
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceConstruction of 4-hydroxycoumarin derivatives with adjacent quaternary and tertiary stereocenters via ternary catalysis†
Mengchu
Zhang
 a,
Tianyuan
Zhang
a,
Sifan
Yu
a,
Huang
Qiu
a,
Abdulla
Yusuf
b,
Xinfang
Xu
a,
Yu
Qian
*a and
Wenhao
Hu
*a
a,
Tianyuan
Zhang
a,
Sifan
Yu
a,
Huang
Qiu
a,
Abdulla
Yusuf
b,
Xinfang
Xu
a,
Yu
Qian
*a and
Wenhao
Hu
*a
aSchool of Pharmaceutical Sciences, Sun Yat-sen University, Guangzhou 510006, China. E-mail: qianyu5@mail.sysu.edu.cn; huwh9@mail.sysu.edu.cn
bCollege of Chemistry and Environmental Science, Laboratory of Xinjiang Native Medicinal and Edible Plant Resources Chemistry, Kashi University, Kashi 844000, China
First published on 16th October 2023
Abstract
4-Hydroxycoumarin derivatives represent one of the most important scaffolds in biologically active substances, pharmaceuticals and functional materials. Herein, we describe an efficient Pd/amine/Brønsted acid ternary-catalytic multicomponent reaction for the rapid construction of substituted 4-hydroxycoumarin derivatives with adjacent quaternary and tertiary stereocenters via convergent assembly of two in situ generated active intermediates. Furthermore, the late-stage transformations of coumarin derivatives and their in vitro trial of antitumor activity successfully demonstrated the potential utilities of the products as platform molecules.
Introduction
4-Hydroxycoumarin derivatives are privileged drug skeletons that are frequently encountered in pharmaceutical molecules,1 such as warfarin,2 dicoumarol,3 bothrioclinin,4 cyclocoumarol,5 pyranocoumarin,6etc. (Scheme 1a). Owing to their diverse biological activities, it is continuingly attractive to develop highly effective synthetic methods for the practical construction of coumarin scaffolds with structural diversity and complexity for drug discovery. | ||
| Scheme 1 (a) Representative examples of bioactive 4-hydroxycoumarin derivatives. (b) Methods for synthesizing pyranocoumarin derivatives. (c) Binary catalysis and ternary catalysis. (d) This work. | ||
In the past few decades, a variety of important advances have made for the construction of dihydropyran-fused coumarin derivatives with commercially available 4-hydroxycoumarin7–13 (Scheme 1b). For example, the Shivashankar group developed a Mitsunobu etherification to provide fused pyranobiscoumarin analogues from 1,2-diols at 80 °C.14 Bezuidenhoudt et al.15 and Abedi-Madiseh et al.16 also synthesized pyrano[3,2-c]coumarins with substituted chalcones at 100 °C, independently. Jeong and coworkers also reported a microwave-promoted reaction with 4-hydroxycoumarins, aldehydes, and acetophenones to afford diverse pyranocoumarins.17 Moreover, formal Friedel–Crafts alkylation between 4-hydroxycoumarins and propargylic alcohols has been reported as an efficient way to furnish multi-substituted pyranocoumarins by different research groups.18–21 However, approaches toward these scaffolds were limited to using bench-stable reagents and usually under harsh conditions, such as high temperature and microware-mediated conditions.22 Considering these inherent drawbacks, highly effective synthetic methods with operational simplicity and mild conditions to construct functionalized coumarin derivatives are still highly desired.
Multicomponent reactions have emerged as a powerful strategy for efficient synthesis of complex heterocycles.23–30 In past decades, an astounding number of binary-catalytic multicomponent reactions via interception of active ylide intermediates or zwitterionic intermediates derived from different carbene precursors have been developed by Gong,31,32 Schneider,33 Wang,34 us35–42 and others,43,44 providing an efficient access to structurally diverse and functionalized heterocyclic scaffolds with one-pot operation under mild conditions (Scheme 1c). However, the development of ternary catalysis systems among higher-order multicomponent reactions is sluggish. The challenges of ternary catalysis are (1) the construction of a compatible reaction system to form two in situ intermediates; (2) the matching issue between two different active intermediates; and (3) the selectivity issue for the convergent assembly of abovementioned two intermediates. In our latest studies,45,46 we reported a novel ternary-catalytic higher-order multicomponent reaction via assembly of in situ generated α,β-unsaturated iminium ions with zwitterionic or ylide intermediates under operationally simple conditions. Inspired by these advances47–50 and as a continuation of our interest in ternary-catalytic higher-order multicomponent reactions, we envisioned that 4-hydrocoumarin might be used as a coupling partner in ternary-catalytic multicomponent reactions to furnish substituted coumarin derivatives via carbene gem-difunctionalization process. Notably, we found that 4-hydroxycoumarins and alkynaldehydes could generate active electrophilic intermediates under catalysis of phosphoric acid and amine catalyst, which provided a fundamental basis to develop an effective and mild protocol for the synthesis of functionalized coumarin derivatives through electrophilic interception of ylide or zwitterionic intermediates. Herein, we report a ternary-catalytic multicomponent reaction through interception of zwitterionic intermediates with active electrophilic intermediates, in situ generated from 4-hydroxycoumarins and alkynaldehydes, enabling convenient access to pyranocoumarins with good yields and diastereoselectivities. More importantly, such coumarin intermediates could also be trapped by oxonium ylide intermediates derived from diazoacetates and alcohols, leading to a valuable and efficient four-component reaction to build diverse coumarin skeletons with adjacent quaternary and tertiary stereocenters.
