moles
Quintero‡
 b,
Carlos J.
Gómez-García
c,
Kazumasa
Suzuki
d,
Chitoshi
Kitamura
a,
Michihisa
Murata
e,
Mercedes
Alonso
f,
Juan
Casado
*b and
Shin-ichiro
Kato
*a
b,
Carlos J.
Gómez-García
c,
Kazumasa
Suzuki
d,
Chitoshi
Kitamura
a,
Michihisa
Murata
e,
Mercedes
Alonso
f,
Juan
Casado
*b and
Shin-ichiro
Kato
*a
aDepartment of Materials Chemistry, School of Engineering, The University of Shiga Prefecture, 2500 Hassaka-cho, Hikone, Shiga 522-8533, Japan. E-mail: kato.s@mat.usp.ac.jp
bDepartament of Physical Chemistry, University of Málaga, Campus de Teatinos s/n, Málaga, 29071, Spain. E-mail: casado@uma.es
cDepartament of Inorganic Chemsitry, University of Valencia, C/ DR. Moliner, 50, 46100 Brujassot, Valencia, Spain
dDepartment of Material Chemistry, Graduate School of Engineering, Nagoya university, Furo-cho, Chikusa-ku, Nagoya 464-8603, Japan
eDepartment of Applied Chemistry, Faculty of Engineering, Osaka Institute of Technology, 5-16-1 Ohmiya, Asahi-ku, Osaka 535-8585, Japan
fEenheid Algemene Chemie (ALGC), Vrije Universiteit Brussel (VUB)Plei nlaan 2, 1050 Brussels, Belgium
First published on 23rd October 2023
Abstract
A new diradical based on diindenocarbazole or difluorenopyrrole was synthesized and experimentally characterized by optical, electrochemical, and magnetic techniques, as well as quantum chemical calculations. The isomerism of these structures tunes the diradical character and the associated properties, representing a unique case of such important modulation. A full study of the electronic structure was carried out considering the perturbative interactions between different canonical forms as well as the anti-aromatic character of the molecular cores. Such a study reveals how we can tune diradical character simply by reorganizing the bonding patterns at constant chemical costs (composition).
Introduction
The two major breakthrough articles on diradicaloid indenoacenes were authored by Haley,1 who described in 2011 the preparation of indeno[1,2-b]fluorene 1, and Tobe,2 who reported in 2013 the synthesis of indeno[2,1-b]fluorene 2, as shown in Scheme 1, and both compounds are members of the subfamily of indenofluorenes. Their births were followed by great activity in the field, with examples showcasing the extension of the acene core,3 with peripheral benzene isomerism,4,5 insertion of heteroatoms,6 and, in some cases, with applications in organic electronics.7–13 Many of these contributions were focused on the search for these molecules as diradicaloids (i.e., optimization of the diradical character) and also as stable anti-aromatic entities.14,15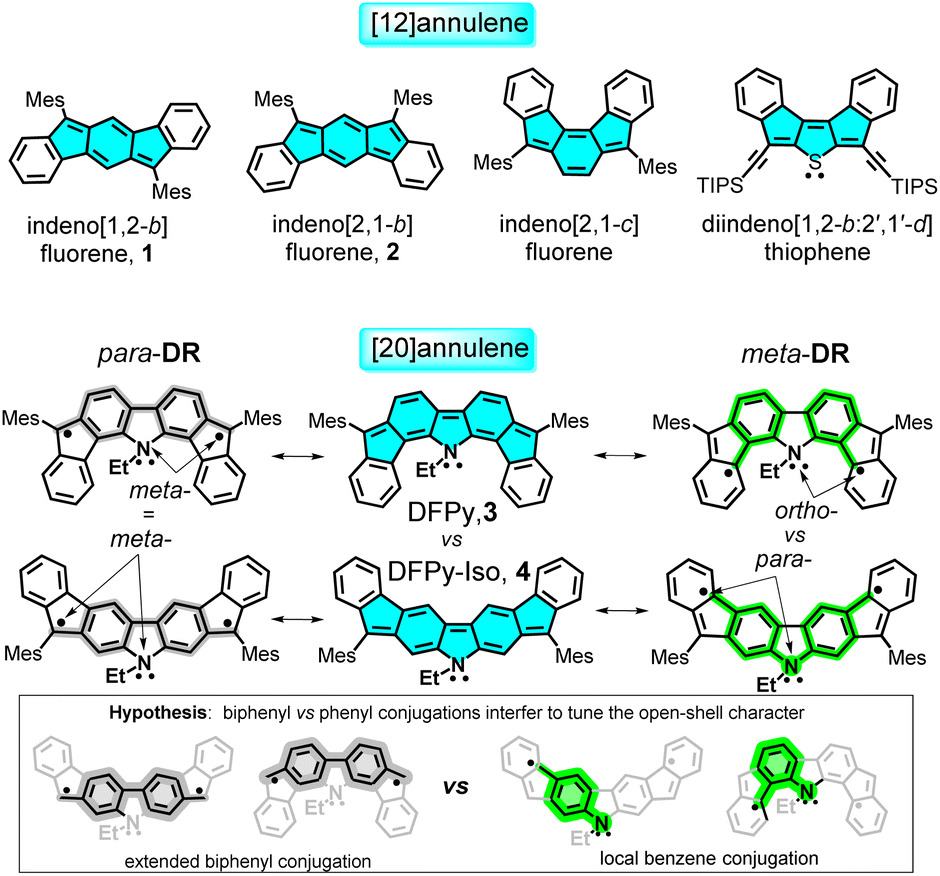 | ||
| Scheme 1 Molecular structures of the most important reported indenofluorenes, together with those of DFPy 3 and its structural isomer, DFPy-Iso 4. The two most relevant canonical forms for 3 and 4 are shown – para-DR (in grey featuring an extended biphenyl conjugation) and meta-DR (in green showing the central biphenyl containing a local benzene conjugation with the N atom). In blue, anti-aromatic 4n π-electron cores are shown. The arrows denote the relative position of the radical and N atom relative to the common benzene ring. Mes: mesityl, TIPS: triisopropylsilyl, Et: ethyl. | ||
Diradicaloids are currently important molecular units for a series of applications in organic electronics, such as in organic field-effect transistor devices,7,8 in thermoelectric and thermopower devices,9,10 in non-linear optical applications,11,12 and in singlet exciton fission.13 In all these studies, the diradical property was theoretically evaluated with the diradical index parameter y0, which varies from 0 to 1 in its movement from closed-shell to full open-shell structures.16–18
Aside from the technological objectives, the first examples of open-shell diradicals were given by Chichibabin,19 Thiele,20 and Müller.21 Academic interest was aimed at understanding the bonding in molecules and how subtle changes in the electronic structure of a given compound can affect the dissociation of their electron bonding Lewis pairs, a subject at the core of understanding the nature of the chemical bond.22–25 In this context, 1 and 2 are 20π-electron anti-aromatic isomers, which strongly differ in diradical character: y0(1) = 0 versus y0(2) = 0.68, with the subsequent impact from their electronic properties. Isomerism thus becomes a powerful strategy that can be used to design diradicals and is an ideal platform for studying open-shell molecules in the search for new and hidden properties.26–29
We recently studied some variants of indenoacenes by substituting the central acene fragment with fluoreno-heteroles, which allows tuning of the diradical character with the heteroatom (O,N-difluoreno[4,3-b:3′,4′-d]pyrrole (DFPy) 3, in Scheme 1, S and SO2), and thus, with the aromatic structure of the central biphenyl.30–33 In this article, we present an electronic and structural isomer of DFPy 3 in which the external benzenes isomerize around the key five-membered rings of the indene fragment to form an electronic isomer of DFPy, difluoreno[2,3-b:3′,2′-d]pyrrole (DFPy-Iso) 4, as shown in Scheme 1. We find properties of 4 that were similar to those of Tobe's 2, but also significant differences when compared with its close relative 3.
