DOI:
10.1039/D3SC03745B
(Edge Article)
Chem. Sci., 2023,
14, 11840-11849
Pd12MnL24 (for n = 6, 8, 12) nanospheres by post-assembly modification of Pd12L24 spheres†
Received
19th July 2023
, Accepted 15th September 2023
First published on 28th September 2023
Abstract
In this contribution, we describe a post-assembly modification approach to selectively coordinate transition metals in Pd12L24 cuboctahedra. The herein reported approach involves the preparation of Pd12L24 nanospheres with protonated nitrogen donor ligands that are covalently linked at the interior. The so obtained Pd12(LH+)24 nanospheres are shown to be suitable for coordinative post-modification after deprotection by deprotonation. Selective formation of tetra-coordinated MB in Pd12MB6L24, tri-coordinated MB in Pd12MB8L24 nanospheres and two-coordinated MB in Pd12MB12L24 nanospheres is achieved as a result of different nitrogen donor ligands. A combination of pulsed EPR spectroscopy (DEER) to measure Cu–Cu distances in the different spheres, NMR studies and computational investigations, support the presence of the complexes at precise locations of the Pd12MB6L24 nanosphere. The general post-assembly modification methodology can be extended using other transition metal precursors or supramolecular systems and can guide precise formation and investigation of novel transition metal-complex containing nanospheres with well-defined composition.
Introduction
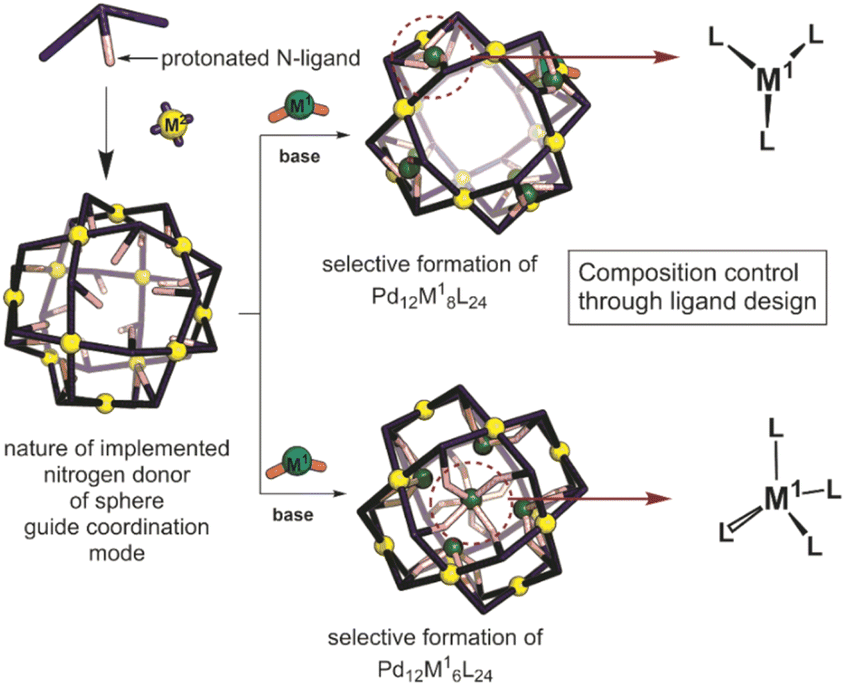
Inspired by the importance of self-assembly processes found in natural systems, supramolecular chemistry has evolved as a strategy to generate functional large molecular objects. In this context, a wide variety of polyhedral coordination cages and nanospheres with the general formula MnL2n, n being 2, 3, 4, 6, 8, 9, 10, 12, 24 and 30 have been reported.1–8 Most of these symmetrical structures can be described as Platonic or Archimedean type solids which are formed from the square-planar complexation of metal ions with ditopic pyridine linkers, most often based on palladium. Next to the highly symmetrical nanospheres, lower symmetry analogues based on two different ditopic ligands can be formed, provided that the ligands are complementary in geometry.9–19 In the same manner, more complex structures have been reported such as stellated20 and “pregnant”21 cuboctahedra. Whereas most nanospheres reported are based on one type of metal center for the construction of the structure, supramolecular nanospheres bearing two different metals also emerged quite recently.23–31 Construction of bimetallic Archimedean solids has been mainly achieved by using non-symmetrical linkers,26 with preferred coordination to the corresponding two different metals used during the assembly process. In a typical procedure, the non-symmetrical building block is first coordinated to the first metal, obtaining a metal complex as the building block that is used in the next step for the formation of supramolecular nanospheres. If the coordination chemistry is sufficiently complementary, all three components can be mixed to yield the desired nanosphere in one step.26 Precisely formed nanospheres based on different metals can display stimuli responsive properties, for example by opening and closing mechanisms using a chemical signal, reminiscent of analogous metal triggered open-closing switches found in spherical capsids.21,32 Several [PdxMyLz] have been reported that have been prepared by a bottom-up approach, i.e., first preparation of MLx complex which is then used for sphere synthesis; however, there are only very few examples of a post-assembly modification approach.20–22 Compatibility issues between the first and the second coordination event complicate such strategies. Typically, the presence of ligands required for the formation of the coordination complex interferes with the sphere formation. Reported approaches can yield unique systems with interesting electrochemical, magnetic and sensing features.27 However, limitations are found when a certain number of metals need to be implemented inside of nanocages and not on the rim/framework making novel post-assembly modification techniques highly desirable.
In a post-assembly modification approach, polyhedral structures can be formed by classical design principles (Fig. 1).33,34 Furthermore, post-assembly modification approaches can be easily implemented into many known coordination-based systems, as the sphere-forming sides remain unchanged. Herein we describe a method for the selective formation of [Pd12M6L24], [Pd12M8L24] and [Pd12M12L24] nanospheres by post-modification of Pd12L24 nanospheres (Fig. 1). The reported procedure provides a new strategy for implementation of transition-metal complexes into supramolecular nanospheres. With our approach, we provide a general methodology for selective coordination post-modification of M12L24 nanospheres which potentially can be applied to other supramolecular systems or other ligand frameworks (e.g., phosphines and N-heterocyclic carbenes). The obtained nanospheres with specific amount of metals are anticipated to be useful for catalytic applications in which location and amount of M in a confined environment within nanospheres can be modified with unprecedented accuracy.
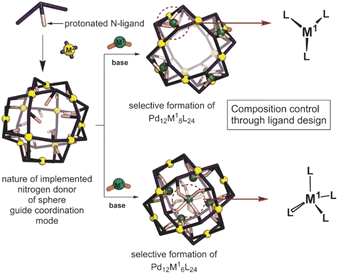 |
| | Fig. 1 Schematic representation showing the herein applied approach for the formation of bimetallic self-assemblies. | |
Results
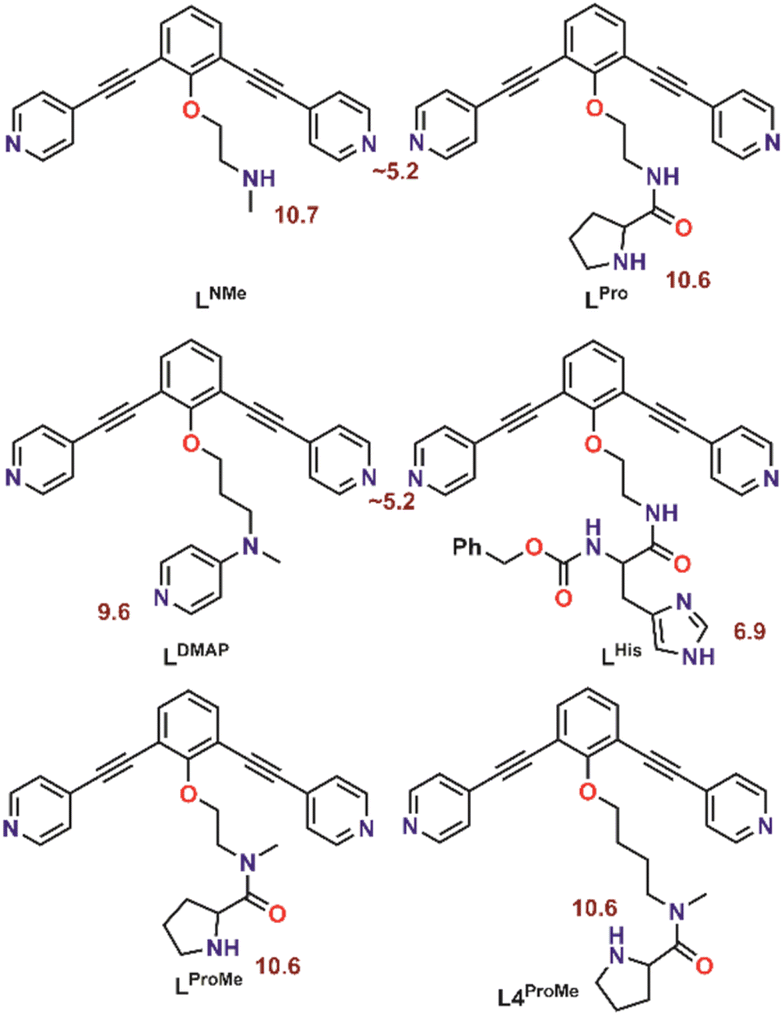
With the goal to prepare nanospheres by self-assembly that can be used for coordinative post-modification, we started with design principles based on earlier reports.35,36 Ligand building blocks that can be used for post-modification by coordination chemistry must contain two functionalities. A sphere forming site, which is typically constructed from two linked pyridine donors with an angle of 120°, and a functional group that can coordinate to a second metal ion. To prevent the second ligand from interfering with the sphere formation, protection is required. Based on our recent work, which describes the formation of Pd12L24 nanospheres with protonated amines as weak acids placed on the endo-position (trialkylamine, NR3H+),36 we envisioned a strategy of positioning protonated nitrogen donor ligands on the inside of Pd12L24 nanospheres. The deprotonation can then be performed after sphere formation, making various nitrogen donor ligands available for further functionalization. In order to study the pKa requirements for such a system, we prepared six different building blocks with nitrogen donor ligands placed at the endo-position. As such, the set includes a 4-dimethylaminopyridine (DMAP) (LDMAP), three pyrrolidine ligands (LPro, LProMe, L4ProMe), a dimethylamine (LNMe) and an imidazole (LHis) donor ligand (Fig. 2), next to the typical two pyridine functions needed for nanosphere formation. All four endo-functionalities represent well known nitrogen donor ligands with different pKa values, that can be used for coordination chemistry after sphere formation. All building blocks were prepared according to standard organic procedures (as summarized in Section SI1†).