Results and discussion
To validate our speculation, we chose commercially available 3-phenylpropiolaldehyde (2a), 4-hydroxy-2H-chromen-2-one (3a), and 2-diazo-N-methyl-N-(p-tolyl)propanamide (1a) as benchmark substrates for the optimization (Table 1). The desired three-component product 4af could be obtained in 85% yield with diastereomeric ratio (dr) > 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 when 1a and 2a were added slowly to [PdCl(η3-C3H5)]2 (5.0 mol%), racemic phosphoric acid (rac-PA) (10.0 mol%), and 4-bromoaniline (50.0 mol%) at −10 °C with 4 Å molecular sieves (MS) as an additive in PhCl for one hour (entry 1). Control experiments revealed that 4-bromoaniline and [PdCl(η3-C3H5)]2 catalyst proved critical to the overall success of the transformation (entries 2 and 3). The absence of rac-PA led the yield of 4af decreasing to 48% (entry 4) and only trace amounts of desired product were observed without 4 Å MS (entry 5). Replacing [PdCl(η3-C3H5)]2 with other metal catalysts such as Rh(II), Cu(I), Ag(I) and Fe(III) provided either lower yields or decreased diastereoselectivities (entries 6 and 7). Solvent-screening (entries 8–10) revealed that PhCl was the best choice, leading to a satisfactory outcome. Moreover, increasing or reducing the reaction temperature was proved to be ineffective and could be detrimental to yield and dr (entries 11 and 12). Importantly, the relative stereochemistry of 4af-major and 4af-minor was unambiguously confirmed by X-ray analysis.
:1 when 1a and 2a were added slowly to [PdCl(η3-C3H5)]2 (5.0 mol%), racemic phosphoric acid (rac-PA) (10.0 mol%), and 4-bromoaniline (50.0 mol%) at −10 °C with 4 Å molecular sieves (MS) as an additive in PhCl for one hour (entry 1). Control experiments revealed that 4-bromoaniline and [PdCl(η3-C3H5)]2 catalyst proved critical to the overall success of the transformation (entries 2 and 3). The absence of rac-PA led the yield of 4af decreasing to 48% (entry 4) and only trace amounts of desired product were observed without 4 Å MS (entry 5). Replacing [PdCl(η3-C3H5)]2 with other metal catalysts such as Rh(II), Cu(I), Ag(I) and Fe(III) provided either lower yields or decreased diastereoselectivities (entries 6 and 7). Solvent-screening (entries 8–10) revealed that PhCl was the best choice, leading to a satisfactory outcome. Moreover, increasing or reducing the reaction temperature was proved to be ineffective and could be detrimental to yield and dr (entries 11 and 12). Importantly, the relative stereochemistry of 4af-major and 4af-minor was unambiguously confirmed by X-ray analysis.
|
|
|||
|---|---|---|---|
| Entry | Deviation from the “standard conditions” | Yieldb (%) | drc |
|
a The reactions were conducted on a 0.1 mmol scale: 1a:2a:3a = 1.5:1.2:1.0, [PdCl(η3-C3H5)]2 (5.0 mol%), racemic phosphoric acid (rac-PA) catalyst (10 mol%), 4-BrC6H4NH2 (50 mol%), 4 Å MS (100 mg). 1a, 2a in 1.0 mL solvent were added into a solution of 3a, [PdCl(η3-C3H5)]2, rac-PA, 4-BrC6H4NH2, and 100 mg 4 Å MS in 1.0 mL solvent via a syringe pump for 1 h, and the resulting mixture was stirred overnight.
b Determined by 1H NMR spectroscopy analyses using 1,3,5-trimethoxybenzene as an internal standard.
c Determined by 1H NMR spectroscopy analyses.
d Isolated yields.