We explore their diradical origin in connection with their isomeric properties, and in the context of anti-aromaticity in reference to their parent [4n]annulene (i.e., [20]annulene). We show that the anti-aromatic fingerprints for 3 and 4 are similar, although with differences arising from the different patterns of local π-electron delocalization of the radical centers in 3 and 4 (see the hypothesis in Scheme 1), which is further related to an underlying captodative mechanism strongly active in the cationic forms.
Results and discussion
The synthesis of 4 is shown in Scheme 2. A Suzuki–Miyaura coupling of bromide 5 (ref. 34) with phenylboronic acid using a catalytic system consisting of Pd2(dba)3·CHCl3 and [(t-Bu)3PH][BF4] along with Cs2CO3 as a base furnished aldehyde 6 in 77% yield. The nucleophilic addition of mesitylmagnesium bromide (MesMgBr) to 6 provided the corresponding secondary alcohol, which was then subjected to a Friedel–Crafts alkylation mediated by BF3·Et2O to afford dihydrodifluorenopyrrole 7 in 52% yield over two steps. Oxidative dehydrogenation of 7 with 2,3-dichloro-5,6-dicyanobenzoquinone (DDQ) provided the deep purple 4 in a nearly quantitative fashion. Unlike 3, compound 4 appeared unstable on common silica gel or neutral alumina; however, 4 was successfully purified with column chromatography on triethylamine-deactivated silica gel. The synthetic details are described in the ESI,† together with detailed chemical characterizations.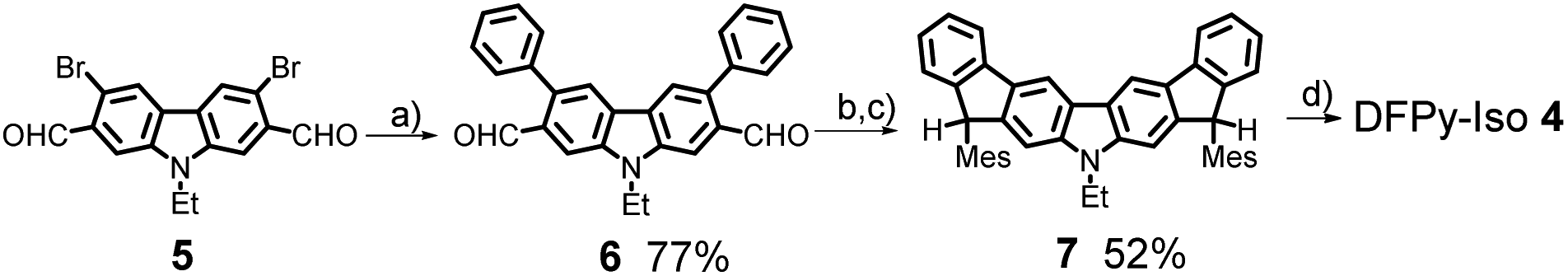 | ||
| Scheme 2 Synthesis of DFPy-Iso, 4. Reagents and conditions: (a) Pd2(dba)3·CHCl3, [(t-Bu)3PH][BF4], Cs2CO3, phenylboronic acid, 1,4-dioxane/H2O, 70 °C. (b) MesMgBr, THF, rt. (c) BF3·Et2O, CH2Cl2, rt. (d) DDQ, toluene, 60 °C. | ||
Fig. 1 displays the optical absorption bands of 3 and 4. These spectra show distinctive optical properties for the two isomers: there is a main band for 3 at 640 nm with low energy shoulders that were assigned as signatures of the diradical character (absorption to the two-photon state). Conversely, the main absorption band for 4 was blueshifted at 577 nm (molar absorptivity, ε = 34![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 100 M−1 cm−1), with a shape similar to that of the main band of 3, accompanied by a broad vibronically resolved absorption peak at 913 nm (ε = 5610 M−1 cm−1) that extends beyond 1100 nm.
100 M−1 cm−1), with a shape similar to that of the main band of 3, accompanied by a broad vibronically resolved absorption peak at 913 nm (ε = 5610 M−1 cm−1) that extends beyond 1100 nm.