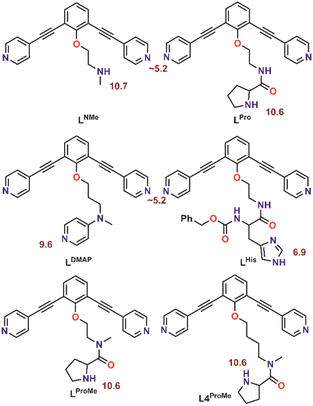 |
| | Fig. 2 Molecular structure of the herein investigated building blocks with dual functionality. The pKa of the corresponding protonated forms is depicted in red. | |
Sphere synthesis
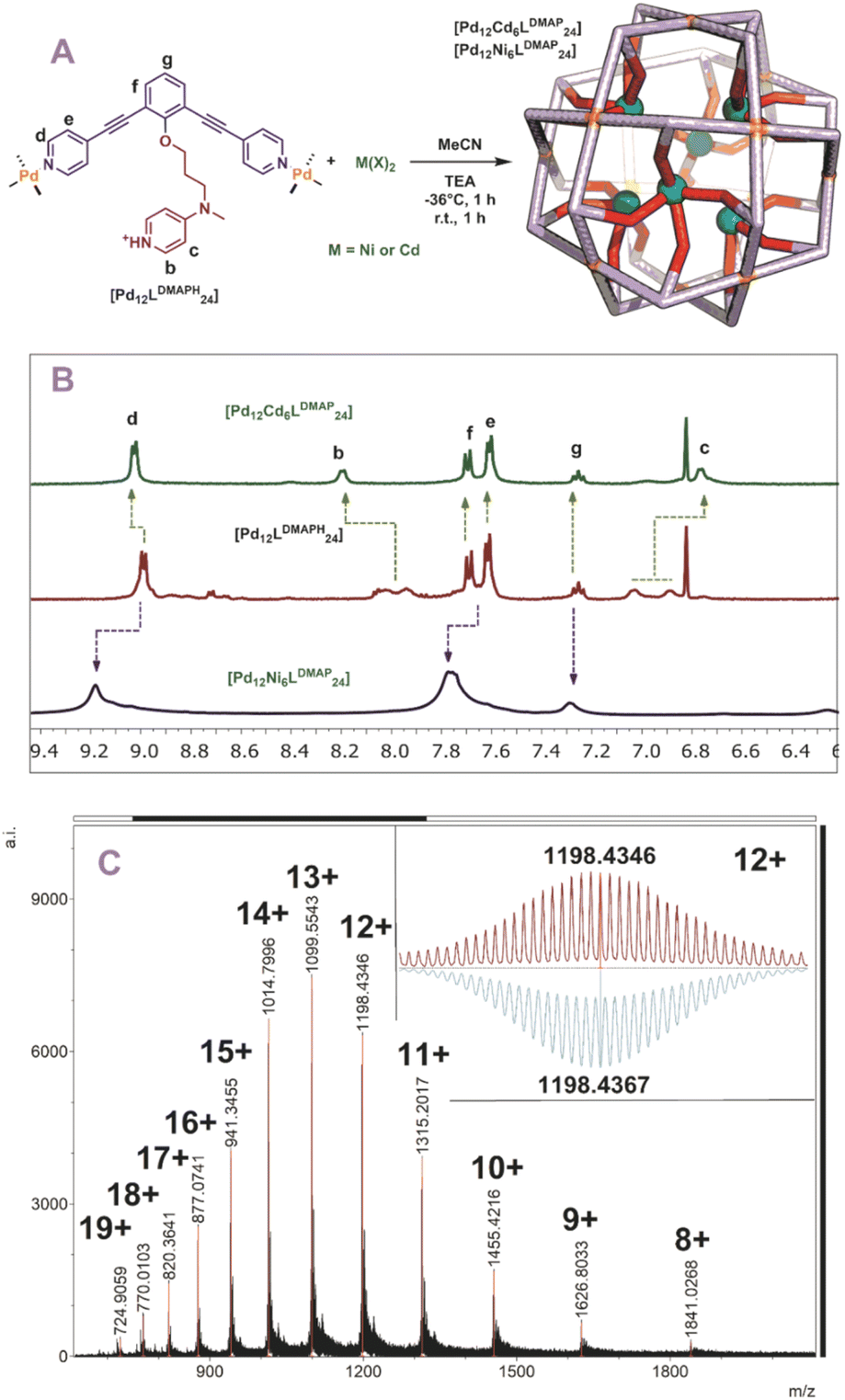
With all six building blocks in hand, sphere formation was attempted to find suitable pKa requirements. Because the presence of an extra coordinating unit (next to the sphere forming pyridines) prohibits formation of well-defined spheres (see formation of ill-defined materials using LDMAP and LProMe without protonation, Fig. S27 and S28†), the difference in pKa between the endo-functionalities and pyridine was used to selectively protonate the inner nitrogen donor ligand. For this purpose, all building blocks were protonated, using pyridinium tetrafluoroborate or pyridinium trifluoromethanesulfonate (Fig. 3A). After successful protonation, the resulting solutions were evaporated to dryness in vacuo to remove the formed pyridine. The resulting solids were dissolved in MeCN. Sphere formation was performed using the so obtained protonated ligands according to the following general procedure described explanatory for LDMAPH. To LDMAPH (10 μmol, 1 eq.) in MeCN-d3 (1 mL), [Pd(MeCN)4](BF4)2 (5.5 μmol, 0.55 eq.) is added and the solution is stirred for 24 h at room temperature. Analysis of the resulting solution shows a downfield shift of the pyridine protons (d and e, Fig. 3C), indicative of coordination to palladium and sphere formation. Interestingly, a downfield shift of the proton located at the DMAP (a, Fig. 3C) is observed upon sphere formation, suggesting that the positively charged sphere has an influence on the pKa of DMAP. Next to 1H-NMR, diffusion ordered NMR (DOSY) shows that a single species with a log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) D value around −9.6 is formed, which translates to a structure with a hydrodynamic diameter of 5.0 nm (Fig. S30†), in line with previously reported Pd12L24 nanospheres.4,35,37,38 ESI-MS analysis of the solutions confirms the formation of [Pd12LDMAPH24] nanosphere as in the MS spectra only signals corresponding to different charged states of these structure ([Pd12LDMAPH24]n+ for 5 ≤ n ≤ 14) are observed (Fig. 3B).
D value around −9.6 is formed, which translates to a structure with a hydrodynamic diameter of 5.0 nm (Fig. S30†), in line with previously reported Pd12L24 nanospheres.4,35,37,38 ESI-MS analysis of the solutions confirms the formation of [Pd12LDMAPH24] nanosphere as in the MS spectra only signals corresponding to different charged states of these structure ([Pd12LDMAPH24]n+ for 5 ≤ n ≤ 14) are observed (Fig. 3B).