|
|||
| 1 | None | 85 (83)d | >20:1 |
| 2 | Without 4-BrC6H4NH2 | <5 | — |
| 3 | Without [PdCl(η3-C3H5)]2 | <5 | — |
| 4 | Without rac-PA | 48 | >20:1 |
| 5 | Without 4 Å MS | 10 | >20:1 |
| 6 | Rh2(OAc)4, Cu(MeCN)4PF6, AgOTf, or FeTPPCl instead of [PdCl(η3-C3H5)]2 | <5 | — |
| 7 | [PdCl(π-cinnyl)]2 instead of [PdCl(η3-C3H5)]2 | 67 | 93:7 |
| 8 | THF instead of PhCl | 53 | 76:24 |
| 9 | DCM instead of PhCl | 67 | >20:1 |
| 10 | Toluene instead of PhCl | 86 | 85:15 |
| 11 | Reaction performed at 0 °C | 63 | 92:8 |
| 12 | Reaction performed at −20 °C | 71 | >20:1 |

With the optimal reaction conditions in hand, we sought to explore the generality of this ternary-catalytic multicomponent reaction. As shown in Scheme 2, a variety of substituted coumarin derivatives was easily obtained with moderate to good yield and up to dr > 20:1. We firstly investigated the scope of diazoacetamides 1 by employing commercially available 2a and 3a as reaction partners. With regard to various R1 groups of diazoacetamides, methyl and ethyl substituents were well tolerated, furnishing corresponding products 4aa and 4ab in 63% and 70% yields, respectively. N-Substituted diazoacetamides tethered with ethyl, isopropyl and benzyl substituents also reacted smoothly to afford 4ac–4ae in high yields (70–80%) with dr > 20:1. Moreover, due to the electronic effect, diazoacetamides with an electron-donating substituent on the aryl group worked better than electron-withdrawing substituents, delivering the desired products with better yields and diastereoselectivities (4af–4ah). We then turned our attention to exploring the scope of substituted coumarins 2. Owing to poor solubility of substituted coumarins in PhCl, the reaction was performed in mixed solvent (PhCl:THF = 1:1). Generally, a wide range of 4-hydroxycoumarins possessing different functional groups, such as 6-F, 6-Cl, 6-Br, 6-NO2, 6-methyl and 7-methyl, were all amenable to the transformation, yielding the desired products (4ba–4bf) with moderate to good yields and diastereoselectivities. Of note, six-membered ring-containing 4-hydroxy-6-methyl-2-pyrone also worked well and gave corresponding product 4bg in 56% yield and dr > 20:1. Lastly, a broad array of alkynaldehydes were checked for their substrate scope. As expected, this catalytic process was well compatible with different alkynaldehydes bearing electron-donating and electron-withdrawing substituents at the para-, ortho-, and meta-positions on the aromatic ring, leading to three-component products (4ca–4cg) with good yields (40–76%) and excellent diastereoselectivities. Importantly, alkynaldehydes bearing electron-donating substituents seemed to be much superior to those bearing electron-withdrawing substituents for the yields of products on account of electronic effect. It is worth mentioning that alkynaldehydes bearing an extended aromatic ring such as biphenyl and 1-naphthyl were readily converted to desired products (4ch and 4ci) in high yields and dr > 20:1. Apart from aromatic alkynaldehydes, a long-chain alkyl with alkynaldehyde substrate was also tolerant to this reaction, producing 4cj in moderate yield and excellent diastereoselectivity.
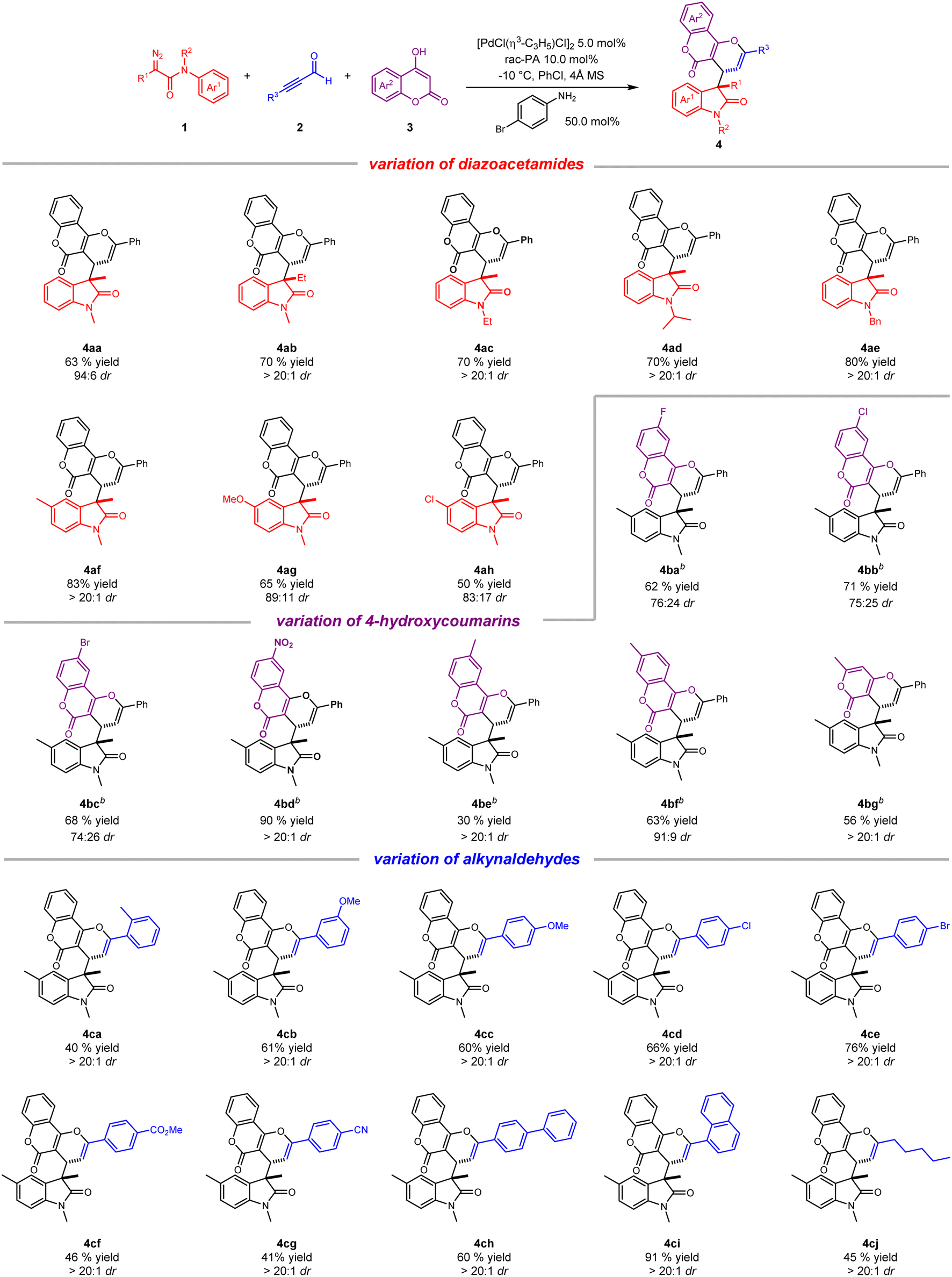 | ||