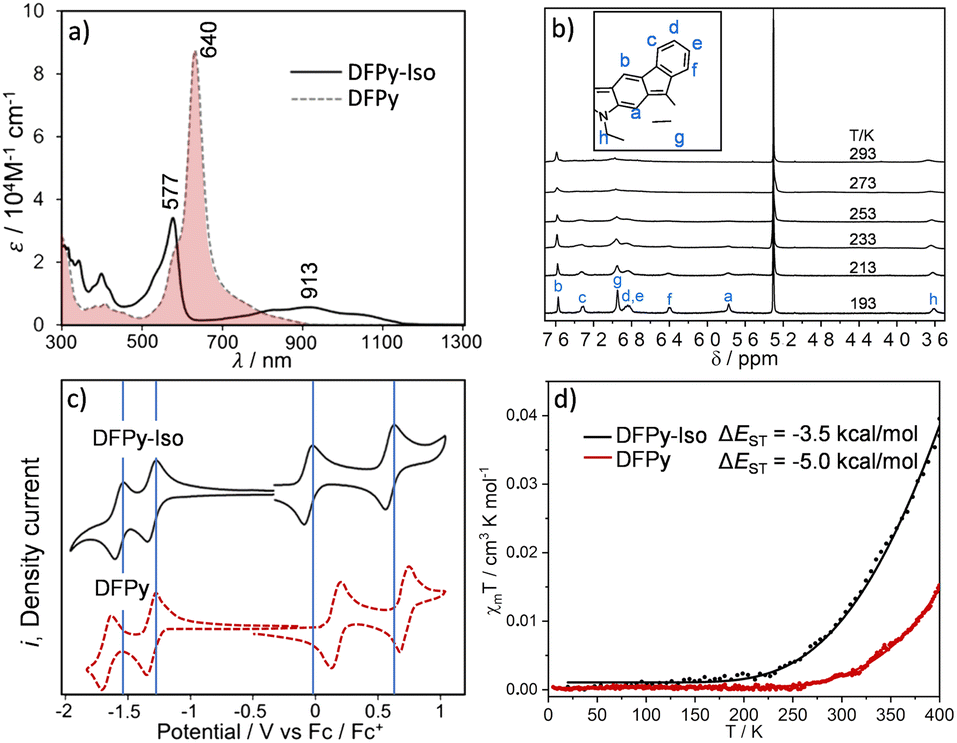 | ||
| Fig. 1 (a) UV-Vis-NIR absorption spectra of 3 (dotted line) and of 4 (solid line) at 298 K in CH2Cl2. (b) 1H NMR spectra of 4 in CD2Cl2 at different temperatures. (c). Cyclic voltammograms of 3 (dotted line) and 4 (solid line) at 298 K in tetrabutyl ammonium hexafluorophosphate 0.1 M electrolyte in CH2Cl2 taken at a scan rate of 100 mV s−1 and represented versus the ferrocene/ferrocenium pair. (d) SQUID measurements (black circles for 4 and red circles for 3) of the magnetic susceptibility, χT, from 4 to 400 K, with the solid lines representing the fitting to the Bleaney–Bowers equation.35 | ||
The spectrum of 4 is very similar to that of 2, which shows the same pattern with a strong well-defined sharp band at 638 nm and a broad band with a maximum peak at 1300 nm, extending up to 2000 nm. This optical fingerprint in 2 was attributed to the anti-aromatic character of the 4n (12) π-electron circuit (shaded blue in Scheme 1).2 In 4, time-dependent density functional theory (TD-DFT) was used to assign the two experimental bands to 484 nm (oscillator strength f = 0.42) and 818 nm (f = 0.11) (Fig. S1 and S2,† respectively).
The cyclic voltammetries of 3 and 4 are displayed in Fig. 1. There are two reversible one-electron reductions; the first one in 3 was at −1.32 V, and in 4 at −1.33 V (Fig. S3, S4, and Table S1†). The second one appears at −1.66 V in 3 and at −1.58 V in 4. Furthermore, in the two compounds, there are two reversible one-electron oxidation waves with the following voltage potentials: +0.17 and +0.71 V in 3 and +0.05 and +0.60 V in 4; that is, 0.12 and 0.11 V lower values in 4 than in 3. This distinctive electrochemical oxidation behaviour between 3 and 4 reveals a role of the nitrogen atom in 4 that does not occur in 3.
To account for this, let us consider two feasible canonical forms for the two neutral diradicals: (i) one with the unpaired electrons placed in the apical five-membered ring carbons (in grey in Scheme 1), where the two radicals are in para-positions (para-DR) regarding the central biphenyl moiety (i.e., extended biphenyl conjugation). In para-DR forms, the nitrogens and the radical centers of 3 and 4 are located in the meta-positions of one benzene of the biphenyl center (i.e., local benzene conjugation). This local meta-substitution disconnects the N/radical electronic communication, and this occurs to the same extent for the two isomers. Therefore, in the para-DR forms, there is little contribution from the N atoms between 3 and 4. (ii) By placing the radicals in the external six-membered rings (in green in Scheme 1), the two radicals are placed in the relative meta-positions of the central biphenyl (meta-DR). In these forms, the radicals and N atoms interact differently by local benzene conjugations in 3 (through the ortho-positions) and in 4 (through the para-positions). We hypothesized that the competition between extended biphenyl and local benzene conjugations will result in the different electronic properties of 3 and 4 diradical isomers.
Oxidation from the neutral meta-DR forms of 3 and 4 in Scheme 3 will occur in the unpaired electrons. Therefore, from this position, there are two possible mechanisms of stabilization of the positive charge of the radical cations: (i) by resonance or delocalization of the lone electron pair of the N (RE in Scheme 3) between the two grey-shaded moieties over the central N-containing red-shaded moiety; and (ii) by captodative effect (CDE in Scheme 3)36 between the positive charge placed in the N-cation containing the quinoidal moiety (i.e., acts as an acceptor, shaded blue in Scheme 3) and the external phenyl-like fragments (acting as a donor, shaded maroon in Scheme 3), with the radical center in-between.