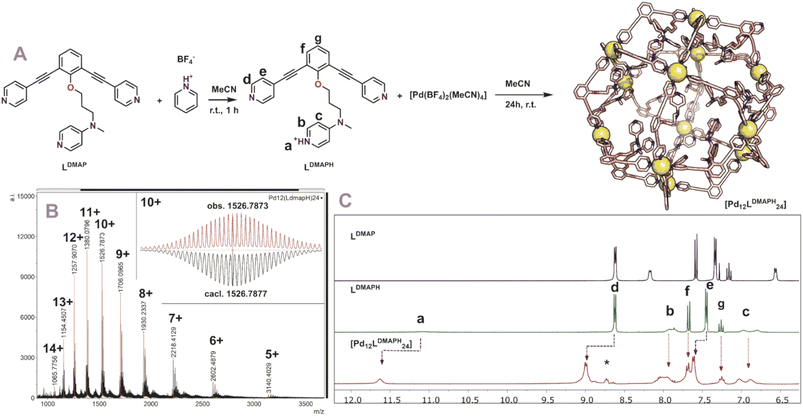 |
| | Fig. 3 (A) Synthetic procedure for the preparation of [Pd12LDMAPH24] and depiction of a molecular model (based on PM3, Pd = yellow spheres); (B) obtained ESI-MS spectrum of the self-assembly (inset: top, measured spectrum; bottom, simulated); (C) 1H-NMR spectra of the building block LDMAP, the protonated building block LDMAPH and the self-assembly [Pd12LDMAPH24]. *Excess of pyridinium BF4 used during the preparation (SI2†). | |
Similar experiments with LPro and LNMe, LProMe and L4ProMe gave similar results, indicating that this protocol also resulted in successful sphere formation with these building blocks (Section SI2†). Nanospheres composed of LDMAPH, LProH, LProMe and L4ProMe display in contrast to reported nanospheres somewhat broader signals in 1H-NMR. We attribute the broadening to different rotamers of the nitrogen containing inner functionalization (see also desymmetrization of building block LDMAPH, duplication of signal b and c, Fig. 3C). Noteworthy, MS analysis of [Pd12LProH24] turned out complicated as a complex spectrum is obtained (due to in cooperation of different solvent). When the amide is alkylated (LProMevs.LPro), straight forward analysis of the MS spectra of [Pd12LProMeH24] (similar to [Pd12LDMAPH24]) is possible as a clear spectrum is obtained without the formation of different clusters.
In contrast to the above-mentioned building blocks, when building block LHis was used under similar conditions, an ill-defined 1H-NMR spectrum was obtained, indicating that pure spheres were not formed. For this building block the difference in pKa between imidazole and pyridine is not sufficiently large for selective sphere formation. Alternatively, the sphere is formed which results in a pKa change similar to that observed for DMAP, leading to a more acidic imidazolium which is capable to protonate the pyridine sites of the building block, thereby decomposing the sphere.
Coordinative post-modification
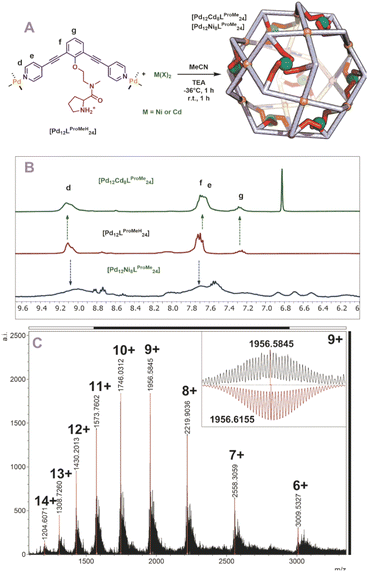
With five novel spheres in hand, the post modification procedure was explored by deprotonation of the encapsulated nitrogen donor ligands in the nanosphere and consecutive coordination to a metal ion/complex of choice. We decided to use cadmium and nickel as the metal for post-modification as analysis of the resulting structures with MS was expected to be straightforward. Keeping in mind the dynamicity of palladium-based nanospheres and the possible coordination of the inner ligands to palladium, all post modification experiments were performed at −36 °C (as the nanosphere decomposes when the deprotonation is performed at room temperature, see Fig. S46 and S47†). At these low temperatures the exchange of the palladium–pyridine bond should be very slow. We first focused on post modification of [Pd12LDMAPH24] as DMAP is known to be an excellent ligand for transition metals. First, 6 eq. of [Cd(OTf)2(MeCN)4] were added to a cooled solution of the [Pd12LDMAPH24] nanosphere in MeCN, in line with an expected four to one coordination to Cd. Subsequently, 26 eq. of triethylamine were added to deprotonate the DMAP while keeping the solution at low temperature for 1 h. The solution was then allowed to warm up to room temperature for 1 h and was analysed using various analytical techniques (Fig. 4A). The 1H-NMR spectrum suggests that the nanosphere remains intact as the position of the pyridine signals remain similar (9 ppm) (Fig. 4B). Moreover, the simplicity of the 1H-NMR spectrum (with single set of DMAP signals b and c, Fig. 4B) shows that the structure remains highly symmetrical. The signals associated to DMAP (b and c) shifted, in line with the formation of a coordination complex with CdII. DOSY NMR shows that the nanosphere has a similar diffusion coefficient as before the post modification, suggesting the structure did not change much in size (ca. 5.0 nm before post-modification; ca. 4.6 nm after post-modification with Cd; Fig. S49†). ESI-MS analysis displayed signals corresponding to different charged species of [Pd12Cd6LDMAP24(−OTf)36−n]n+ for 5 ≤ n ≤ 13 (Fig. S50†). Importantly, the MS spectra did not contain any signals that could be attributed to nanosphere structures with a different palladium/cadmium stochiometry. The combination of all analytical techniques provides good support for the selective post modification of the DMAP containing palladium assembly to incorporate exactly 6 cadmium ions.
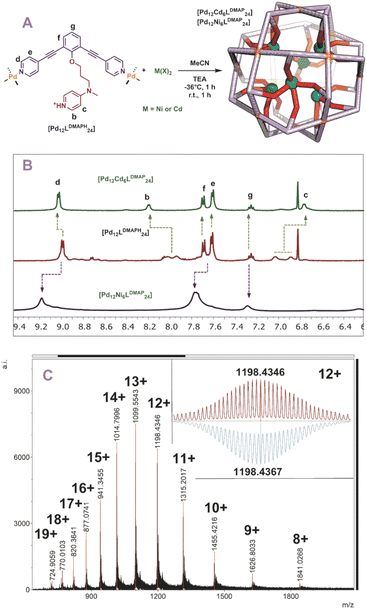 |
| | Fig. 4 (A) Post-modification of [Pd12LDMAPH24] with Ni2+ and Cd2+; (B) 1H-NMR of the self-assembly before (middle) and after (top and bottom) post-modification; (C) measured ESI-MS spectra of [Pd12Ni6LDMAP24] (inset: top, measured spectrum; bottom, simulated). | |
When an identical procedure was performed using [Ni(BF4)2(MeCN)4] as a precursor, ESI-MS analysis displayed signals only corresponding to different charged species of [Pd12Ni6LDMAP24(BF4−)36−n]n+ for 8 ≤ n ≤ 19 (Fig. 4C). Due to the paramagnetic character of nickel, all DMAP signals were no longer visible in the 1H-NMR spectrum as a result of coordination to paramagnetic NiII. The outer proton signals remain visible (although somewhat broader, Fig. 4B) and are located at similar shifts as found for the nanosphere structure before the post-modification. The presence of the sphere was further supported by DOSY NMR which displayed a similar diffusion coefficient as for the cadmium analogue (logDNi = −9.6; logDCd = −9.54), providing evidence that the developed technique is not only applicable for cadmium but also for nickel.