| Scheme 2 Substrate scope for the synthesis of 4-hydroxycoumarin derivativesa. a Reaction conditions: 1 (0.15 mmol), 2 (0.12 mol), 3 (0.10 mol), [PdCl(η3-C3H5)]2 (5.0 mol%), rac-PA (10 mol%), 4-BrC6H4NH2 (50 mol%), 4 Å MS (100 mg), PhCl (2 mL), −10 °C, 12 h. Isolated yields. The dr was determined by 1H NMR spectroscopy analyses of the crude reaction mixture. b Mixed solvent (PhCl:THF = 1:1, 2 mL) was used. | ||
In an effort to exploit the generality of this ternary-catalytic higher-order multicomponent reaction to synthesize 4-hydrocoumarin derivatives, we next pursued the development of an efficient four-component reaction to directly trap oxonium ylides in situ generated from diazoacetates and alcohols by coumarin intermediates (Table 2). Encouragingly, a desirable four-component reaction with diazoacetate 5a, 4-bromobenzyl alcohol 6a, phenylpropiolaldehyde 2a and 4-hydroxycoumarin 3a was accomplished by skillfully altering the metal catalyst and amine catalyst as well as modifying the loading of reaction substrates, leading to functionalized 4-hydroxycoumarin derivatives with newly formed quaternary and tertiary stereocenters (Table 2). The best result was obtained under the catalysis of Rh2(OAc)4, 4-nitroaniline, and rac-PA, leading to the corresponding product 7a with 78% isolated yield and dr > 20:1 (entry 1). The structure of 7a was unambiguously confirmed by a later X-ray diffraction study. Control experiments showed that 4-nitroaniline, Rh2(OAc)4, rac-PA and 4 Å MS were indispensable for the higher-order multicomponent reaction (entries 2–5). In particular, replacing Rh2(OAc)4 with [PdCl(η3-C3H5)]2 gave lower yield (entry 6). This protocol enabled operationally facile access to valuable coumarin derivatives with adjacent quaternary and tertiary stereocenters using common, bench-stable, and readily accessible starting materials.
|
|
|||
|---|---|---|---|
| Entry | Deviation from the “standard conditions” | Yieldb (%) | drc |
|
a The reactions were conducted on a 0.1 mmol scale: 5a:6a:2a:3a = 1.5:3.0:3.0:1.0, Rh2(OAc)4 (5.0 mol%), rac-PA (10 mol%), 4-NO2C6H4NH2 (50 mol%) and 4 Å MS (100 mg). 5a, 2a in 1.0 mL DCM were added into a solution of 6a, 3a, Rh2(OAc)4, rac-PA, 4-NO2C6H4NH2, and 100 mg 4 Å MS in 1.0 mL DCM via a syringe pump for 1 hour, and the resulting mixture was stirred for another 2 hours.
b Determined by 1H NMR spectroscopy analyses using 1,3,5-trimethoxybenzene as an internal standard.
c Determined by 1H NMR spectroscopy analyses.
d Isolated yield.
|
|||
| 1 | None | 80 (78)d | >20:1 |
| 2 | Without 4-NO2C6H4NH2 | <5 | — |
| 3 | Without Rh2(OAc)4 | <5 | — |
| 4 | Without rac-PA | <5 | — |
| 5 | Without 4 Å MS | 49 | >20:1 |
| 6 | [PdCl(η3-C3H5)]2 in place of Rh2(OAc)4 | 36 | >20:1 |

Late-stage modifications of bioactive molecules have become an efficient tool for rapid access to new drug candidates. To showcase the practicability of the novel multicomponent method for late-stage synthesis, a series of drugs and their derivatives were evaluated (Scheme 3). For example, natural alcohols, such as piperonyl alcohol, perilla alcohol, citronellol, geraniol and phytol, could be easily transformed into desired products (7b–7f) with satisfactory yields and good diastereoselectivities. Furthermore, drug derived substrate like 1,2:3,4-di-O-isopropylidene-D-galactopyranose was compatible with this protocol to furnish the corresponding product 7g with good yield and moderate diastereoselectivity. These results highlighted the excellent scalability of our newly developed ternary-catalytic method.