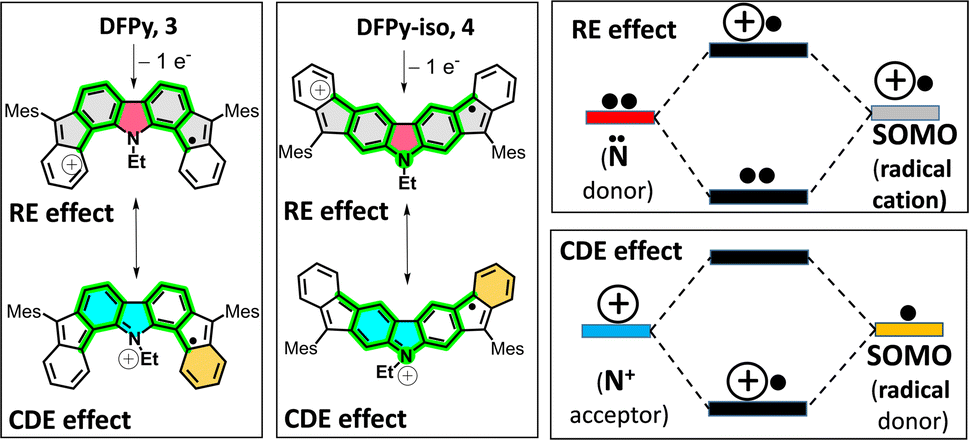 | ||
| Scheme 3 Stabilization mechanisms acting on the radical cations of 3 and 4 by resonant delocalization (RE, between the grey and red shaded moieties) and captodative effects (CDE, between the blue- and maroon-shaded moieties). Right: RE and CDE effects interpreted from molecular orbital theory where the colour codes of the energy levels correspond to those of the moieties in the valence bond forms. | ||
All these mechanisms are much more efficient through the relative para-positions of the central benzene rings of 4 than by the ortho-positions of the same benzene of 3, resulting in greater stabilization and subsequent lowering of the oxidation potentials from 3 to 4. Conversely, the formation of the radical anions is better accounted from the para-DR canonical forms (a larger repulsion of the extra charge and the N lone electron pair would minimize the role of the meta-DR forms), yielding similar reduction potentials as already stated for the similar role of the para-DR forms in 3 and 4.
The diradical character of 4 is anticipated by the appearance of the 1H NMR spectrum in Fig. 1 recorded at 293 K in CD2Cl2/Et3N, which presents severe line broadening in stark contrast to the sharp signals of the 1H NMR spectrum of 3 at 293 K (Fig. S5†). The cooling of 4 to 193 K was accompanied by progressive line sharpening (Fig. 1b and Table S2†). This effect was ascribed to the significant population at 293 K of paramagnetic triplet species in 4, which were absent in 3, and was in agreement with a smaller energy gap between the ground electronic singlet state and the first excited state triplet (i.e., ΔEST) in 4 than in 3. The ΔEST estimated from superconducting quantum interference device (SQUID) measurements of 4 is ≈−3.5 kcal mol−1, and was in agreement with the value theoretically calculated for 4: −2.1 kcal mol−1 (Table 1).
| Compound | ΔEST (kcal mol−1) | y 0 = nLUNO |
|---|---|---|
| 3 (UDFT/B3LYP/6-31G**) | −5.3 (exp: −5.0) | y 0 = 0.353 |
| 4 (UDFT/B3LYP/6-31G**) | −2.1 (exp: −3.5) | y 0 = 0.596 |
| (RAS-CI-SF [4,4]) | y 0 (3) = 0.283 | y 0 (4) = 0.394 |
These values are much smaller than those measured (−5.0 kcal mol−1) and calculated (−5.3 kcal mol−1) in 3 in Table 1. This trend is consistent with the diradical index values calculated for 3 and 4, which amount to y0 = 0.353 and y0 = 0.596, respectively, at the UB3LYP/6-31G** level (Table 1 also includes y0 for RAS-CI-SF [4,4] calculations).
The larger diradical character in 4 as compared to 3 can be assessed by considering the canonical forms in Scheme 1. The para-DR form is the most relevant contributor to the open-shell description of the two isomers because it presents four benzene rings with Clar aromatic sextets. Because there will be comparable stabilizations in 3 and in 4 for the para-DR form, these should not distinctively impact the diradical property in the two isomers. The situation drastically changes in the meta-DR forms created by breaking the aromaticity of the external benzenes (i.e., by which the meta-DR forms will play a secondary role). However, the placement of the radical centers in the six-membered rings in 4 enables location of the radicals and N atoms in local benzene para-conjugation. A similar situation was described in the radical cation of 4.
We recently reported the modulation of y0 and of ΔEST in anti- and syn-IIDBT (Scheme 4) isomers,4,29 where larger y0 and smaller ΔEST were found for the syn-IIDBT isomer due to the cross-conjugation situation of the electron radical and the lone electron sulfur pair (see the yellow path in Scheme 4) that disconnects them from mutual π-delocalization. Conversely, in anti-IIDBT, radical/lone electron pair linear conjugation of the three bodies reduces electron–electron repulsion by configuration interactions. The enhanced repulsion in syn-IIDBT enlarges y0 and reduces ΔEST.
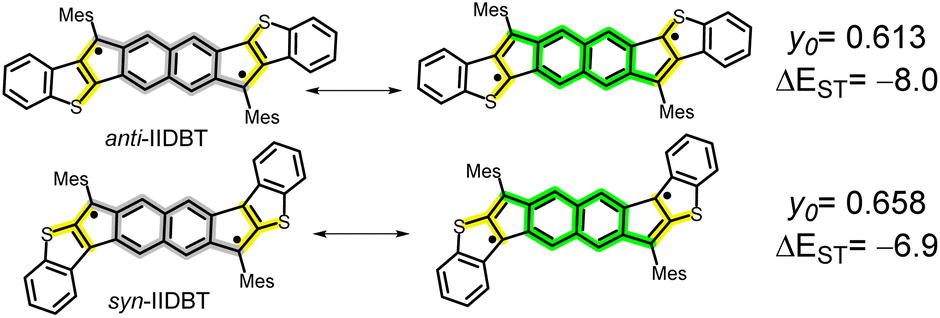 | ||
| Scheme 4 Diradical forms contributing to the stabilization of anti- and syn-indenoindenodibenzothiophes (anti-IIDBT and syn-IIDBT). y0 and experimental SQUID ΔEST in kcal mol−1 are shown. | ||
Hence, in the meta-DR forms of 3 and 4, their local benzene conjugation between the radical and the N lone electron pair minimizes their electron repulsion through the relative para-positions, thus favoring this delocalization path. This delocalization effect is driven by the formation of an ionic/zwitterionic structure that becomes stabilized by electrostatic effects,37 as depicted in Scheme 5 for 4. This preferred local path of para-conjugation in 4 works by interfering and diminishing the radical–radical coupling and overlap between the two unpaired electrons in the para-DR form in biphenyl conjugation, which increases y0 and narrows ΔEST.