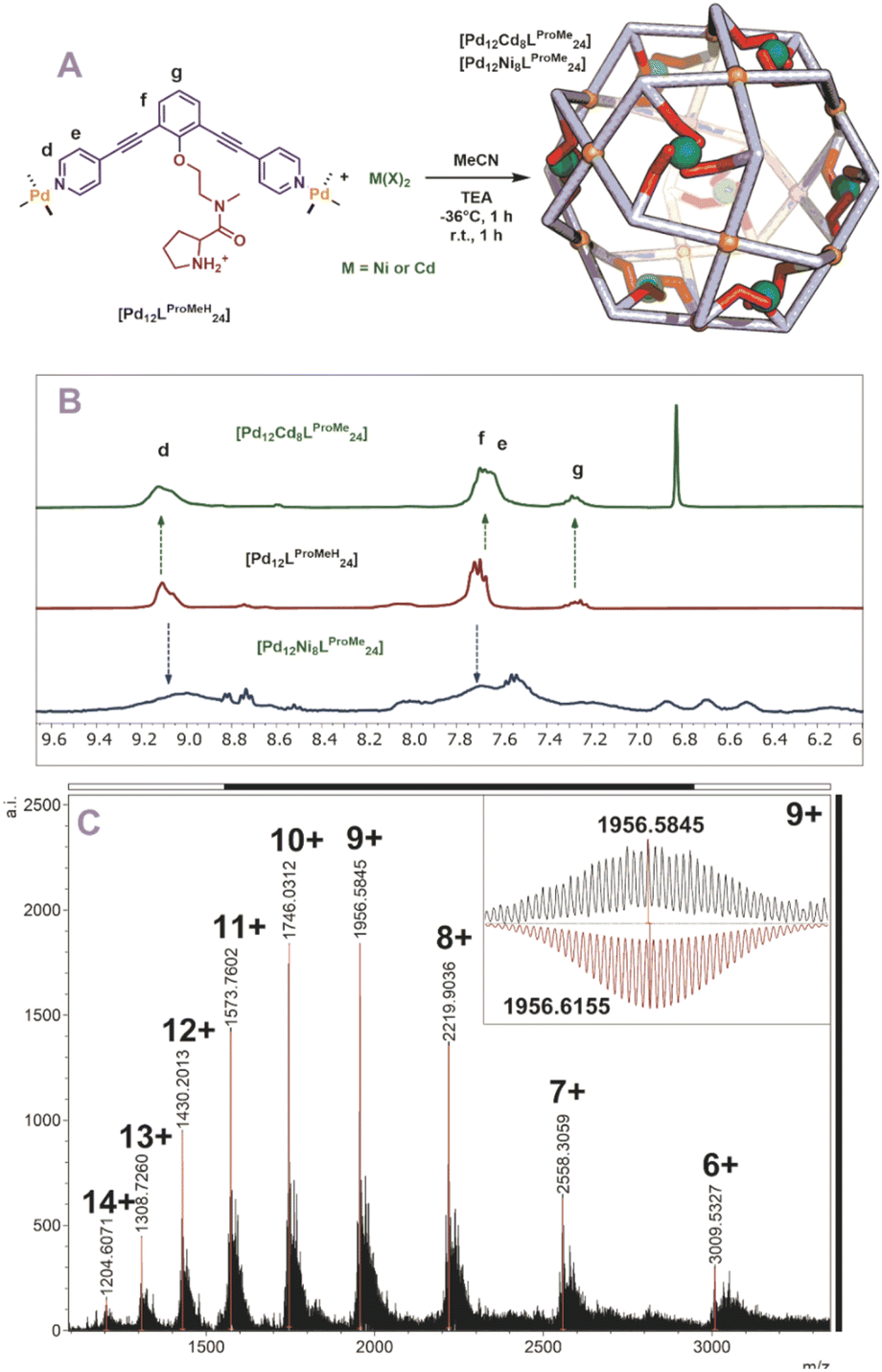
Next, we studied the post modification of [Pd12LProH24] using the same methodology. A similar protocol for post modification was used, in which different equivalents of cadmium were added to a cold solution of [Pd12LProH24] (Fig. S62 and S63†). 1H-NMR showed no changes of the sphere framework protons after the addition of cadmium, suggesting that the spherical structure remains intact (Fig. S62 and S63†). The DOSY spectrum shows that the species has a similar diffusion as before coordination to cadmium (before and after modification d = 4.4 nm, Fig. S63†). MS analysis of the cadmium containing sphere and sphere precursor was not successful, which we attribute to difficulties in MS analysis already for the parent nanocage [Pd12LProH24]. As MS analysis is required for identification of metal–metal stochiometries, we focused on the alkylated variant of the nanocage [Pd12LProMeH24] for post modification. After initial attempts using different amount (6, 8, 12 eq.) of cadmium for the post modification of [Pd12LProMeH24] in analogy to the DMAP containing nanocage (Fig. 5A), we observe from MS analysis that 8 cadmium complexes are in cooperated into the nanocage. Therefore, we performed the post modification by addition of 8 eq. [Cd(OTf)2(MeCN)4] to a cooled solution of [Pd12LProMeH24], followed by addition of base. The so obtained mixture displays presence of the nanocage (similar shifts of framework in 1H-NMR and DOSY, Fig. 5B). MS analysis of the solution provides evidence that only nanocage of the composition [Pd12LProMe24Cd8] is formed after applying our protocol (Fig. 5C). In analogy, also nickel can be in cooperated as supported by equivalent analysis techniques (although 1H-NMR and DOSY are broader due to the paramagnetic nature of Ni) (Fig. 5B, S67 and S68†).
 |
| | Fig. 5 (A) Post-modification of [Pd12LProMeH24] with Ni2+ and Cd2+; (B) 1H-NMR of the self-assembly before (middle) and after (top and bottom) post-modification, (C) measured ESI-MS spectra of [Pd12Cd6LProMe24] (inset: top, measured spectrum; bottom, simulated). | |
Because the sphere containing LNMeH has a short length between sphere forming site and nitrogen donor for post-modification, we performed the post-modification of [Pd12LNMeH24] with 12 eq. of [Ni(BF4)2(MeCN)4]. 1H-NMR showed signals typical for the nanosphere indicating it again remains intact after the post modification (Fig. S74†). The DOSY spectrum supports that the sphere remains of the same size, displaying only one species in solution with a similar diffusion coefficient as the starting material (d = 4.2 nm before functionalization; paramagnetic DOSY after modification d = 5.0 nm). MS analysis showed the sphere being intact (Fig. S76†). However, only signals of nanospheres with protonated and deprotonated ligands were observed, and nickel coordination could not be confirmed by MS analysis. We anticipate that the formed nickel complexes via the amine ligands are too labile.
Extension of the scope
After the post-modification with nickel and cadmium, we wondered if our post-modification procedure is also applicable to other transition metals. In a small exemplary scope we post-modified [Pd12LDMAPH24] nanospheres with two different first row transition metals such as Cu(BF4)2 and Co(BF4)2(H2O)6. The two investigated metals formed selectively the M6Pd12LDMAP24 nanospheres when the same procedure was applied, as supported by MS analysis (Section SI3†). Relatively broad 1H-NMRs and inconclusive DOSY were obtained due to the paramagnetic character of the applied metals. Next to those bimetallic nanospheres, also one nanospheres containing three different metals [Pd12LDMAP24Cd6−nNin] (for n = 0–4) was prepared using a mixture of [Cd(OTf)2(MeCN)4] and Ni(OTf)2. In this experiment, a statistic distribution of the two metals is obtained with the total number of Cd and Ni being 6 (see ESI for details, Fig. S79–S82†).
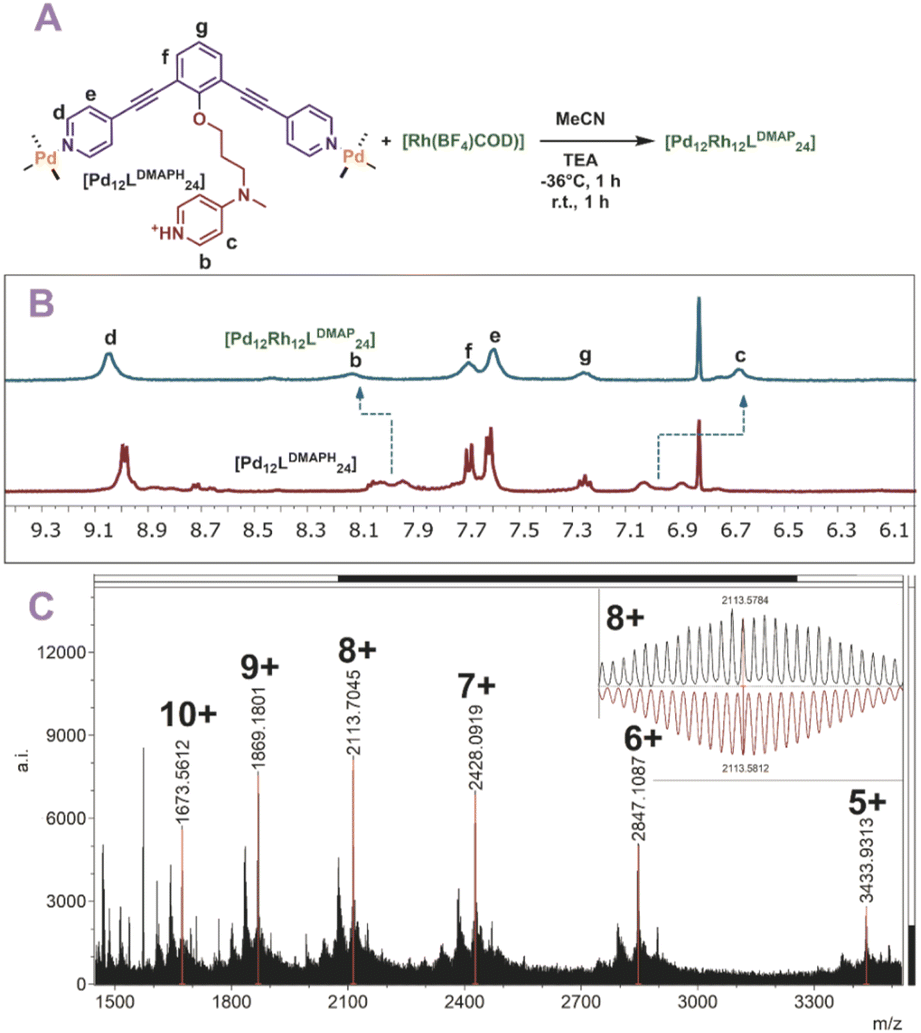
Finally, also [Rh(COD)(MeCN)2(BF4)] was used for post-modification (Fig. 6A). The rhodium precursor requires only two extra ligands for formation of a tetracoordinated complex, as the COD occupies the other sites. As such, 12 of these transition metals can theoretically coordinate via post-modification of spheres. Indeed, when rhodium is used for post-modification of [Pd12LDMAPH24] the [Pd12[Rh(COD)]12LDMAP24] nanosphere was selectively formed as supported by 1H-NMR, DOSY and MS analysis (Fig. 6B and C).