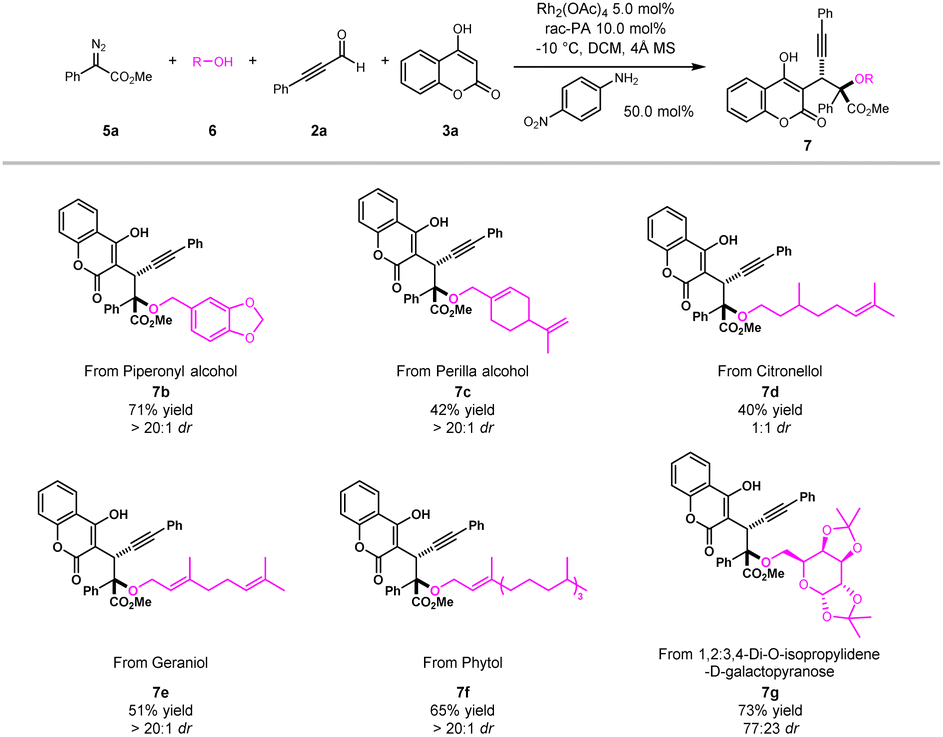 | ||
| Scheme 3 Late-stage functionalization of complex and biorelevant moleculesa. a Reaction conditions: 5a (0.30 mmol), 6 (0.60 mol), 2a (0.60 mol), 3a (0.20 mol), Rh2(OAc)4 (5.0 mol%), rac-PA (10 mol%), 4-NO2C6H4NH2 (50 mol%), 4 Å MS (100 mg), DCM (4 mL), −10 °C, 3 h. Isolated yields. The dr was determined by 1H NMR spectroscopy analyses. | ||
To demonstrate the practicability of this reaction, we carried out a large-scale synthesis (Scheme 4a). The desired product 4ae was obtained in 79% yield (0.41 g) with dr > 20:1. Under the catalysis of Pd/C and H2, the reduction of 4ae could be realized, providing unexpected product 8 in 85% yield with maintained stereoselectivity (Scheme 4b). The relative configuration of 8 was determined by X-ray diffraction analysis.
 | ||
| Scheme 4 Synthetic transformations and preliminary mechanistic investigation. | ||
Later, representative compounds (4ab, 4ae, 4af, 4bc, 4be, 4cc, 4cd, and 4cf) were subjected to an anticancer activity test using cell viability via the CCK8 assay for HCT116 (colon cancer), MCF-7 (breast cancer) and SJSA-1 (osteosarcoma) human cancer cell lines (see the ESI† for details). The results showed that these pyrano[3,2-c]coumarin scaffolds exhibited significant and broad anticancer potency, especially compound 4ab (HCT116, IC50 < 10 μM).51–53 Furthermore, to shed light on the reaction mechanism, a series of mechanistic exploration experiments were performed. At first, when alkynaldehyde 2a was treated with 4-hydroxycoumarin 3a for 1 h, the subsequent addition of diazoacetamides 1a was unable to afford the Michael-type addition product 4af which cast doubt on the existence of intermediate D (Scheme 4c). This confusing result prompted us to identify the intermediate derived from alkynaldehyde 2a and 4-hydroxycoumarin 3a. Interestingly, the reaction of 4-hydroxycoumarin 3a with the same equivalent of alkynaldehyde 2a without any other additive could rapidly afford the adduct 9 with 50% yield, which showed the strong nucleophilicity of 3a and proved the occurrence of intermediate D (Scheme 4d). Furthermore, the reaction of diazoacetamides 1a with the adduct 9 failed to afford the desired product 4af (Scheme 4e), indicating that the adduct 9 was not the intermediate during the whole reaction time. We then deliberately prepared other envisioned intermediates: 2-phenyl-4H,5H-pyrano[3,2-c]chromen-5-one (10) and 1,3,5-trimethylindolin-2-one (11). The reaction of 1a with 10 under standard conditions did not give targeted product 4af (Scheme 4f). Moreover, the reaction of 11 in the presence of alkynaldehyde 2a and 4-hydroxycoumarin 3a was monitored and no formation of 4af was observed (Scheme 4g). These results implied that neither 10 nor 11 was the intermediate of this multicomponent reaction and proved that a synergetic process involving convergent assembly of two in situ generated active intermediates might be more plausible.
On the basis of the aforementioned results and previous reports,45,46 a plausible reaction pathway was proposed as shown in Scheme 5. Initially, the imine intermediate Ain situ generated from alkynaldehydes 3 in the presence of rac-PA and amine catalysts could easily react with 4-hydroxycoumarins 2 to deliver intermediate B. Under amine-PA catalysis, the phosphate anion-coordinated species D was derived from the intermediate C after releasing the amine catalyst. Additionally, the existence of intermediate D (calculated m/z 275.0703) was confirmed through ESI-HRMS analyses of the MCR mixture (observed m/z 275.0714) (for more details see ESI†).