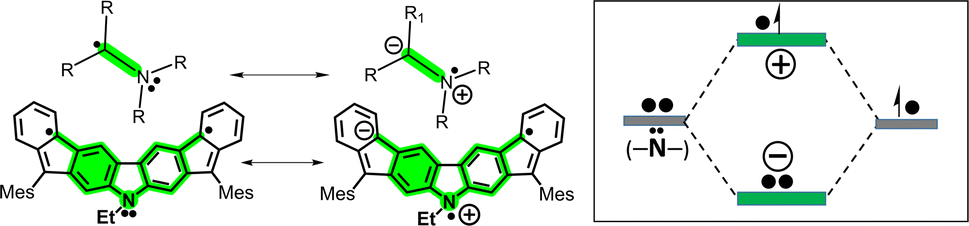 | ||
| Scheme 5 Left: Electrostatic stabilization by formation of ionic forms in amino radical compounds37 and in the case of 4. Right: Orbital hybridization of the N and radical to originate the ionic forms. | ||
In the case of 3, a similar description can be produced, with the difference that the local radical ↔ N benzene conjugation and formation of ionic form takes place over the ortho-positions. This also disrupts the extended biphenyl radical–radical overlap, but to a lesser extent. Notably, this particular interference effect of local (i.e., para- and ortho-) conjugations over the extended biphenyl radical–radical conjugation is exclusively mediated by N atoms, revealing new venues for the design of nitrogen-doped indenofluorenes.
Fig. 2 displays the spin-density distributions for 3 and 4. It is clearly seen that there are minor contributions for 3 for the peripheral rings, whereas these contributions are much greater in 4, which is in agreement with the role played by these peripheral or outermost benzene rings in the meta-DR forms. The establishment of ionic canonical forms provokes the shortening of the CN bonds in 4 as compared to 3, and is observed in the optimized geometries of both compounds in Fig. 2 and S6:† the CN bond distance in 3 is 1.411 Å, whereas in 4, it is 1.391 Å.
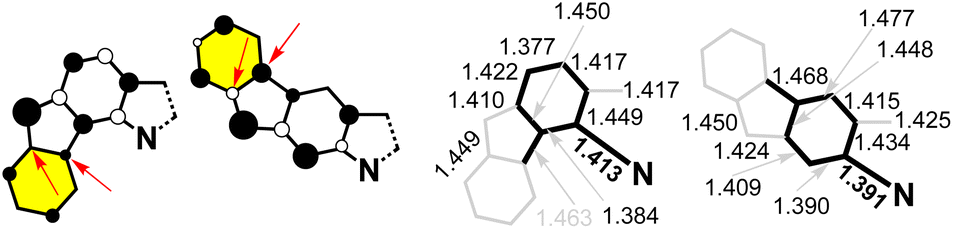 | ||
| Fig. 2 The data for 3versus4 at the UB3LYP/6-311G* level of theory. Left: spin-density distributions (arrows denote key values on the peripheral benzenes). Right: main bond distances from the optimized geometries (in bold, key values of local benzene ortho- and para-conjugations). | ||
Stability studies of diradcials 3 and 4 carried out in CH2Cl2 solutions at 298 K (Fig. S7 and S8†) show a half-life (τ1/2) for 4 of 630 hours (≈26 days) and a much longer stability for 3, which survives for much longer periods of time under the same conditions (after 30 days, only 10% disappeared). This difference in stability is correlated with the larger y0 value of 4 and with the smaller ΔEST. Compared to other indenoacene diradicals such as diindenoanthracene (DIAn)38,39 the half-life of 4 is much less than that of DIAn (τ1/2 = 64 days).
The electronic structures of the pioneering indenofluorenes 1 and 2 have been interpreted as invoking the anti-aromatic character of their π-electronic cores, with 12π-electrons in the internal circuits (shaded blue in Scheme 1).1,2,14,16 Indeed, from the 1H NMR spectra of 4 in Fig. 1, the chemical shift of the proton Ha (5.78 ppm) is located in the high-field region. This seems to reveal the existence of a paratropic ring current that could be related to the presence of a 20π-antiaromatic circuit in 4 (shaded blue in Scheme 1, where the N atom is assumed to contribute with two electrons – the lone electron pz – pair).
To understand the anti-aromaticity in 1vs.2 at the Hückel level, we begin with the construction of the singly occupied molecular orbital (SOMO) orbital (common to 3 and 4) following the Frost diagram for [20]annulene in Fig. 3.40 From this anti-aromatic SOMO orbital, we applied a first order perturbation approach to construct 3 and 4, for which two steps are needed: (i) in both isomers, the N atom participates with two electrons (its lone electron pair; see the box in Fig. 3), thus replacing two carbon atoms of the [20]annulene; and (ii) we include in Fig. 3 the bridging bonds between C atoms (note the yellow lines that denote the different connectivity patterns in the [20]annulene circuit) that produce either 3 or 4.