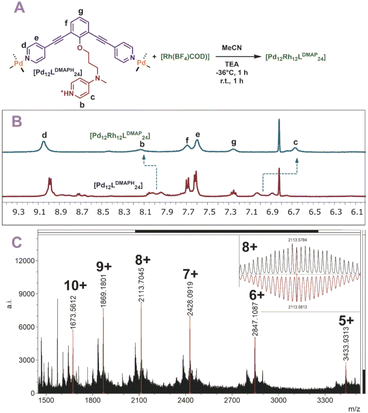 |
| | Fig. 6 (A) Post-modification of [Pd12LDMAPH24] with Rh1+; (B) 1H-NMR of the self-assembly before (bottom) and after (top) post-modification; (C) measured ESI-MS spectra of [Pd12Rh12LDMAPH24] (inset: top, measured spectrum; bottom, simulated). | |
Kinetic properties of the post modified nanospheres
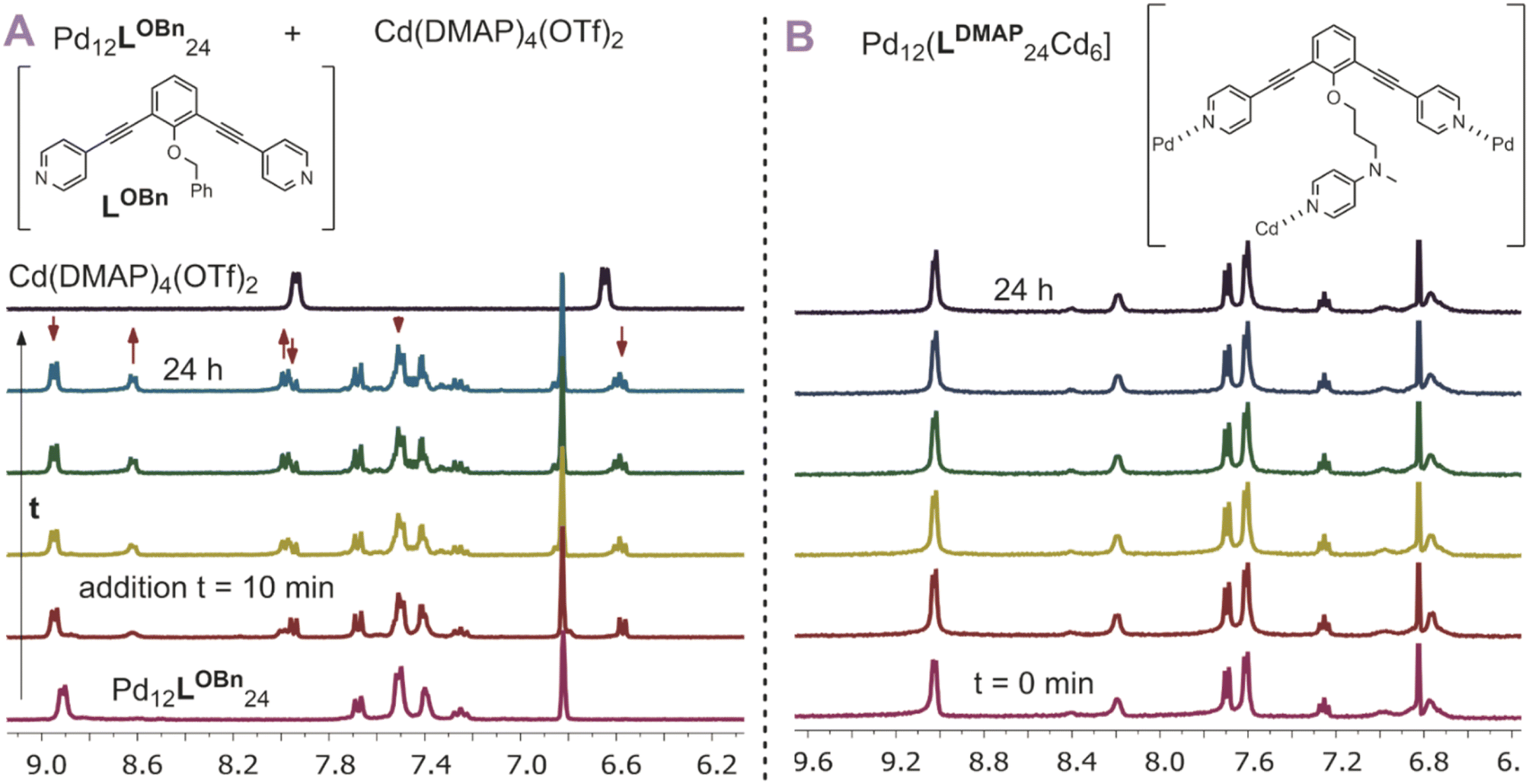
After having established post-assembly modification techniques for the incorporation of a different number of transition-metal complexes into Pd12L24 nanospheres, we studied briefly the properties of the so formed complexes. In particular, we were wondering if the incorporated complexes behave differently from the molecular analogues. As we expected the rim of the nanocage to act as an extended linker between the inner nitrogen donors, we anticipated the inner nitrogen donors to act like multi-dentate ligands (opposed to monodentate, highly dynamic ligands without the nanosphere). To investigate our hypothesis, we followed the stability of [Pd12Cd6LDMAP24] over the course of 24 h at 300 K using 1H-NMR with mesitylene as an internal standard (Fig. 7B). During the course of the experiment, all chemical shifts remained the same (quantity and position), indicating that the nanocage with internal Cd(DMAP)4 complexes is stable. In contrast to that, when a molecular Cd(DMAP)4 complex is added to a model nanosphere ([Pd12LOBn24]) (Fig. 7A), we observe quick decomposition of the nanosphere. Therefore, we conclude that whereas molecular Cd(DMAP)4 complexes are highly dynamic in nature (leading to exchange with palladium corner stones), once implemented into the sphere through post-modification of [Pd12LDMAPH24], the dynamicity is significantly reduced (indicative of multidentate character of the inner nitrogen donors).
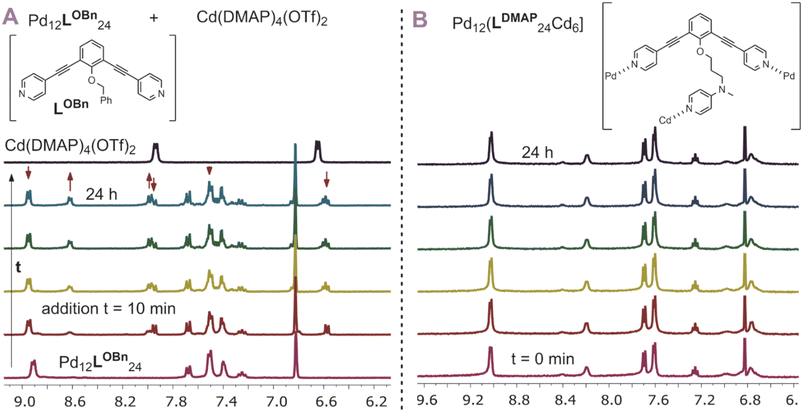 |
| | Fig. 7 (A) Stability of a model nanocage Pd12LOBn24 in presence of a model cadmium complex showing decomposition of the nanocage by exchange of the DMAP ligand from cadmium to palladium; (B) stability of [Pd12Cd6LDMAP24] over time showing no significant changes over 24 h. | |
Localization of TMC within the nanospheres – metal ion-metal ion distance measurements
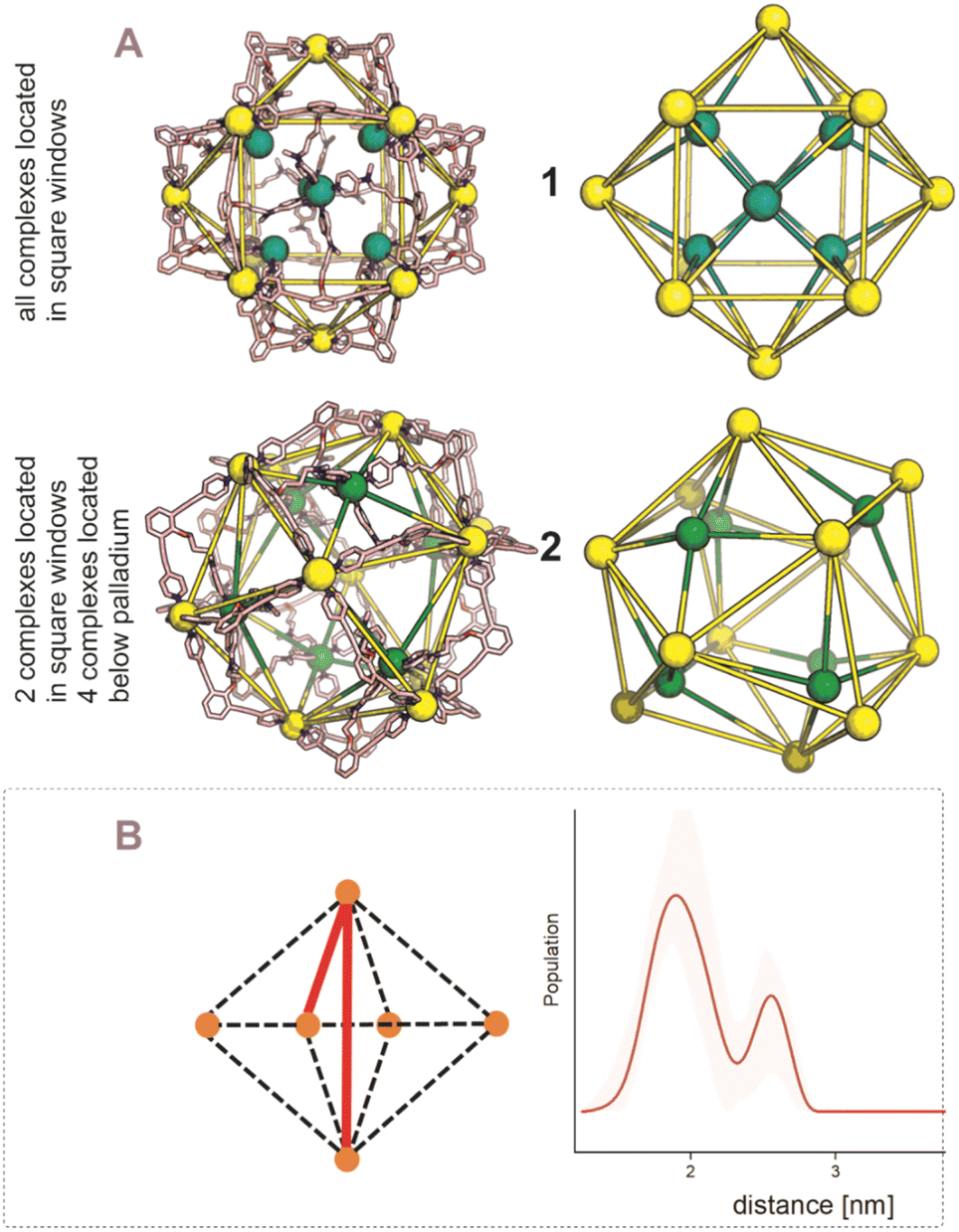
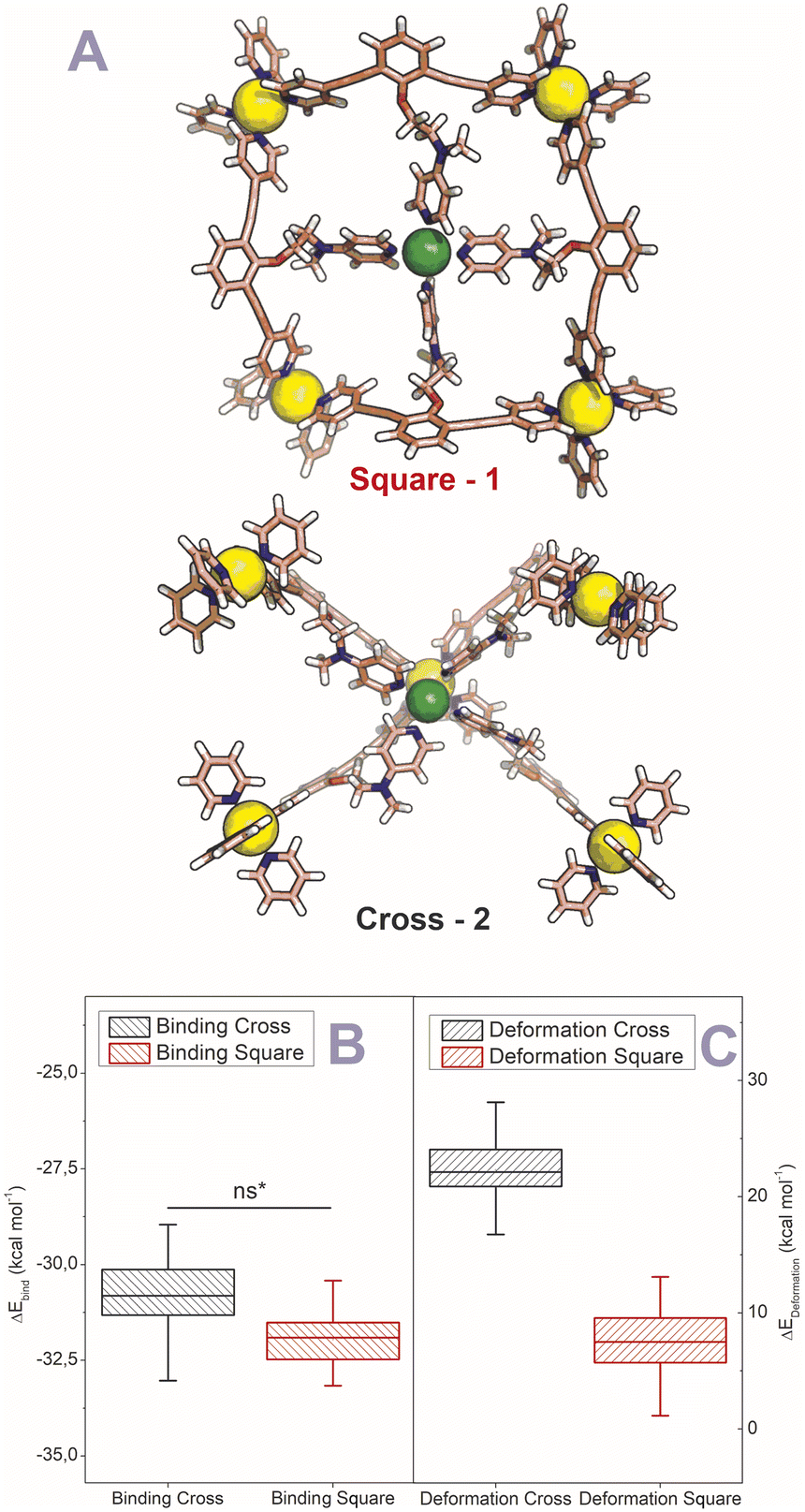
After establishing the precise formation of heterometallic nanospheres and supported a unique coordination environment around the internally formed complexes for [Pd12LDMAP24Cd6] and [Pd12LProMe24Cd8], we wanted to identify the location of the interior complexes. Because growing crystals of the large self-assembled M12L24 spheres is challenging, we were not able to obtain suitable crystals for X-ray analysis. We therefore further studied the location of the transition-metal ions in the [Pd12M6L24] and [Pd12M8L24] nanospheres via their characteristic distances. Our post-modified nanospheres represent structures related to mathematical stellated cuboctahedra (e.g., Fig. 8A). Therefore [Pd12M6L24], have defined distances between the inner metal-nodes M (1 or 2, Fig. 8A). In structure 1, all complexes are located in the six square windows of the nanosphere. In contrast, in structure 2, four complexes are located below a palladium center and two in the square windows (Fig. 8A). In order to support the location of the transition metal complexes M, we measured the Cu(II)–Cu(II) distances within the [Pd12Cu6LDMAP24] nanosphere using EPR spectroscopy. First, regular EPR was recorded of [Pd12Cu6LDMAP24] and [Pd12Cu8LPro24] (Fig. S83 and S84†). Fitting of the obtained EPR spectra reveals as expected Cu(II) species with 4 surrounding nitrogen ligands (in the case of LPro, the 4th vacant site is occupied by acetonitrile). Furthermore, tetrahedral ligand orientation is