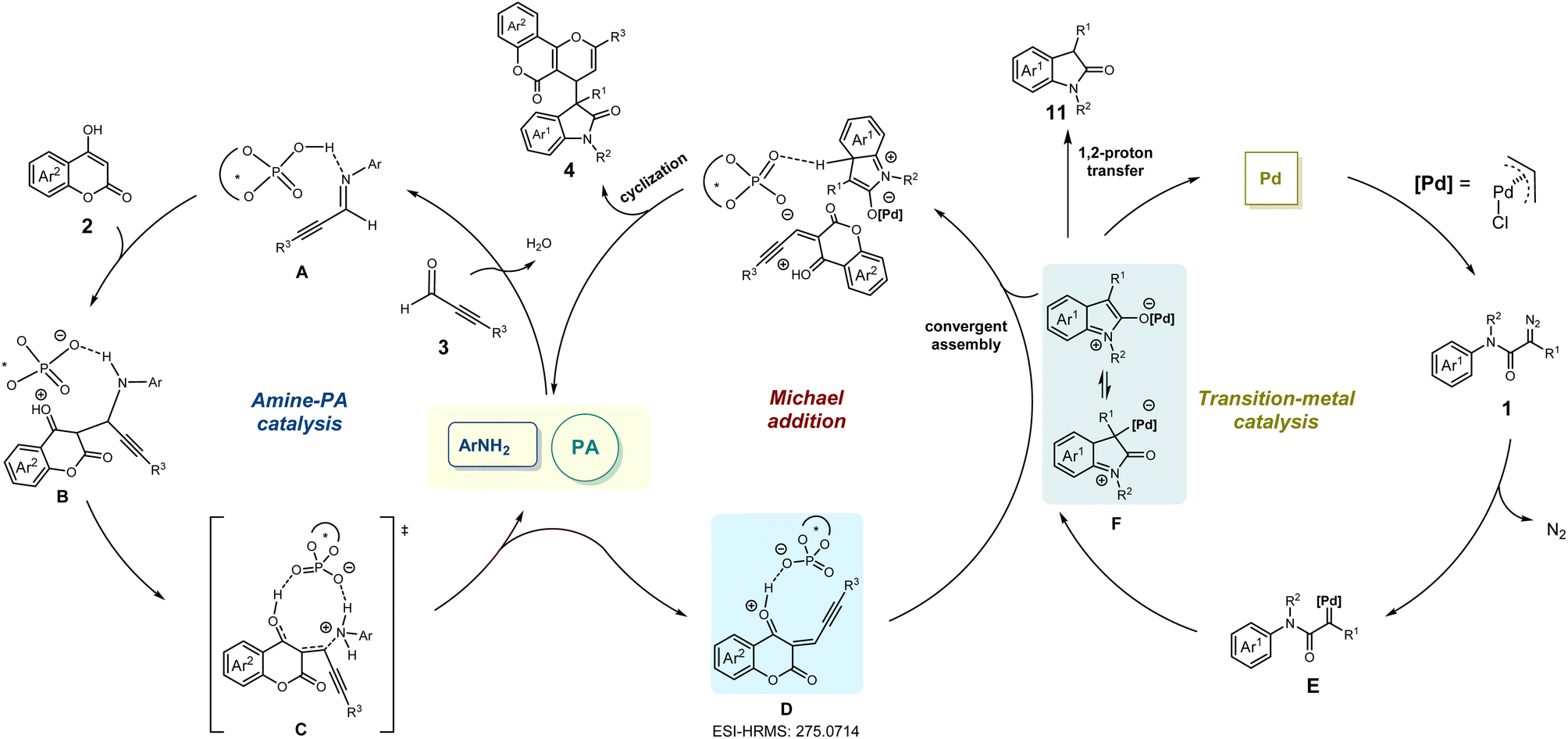 | ||
| Scheme 5 Proposed mechanism. | ||
Meanwhile, a palladium carbene intermediate E was obtained by extraction of nitrogen from diazoacetamides 1 under the catalysis of palladium(II) complex, which could convert into zwitterionic intermediate or enolate form Fvia an intramolecular interception process. Finally, such zwitterionic intermediate or enolate form F reacted with active electrophilic intermediate D, followed by intramolecular cyclization to furnish the multi-component product 4.
Conclusions
In conclusion, we have developed a straightforward protocol for building 4-hydroxycoumarin scaffolds via coupling of two in situ generated active intermediates, leading to an array of coumarin derivatives bearing adjacent quaternary and tertiary stereocenters with moderate to good yields and diastereoselectivities. The method features ternary catalysis, broad substrate scope, and mild reaction conditions. This strategy could inspire more efforts to exploit the in situ generated active intermediate for the synthesis of polyfunctionalized skeletons for drug discovery.Data availability
Further experimental details, synthetic procedures, characterization data, copies of NMR spectra and X-ray crystallographic data are available in the ESI.†Author contributions
M. Z., S. Y., H. Q., Y. Q. and W. H. conceived and designed the project; M. Z. conducted most of the experiments; T. Z. performed the anticancer activity test on cell viability with most compounds; M. Z., A. Y. and X. X. analysed the data and contributed to the preparation of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Support for this research from the National Natural Science Foundation of China (92056201, 22171294, 81973176, 22001268), the Natural Science Foundation of Guangdong Province, China (2021A1515010401), Key-Area Research and Development Program of Guangdong Province (2022B1111050003), Guangdong Provincial Key Laboratory of Construction Foundation (2023B1212060022), Guangdong Natural Science Fund (2023A1515011486) and the Key Laboratory Open Project of Xinjiang Uygur Autonomous Region (2021D04018) is gratefully acknowledged.Notes and references
- D. Cao, Z. Liu, P. Verwilst, S. Koo, P. Jangjili, J. S. Kim and W. Lin, Chem. Rev., 2019, 119, 10403–10519 CrossRef CAS PubMed.