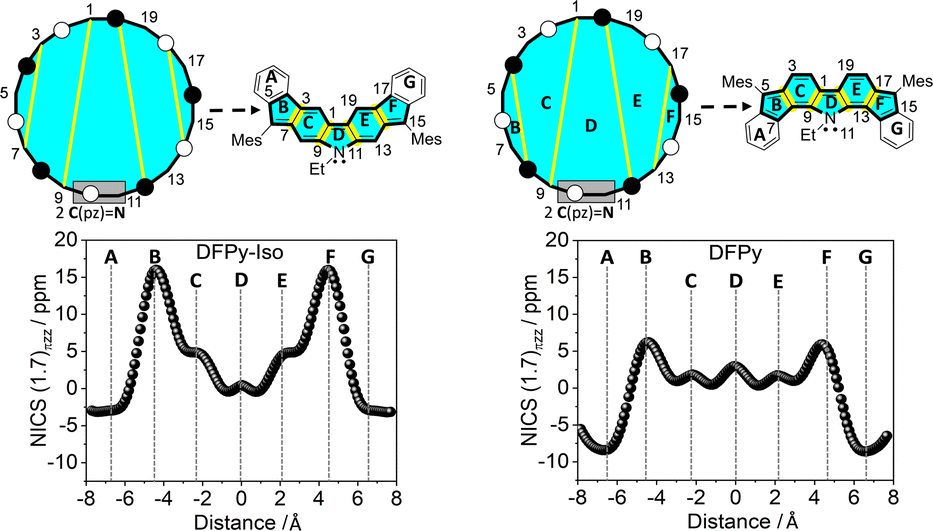 | ||
| Fig. 3 Top: Construction of the 20π-electron central anti-aromatic core of DFPy-Iso (4) and DFPy (3) from the Frost circle at the Hückel level of theory. The grey boxes represent the replacement of two pz carbon electrons by the lone electron pair of the N atoms. Rings are denoted as capital letters to emphasize the correspondence between the Frost circles and the actual cores of 3 and 4. Bottom: NICS πzz-XY-scan plots calculated for 4 and 3 at the UB3LYP/6-31G* level of theory on the singlet open-shell configuration. | ||
In this manner, the B and F rings in Fig. 3 for both compounds merge from different connections of atoms in the periphery (i.e., connecting atoms 3/7 and 14/18 in 4 and 4/8 and 15/19 in 3). These distinctive internal bonds in 3 and 4 have at the Hückel level the same path of perturbative coupling of CC bonds (i.e., in both compounds, there are 2 CC bonds between non-bonding atoms and 2 CC bonds between bonding atoms). This might be indicating that the presence of an anti-aromatic character in 3 and 4 is similar (same 20 periphery with perturbative internal bonds of the same nature).
Consequently, the difference in the electronic properties of these two isomers is related to the different impact on the conjugative effects on the para-DR and meta-DR, as discussed above. Therefore, the paratropic ring current (Fig. S9†) accounting for the displacement at high fields of the Ha atom in the 1H NMR spectrum might result from the diradical character. The connection between diradical character and paratropic current originates from the configurational (partial) mixing of the closed-shell highest occupied molecular orbital (HOMO)2 and the HOMO, HOMO→lowest occupied molecular orbital (LUMO), LUMO configurations, which results in inbalance of the total orbital angular momentum of the molecules. This produces a paratropic ring current in the presence of an external magnetic field.
Given the local character of the conjugative effects on the meta-DR forms, the electronic structures of 3 and 4 can be understood in terms of local five- and six-membered ring currents (i.e., shaded blue in Fig. 3), for which we have calculated the NICS πzz-XY-scan plots.41 There is less aromatic character for external rings A and G in 4 than in 3, in accordance with the contributions of the meta-DR structures. Furthermore, five-membered rings B and F are more anti-aromatic in 4 than in 3, which is in parallel with the larger positive NICS πzz values for rings C and E in 4 than in 3 as a result of the increased quinoidal character of these six-membered rings in the compound with larger diradical character (i.e., greater contribution of the interaction between the radical and the lone electron pair of the N atoms).
Because NICS-scan plots exhibit certain limitations in polycyclic compounds due to the superposition of local magnetic responses,42 we have performed advanced magnetically induced current density calculations43 with the GIMIC program44 for the open-shell singlet configuration of 3 and 4 (Fig. 4). The integrations around different planes located perpendicular to selected bonds of the ring structure (Fig. S13†) allowed us to obtain the ring current strength (RCS) values for the different rings. The current density vector plots at 1.0 Å above the molecular plane show intense paratropic ring currents within the rings B–F composing the 20π-electron core. By contrast, the external six-membered rings A and G exhibit a local diatropic current, which has a larger RCS in 3 (9.0 nA T−1) than in 4 (7.0 nA T−1), in accordance with the NICS scan analysis. Although the intensity of the paratropic ring currents inside the five-membered rings B and F was higher in 4 than in 3, overall similar net RCS values were obtained, which indicates a similar aromaticity pattern in both isomers.
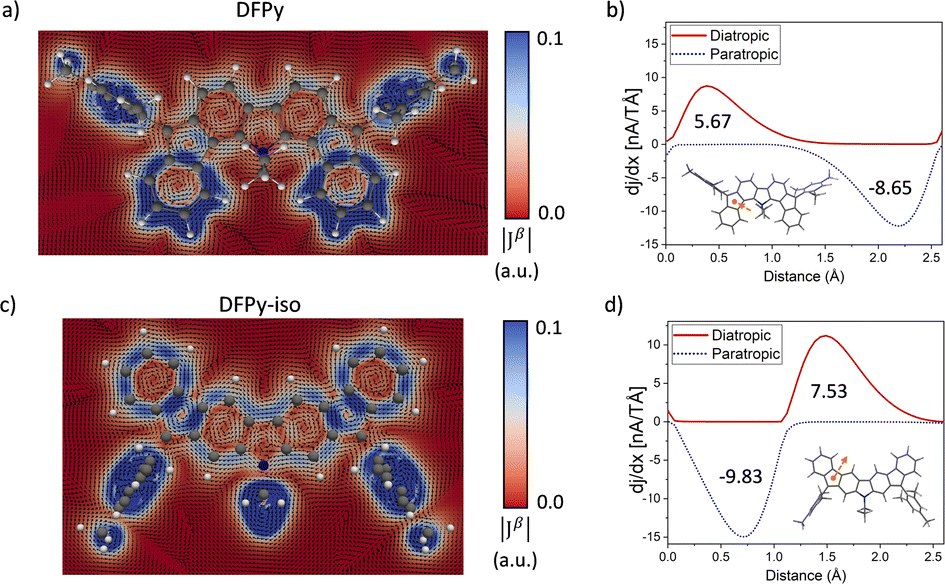 | ||
| Fig. 4 Current density analysis of 3 and 4. On the left, current density vector plots at 1.0 Å above the molecular plane highlight the π-current circuits. On the right, current strength profiles corresponding to the selected plane are indicated in the inset. | ||
Conclusions
We report the new diradical compound difluoreno[2,3-b:3′,2′-d]pyrrole (DFPy-Iso) 4, which is an electronic isomer of difluoreno[4,3-b:3′,4′-d]pyrrole (DFPy) 3. Despite their roles as isomeric parents, both molecules present substantially different diradical properties due to the interplay of secondary resonance forms, which interfere with the main canonical structures. We highlight the presence of secondary forms with local conjugation between the radical and the lone electron pair of the N either in para- or ortho-conjugation. The different extent of the competition between para-DR and meta-DR is at the origin of the different optical, magnetic, and diradical properties.The same impact of these contributions to the electronic structure was observed in the radical cations of 3 and 4, in which the meta-DR conjugation mutes into a captodative effect that stabilizes the oxidation processes in 4 as compared to 3. Diradicaloids 3 and 4 contain the same anti-aromatic 4n Hückel structures, which are equally perturbed by the formation of internal CC bonds, resulting in 20π-electron anti-aromatic characteristics that are very similar overall at the Hückel level. Despite their primordially similar Hückel anti-aromatic shapes, they develop greatly different open-shell properties. This paper highlights the importance of obtaining a detailed understanding of the electronic structure of isomer diradicals because it might become a powerful tool for the modulation of diradical character and magnetic properties without a great deal of additional chemical costs.