observed, building the fundament for computational investigation (vide infra). Next, we used DEER (double electron electron resonance), a pulsed EPR method that is specific to the dipolar interaction of paramagnetic centers and thereby their distance.39,40 Although DEER is established and perhaps most well-known in the biochemistry field for nitroxide spin labels41 it is used since 2003 to determine Cu(II)–Cu(II) distances in the nanometer range.42–45 For detailed information on the method, the setup and the analysis involved to obtain the distances, see Section SI5.† The DEER data show that [Pd12Cu6LDMAP24] has two Cu–Cu distances (Fig. 8 B, red trace). A shorter distance, r1, at 1.9 nm and a longer one, r2, at 2.7 nm (Table 1). The intensity of the peak at r2 is smaller than that of r1. Validation with DEER analysis44 shows that both peaks are essential to describe the data. The observed distances are compared to those based on the PM3 modeled (Table 1) structures 1 and 2 of [Pd12Cu6LDMAP24] (Fig. 8 A). For Pd12Cu6LDMAP24, the observed distances match the ones from PM3 model of both model 1 or 2 (with slight better fit for the longer distance of 1), supporting incorporation of six copper complexes inside the sphere. From solely distance measurements, a clear differentiation of the two possible structures 1 or 2 (Fig. 8A) is not possible. However, as we have shown, dynamic exchange of the coordinated DMAP is very slow inside the nanocage. The simple 1H-NMR of the Cd containing nanocage suggest therefore, with is single set of DMAP signals, that all complexes experience the same chemical environment. As depicted (Fig. 8A), only structure 1 provides the same chemical environment for the TSM located inside the nanosphere (i.e., all complexes are located in the square windows). Therefore, our analytical methods suggest the location of the TSM after post-assembly modification in the square windows (structure 1, Fig. 8A). To further support this, both potential structures 1 and 2 (Fig. 8A) were optimized in silico. To estimate the binding free energy of the copper ion (Cu2+) we constructed a series of models representing association at the cross (Fig. 9A) Cu1Pd5(LDMAP)4(py)12 or square face (Fig. 9B) Cu1Pd4(LDMAP)4(py)8. The two model structures were first optimized with a GFN2-xTB Hamiltonian and analytical linearized Poisson–Boltzmann (ALPB) implicit solvation parameterized for acetonitrile. Afterwards, these optimized models were used for molecular dynamics calculations with harmonic restraints between the outermost Pd2+ centers to preserve the overall structure of the cage. In the specific cases of the intersection binding site (Fig. 9A), Cu1Pd5(LDMAP)4(py)12 and Pd5(LDMAP)4(py)12, the Pd2+ center was not restrained. A total of 64 frames were extracted from the 1 ns molecular dynamics trajectory for which the single-trajectory binding free energies were computed from the single point energy difference for each frame with and without Cu2+ at a GFN2-xTB level of theory with ALPB implicit acetonitrile solvation. We observe that the binding of the copper ion in either of the two positions has non-significant (ns*) difference in energy (Fig. 9B). However, the structural changes at the ligand framework have significant energetic penalty for the nanosphere (Fig. 9C). Whereas the deformation of the nanocage remains relatively small when the copper is located in the square windows (Fig. 8A, 1), a significantly higher deformation penalty is observed when the copper is located below the palladium (Fig. 8A, 2). Therefore, all analytical methods (DEER, H-NMR) and theoretical calculations support location of all transition metal complexes at the square windows of Pd12LDMAP24, suggesting that structure 1 (Fig. 8A) is the most likely one.