- T. Nisar, H. Sutherland-Foggio and W. Husar, Lancet Neurol., 2020, 19, 35 CrossRef PubMed.
- C. Sun, W. Zhao, X. Wang, Y. Sun and X. Chen, Pharmacol. Res., 2020, 160, 105193 CrossRef CAS PubMed.
- C. Wang, Q. Zheng, Y. Lu, Y. Xiao, J. Li and Y. Ding, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2003, 59, O593–O595 CrossRef PubMed.
- A. M. Rayar, N. Lagarde, F. Martin, F. Blanchard, B. Liagre, C. Ferroud, J. F. Zagury, M. Montes and M. Sylla-Iyarreta Veitia, Eur. J. Med. Chem., 2018, 146, 577–587 CrossRef CAS PubMed.
- M. T. Khandy, A. K. Sofronova, T. Y. Gorpenchenko and N. K. Chirikova, Plants, 2022, 11, 3135–3167 CrossRef CAS PubMed.
- Y. Li, Y.-C. Hu, H. Zheng, D.-W. Ji, Y.-F. Cong and Q.-A. Chen, Eur. J. Org Chem., 2019, 2019, 6510–6514 CrossRef CAS.
- J. Zhang, G. Yin, Y. Du, Z. Yang, Y. Li and L. Chen, J. Org. Chem., 2017, 82, 13594–13601 CrossRef CAS PubMed.
- C. Ren, F. Wei, Q. Xuan, D. Wang and L. Liu, Adv. Synth. Catal., 2016, 358, 132–137 CrossRef CAS.
- Z. W. Qiu, X. T. Xu, H. P. Pan, Z. S. Jia, A. J. Ma, J. B. Peng, J. Y. Du, N. Feng, B. Q. Li and X. Z. Zhang, J. Org. Chem., 2021, 86, 6075–6089 CrossRef CAS PubMed.
- S. Mahato, S. Santra, R. Chatterjee, G. V. Zyryanov, A. Hajra and A. Majee, Green Chem., 2017, 19, 3282–3295 RSC.
- R. Sarkar, S. Mitra and S. Mukherjee, Chem. Sci., 2018, 9, 5767–5772 RSC.
- C. Theunissen, J. Wang and G. Evano, Chem. Sci., 2017, 8, 3465–3470 RSC.
- N. Jagadishbabu and K. Shivashankar, J. Heterocycl. Chem., 2017, 54, 1543–1549 CrossRef CAS.
- M. Gohain, J. H. van Tonder and B. C. B. Bezuidenhoudt, Tetrahedron Lett., 2013, 54, 3773–3776 CrossRef CAS.
- M. Lashkari, M. Ghashang and A. Abedi-Madiseh, Org. Prep. Proced. Int., 2020, 53, 52–58 CrossRef.
- M. Veeranarayana Reddy, B. Siva Kumar, K. T. Lim, B. G. Cho and Y. T. Jeong, Tetrahedron Lett., 2016, 57, 476–478 CrossRef CAS.
- X. Lin, X. Dai, Z. Mao and Y. Wang, Tetrahedron, 2009, 65, 9233–9237 CrossRef CAS.
- Z. Yue, Z. Wang, Y. Zhang, X. Chen, P. Li and W. Li, Org. Biomol. Chem., 2022, 20, 6334–6338 RSC.
- H. M. Tanuraghaj and M. Farahi, RSC Adv., 2018, 8, 27818–27824 RSC.
- Q. Ren, J. Kang, M. Li, L. Yuan, R. Chen and L. Wang, Eur. J. Org. Chem., 2017, 2017, 5566–5571 CrossRef CAS.
- B. Borah, K. Dhar Dwivedi and L. R. Chowhan, Asian J. Org. Chem., 2021, 10, 3101–3126 CrossRef CAS.
- X. Guo and W. Hu, Acc. Chem. Res., 2013, 46, 2427–2440 CrossRef CAS PubMed.
- D. Zhang and W. Hu, Chem. Rec., 2017, 17, 739–753 CrossRef CAS PubMed.
- Y. Xia, D. Qiu and J. Wang, Chem. Rev., 2017, 117, 13810–13889 CrossRef CAS PubMed.
- S. F. Zhu and Q. L. Zhou, Acc. Chem. Res., 2012, 45, 1365–1377 CrossRef CAS PubMed.
- M. Zhang, X. Xu and W. Hu, in Transition Metal-Catalyzed Carbene Transformations, ed. C. M. Chi, J. Wang, and M. P. Doyle, Wiley-VCH GmbH, 2022, ch. 11, pp. 325–369, DOI:10.1002/9783527829170.ch11.