Data availability
The data supporting this article have been uploaded as part of the ESI.†Author contributions
J. C. and S. K. conceived the project. R. M., K. S., C. K., and M. M. performed the synthesis and chemical characterization. S. M. Q. performed the spectroscopic and electrochemical characterization. C. J. G. G. performed the SQUID measurements. M. A. performed and interpreted part of the quantum chemical calculations. J. C. and S. K. wrote and edited the manuscript. All authors reviewed and edited the manuscript. J. C., M. A., and S. K. supervised the project.Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
We thank MINECO/FEDER of the Spanish Government (project reference PID2021-127127NB-I00 and PID-2021-125907NB-I00) and the Junta de Andalucía (project reference PROYEXCEL-0328). We acknowledge JSPS KAKENHI grant (21K05042) and Research Central Services (SCAI) of the University of Málaga for the access to the facilities. S.-i. K. gratefully acknowledges the Yashima Environment Technology Foundation and the Asahi Glass Foundation for financial support. This work was partially supported by the Cooperative Research Program “Network Joint Research Center for Materials and Devices” (Kyushu University) and the Grant-in-Aid for the promotion and enhancement of education and research from the University of Shiga Prefecture (2023). Mass spectrometric data were collected at Hiroshima University (N-BARD: Ms Tomoko Amimoto). M. A. thanks the Vrije Universiteit Brussel (VUB) for financial support through a Strategic Research Program awarded to the ALGC research group.Notes and references
- D. T. Chase, B. D. Rose, S. P. McClintock, L. N. Zakharov and M. M. Haley, Angew. Chem., Int. Ed., 2011, 50, 1127–1130 ( Angew. Chem. , 2011 , 123 , 1159–1162 ) CrossRef CAS PubMed.
- A. Shimizu, R. Kishi, M. Nakano, D. Shiomi, K. Sato, T. Takui, I. Hisaki, M. Miyata and Y. Tobe, Angew. Chem., Int. Ed., 2013, 52, 6076–6079 ( Angew. Chem. , 2013 , 125 , 6192–6195 ) CrossRef CAS PubMed.
- J. J. Dressler, A. C. Valdivia, R. Kishi, G. E. Rudebusch, A. M. Ventura, B. E. Chastain, C. J. Gómez-García, L. N. Zakharov, M. Nakano and J. Casado, Chem, 2020, 6, 1353–1368 CAS.
- J. J. Dressler, M. Teraoka, G. L. Espejo, R. Kishi, S. Takamuku, C. J. Gómez-García, L. N. Zakharov, M. Nakano, J. Casado and M. M. Haley, Nat. Chem., 2018, 10, 1134–1140 CrossRef CAS PubMed.
- G. E. Rudebusch, A. G. Fix, H. A. Henthorn, C. L. Vonnegut, L. N. Zakharov and M. M. Haley, Chem. Sci., 2014, 5, 3627–3633 RSC.
- A. G. Fix, P. E. Deal, C. L Vonnegut, B. D. Rose, L. N. Zakharov and M. M. Haley, Org. Lett., 2013, 15, 1362–1365 CrossRef CAS PubMed.
- (a) T. Jousselin-Oba, M. Mamada, A. Okazawa, J. Marrot, T. Ishida, C. Adachi, A. Yassar and M. Frigoli, Chem. Sci., 2020, 11, 12194–12205 RSC; (b) S. Dong and Z. Li, J. Mater. Chem. C, 2022, 10, 2431–2449 RSC.
- D. T. Chase, A. G. Fix, S. J. Kang, B. D. Rose, C. D. Weber, Y. Zhong, L. N. Zakharov, M. C. Lonergan, C. Nuckolls and M. M. Haley, J. Am. Chem. Soc., 2012, 134, 10349–10352 CrossRef CAS PubMed.
- D. Yuan, D. Huang, S. M. Rivero, A. Carreras, C. Zhang, Y. Zou, X. Jiao, C. R. McNeill, X. Zhu, C.-a. Di, D. Zhu, D. Casanova and J. Casado, Chem, 2019, 5, 964–976 CAS.
- Z. X. Chen, Y. Li and F. Huang, Chem, 2021, 7, 288–332 CAS.
- K. Kamada, S. I. Fuku-En, S. Minamide, K. Ohta, R. Kishi, M. Nakano, H. Matsuzaki, H. Okamoto, H. Higashikawa, K. Inoue, S. Kojima and Y. Yamamoto, J. Am. Chem. Soc., 2013, 135, 232–241 CrossRef CAS PubMed.
- H. Koike, M. Chikamatsu, R. Azumi, J. y. Tsutsumi, K. Ogawa, W. Yamane, T. Nishiuchi, T. Kubo, T. Hasegawa and K. Kanai, Adv. Funct. Mater., 2016, 26, 277–283 CrossRef CAS.
- S. Lukman, J. M. Richter, L. Yang, P. Hu, J. Wu, N. C. Greenham and A. J. Musser, J. Am. Chem. Soc., 2017, 139, 18376–18385 CrossRef CAS PubMed.
- (a) C. K. Frederickson, L. N. Zakharov and M. M. Haley, J. Am. Chem. Soc., 2016, 138, 16827–16838 CrossRef CAS PubMed; (b) J. L. Marshall, K. Uchida, C. K. Frederickson, C. Schutt, A. M. Zeidell, K. P. Goetz, T. W. Finn, K. Jarolimek, L. N. Zakharov, C. Risko, R. Herges, O. D. Jurchescu and M. M. Haley, Chem. Sci., 2016, 7, 5547–5558 RSC.