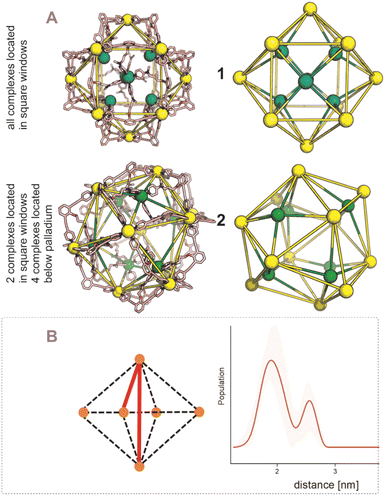 |
| | Fig. 8 (A) PM3 minimized structured of two possible structures 1 and 2 for [Pd12M6LDMAP24] assemblies; extraction of the coboctrahedra-forming palladium centers and the implemented second metal (Pd = yellow; Cd, Ni, Co, Cu = green, tetrahedral orientation of ligands around Cd, Ni, Co, Cu, vide infra). (B) Display of implemented metal centers and the unique pair–wise distances. Determination of distances between the copper centers with a pulsed EPR method, DEER, for [Pd12Cu6LDMAP24]. The shaded areas represent the confidence interval after validation. For raw data, details and interpretation, see Section SI5 in ESI.† | |
Table 1 Experimental and PM3 model-derived distance parameters obtained for Pd12Cu6LDMAP24 and Pd12Cu8LPro24. The DEER data were analyzed by means of Tikhonov regularization. 〈r〉 is the center of the distribution distance in nm, S(r) the width, for details, see SI5 in SI
| Pd12Cu6LDMAP24 distances from DEER |
Model 1 |
Model 2 |
Pd12Cu8LPro24 distances from DEER |
Model 1 |
Model 2 |
| <r> (nm) |
S(r) |
r (nm) |
r (nm) |
〈r〉 (nm) |
S(r) |
r (nm) |
r (nm) |
| 1.9 |
0.18 |
1.9 |
1.9 |
1.8 |
0.18 |
2.0 |
2.1 |
| 2.5 |
0.12 |
2.7 |
2.9 |
|
|
2.8 |
2.4 |
|
|
|
|
|
|
|
3.4 |
2.8 |
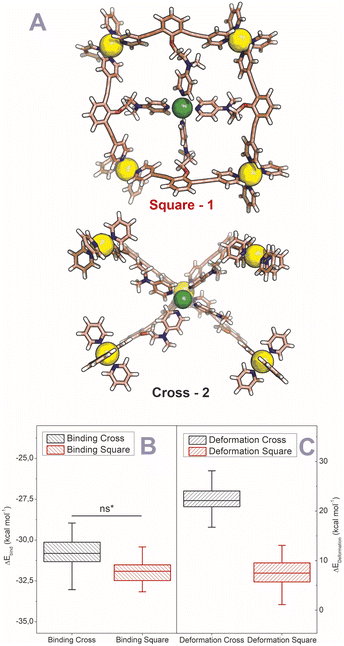 |
| | Fig. 9 (A) GFN2-xTB Hamiltonian and analytical linearized Poisson–Boltzmann (ALPB) optimized model structures of two possible environments around copper (square and cross representing 1 and 2 respectively) (Pd = yellow; Cu = green (tetrahedral orientation of ligands)). (B) Binding energies for the square and cross fragments displayed in (A). (C) Deformation energies for the construction of 1 or 2 (Fig. 8A) with the copper located in the squares and in crosses respectively. | |
For the [Pd12M8LPro24], identification of the exact location of M was found to be more complicated. The distance distribution of [Pd12Cu8LPro24] has one broad peak centered at 1.8 nm, with a tail in the distribution that extends to approximately 3 nm (Fig. 10B, blue trace, Table 1). Because of short evolution times (see SI5†), for distances above 2.8 nm, the shape of features in the distance distribution is not reliable. Therefore, in [Pd12Cu8LPro24], validation shows that the part of the distance distribution from 2.2 nm to longer distances is not reliable, i.e. the confidence interval (green shaded area) extends as far as the baseline, showing that this part of the distance distribution is not essential to fit the data. Also, for the [Pd12Cu8LPro24] nanosphere the observed distance of 1.8 nm matches rather well that of the PM3 calculated structure 2.0 nm (expected in both potential models of the nanosphere, 1 and 2, Fig. 10A and Table 1). Next to this, also two longer distances at 2.8 and 3.4 nm are expected based on the calculated structure for 1. Although the distribution (Fig. 10B) shows some intensity in this distance region, such distances cannot be reliably determined given the short evolution time of the DEER traces (see S8†). Structure 2 (Fig. 10A) should also display a distance of 2.4 nm in similar intensity as 2.1 nm. The missing of a feature for 2.4 nm in the measurement, hints towards a higher probability of 1 over 2 for proline functionalized nanospheres. Due to the broadness of the 1H-NMR, a certain determination of TSM location was not possible without X-ray structure analysis. However, missing features of 2 in the DEER measurement provide support for structure 1 (Fig. 10A).
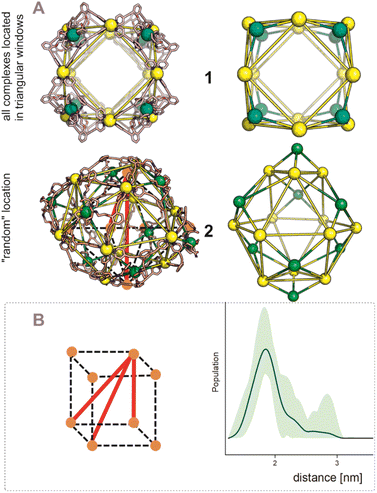 |
| | Fig. 10 (A) PM3 minimized structured of two possible structures 1 and 2 for [Pd12M8LProMe24] assemblies; extraction of the coboctrahedra-forming palladium centers and the implemented second metal (Pd = yellow; Cd, Ni, Co, Cu = green). (B) Display of implemented metal centers and the unique pair–wise distances for 1. Determination of distances between the copper centers with a pulsed EPR method, DEER, for [Pd12Cu8LPro24]. The shaded areas represent the confidence interval after validation. For raw data, details and interpretation, see Section SI5 in ESI.† | |
Conclusion
In summary, we described a new procedure for the selective post-assembly modification of Pd12L24 nanospheres by metal coordination. Pd12L24 nanospheres with different types of protonated nitrogen-donor ligands within the interior can be prepared, provided that a sufficient pKa difference exist between the endo-functionality and the pyridines of the sphere forming building block. The selective protonation protects the inner nitrogen-donor ligands for coordination to palladium during sphere formation. Novel spheres can be obtained bearing exactly 6, 8 or 12 transition metal complexes after addition of a second metal of choice and deprotection by deprotonation. The post-assembly modification strategy used here on Pd12L24 assemblies establishes guidelines for post-modification of other supramolecular systems, leading to new opportunities to make materials with unique coordination motifs, compositions and geometrical features. Given the specific number of metals present in catalytic centers of enzymes, such as four copper atoms in laccase,46 this methodology may enable to make more precise models for such enzymes by selective formation of nanostructures with a different number of metal centers at a well-defined location. As such, these nanospheres provide an exciting opportunity for further investigations.
Data availability
Data that support the findings of this study are present in the article and its ESI.† The corresponding author will provide additional data not available in these documents upon reasonable request.
Author contributions
Conceptualization: EOB, JNHR; formal analysis: EOB, LP, FZ, DAP; funding acquisition: JNHR, BdB, MH; investigation: EOB, LP, FZ, DAP, SM; supervision: JNHR; validation: EOB, LP, FZ, DAP, SM, MH, BdB, JNHR; visualization: EOB; writing – original draft: EOB; writing – review & editing: EOB, LP, FZ, DAP, SM, MH, BdB, JNHR.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We kindly acknowledge the University of Amsterdam for financial support to RPA sustainable chemistry. The work by L. P. was supported by the Netherlands Organization of Scientific Research (NWO/OCW), as part of the Frontiers of Nano-Science program.
References
- K. Chand, K. Biradha, M. Kawano, S. Sakamoto, K. Yamaguchi and M. Fujita, Chem.–Asian J., 2006, 1, 82–90 CrossRef PubMed.