- D. N. Huh, Y. Cheng, C. W. Frye, D. T. Egger and I. A. Tonks, Chem. Sci., 2021, 12, 9574–9590 RSC.
- K. Li, Y. Lv, Z. Lu, X. Yun and S. Yan, Green Synth. Catal., 2022, 3, 59–68 CrossRef.
- M. Wang, X. Meng, C. Cai, L. Wang and H. Gong, Green Synth. Catal., 2022, 3, 168–174 CrossRef.
- L. Ren, X. L. Lian and L. Z. Gong, Chem, 2013, 19, 3315–3318 CrossRef CAS PubMed.
- D.-F. Chen, C.-l. Zhang, Y. Hu, Z.-Y. Han and L.-Z. Gong, Org. Chem. Front., 2015, 2, 956–960 RSC.
- S. K. Alamsetti, M. Spanka and C. Schneider, Angew. Chem., Int. Ed., 2016, 55, 2392–2396 CrossRef CAS PubMed.
- X. S. Liang, R. D. Li and X. C. Wang, Angew. Chem., Int. Ed., 2019, 58, 13885–13889 CrossRef CAS PubMed.
- D. Zhang, X. Wang, M. Zhang, Z. Kang, G. Xiao, X. Xu and W. Hu, CCS Chem., 2020, 2, 432–439 CrossRef CAS.
- K. Hong, S. Dong, X. Xu, Z. Zhang, T. Shi, H. Yuan, X. Xu and W. Hu, ACS Catal., 2021, 11, 6750–6756 CrossRef CAS.
- S. Zhou, Q. Liu, M. Bao, J. Huang, J. Wang, W. Hu and X. Xu, Org. Chem. Front., 2021, 8, 1808–1816 RSC.
- K. Hong, J. Shu, S. Dong, Z. Zhang, Y. He, M. Liu, J. Huang, W. Hu and X. Xu, ACS Catal., 2022, 12, 14185–14193 CrossRef CAS.
- X. Yang, K. Hong, S. Zhang, Z. Zhang, S. Zhou, J. Huang, X. Xu and W. Hu, ACS Catal., 2022, 12, 12302–12309 CrossRef CAS.
- S. Dong, K. Hong, Z. Zhang, J. Huang, X. Xie, H. Yuan, W. Hu and X. Xu, Angew. Chem., Int. Ed., 2023, e202302371, DOI:10.1002/anie.202302371.
- M. Zhang, Y. Li, Y. Wang, J. Shu, T. Zhang, D. Zhang, S. Cai, T. Shi and W. Hu, Green Synth. Catal., 2023 DOI:10.1016/j.gresc.2023.04.006.
- X. Tian, X. Xu, T. Jing, Z. Kang and W. Hu, Green Synth. Catal., 2021, 2, 337–344 CrossRef.
- Z. Y. Cao, Y. L. Zhao and J. Zhou, Chem. Commun., 2016, 52, 2537–2540 RSC.
- C. Ma, J. Y. Zhou, Y. Z. Zhang, Y. Jiao, G. J. Mei and F. Shi, Chem.–Asian J., 2018, 13, 2549–2558 CrossRef CAS PubMed.
- R. Y. Hua, S. F. Yu, X. T. Jie, H. Qiu and W. H. Hu, Angew. Chem., Int. Ed., 2022, 61, e202213407 CrossRef CAS PubMed.
- S. Yu, W. Chang, R. Hua, X. Jie, M. Zhang, W. Zhao, J. Chen, D. Zhang, H. Qiu, Y. Liang and W. Hu, Nat. Commun., 2022, 13, 7088 CrossRef CAS PubMed.
- R. Gurubrahamam, B. F. Gao, Y. M. Chen, Y. T. Chan, M. K. Tsai and K. Chen, Org. Lett., 2016, 18, 3098–3101 CrossRef CAS PubMed.
- D. Hack, P. Chauhan, K. Deckers, G. N. Hermann, L. Mertens, G. Raabe and D. Enders, Org. Lett., 2014, 16, 5188–5191 CrossRef CAS PubMed.
- V. Modrocka, E. Veverkova, M. Meciarova and R. Sebesta, J. Org. Chem., 2018, 83, 13111–13120 CrossRef CAS PubMed.
- F. F. Wolf, H. Klare and B. Goldfuss, J. Org. Chem., 2016, 81, 1762–1768 CrossRef CAS PubMed.
- M. Alam, A. Khan, A. Wadood, A. Khan, S. Bashir, A. Aman, A. K. Jan, A. Rauf, B. Ahmad, A. R. Khan and U. Farooq, Front. Pharmacol., 2016, 7, 1–6 Search PubMed.
- Y. Sugiyama, S. Nakamura, Y. Tokuda, M. Nakano, Y. Hattori, H. Nishiguchi, Y. Toda, S. Hosogi, M. Yamashita, K. Tashiro and E. Ashihara, Biochem. Biophys. Res. Commun., 2023, 638, 200–209 CrossRef CAS PubMed.
- M. H. Lin, C. H. Cheng, K. C. Chen, W. T. Lee, Y. F. Wang, C. Q. Xiao and C. W. Lin, Chem.-Biol. Interact., 2014, 218, 42–49 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2264577, 2264587, 2264580, 2269933. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3sc03452f |
| This journal is © The Royal Society of Chemistry 2023 |