- T. Xu, Y. Han, Z. Shen, X. Hou, Q. Jiang, W. Zeng, P. Wen Ng and C. Chi, J. Am. Chem. Soc., 2021, 143, 20562–20568 CrossRef CAS.
- M. Abe, Chem. Rev., 2013, 113, 7011–7088 CrossRef CAS PubMed.
- M. Nakano and B. Champagne, J. Phys. Chem. Lett., 2015, 6, 3236–3256 CrossRef CAS.
- Diradicaloids, ed. J. Wu, Jenny Stanford Publishing, New York, 2022 Search PubMed.
- A. E. Tschitschibabin, Ber. Dtsch. Chem. Ges., 1907, 40, 1810–1819 CrossRef CAS.
- J. Thiele and H. Balhorn, Ber. Dtsch. Chem. Ges., 1904, 37, 1463–1470 CrossRef.
- E. Müller and H. Pfanz, Ber. Dtsch. Chem. Ges., 1941, 74, 1051–1074 CrossRef.
- Z. Zeng, X. Shi, C. Chi, J. T. L. Navarrete, J. Casado and J. Wu, Chem. Soc. Rev., 2015, 44, 6578–6596 RSC.
- J. Casado, Top. Curr. Chem., 2017, 375, 209 CrossRef PubMed.
- Y. Tobe, Top. Curr. Chem., 2018, 376, 12 CrossRef PubMed.
- T. Kubo, Chem. Lett., 2015, 44, 111–122 CrossRef.
- J. E. Barker, T. W. Price, L. J. Karas, R. Kishi, S. N. MacMillan, L. N. Zakharov, C. J. Gómez-GarcÍa, J. I. Wu, M. Nakano and M. M. Haley, Angew. Chem., Int. Ed., 2021, 60, 22385–22392 ( Angew. Chem. , 2021 , 133 , 22559–22566 ) CrossRef CAS PubMed.
- X. Tian, J. Guo, W. Sun, L. Yuan, C. Dou and Y. Wang, Chem.–Eur. J., 2022, 28, e2022000 Search PubMed.
- A. S. Hacker, M. Pavano, J. E. Wood, H. Hashimoto, K. M. D'Ambrosio, C. K. Frederickson, J. L. Zafra, C. J. Gómez-García, V. Postils, A. R. McDonald, D. Casanova, D. K. Frantz and J. Casado, Chem. Commun., 2019, 55, 14186–14189 RSC.
- J. E. Barker, J. J. Dressler, A. Cárdenas Valdivia, R. Kishi, E. T. Strand, L. N. Zakharov, S. N. MacMillan, C. J. Gómez-García, M. Nakano, J. Casado and M. M. Haley, J. Am. Chem. Soc., 2020, 142, 1548–1555 CrossRef CAS PubMed.
- S. Mori, S. Moles Quintero, N. Tabata, R. Kishi, R. González Núñez, A. Harbuzaru, R. Ponce Ortiz, J. Marín-Beloqui, S. Suzuki, C. Kitamura, C. J. Gómez-García, Y. Dai, F. Negri, M. Nakano, S.-i. Kato and J. Casado, Angew. Chem., Int. Ed., 2022, 61, e202206680 ( Angew. Chem. , 2022 , 134 , e202206680 ) CrossRef CAS PubMed.
- S. Mori, M. Akita, S. Suzuki, M. S. Asano, M. Murata, T. Akiyama, T. Matsumoto, C. Kitamura and S.-i. Kato, Chem. Commun., 2020, 56, 5881–5884 RSC.
- S.-i. Kato, Adv. Phys. Org. Chem., 2021, 55, 41–66 CrossRef CAS.
- N. Tabata, T. Uchino, C. Kitamura, K. Yoshizawa, Y. Shiota and S.-i. Kato, Chem. Sci., 2023, 14, 5974–5982 RSC.
- D. Myśliwiec and M. Stępień, Angew. Chem., Int. Ed., 2013, 52, 1713–1717 ( Angew. Chem. , 2013 , 125 , 1757–1761 ) CrossRef PubMed.
- B. Bleaney and K. D. Bowers, Proc. R. Soc. London, Ser. A, 1952, 214, 451–465 CAS.
- H. G. Viehe, Z. Janousek, R. Merenyi and L. Stella, Acc. Chem. Res., 1985, 18, 148–154 CrossRef CAS.
- F. G. Bordwell, X.-M. Zhang and M. S. Alnajjar, J. Am. Chem. Soc., 1992, 114, 7623–7629 CrossRef CAS.
- G. E. Rudebusch, J. L. Zafra, K. Jorner, K. Fukuda, J. L. Marshall, I. Arrechea-Marcos, G. L. Espejo, R. P. Ortiz, C. J. Gómez-García, L. N. Zakharov, M. Nakano, H. Ottosson, J. Casado and M. M. Haley, Nat. Chem., 2016, 8, 753–759 CrossRef CAS PubMed.
- G. E. Rudebusch, G. L. Espejo, J. L. Zafra, M. Peña-Alvarez, S. N. Spisak, K. Fukuda, Z. Wei, M. Nakano, M. A. Petrukhina, J. Casado and M. M. Haley, J. Am. Chem. Soc., 2016, 138, 12648–12654 CrossRef CAS PubMed.
- S. Moles Quintero, M. M. Haley, M. Kerstez and J. Casado, Angew. Chem. Int. Ed., 2022, 61, e202209138 ( Angew. Chem. , 2022 , 134 , e202209138 ) CrossRef CAS PubMed.
- R. Gershoni-Poranne and A. Stanger, Chem.–Eur. J., 2014, 20, 5673–5688 CrossRef CAS PubMed.
- M. Orozco-Ic, M. Dimitrova, J. Barroso, D. Sundholm and G. Merino, J. Phys. Chem. A, 2021, 125, 5753–5764 CrossRef CAS PubMed.
- D. Sundholm, H. Fliegl and R. J. F. Berger, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2016, 6, 639–678 CAS.
- J. Jusélius, D. Sundholm and J. Gauss, J. Chem. Phys., 2004, 121, 3952–3963 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sc03297c |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2023 |