- D. Fujita, A. Takahashi, S. Sato and M. Fujita, J. Am. Chem. Soc., 2011, 133, 13317–13319 CrossRef CAS PubMed.
- D. Fujita, Y. Ueda, S. Sato, H. Yokoyama, N. Mizuno, T. Kumasaka and M. Fujita, Chem, 2016, 1, 91–101 CAS.
- S. Sato, Y. Ishido and M. Fujita, J. Am. Chem. Soc., 2009, 131, 6064–6065 CrossRef CAS PubMed.
- Q.-F. Sun, J. Iwasa, D. Ogawa, Y. Ishido, S. Sato, T. Ozeki, Y. Sei, K. Yamaguchi and M. Fujita, Science, 2010, 328, 1144–1147 CrossRef CAS PubMed.
- K. Suzuki, M. Tominaga, M. Kawano and M. Fujita, Chem. Commun., 2009, 1638 RSC.
- M. Han, D. M. Engelhard and G. H. Clever, Chem. Soc. Rev., 2014, 43, 1848–1860 RSC.
- E. O. Bobylev, D. A. Poole III, B. de Bruin and J. N. H. Reek, J. Am. Chem. Soc., 2022, 144, 15633–15642 CrossRef CAS PubMed.
- S. Pullen, J. Tessarolo and G. H. Clever, Chem. Sci., 2021, 12, 7269–7293 RSC.
- D. Preston, J. E. Barnsley, K. C. Gordon and J. D. Crowley, J. Am. Chem. Soc., 2016, 138, 10578–10585 CrossRef CAS PubMed.
- J. Tessarolo, H. Lee, E. Sakuda, K. Umakoshi and G. H. Clever, J. Am. Chem. Soc., 2021, 143, 6339–6344 CrossRef CAS PubMed.
- L. R. Holloway, P. M. Bogie and R. J. Hooley, Dalton Trans., 2017, 46, 14719–14723 RSC.
- A. M. Johnson and R. J. Hooley, Inorg. Chem., 2011, 50, 4671–4673 CrossRef CAS PubMed.
- S. Sudan, R.-J. Li, S. M. Jansze, A. Platzek, R. Rudolf, G. H. Clever, F. Fadaei-Tirani, R. Scopelliti and K. Severin, J. Am. Chem. Soc., 2021, 143, 1773–1778 CrossRef CAS PubMed.
- R. Li, F. Fadaei-Tirani, R. Scopelliti and K. Severin, Chem. – Eur. J., 2021, 27, 9439–9445 CrossRef CAS PubMed.
- A. Kumar and P. S. Mukherjee, Chem. – Eur. J., 2020, 26, 4842–4849 CrossRef CAS PubMed.
- W. M. Bloch and G. H. Clever, Chem. Commun., 2017, 53, 8506–8516 RSC.
- K. Acharyya, S. Bhattacharyya, H. Sepehrpour, S. Chakraborty, S. Lu, B. Shi, X. Li, P. S. Mukherjee and P. J. Stang, J. Am. Chem. Soc., 2019, 141, 14565–14569 CrossRef CAS PubMed.
- B. Olenyuk, M. D. Levin, J. A. Whiteford, J. E. Shield and P. J. Stang, J. Am. Chem. Soc., 1999, 121, 10434–10435 CrossRef CAS.
- I. A. Bhat, D. Samanta and P. S. Mukherjee, J. Am. Chem. Soc., 2015, 137, 9497–9502 CrossRef CAS PubMed.
- Q.-F. Sun, S. Sato and M. Fujita, Nat. Chem., 2012, 4, 330–333 CrossRef CAS PubMed.
- M. T. Yong, O. M. Linder-Patton and W. M. Bloch, Inorg. Chem., 2022, 61, 12863–12869 CrossRef CAS PubMed.
- S. Li, C. Liu, Q. Chen, F. Jiang, D. Yuan, Q.-F. Sun and M. Hong, Chem. Sci., 2022, 13, 9016–9022 RSC.
- W. J. Ramsay, F. T. Szczypiński, H. Weissman, T. K. Ronson, M. M. J. Smulders, B. Rybtchinski and J. R. Nitschke, Angew. Chem., Int. Ed., 2015, 54, 5636–5640 CrossRef CAS PubMed.
- M. Hardy, N. Struch, J. J. Holstein, G. Schnakenburg, N. Wagner, M. Engeser, J. Beck, G. H. Clever and A. Lützen, Angew. Chem., Int. Ed., 2020, 59, 3195–3200 CrossRef CAS PubMed.
- M. Hardy and A. Lützen, Chem. – Eur. J., 2020, 26, 13332–13346 CrossRef CAS PubMed.
- R. A. S. Vasdev, J. A. Findlay, D. R. Turner and J. D. Crowley, Chem.–Asian J., 2020, 16, 39–43 CrossRef PubMed.
- D. Yang, J. L. Greenfield, T. K. Ronson, L. K. S. von Krbek, L. Yu and J. R. Nitschke, J. Am. Chem. Soc., 2020, 142, 19856–19861 CrossRef CAS PubMed.
- N. Singh, S. Jang, J.-H. Jo, D. H. Kim, D. W. Park, I. Kim, H. Kim, S. C. Kang and K.-W. Chi, Chem. – Eur. J., 2016, 22, 16157–16164 CrossRef CAS PubMed.
- J. Guo, Y.-W. Xu, K. Li, L.-M. Xiao, S. Chen, K. Wu, X.-D. Chen, Y.-Z. Fan, J.-M. Liu and C.-Y. Su, Angew. Chem., Int. Ed., 2017, 56, 3852–3856 CrossRef CAS PubMed.
- H. Li, Y.-F. Han, Y.-J. Lin, Z.-W. Guo and G.-X. Jin, J. Am. Chem. Soc., 2014, 136, 2982–2985 CrossRef CAS PubMed.
- L. S. Lisboa, J. A. Findlay, L. J. Wright, C. G. Hartinger and J. D. Crowley, Angew. Chem., Int. Ed., 2020, 59, 11101–11107 CrossRef CAS PubMed.
- F. J. Rizzuto, W. J. Ramsay and J. R. Nitschke, J. Am. Chem. Soc., 2018, 140, 11502–11509 CrossRef CAS PubMed.
- S. C. Bete and M. Otte, Angew. Chem., Int. Ed., 2021, 60, 18582–18586 CrossRef CAS PubMed.
- K. Harris, Q.-F. Sun, S. Sato and M. Fujita, J. Am. Chem. Soc., 2013, 135, 12497–12499 CrossRef CAS PubMed.
- R. Zaffaroni, N. Orth, I. Ivanović-Burmazović and J. N. H. Reek, Angew. Chem., Int. Ed., 2020, 59, 18485–18489 CrossRef CAS PubMed.
- S. Gonell, X. Caumes, N. Orth, I. Ivanović-Burmazović and J. N. H. Reek, Chem. Sci., 2019, 10, 1316–1321 RSC.
- C. Liu, E. O. Bobylev, Y. Fu, D. A. Poole III, K. Robeyns, C. Fustin, Y. Garcia, J. N. H. Reek and M. L. Singleton, Chem. – Eur. J., 2020, 26, 11960–11965 CrossRef CAS PubMed.
- A. D. Milov, A. B. Ponomarev and Yu. D. Tsvetkov, Chem. Phys. Lett., 1984, 110, 67–72 CrossRef CAS.
- M. Pannier, S. Veit, A. Godt, G. Jeschke and H. W. Spiess, J. Magn. Reson., 2000, 142, 331–340 CrossRef CAS PubMed.
- O. Schiemann, C. A. Heubach, D. Abdullin, K. Ackermann, M. Azarkh, E. G. Bagryanskaya, M. Drescher, B. Endeward, J. H. Freed, L. Galazzo, D. Goldfarb, T. Hett, L. Esteban Hofer, L. Fábregas Ibáñez, E. J. Hustedt, S. Kucher, I. Kuprov, J. E. Lovett, A. Meyer, S. Ruthstein, S. Saxena, S. Stoll, C. R. Timmel, M. Di Valentin, H. S. Mchaourab, T. F. Prisner, B. E. Bode, E. Bordignon, M. Bennati and G. Jeschke, J. Am. Chem. Soc., 2021, 143, 17875–17890 CrossRef CAS PubMed.
- M. J. Lawless, J. L. Sarver and S. Saxena, Angew. Chem., Int. Ed., 2017, 56, 2115–2117 CrossRef CAS PubMed.
- F. D. Breitgoff, K. Keller, M. Qi, D. Klose, M. Yulikov, A. Godt and G. Jeschke, J. Magn. Reson., 2019, 308, 106560 CrossRef CAS PubMed.
- I. M. C. van Amsterdam, M. Ubbink, G. W. Canters and M. Huber, Angew. Chem., Int. Ed., 2003, 42, 62–64 CrossRef CAS PubMed.
- G. Jeschke, V. Chechik, P. Ionita, A. Godt, H. Zimmermann, J. Banham, C. R. Timmel, D. Hilger and H. Jung, Appl. Magn. Reson., 2006, 30, 473–498 CrossRef CAS.
- D. M. Mate and M. Alcalde, Biotechnol. Adv., 2015, 33, 25–40 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Leonardo
Passerini
b,
Felix J.
de Zwart
a,
Leonardo
Passerini
b,
Felix J.
de Zwart